

Abstract
Circulating low‐density lipoprotein cholesterol (LDLc) is regulated by membrane‐bound LDL receptor (LDLr). Upon LDLc and LDLr interaction the complex is internalized by the cell, leading to LDLc degradation and LDLr recycling back to the cell surface. The proprotein convertase subtilisin/kexin type 9 (PCSK9) protein regulates this cycling. PCSK9 is secreted from the cell and binds LDLr. When the complex is internalized, PCSK9 prevents LDLr from shuttling back to the surface and instead targets it for degradation. PCSK9 is a serine protease expressed as a zymogen that undergoes autoproteolysis, though the two resulting protein domains remain stably associated as a heterodimer. This PCSK9 autoprocessing is required for the protein to be secreted from the cell. To date, direct analysis of PCSK9 autoprocessing has proven challenging, as no catalytically active zymogen has been isolated. A PCSK9 loss‐of‐function point mutation (Q152H) that reduces LDLc levels two‐fold was identified in a patient population. LDLc reduction was attributed to a lack of PCSK9(Q152H) autoprocessing preventing secretion of the protein. We have isolated a zymogen form of PCSK9, PCSK9(Q152H), and a related mutation (Q152N), that can undergo slow autoproteolysis. We show that the point mutation prevents the formation of the mature form of PCSK9 by hindering folding, reducing the rate of autoproteolysis, and destabilizing the heterodimeric form of the protein. In addition, we show that the zymogen form of PCSK9 adopts a structure that is distinct from the processed form and is unable to bind a mimetic peptide based on the EGF‐A domain of the LDLr.
Keywords: PCSK9, autoprocessing, autoproteolysis, zymogen, point mutations, serine protease, low‐density lipoprotein receptor, LDLR, protein refolding
Abbreviations
- PCSK9
proprotein convertase subtilisin/kexin type 9
- LDLc
low density lipoprotein cholesterol
- LDLr
low density lipoprotein receptor
- WT
wild‐type
Introduction
Circulating low‐density lipoprotein cholesterol (LDLc) is a major risk factor for cardiovascular disease. High concentrations of LDLc are associated with increased probability of developing myocardial infarctions and strokes, whereas low concentrations are associated with reduced risk. LDLc in the circulation is regulated via its interaction with the LDL receptor (LDLr). Upon interaction, the complex is internalized into the cell, where LDLc is broken down and LDLr is recycled back to the cell surface. Proprotein convertase subtilisin/kexin type 9 (PCSK9) protein is a key post‐translational regulator of LDLr levels. PCSK9 binds LDLr and directs it to the lysosome where it is degraded.1, 2 Analysis of PCSK9 from patient populations has identified gain‐of‐function and loss‐of‐function point mutations that give rise to elevated and reduced levels of LDLc, respectively.3 As a result, there is significant interest in developing therapeutics that inhibit PCSK9 function as a treatment for cardiovascular disease.
PCSK9 is the ninth member of a family of subtilisin‐like serine proteases. Similar to the other members of this family, PCSK9 contains a signal sequence, a prodomain, a catalytic domain that contains the catalytic triad typical of serine proteases, and a C‐terminal domain, which varies in function among family members (Fig. 1).4 Members of this family are synthesized in an inactive zymogen form that, after removal of the signal peptide, typically undergo two cleavage steps in order to form an active protease. In the first step, the peptide backbone between the prodomain and the catalytic domain is cleaved. The prodomain remains bound to the catalytic domain, inhibiting its activity, until a second cleavage event liberates the prodomain and leads to an active protease. PCSK9 undergoes the first cleavage step to form a heterodimer between the prodomain and the rest of the protein. It does not, however, undergo the second step. In fact, once‐cleaved the proteolytically inactive heterodimer executes the physiological role of LDLc regulation.5, 6 The PCSK9 heterodimer is secreted from the cell and directly interacts with LDLr, targeting it for lysosomal degradation. The decreased surface levels of LDLr result in reduced LDLc clearance and therefore elevated circulating LDLc.7 Inhibiting the interaction between PCSK9 and LDLr has therefore been a prime therapeutic strategy for lowering LDLc levels.8
Figure 1.
