

Abstract
Recombinant protein production is at the core of biotechnology and numerous molecular tools and bacterial strains have been developed to make the process more efficient. One commonly used generic solution is to supply extra copies of low‐abundance tRNAs to compensate for the presence of complementary rare codons in genes‐of‐interest. Here we show that such extra tRNA, supplied by the commonly used pLysSRARE2 plasmid, can cause two side effects: (1) growth and gene expression can be impaired, and (2) apparent positive effects can be caused by differential expression of the lysozyme gene encoded on the same plasmid and not the tRNAs per se. These phenomena seem to have been largely overlooked despite the huge popularity of the T7/pET‐based systems for bacterial protein production.
Keywords: protein production, codon optimization, tRNA complementation, pRARE, BL21, Rosetta
Introduction
The genetic code is degenerate and between one and six different codons can encode a single amino acid. The frequency for using a specific codon varies between different organisms — a phenomenon referred to as codon bias. This has long been recognized as a major challenge in heterologous gene expression, as the host might not transcribe and/or translate the foreign DNA sequence in an optimal manner. Partly, this may be caused by a parallel low abundance of tRNAs cognate to codons that are low in frequency — these codons are commonly referred to as rare codons.1
Numerous studies have shown that synonymous codon mutations can have drastic consequences for gene expression. Factors such as mRNA stability and structure can be affected, but also initiation and elongation of translation, as well as protein folding.2, 3, 4, 5 It is possible that the redundancy in the genetic code have evolved as an extra layer of information stored in DNA — for example as a method to preserve structural information of proteins within the nucleotide content.6 In this context it is important to note that rare codons can be an advantage; for example the hypothesis that rare codons in the 5′ end of genes may positively influence translation efficiency7 was recently corroborated by assaying the expression of 14.000 synthetic reporter genes with codon usage variation in the 5′ end.8
Several strategies have been developed to optimize genes for expression (for reviews on the topic see Refs. 9, 10, 11, 12, 13, 14), but the success rate is hard to gauge because of the inherent bias against negative results in scientific literature.15 A seemingly attractive alternative to DNA re‐coding is overexpression of low‐abundance tRNA species. This way, the host, rather that the individual genes, is harmonized towards an ideal gene expression chassis. In one example, Brinckmann and co‐workers coexpressed dnaY (now known as argU) with eukaryotic genes known to express poorly in E. coli.16 The genes had a high content of AGA and AGG codons, which are recognized by the tRNAAGG/AGA produced from dnaY. Upon supplying dnaY on a plasmid, improved production of the recombinant protein was observed (up to 30% of total cellular protein).16 A variant of the approach was later used for expression of parasite genes, which are known to have a high AT‐content, and addition of a plasmid supplying minor tRNAs cognate to rare arginine‐, isoleucine‐, and glycine‐codons, improved the expression levels significantly.17 The concept of tRNA overexpression has been further developed and commercialized, and several bacterial strains are available for “universal expression” of eukaryotic genes.18 In a recent large‐scale protein production project, the Human Protein Atlas Project, changing the bacterial expression strain from the traditionally applied BL21 to the tRNA‐supplying Rosetta™ improved the overall yields of recombinant proteins.19 Following up on this work, the effect of different promoters in the two expression strains was tested, suggesting that Rosetta generally supported higher expression, regardless of promoter choice.20
As with codon optimization, bias in the way scientific results are published, makes it hard to evaluate the global success rate of an approach such as (rare) tRNA complementation. We have searched for examples where there was direct proof a positive effect of tRNA supply on expression of a specific gene, but without success. For the record, here we report on different examples where a tRNA encoding plasmid has a negative effect on gene expression, and examples where other factors in the tRNA supplying plasmids are causing increased expression — giving the false impression that tRNA complementation works as intended.