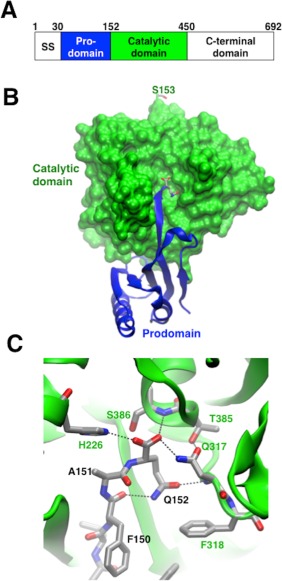
Overview of PCSK9 and the interactions of Q152. (A) Cartoon representation of the regions of PCSK9. SS refers to the signal sequence. (B,C) Details of the prodomain interaction with the catalytic domain, focusing on the Q152 interactions. (B) Space‐filling model of the catalytic domain (green) and a cartoon representation of the prodomain (blue). Residues S153 (at top) and Q152 (at center) are shown in a stick representation. (C) Close‐up of the interactions made by Q152.
Recently, a novel PCSK9 loss‐of‐function mutation that leads to an almost 2‐fold reduction in LDLc levels was identified within a French‐Canadian family.9 The point mutation, Q152H, is located at the P1 position of the proposed recognition sequence for PCSK9, which cleaves itself between residues Gln152 and Ser153. In the crystal structure of processed PCSK9, Gln152 is at the end of an extended stretch of amino acids that contributes a strand to a 2‐stranded anti‐parallel β‐sheet (Fig. 1).10, 11 Gln152 is located deep in the catalytic domain at the catalytic site and makes extensive packing interactions with the neighboring residues from the catalytic domain. The Q152H point mutation was shown in cells to cause a deficiency in the processing of PCSK9, which is required for secretion and subsequent interaction with the LDL receptor.9, 12, 13 There have been no previous reports of a method to produce catalytically active PCSK9 zymogen, preventing a detailed molecular characterization of the effect of this mutation on PCSK9 autoprocessing.
We have succeeded in producing the first purified forms of PCSK9 zymogen capable of undergoing autoproteolysis. The proteins contain the prodomain and catalytic domain (ProCat) with the point mutation Q152H, and a related point mutation Q152N. We have also developed a refolding protocol for the wild type and mutant ProCat constructs that enables us to observe maturation of wild type PCSK9 and to determine the effect of the point mutations on protein folding. The in vitro assays may also serve as a useful tool in the identification of small molecule inhibitors of PCSK9 autoprocessing. Our observations of the PCSK9 zymogen indicate that it forms a structure distinct from the processed form, and may therefore provide alternative binding sites for small molecules.
Results and Discussion
Point mutations at Q152 affect the rate of autoproteolysis
Soluble ProCat(WT), ProCat(Q152H), and ProCat(Q152N) were expressed in E. coli as maltose binding protein (MBP) fusions, and purified through affinity and size exclusion chromatography. The MBP fusion was removed during purification. All three constructs gave milligram quantities of purified protein per liter of cell media. The wild type ProCat fully matured prior to purification and will be referred to as ProCat(WT)processed. At the beginning of purification, ProCat(Q152H) and ProCat(Q152N) existed primarily in their unprocessed form. Over time they were observed to undergo autoproteolysis. By minimizing the steps taken between lysis and storage of the purified protein the zymogen form could be isolated. These forms of the two mutants will be denoted with the superscript “zymogen”. Comparison of the autoproteolysis of the two ProCat mutants showed similar yields after incubation at 18˚C for 48 hours, with 66% and 64% of the total amount of ProCat(Q152H)zymogen and ProCat(Q152N)zymogen being processed, respectively (Fig. 2). The processing of ProCat(Q152N)zymogen proceeded more rapidly with half maximal processing occurring at 2 hrs, compared to 6 hrs for ProCat(Q152H)zymogen. The slower rate for ProCat(Q152H)zymogen suggests that this mutation hinders the positioning of the substrate peptide sequence in the catalytic site to a greater degree than the Q152N mutation. The difference in autoproteolysis can be rationalized by the tight packing interactions made by Q152 in the crystal structure of PCSK9 (Fig. 1). The Q152H mutation would be predicted to result in a greater number of steric clashes with nearby residues from the catalytic domain compared to Q152N.
Figure 2.
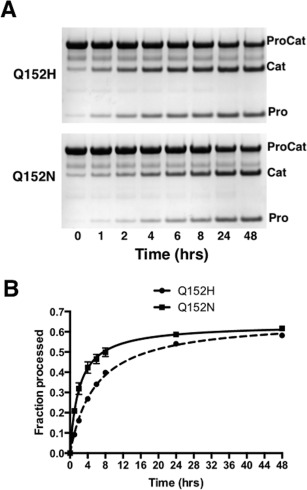
Autoproteolysis of the ProCat mutants. (A) An example of SDS‐PAGE gels of a time course analysis of the proteolysis of ProCat(Q152H) (Top) and ProCat(Q152N) (bottom) over 48 hrs. “ProCat”, “Cat”, and “Pro” indicate the unprocessed ProCat, the catalytic domain, and the prodomain, respectively. (B) A graphical representation of this data with a plot of the fraction of ProCat(Q152H) (Squares, solid line) and ProCat(Q152N) (circles, dashed line) processed versus time.