Results and Discussion
In searching for genes that could be influenced by tRNA complementation, we came across six synonymous codon variants of DasherGFP (which varied in nucleotide sequence but encoded the same protein). These variants were designed to explore the effect of rare codons on protein production (Claes Gustafsson and Mark Welch, DNA2.0, personal communication). We transformed DasherGFP constructs into E. coli strains, with either pLysS or pLysSRARE2, a rare tRNA‐supplementing plasmid derived from pLysS [Fig. 1(A)], and assayed protein production by whole cell fluorescence in a 96 deep‐well format. Whole cell fluorescence has previously been shown to correlate strongly with total protein produced with a similar fluorescent protein.2 Both pLysS and pLysSRARE2 contain the T7 lysozyme encoding gene “3.5” included to counteract toxic effects of the T7 polymerase when this is present.21 Contrary to our expectations, several of the tested DasherGFP variants showed a remarkable drop in production in the presence of pLysSRARE2 in different strains and under different growth conditions (data not shown).
Figure 1.
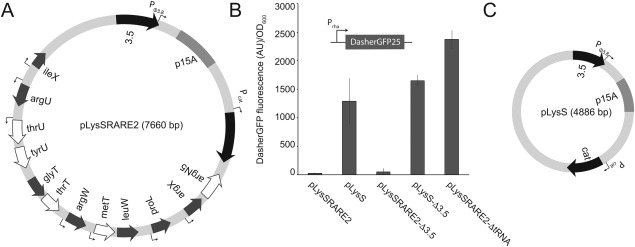
Different effects of the pLysSRARE2 and the pLysS plasmids on bacterial production of the fluorescent protein DasherGFP25. A: Illustration of the pLysSRARE2 plasmid (not drawn to scale). B: Whole cell fluorescence (arbitrary units)/OD600nn from E. coli BL21 (DE3) producing DasherGFP in the presence of pLysSRARE2, pLysS or derivatives were either the 3.5 gene or the extra tRNA genes were deleted. C: Illustration of the pLysS plasmid (not drawn to scale).
To understand this phenomenon better we focused on the DasherGFP25 variant that contains a total of 13 rare codons evenly distributed in the coding sequence (out of 258 codons in total). In a representative experiment, we co‐transformed the DasherGFP25 construct together with pLysSRARE2 or the pLysS control into BL21(DE3). By diluting an overnight culture, we started all expression cultures at OD600 0.05 and grew them at 37°C to exponential phase (OD 0.3–0.8) followed by induction with 2 mM L‐rhamnose. Four hours later, protein production levels were estimated from whole cell GFP fluorescence in a plate reader. As observed in our pilot experiments, the presence of pLysSRARE2 had a major inhibitory effect on production of DasherGFP25, showing approximately 15‐fold less production under these conditions [Fig. 1(B)]. Frequently, we also observed that the growth rate of the expression strain was affected, suggesting a fitness cost caused by the presence of the pLysSRARE2 plasmid (data not shown).
Despite the overall similarity with pLysS [Fig. 1(A,C)], apart from the tRNA genes, pLysSRARE2 contains DNA included in the tRNA‐encoding region of the plasmid that could influence heterologous gene expression or bacterial physiology. As a control, we deleted all tRNA coding sequences from the plasmid, creating a pLysS‐like plasmid (pLysSRARE2‐ΔtRNA) that we co‐transformed with the DasherGFP25 construct. In the absence of all tRNAs, no inhibitory effect on protein production was observed [Fig. 1(B)]. To test the effect of the 3.5 gene, we also deleted 3.5 from pLysSRARE2 and pLysS (pLysSRARE2‐Δ3.5 and pLysS‐Δ3.5), but no significant differences were observed [Fig. 1(B)]. We conclude that the presence of additional tRNA copies can have a negative effect on heterologous protein production.