Point mutations at Q152 directly influence folding of ProCat
When wild type ProCat was expressed in the absence of the N‐terminal MBP fusion it was shuttled into inclusion bodies before any processing could occur. We were able to purify the denatured wild type zymogen by affinity and size exclusion chromatography under denaturing conditions. Screening of 165 refolding conditions that explored pH, salt content, additives, and redox state revealed that the denatured ProCat(WT)zymogen could be refolded by dilution into a solution containing the nondetergent sulfobetain NDSB201, and a 9:1 ratio of reduced to oxidized glutathione. Successful refolding was monitored over time by the cleavage of the zymogen into the prodomain and catalytic domain, visualized by SDS‐PAGE (Fig. 3). Of the refolded protein ProCat(WT)zymogen, 56% was cleaved overall, with half occurring within 4 h. Although the rate of autoproteolysis of the refolded ProCat(WT)zymogen under these conditions is similar to that of the soluble Q152X mutations, it does not necessarily reflect the rate of folding in the cell. The refolding procedure does; however, provide an approach to directly compare the effect of the Q152 point mutations on the rate of protein folding and autoprocessing to the wild type protein. The two soluble mutants were initially subjected to the same denaturing and reducing conditions as the wild type protein. The three proteins were diluted into the refolding buffer and samples were removed over a 48‐hour period to monitor refolding and autoproteolysis by SDS‐PAGE. Refolded ProCat(Q152H)zymogen and ProCat(Q152N)zymogen gave similar levels of processing, with 58% and 59% of the total protein being processed, respectively. The two mutants were processed more slowly than wild type, with half of the total protein cleaved within 8 hours for refolded ProCat(Q152N)zymogen and 14 hours for refolded ProCat(Q152H)zymogen. Interestingly, the data for the two mutants fit best to a sigmoid response curve, suggesting a delay in initiation of the autoprocessing (Fig. 3). The apparent delay implies that the point mutations, Q152H in particular, affect the folding of the PCSK9 in addition to affecting the rate of autoproteolysis. Prodomains have long been implicated in the folding of secreted proteases.14 The effect on protein folding we observed could be related to the point mutation hindering the optimal interaction between the two domains that is required for correct folding of the ProCat region of PCSK9.
Figure 3.
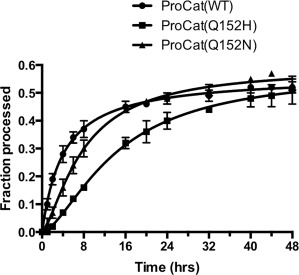
Comparison of the autoproteolysis of ProCat constructs upon refolding. Plot of the fraction of ProCat(WT) (circles), ProCat(Q152H) (squares), and ProCat(Q152N) (triangles) processed over a 48‐hour period after diluting denatured protein into refolding/autoproteolysis solution. The plot shows the average of three experiments.
Autoproteolysis of the Q152H mutation is significantly affected by solution conditions
As with many in vitro enzymatic assays, solution conditions may affect activity. The ability to directly measure autoproteolysis by the PCSK9 mutants allowed for the study of solution conditions and their effect on the cleavage rate. The zymogens were incubated with varying pH, NaCl concentration, and glycerol concentration for 24 hours and the amount of cleavage analyzed by SDS‐PAGE (Fig. 4). Both ProCat(Q152H)zymogen and ProCat(Q152N)zymogen gave optimal processing at a pH range of 8 – 9 and exhibited a significant reduction in processing at pH 5. This is in agreement to what has been observed for a related protein, Proteinase K, which shows optimal activity under more basic conditions.15 Low concentrations of sodium chloride reduced the level of processing for ProCat(Q152H)zymogen but had a negligible effect on ProCat(Q152N). Reducing the glycerol concentration from 10% to 1% led to a 2‐fold loss in the amount of processed protein for ProCat(Q152H)zymogen. There was a slight decrease in processing for ProCat(Q152N)zymogen. The variation in behavior for the two mutants suggests that their activities and possibly structures are different, with ProCat(Q152H)zymogen significantly more sensitive to solution conditions than ProCat(Q152N)zymogen and, presumably, the wild type protein. From these studies it is clear that the processing of PCSK9(Q152H) under physiologically relevant solution conditions is less favorable than for wild type PCSK9.
Figure 4.
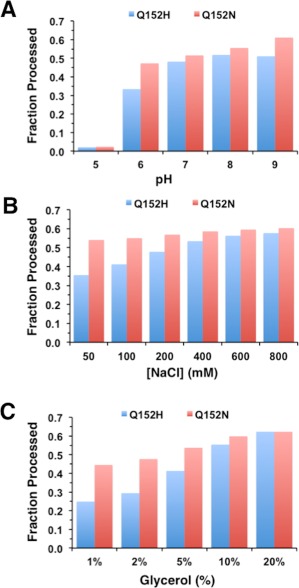
Effect of solution conditions on autoproteolysis of the ProCat mutants. The plots shows the amount of ProCat(Q152H)(blue columns) and ProCat(Q152N) (red columns) processing that occurred after a 24 hour time period for solution conditions that varied in (A) pH, (B) NaCl concentration, and (C) glycerol concentration.
Mutations at Q152 destabilize the interaction between the catalytic domain and prodomain
The specific identity of the amino acid at position 152 in PCSK9 clearly has a significant impact on PCSK9 autoproteolysis. To understand the effect the point mutation had on the stability of PCSK9 after processing, attempts were made to isolate the processed forms of ProCat(Q152H) and ProCat(Q152N). Separating the processed form of ProCat(Q152H) from the zymogen form was unsuccessful as a result of aggregation of the cleaved protein. In contrast, ProCat(Q152N)Processed could be isolated, although it also exhibited a propensity to aggregate over time (data not shown). To analyze the effect of Q152N on the stability of the processed protein, the melting temperatures of ProCat(WT)Processed and ProCat(Q152N)Processed were determined by differential scanning calorimetry (DSC). ProCat(WT)processed and ProCat(Q152N)processed melted with Tm values of 51.9 ± 0.1˚C and 43.5 ± 0.4˚C, respectively. The 8˚C reduction in melting temperature caused by the Q152N mutation could be explained by the loss of the hydrogen bond made by Gln152 and a poor fit to the Gln‐binding pocket by the Asn, which would lead to an instability between the prodomain and the catalytic domain (Fig. 1).
Autoproteolysis of ProCat(Q152H) and ProCat(Q152N) are predominantly intramolecular, but low levels of intermolecular cleavage occur
PCSK9 is believed to exist as an active protease only when it is in its zymogen state. After autoproteolysis, its protease activity is proposed to be inhibited by the binding of the prodomain.13, 16, 17 Mechanistically, the catalytically active zymogen could cleave the linker between the prodomain and the catalytic domain internally or act on a second PCSK9 molecule. With isolation of the zymogen forms of ProCat(Q152H) and ProCat(Q152N) the intramolecular versus intermolecular mechanism could be directly addressed in vitro. ProCat(S386A) was used as a substrate for this reaction as it is incapable of autoproteolysis. To visualize any cleavage that occurred, ProCat(S386A) was produced with a C‐terminal Avi‐tag that was biotinylated in vitro. Upon cleavage, the catalytic domain of ProCat(S386A) would run at a lower mass than the ProCat(S386A)zymogen when visualized on a western probed with a streptavidin‐HRP conjugate. ProCat(S386A) was incubated alone and in the presence of the other PCSK9 ProCat constructs discussed, and samples removed after 48 hours (Fig. 5). No cleavage was observed for ProCat(S386A) on its own, as expected. In the presence of ProCat(Q152H)zymogen and ProCat(Q152N)zymogen a clear, but still minor, band for the catalytic domain of ProCat(S386A) was evident by western blotting. This confirms that the two zymogens can promote intermolecular cleavage of the substrate, but at exceptionally low levels compared to what was observed for autoproteolysis of the proteins themselves.
Figure 5.
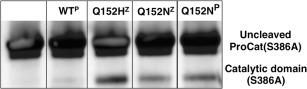
Minor amounts of intermolecular proteolysis are observable in vitro. Western blot of C‐terminally biotinylated ProCat(S386A), which was incubated alone, and in the presence of ProCat(WT)Processed, ProCat(Q152H)zymogen, ProCat(Q152N)zymogen, and ProCat(Q152N)Processed, denoted by “WTP”, “Q152HZ”, “Q152NZ”, and “Q152NP”, respectively. The western was visualized using a streptavidin‐HRP conjugate.