We broadened our search for genes relying on pLysSRARE2 for proper expression and noticed that several plant cytochrome P450 genes and a human breast cancer‐related gene fragment BRCA1 (Alex Toftgaard Nielsen and Ariane Zutz, personal communication) seemed to be positively affected in expression by the pLysSRARE2 plasmid compared to pLysS. Here, we focus on the P450 CYP720B4 from the plant Pinus sitchensis involved in the biosynthesis of resin acid, a secondary metabolite involved in the defense against insects22 and a fragment of BRCA1, a protein that plays a role in maintaining genomic stability and acts as a tumor suppressor.23
Both expression constructs contain the common T7 promoter, and the CYP720B4 membrane protein is fused to GFP [Fig. 2(A)], whereas BRCA1 translation can be followed by coupling with a red fluorescent protein [Fig. 2(B)]. Our initial experimental setup confirmed the apparent positive effect of pLysSRARE2; CYP720B4 and BRCA1 expressed to 4‐ and 130‐fold higher levels, respectively, compared to the pLysS‐controls [Fig. 2(A,B)]. However, with the pLysSRARE2‐ΔtRNA, both CYP720B4 and BRCA1 still expressed to significantly higher levels than with pLysS. Thus, it appears that the positive effect of pLysSRARE2 could not solely be attributed to the extra tRNA copies. T7 lysozyme, encoded by the 3.5 gene on pLysSRARE2, pLysSRARE2‐ΔtRNA and pLysS, inhibits T7 polymerase24 and thus could be a contributing factor in the differential expression of genes controlled by the T7 promoter. To gain insight into the role of T7 lysozyme we monitored expression of CYP720B4 and BRCA1 with pLysSRARE2‐Δ3.5 and pLysS‐Δ3.5. We found that CYP720B4 expressed at even higher levels than with pLysSRARE2 suggesting that the major difference between pLysSRARE2 and pLysS was related to the lysozyme encoding gene [Fig. 2(A)]. With BRCA1, the effect of 3.5 was also apparent as the expression increased more than 150‐fold upon removing 3.5 from pLysS [Fig. 2(B)].
Figure 2.
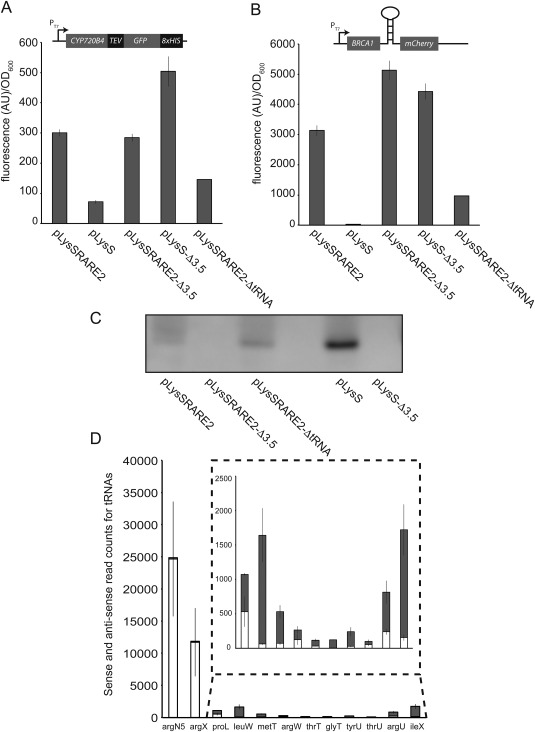
Side effects of the pLysSRARE2 plasmid may lead to the false conclusion that the tRNA genes stimulate expression. A: Whole cell fluorescence (arbitrary units)/OD600nm) from E. coli BL21 (DE3) expressing CYP720B4 fused with gfp and B: BRCA1 translationally coupled with the mCherry red fluorescent protein. In both cases expression are in the presence of pLysSRARE2, pLysS or derivatives were either the 3.5 gene or the extra tRNA genes were deleted. C) Semi‐quantitative immunoblot detection of T7 lysozyme from BL21 (DE3) harboring the pLysSRARE2 or the pLysS plasmids or derivatives were either the 3.5 gene or the extra tRNA genes were deleted. D) Sense (dark grey) and anti‐sense (white) read counts from RNA originating from the tRNA regions in the pLysSRARE2 plasmid isolated from BL21 (DE3) harboring the plasmid.