Curiously, ProCat(Q152H)zymogen had greater intermolecular proteolytic activity than ProCat(Q152N)zymogen despite the same concentration of zymogen being present. In addition, incubation with ProCat(Q152N)processed gave the same level of processing as ProCat(Q152N)zymogen. The source of this cleavage is likely the catalytic domain free in solution, which would occur through dissociation of ProCat to the prodomain and catalytic domain. The very low levels of free‐catalytic domain‐mediated cleavage suggest a stable ProCat complex in which very little catalytic domain dissociates. Taken together these results argue that the processed catalytic domain is likely to be the catalyst of the intermolecular cleavage events rather than the zymogen. Almost no cleavage was evident with the ProCat(WT) as the catalytic domain and prodomain form too stable a complex. In contrast, the point mutations at Q152 have been shown to destabilize the interaction between the prodomain and catalytic domain, likely leading to a greater amount of catalytic domain free in solution. The increased level of cleavage observed with the Q152H mutation agrees well with the studies that show this mutation has a greater destabilizing effect than Q152N, and therefore would lead to more ProCat(Q152H) catalytic domain free in solution. These interpretations are supported by recent cellular expression studies in which overexpressed PCSK9 showed only trace amounts of processing of PCSK9(S386A), but the cleavage was increased by overexpression of a construct of PCSK9 that was missing the prodomain.13
Zymogen and processed ProCat adopt distinct structures
During purification of the ProCat proteins the mutant zymogens eluted earlier than ProCat(WT)processed on a size exclusion column, suggesting that they had a significantly different shape or mass than the wild type (Fig. 6). Size exclusion chromatography with multi‐angle light scattering analysis of 20 µM samples of the mutants showed they primarily formed monomers in solution and that the change in elution time was therefore not due to a change in stoichiometry (Table 1). The change in elution therefore suggests that either the point mutations induce a change in overall shape of the protein or that the zymogen structure is distinct from the final processed form. While it is conceivable that the Q152X mutations might have an effect on the positioning of the prodomain relative to the catalytic domain, thus leading to a change in overall shape, the S386A mutation, which also elutes comparably to the mutant zymogens, is unlikely to perturb the structure of ProCat in a significant manner. In addition, the processed form of ProCat(Q152N) elutes at a similar location as the processed form of ProCat(WT), arguing that the mutation has minimal effect on the shape of the protein. We therefore infer that the difference in elution occurs as a result of the ProCat zymogens, both WT and mutant, adopting a distinct shape relative to the shape of processed ProCat proteins.
Figure 6.
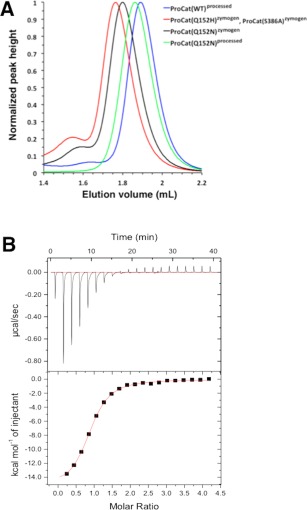
Zymogen and processed ProCat are structurally and functionally distinct. (A) Size exclusion analysis of the ProCat constructs. Plot of the elution volume and the normalized UV absorption at 280 nm for ProCat(WT)processed(blue), ProCat(Q152H)zymogen(red), ProCat(Q152N)zymogen(black), and ProCat(Q152N)processed(green). ProCat(S386A)processed eluted at the same location as ProCat(Q152H)zymogen and has been left out of this figure for clarity. (B) ITC measurements showing the background‐subtracted heats per injection (top panel) and binding isotherms (bottom panel) for the titration of WT‐ProCat PCSK9 with a peptide mimetic of the EGF‐A domain of the LDLr . Integrated heats were fit by non‐linear regression to a model for a single set of sites. The three zymogen constructs did not show binding to peptide (data not shown).
Table 1.
Summary of the SEC‐MALS and DSC analysis of the ProCat constructs
| Theoretical Mass (kDa) | SEC‐MALS Mass (kDa) | T m (˚C) | ΔT m a (˚C) | |
|---|---|---|---|---|
| ProCat(WT)processed | 45.2 | 47.4 ± 0.2 | 51.9 ± 0.1 | 1.6 |
| ProCat(Q152H)zymogen | 46.3 | 48.5 ± 2.0 | 52.8 ± 0.2 | −0.5 |
| ProCat(Q152N)zymogen | 45.2 | 49.0 ± 0.2 | 51.4 ± 0.3 | −0.4 |
| ProCat(Q152N)processed | 45.2 | 46.1 ± 0.7 | 43.5 ± 0.4 | 1.7 |
| ProCat(S386A)zymogen | 47.3 | 49.2 ± 2.3 | 53.2 ± 0.1 | −0.5 |
ΔT m represents the difference in melting temperature between the protein in the absence and presence of peptide.