The pLysSRARE2 plasmid seems to be directly derived from pLysS (the exact design is not revealed by the commercial vendor), preserving the entire 3.5 gene and surrounding sequence, including the Φ3.8 promoter that drives expression of 3.5.25 However the Φ3.8 promoter is placed unconventionally downstream from the 3.5 gene and thus T7 RNA polymerase will only transcribe 3.5 after having transcribed the entire plasmid sequence present upstream from 3.5.25 In the case of pLysSRARE2, this includes all the tRNA sequences [Fig. 1(A)] and a total of 2774 nucleotides in addition to those present in the corresponding region in pLysS. To confirm this potential difference in expression of 3.5 from pLysS and pLysSRARE2, we probed the presence of T7 lysozyme by western blotting and observed that it was nearly completely absent from cells harboring the pLysSRARE2 plasmid compared to pLysS [Fig. 2(C)]. Upon removal of the tRNAs from pLysSRARE2, expression of 3.5 increased slightly, but did not reach the same level as with pLysS. To probe the functionality and consequences of the pLysSRARE2 plasmid more broadly, we expressed a DasherGFP construct for 4 h in BL21(DE3) with pLysSRARE2 or pLysS, and quantified the total RNA from these two strains with transcriptomics. Although many genes appeared to express differently with the two different plasmids, we were unable to identify clear trends of differentially expressed genes characteristic for a specific physiological response such as stress or starvation (Supporting Information Table S1). Since T7 RNA polymerase needs to transcribe the majority of the pLysSRARE2 plasmid to reach the 3.5 gene, it could have a major impact on the way the tRNAs are transcribed from the plasmid too. Although our method for RNA extraction did not allow us to detect accurate tRNA concentrations, with pLysSRARE2, the RNA sequencing analysis identified very high levels of RNA originating from regions immediately downstream from the Φ3.8 promoter and the cam resistance gene (in argX and argN5, [Fig. 1(A)]). However, when analyzing the strand specificity of the sequencing, more than 99% of this RNA sequence originated from the antisense strand of argX and argN5 and, thus, will not lead to functional tRNA molecules [Fig. 2(D), Table S2]. RNA sequence originating from regions downstream from argX and argN5 were not at the same high levels and the strand specificity was not as prominent, although again a significant proportion of sequence from the antisense strand of several of the other tRNAs is worth noting [Fig. 2(D)]
Materials and Methods
Bacterial strains
Escherichia coli strain NEB 5‐alpha (New England BioLabs, Ipswich, USA) was used for cloning of PCR products and propagation of plasmids. E. coli BL21(DE3) was used for gene expression.