To investigate the change in shape further, small angle X‐ray scattering (SAXS) data were collected on ProCat(WT)processed and ProCat(S386A)zymogen. SAXS provides a low resolution model of a protein that gives insight into its average particle size and shape.18 Similar to the MALS experiments, the proteins were passed over a size exclusion column immediately before SAXS analysis to separate any oligomeric species present. ProCat(WT)processed eluted as a single monomeric species and gave data that was consistent with a radius of gyration (Rg) of 2.4 ± 0.1 nm and a maximum particle dimension (Dmax) of 8.7 ± 0.9 nm. In addition, fitting the crystal structure of ProCat(WT) to the SAXS‐model gave very good agreement, with a χ2 value of 1.46. The major species of ProCat(S386A)zymogen eluted from the size exclusion column gave SAXS‐data that suggested it adopted a different shape, with a larger radius of gyration (Rg) of 2.8 ± 0.1 nm and a larger maximum particle dimension (Dmax) of 10.5 ± 1.0 nm. Attempts to fit the crystal structure of ProCat(WT) were unsuccessful, thus indicating a clear difference between the processed ProCat(WT) structure and the ProCat(S386A) zymogen. The reported crystal structures of processed apo‐PCSK9 show residues Gln 152 and Ser 153 separated by ∼25 Å after autoprocessing,10 which is also consistent with the zymogen form being structurally distinct from the processed form. The SEC‐MALS and SAXS experiments described here provide direct experimental evidence supporting the formation of distinct structures for PCSK9 before and after processing.
The binding site for the LDLr is disrupted in the ProCat zymogens
The most likely structural difference to occur between the zymogen and processed form of ProCat is the position of the prodomain relative to the catalytic domain. Such a change would likely have consequences for the interaction between the PCSK9 and the LDLr as Ser153, Ile154, and Pro155 form a portion of the LDLr binding site in PCSK9.16 Movement of the prodomain would likely change the position of these residues and therefore disrupt this interaction. To test this hypothesis, we performed DSC on the wild type and mutant ProCat proteins in the absence and presence of a peptide mimetic based on the EGF‐A domain of the LDLr that binds the LDLr binding site on PCSK9 (Table 1).19 ProCat(WT)processed showed a 1.6˚C increase in thermal stability in the presence of the peptide in contrast to the three zymogens, which showed a negative change in the melting temperature. This confirmed that the peptide bound and stabilized ProCat(WT), but not the zymogens. We also performed direct binding studies of the peptide to ProCat(WT) and the zymogens with isothermal titration calorimetry. ProCat(WT) gave an equilibrium dissociation constant (Kd) of 1.9 µM and an N value of 0.86, which is in agreement with previously published data19 (Fig. 6). The three zymogens showed no evidence of binding upon titration of the peptide into the proteins (data not shown). The inability of the zymogens to interact with the peptide therefore supports a model in which the relative orientation of the prodomain and catalytic domain is different compared to the processed form of PCSK9. This is supported by the observation that full length PCSK9(S386A) zymogen that was secreted from mammalian cells does not interact with the LDLr (6). Previous studies have suggested that the zymogen form of PCSK9 can interact with intracellular LDLr.20 Our studies show that this is unlikely to occur through contacts made via the ProCat region of PCSK9, but it does not discount the possibility that the C‐terminal domain of PCSK9 could be responsible for this interaction.
Conclusions
The PCSK9(Q152H) point mutation leads to an increased clearance of LDLc in the circulation, as a result of a reduced PCSK9‐mediated degradation of the LDLr. The proposed cause of this phenotype is the inability of the PCSK9(Q152H) zymogen to undergo autoproteolysis to its mature form, which is required for secretion from the cell. Our studies show that the lack of processed PCSK9(Q152H) can be attributed to the mutation having a direct effect on the folding of PCSK9, the rate of intramolecular cleavage between the prodomain and the catalytic domain, and the stability of the interaction between these two domains post cleavage. Our studies may also enable new approaches to identify small molecule inhibitors of PCSK9. Targeting the active site of PCSK9 in vitro has proven to be challenging because only the processed form of PCSK9, where the binding site of the prodomain occludes the active site, has been available. Our observations of PCSK9 before and after autoproteolysis suggest the zymogen adopts a structure that is distinct from the processed form. These observations together with the ability to isolate milligram quantities of PCSK9 zymogen forms that undergo autoproteolysis may provide a unique approach to identify small molecules that inhibit PCSK9 folding and/or autoproteolysis and prevent secretion from the cell.
Materials and Methods
Expression of soluble ProCat proteins
The gene encoding PCSK9 was codon‐optimized for bacterial expression and synthesized commercially (GeneArt, Life Technologies). PCSK9(31‐447) was PCR‐amplified from the synthesized gene and cloned into a derivative of pET21b that was designed to append an N‐terminal fusion tag comprised of 6 histidines, maltose binding protein, and a TEV protease cleavage site. PCSK9(31‐447,Q152H), PCSK9(31‐447,Q152N), and PCSK9(31‐447,S386A), were obtained by site‐directed mutagenesis of the ProCat(WT) vector using the Quikchange kit (Agilent, Santa Clara, CA). PCSK9(31‐447,Q152H) was also PCR‐amplified to include a C‐terminal FLAG sequence and then reinserted back into the same vector backbone. Similarly, PCSK9(31‐447,S386A) was PCR‐amplified in order to append a C‐terminal Avi‐tag sequence and then reinserted back into the same vector backbone. The construct used for generating the insoluble wild type ProCat was PCR‐amplified from the codon‐optimized gene to include a C‐terminal polyhistidine sequence and inserted into a pET21b vector. Correct insertion of the genes into the expression vectors was confirmed by sequencing. The proteins were expressed in either SHuffle T7 (DE3) (New England Biolabs, Ipswich, MA) or Origami B (DE3) competent cells (EMD‐Millipore, Billerica, MA). The cells were grown at 37˚C before inducing with IPTG and were then left to grow overnight at 18˚C. The cells were pelleted by centrifugation and stored at −80˚C until use.
Purification of soluble ProCat protein constructs
Cell pellets were resuspended in 50 mM Tris‐HCl pH 8, 500 mM NaCl, 10% glycerol, and 20 mM imidazole. Cells were lysed by addition of Bugbuster and Lysonase (EMD‐Millipore) and incubation for 1 h at 4˚C. The soluble protein was extracted by centrifugation at 10,000 rpm and filtered before loading onto 2 x 5 stacked 5 mL Ni‐charged HisTrap columns (GE Healthcare). Running buffers for the column were as follows: Buffer A contained 50 mM Tris‐HCl pH 8, 500 mM NaCl, 10% glycerol, and buffer B contained the same as Buffer A but with 1M imidazole. The column was washed with 2% of buffer B before applying a gradient of 2 to 52% buffer B over 10 column volumes. Combined fractions were loaded directly onto a Superdex 200 26/600 column (GE Healthcare), which was equilibrated in the same solution as Buffer A for the HisTrap column. Eluted fractions were concentrated and the His(6)‐MBP fusion cleaved by incubation with TEV protease overnight at 4˚C. The His(6)‐MBP and polyhistidine‐tagged TEV protease was removed by passing the solution over the HisTrap column a second time. The flow through of the HisTrap column was concentrated and passed over a Superdex 75 26/600 column equilibrated in 20 mM Hepes pH 8, 500 mM NaCl, and 10% glycerol. For the glutamine mutants the lysis and purification were performed within two days in order to minimize autoproteolysis of the two mutants, with the TEV proteolysis step being performed at the end of day 1. The processed form of ProCat(Q152N) was obtained by incubating the protein at room temperature for 24 hours. The protein was separated from the uncleaved and aggregated material by passing over a Superdex 75 column twice. The proteins were concentrated and stored at −80˚C until use.
Purification of denatured ProCat protein
Cells containing the insoluble ProCat with a C‐terminal His(6) sequence were lysed in a similar manner to that described previously. After centrifugation to remove the soluble fraction, the pellet was resuspended with 50 mM Tris‐HCl, 500 mM NaCl, 20 mM Imidazole, 6M Urea, and 10 mM DTT at pH 8.0 then incubated overnight at 4˚C with mild agitation. Following centrifugation, the solubilized protein in the supernatant was filtered and purified on a 5 mL Ni‐charged HisTrap HP column (GE Healthcare) as described previously except the running buffers also contained 6M urea. The eluted protein was concentrated and passed over a Sephacryl S400 column (GE Healthcare) equilibrated with 50 mM Tris‐HCl, 500 mM NaCl, 6M urea, 1 mM DTT, at pH 8. The protein was concentrated and stored at −80˚C until use.
Biotinylation of ProCat(S386A)‐avi
The Avi‐tagged ProCat(S386A) was transferred into a solution containing 50 mM bicine pH 8.3, 50 mM NaCl, 10 mM MgOAc, 10 mM ATP, and 50 µM biotin by dilution and spin concentration to give a final protein concentration of 40 µM. BirA ligase was added to give a final concentration of 0.5 µM and the solution was incubated at room temperature. After 2 h the protein was diluted 10‐fold with 50 mM Hepes pH 8, 500 mM NaCl, 10% glycerol, concentrated, and passed over a Superdex 200 10/300 column (GE Healthcare) equilibrated with the storage buffer. The protein was concentrated and stored at −80˚C until use.