DNA constructs
The plasmid pLysSRARE2 was isolated from RosettaTM2(DE3)pLysS and pLysS from BL21(DE3) pLysS (Novagen, Merck Millipore, Germany). pLysSRARE2‐ΔtRNA was constructed in several steps: first the region containing argN5, argX, proL, leuW, metT, and argW; and another region containing argU was deleted. This was accomplished by amplifying the backbone with the oligonucleotides 5'‐AGTCGGTGUCGTTGATACCGCAATGCGGTGTAAT C‐3' and 5'‐ATCGATGAUATTTTGAACCCCGCTTCG GCGGGGTTTTTTG‐3' and the product with a fragment containing the region encoding thrU, tyrU, glyT, and thrT amplified with the oligonucleotides 5'‐ACACCGACUTTACAATTCAATCAGTTACGCCTT CTTTATATCCTC‐3' and 5'‐ATCATCGAUCAATGCG GCATGAGTATACCCGCTAATG‐3'. Next, glyT was removed using the oligonucleotides 5'‐AGTCGGTG UCATCCTGGAACTCGGCTACCTGATTTTC‐3' and 5'‐ACACCGACUAAGATGTGCTGATATAGCTCAGTT GGTAGAG‐3'. In the third step, ileX was removed using the oligonucleotides 5'‐ATCGATUCCAGCG GCCGCAAAGGCT‐3' and 5'‐AATCGAUTGGATTAT AAAGTAACTCCGTGTC‐3' and finally the region containing thrU, tyrU, and thrT was deleted using the oligonucleotides 5'‐AGTGGAUCAAGTAGCGCG CACTCTA‐3' and 5'‐ATCCACUTCTTTTCTCCTCC CTGTTTTTTCC‐3'. All deletions were made by PCR followed by assembly with uracil excision as previously described.26, 27
PCR and DNA assembly
PCR products were amplified in 50 µL reactions containing:27 1 µL PfuX7 DNA polymerase {Norholm:2010hs}, 0.2 mM dNTPs (Thermo Scientific, Waltham, USA), 1 mM MgCl2, 1 mM DMSO, 0.5 µM forward oligonucleotide, 0.5 µM reverse oligonucleotide, Phusion® HF Reaction Buffer (New England BioLabs, Ipswich, USA) and 50 ng plasmid template. A touch‐down PCR program was used for amplification: Step 1: 2 min 98°C; step 2: 15 sec 98°C, 20 sec 65°C (−1°C per cycle), 45 sec per kb at 72°C (step 2 repeated nine times until 55°C, then repeated 20 cycles at the annealing temperature 55°C); step 3: 5 min 72°C; step 4: hold at 10°C. PCR products were gel purified using NucleoSpin Gel and PCR Clean‐up (Macherey‐Nagel, Düren, Germany) and eluted in 10% TE buffer. A Nanodrop spectrophotometer 2000 (Thermo Scientific, Waltham, USA) was used for estimation of PCR product and vector concentration. Purified PCR products were incubated with 1 µL USERTM enzyme (New England BioLabs, Ipswich, USA) for 30 min at 37°C and subsequently mixed with linearized vector backbone. 3 µL of the assembled PCR product:vector solution was transformed into NEB 5‐alpha chemically competent cells according to the manufacturer's protocol. Transformants were selected on Luria Bertoni (LB) agar plates supplemented with 25 μg/mL kanamycin, 17 μg/mL chloramphenicol. Colonies were screened for gene insert by colony PCR using OneTaq 2X Master mix (New England BioLabs, Ipswich, USA). Vectors were extracted and purified using QIAprep Spin Kit (Qiagen) and verified by sequencing.
Culture medium and expression conditions
All experiments were performed with biological triplicates with BL21(DE3) as expression host. Overnight (ON) cultures in 96‐deep well plates: 500 μL (DasherGFP variants BRCA1) or 800 μL (CYP720B4) terrific broth (TB) medium supplemented with 25 μg/mL kanamycin and 17 μg/mL chloramphenicol was inoculated with single colonies and grown overnight at 37°C (DasherGFP variants, BRCA1) or 30°C (CYP720B4), at 300 rpm in a 5 cm orbital shaking incubator. ON cultures were diluted to a start OD600nm of 0.05 in fresh TB medium including antibiotics. The expression culture volume in 96‐deep well plates was 500 μL (Dashers, BRCA1), in 24‐deep well plates it was 5 mL (CYP720B4); all grown to an OD600nm of 0.3–0.5. After induction with 2 mM l‐rhamnose (DasherGFP variants) or 0.4 mM IPTG (BRCA1, CYP720B4), growth was continued for 4 h at 37°C, 300 rpm (DasherGFP variants) or overnight at 25°C, 300 rpm (BRCA1) or 3 h 25°C, 150 rpm (CYP720B4).
Whole cell fluorescence measurements
Whole cell fluorescence was measured using 200 μL (Dashers and BRCA1) or 2 mL (CYP720B4) induced culture. CYP720B4 cultures were harvested (2500g, 20 min) and the pellets resuspended in a total volume of 100 μL PBS buffer. The GFP fluorophore was allowed to form for 2 h at room temperature (RT). The cultures harboring the DasherGFP variants or BRCA1 were measured directly after culturing. Fluorescence was detected by using excitation/emission wave length depending on the construct: 505 nm/525 nm for the DasherGFP variants, 580 nm/610 nm for BRCA1‐mCherry and 485 nm/512 nm for CYP720B4‐GFP with a window of +/− 9 nm, gain value 50 (CYP720B4) or 80 (BRCA1), using plate reader SynergyMx SMATLD (BioTek, Winooski, USA).
SDS‐page
Cell pellets from 450 μL expression culture were resuspended in 20 mM Tris‐HCL pH 8.0 including benzonase (0.5 μL/mL) and adjusted to equal OD values. Cell lysis was done by freeze‐thawing and the cell lysate was subsequently mixed 1:1 with SDS‐sample buffer (125 mM Tris‐HCl pH 6.8, 20% glycerol, 4% SDS, 100 mM DTT, bromphenol blue) and boiled at 99°C for 5 minutes. For SDS‐PAGE, equal volumes of cell lysate‐sample buffer mixes were loaded and run in parallel with PageRuler™ Prestained Protein Ladder 10‐170K (Thermo Scientific, Waltham, USA) on RunBlue 4–20% gradient SDS gels (Expedeon, USA) with RunBlue Rapid running buffer (Expedeon, USA) at 125 kV for 60 mins.
Immunoblotting
Proteins were transferred from SDS‐PAGE onto nitrocellulose sheets (iBlot transfer stacks with Novex mini nitrocellulose membranes and iBlot Dry blotting system, ThermoFischer Scientific, USA). Blocking was done ON at 4°C with 5% (w/v) dried milk in TBS‐T buffer, followed by 4 × 5 mins washing with excessive TBS‐T volumes. Incubation with the 1° antibody (anti‐T7 lysozyme from rabbit, 1:5000 dilution) for 1 h at RT in TBS‐T solution was followed by excessive washing with TBS‐T, 1 h incubation with 2° antibody (anti‐rabbit from donkey, 1:2500 dilution) in TBS‐T and again excessive washing. Detection was performed with Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences, UK).
RNA isolation
One mL samples in triplicates were harvested after 4 h induction and snapfreezed at −80°C after STOP solution (5% Phenol for RNA, 100% EtOH) treatment. Total RNA isolation was performed using Qiagen RNAeasy kit according to the manufacturers' instructions. The protocol includes DNase treatment.
RNA library preparation and sequencing
The sequencing libraries were prepared in triplicates using a TruSeq RNA Stranded Sample Preparation kit (Illumina, San Diego CA) and pooled together prior to sequencing. An average cDNA library size was determined using the Agilent DNA 1000 kit on an Agilent 2100 Bioanalyzer. Libraries were normalized and pooled in 10 mM Tris‐Cl, pH 8.0, plus 0.05% Tween 20 to the final concentration of 10 nM. Denaturated in 0.2N NaOH, 10 pm pool of six libraries in 600 μl ice‐cold HT1 buffer was loaded onto the flow cell provided in the MiSeq Reagent kit v2 300 cycles and sequenced on a MiSeq® (Illumina Inc., San Diego CA) platform with a paired‐end protocol and read lengths of 151 nt. On a regular basis, concentration and quality of the nucleic acids were determined by using a Qubit 2.0 fluorometer (Invitrogen) or an Agilent 2100 Bioanalyzer (Agilent Technologies).
Analysis of RNA‐sequencing data
TopHat (2.1.0) and Cufflinks (2.2.1) suite was used for RNA‐seq‐based differential gene expression analysis as described by Trapnell et al.28 Reference genome and annotations for BL21(DE3) strain were retrieved from NCBI Reference Sequence Database (Version (carries DasherGFP) NC_012971.2). Features present on the pDR861‐SR, pLysS and pRARE plasmids are added to reference manually. Sense and antisense read counts for tRNAs present on pRARE are determined using HTSeq library described by Anders et al.29 over reads mapped to plasmids alone.
Conclusion
It has previously been noted that tRNAs are post‐transcriptionally modified and amino‐acetylated. These processes must be essential for complementation of low‐abundance tRNAs and immature tRNA could have metabolic side effects in the cell.30 Indeed, Fedyunin and co‐workers showed that when expression of specific tRNAs was upregulated between 5‐ and 27‐fold, several proteins in the cell suffered from altered solubility, making them aggregation‐prone.31 It is likely that the apparent negative impact of pLysSRARE2 on cell growth and on expression of genes encoding DasherGFP is related to such metabolic side effects, although our RNAseq analysis gave no specific clues in this direction. Instead, our analysis seems to point to an apparent design flaw in the pLySRARE plasmid: T7 RNA polymerase transcribes major parts of the plasmid from the Φ3.8 promoter, leading to antisense RNA of several of the encoded tRNAs. ArgX recognizes CGA and CGG — two of the most rarely occurring codons in E. coli 32—and it seems likely that over expression of antisense RNA of this tRNA can only make the situation worse. As shown here, another consequence of the pLysSRARE2 design is that the 3.5 lysozyme‐encoding gene is expressed at much lower levels than in pLysS. This is probably a direct consequence of the extra DNA added between the Φ3.8 promoter and 3.5 in pLysSRARE2. In line with this, we observe in the RNAseq data that the levels of identified RNA from these regions decrease with the distance from the Φ3.8 promoter. Given that T7 lysozyme directly inhibits T7 RNA polymerase, it is therefore no surprise that genes expressed from the T7 promoter express very differently when combined with either pLysS or pLysSRARE2 — as shown here for two very different genes and expression constructs. Given the huge popularity of the T7/pET‐based expression systems, it is obviously important to know if the developed tools work as intended. There still seems to be very little direct proof available for the efficiency of tRNA overexpression in providing “universal expression”.
Supporting information
Supporting Information Table 1.
Supporting Information Table 2.
Acknowledgments
Tonja Wolff is thanked for technical assistance and Anna Koza and Emre Özdemir for their assistance with RNAseq. Jan‐Willem de Gier is thanked for sharing the T7 lysozyme antibody. Dan Daley is thanked for comments on the manuscript. This work was supported by The Novo Nordisk Foundation.
References
- 1. Jakubowski H, Goldman E (1984) Quantities of individual aminoacyl‐tRNA families and their turnover in Escherichia coli. J Bacteriol 158:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kudla G, Murray AW, Tollervey D, Plotkin JB (2009) Coding‐sequence determinants of gene expression in Escherichia coli. Science 324:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marin M (2008) Folding at the rhythm of the rare codon beat. Biotechnol J 3:1047–1057. [DOI] [PubMed] [Google Scholar]
- 4. Tsai C‐J, Sauna ZE, Kimchi‐Sarfaty C, Ambudkar SV, Gottesman MM, Nussinov R (2008) Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J Mol Biol 383:281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fredrick K, Ibba M (2010) How the sequence of a gene can tune its translation. Cell 141:227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zull JE, Smith SK (1990) Is genetic code redundancy related to retention of structural information in both DNA strands? Trends Biochem Sci 15:257–261. [DOI] [PubMed] [Google Scholar]
- 7. Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y (2010) An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141:344–354. [DOI] [PubMed] [Google Scholar]
- 8. Goodman DB, Church GM, Kosuri S (2013) Causes and effects of N‐terminal codon bias in bacterial genes. Science 342:475–479. [DOI] [PubMed] [Google Scholar]
- 9. Makrides SC (1996) Strategies for achieving high‐level expression of genes in Escherichia coli. Microbiol Rev 60:512–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baneyx F (1999) Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol 10:411–421. [DOI] [PubMed] [Google Scholar]
- 11. van de Linde S, Aufmkolk S, Franke C, Holm T, Klein T, Löschberger A, Proppert S, Wolter S, Sauer M (2013) Investigating cellular structures at the nanoscale with organic fluorophores. Chem Biol 20:8–18. [DOI] [PubMed] [Google Scholar]
- 12. Jana S, Deb JK (2005) Strategies for efficient production of heterologous proteins in Escherichia coli. Appl Microbiol Biotechnol 67:289–298. [DOI] [PubMed] [Google Scholar]
- 13. Sørensen HP, Mortensen KK (2005) Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol 115:113–128. [DOI] [PubMed] [Google Scholar]
- 14. Rosano GL, Ceccarelli EA (2014) Recombinant protein expression in Escherichia coli: Advances and challenges. Front Microbiol 5:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nørholm M, Light S, Virkki M, Elofsson A, Heijne von G, Daley D (2011) Manipulating the genetic code for membrane protein production: What have we learned so far? Biochim Biophys Acta provisionally accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brinkmann U, Mattes RE, Buckel P (1989) High‐level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene 85:109–114. [DOI] [PubMed] [Google Scholar]
- 17. Baca AM, Hol WGJ (2000) Overcoming codon bias: A method for high‐level overexpression of Plasmodium and other AT‐rich parasite genes in Escherichia coli. Int J Parasitol 30:113–118. [DOI] [PubMed] [Google Scholar]
- 18. Novy R, Drott D, Yaeger K, Mierendorf R (2001) Overcoming the codon bias of E. coli for enhanced protein expression. inNovations :4–6. [Google Scholar]
- 19. Tegel H, Tourle S, Ottosson J, Persson A (2010) Increased levels of recombinant human proteins with the Escherichia coli strain Rosetta(DE3). Prot Express Purif 69:159–167. [DOI] [PubMed] [Google Scholar]
- 20. Tegel H, Ottosson J, Hober S (2011) Enhancing the protein production levels in Escherichia coli with a strong promoter. FEBS J 278:729–739. [DOI] [PubMed] [Google Scholar]
- 21. Studier FW (1991) Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol 219:37–44. [DOI] [PubMed] [Google Scholar]
- 22. Hamberger B, Ohnishi T, Hamberger B, Séguin A, Bohlmann J (2011) Evolution of diterpene metabolism: Sitka spruce CYP720B4 catalyzes multiple oxidations in resin acid biosynthesis of conifer defense against insects. Plant Physiol 157:1677–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deng C‐X (2006) BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 34:1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moffatt BA, Studier FW (1987) T7 lysozyme inhibits transcription by T7 RNA polymerase. Cell 49:221–227. [DOI] [PubMed] [Google Scholar]
- 25. Studier FW (1991) Use of bacteriophage T7 lysozyme to improve an inducible T7 expression System. J Mol Biol 219:37–44. [DOI] [PubMed] [Google Scholar]
- 26. Nour‐Eldin HH, Hansen BG, Nørholm MHH, Jensen JK, Halkier BA (2006) Advancing uracil‐excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res 34:e122–e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nørholm MHH (2010) A mutant Pfu DNA polymerase designed for advanced uracil‐excision DNA engineering. BMC Biotechnol 10:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anders S, Pyl PT, Huber W (2014) HTSeq A—Python framework to work with high‐throughput sequencing data. Bioinformatics 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C (2009) Design parameters to control synthetic gene expression in Escherichia coli. PLoS One 4:e7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fedyunin I, Lehnhardt L, Böhmer N, Kaufmann P, Zhang G, Ignatova Z (2012) TRNA concentration fine tunes protein solubility. FEBS Lett 586:3336–3340. [DOI] [PubMed] [Google Scholar]
- 32. Nakamura Y, Gojobori T, Ikemura T (2000) Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res 28:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table 1.
Supporting Information Table 2.