Autoproteolysis assay
The ProCat proteins were diluted to 5 µM with 20 mM Hepes pH 8, 500 mM NaCl, 10% glycerol at room temperature. Samples were removed after 0, 1, 2, 4, 6, 8, 24, and 48 h and the reaction stopped by addition of 4xLDS sample buffer (Invitrogen) containing TCEP reducing reagent. 20 µL of the solutions were loaded onto a NuPAGE 4‐12% Bis‐Tris protein gel (Invitrogen). For analysis of the effect of pH on the autoproteolysis of the mutants the proteins were diluted to 5 µM with solutions containing 500 mM NaCl and 10% glycerol, and buffered with one of the following: 50 mM citrate buffer pH 5, 50 mM MES pH 6, 50 mM phosphate buffer pH 7, 50 mM HEPES pH 8, and 50 mM bicine pH 9. For analysis of the effect of NaCl, the proteins were diluted with solutions containing 50 mM Hepes pH 8, 10% glycerol, and varying [NaCl] to give a final concentration of 50, 100, 200, 400, 600, and 800 nM. Similarly, for the analysis of the effect of glycerol, the proteins were diluted with solutions containing 50 mM Hepes pH 8, 500 mM NaCl, and varying concentrations of glycerol to give 1, 2, 5, 10, and 20%. In all three sets of experiments, the solutions were left to incubate at room temperature and samples taken and run on an SDS‐PAGE gel after 24 h. The gels were quantified using the Syngene imager GeneTools software. The plots were analyzed using the Graphpad Prism software.
Refolding assay monitored by autoproteolysis
The proteins were dialyzed into 50 mM Hepes pH 8, 500 mM NaCl, 10% glycerol, 6M urea, and 1 mM DTT. After dialysis the concentrations were adjusted to give 2 mg/mL. The refolding/autoproteolysis assay was performed by diluting the proteins 10‐fold with 50 mM Hepes pH 8, 500 mM NaCl, 10% glycerol, 1M NDSB201, 9 mM GSH, and 1 mM GSSG and incubating at RT. Samples were removed after 0, 1, 2, 4, 6, 8, 16, 20, 24, 32, 40, 44, and 48 h and analyzed as described for the autoproteolysis assay.
Intermolecular processing assay
Biotinylated ProCat(S386A) was diluted to 5 µM with 50 mM Hepes pH 7, 500 mM NaCl, 10% glycerol in the absence and presence of 5 µM ProCat(WT)processed, ProCat(Q152H)zymogen, ProCat(Q152N)zymogen, and ProCat(Q152N)processed. The solutions were incubated at room temperature for 48 h before addition of LDS loading buffer containing TCEP. 5 µL of the solutions were run on a NuPAGE 4 − 12% Bis‐Tris protein gel (Invitrogen). The bands were transferred to a nitrocellulose membrane using an iBlot dry blotting system (Life Technologies) and were incubated with a streptavidin‐HRP conjugate (Life Technologies). The bands were visualized using SuperSignal west pico chemiluminescent substrate (Thermo Scientific).
Differential scanning calorimetry (DSC) measurement
Thermal denaturation experiments were performed with a VP‐DSC microcalorimeter (Malvern Instruments, Malvern, UK). The DSC scans were run between 25°C and 90°C at a rate of 120°C/h. The experiments were performed using 0.4 mg/mL concentration of wild‐type or mutant protein (8.5–8.9 µM), with and without a mimetic peptide based on the EGF‐A domain from the LDL‐r at 100 µM.19 The respective reference scans were run under identical DSC set up conditions and subtracted from each sample scan. The heat capacity curves and midpoint temperature (T m) were analyzed using Origin 7.0 software.
Isothermal titration calorimetric (ITC) analysis
Assessments of the EGF‐A domain peptide mimetic19 binding to wild‐type and mutant ProCat proteins were conducted on an Auto‐iTC200 microcalorimeter (Malvern). The protein was dialyzed extensively against a buffer comprised of 25 mM HEPES (pH 7.4), 150 mM NaCl and 0.1 mM CaCl2, and the dialysate was used to prepare fresh working solutions of protein (20 µM) and peptide (400 µM). Titrations were performed at 25°C and consisted of a single initial injection of 0.4 µL, followed by 19 injections of 2 µL of peptide into a sample cell containing wild‐type or mutant PCSK9, with an interval of 120 s between successive injections. The reference power and stirring speed were set at 10 µCal/s and 1000 RPM, respectively. Heat of dilution and mixing were obtained from injections of peptide into ITC buffer lacking protein and subtracted from the integrated data before curve fitting. Thermodynamic data were analyzed using Origin 7.0 software, and fit by non‐linear regression to the model for a single binding site.
SEC‐MALS analysis
Size exclusion chromatography with multi‐angle light scattering (SEC‐MALS) was performed with 50 µL samples of 20 µM ProCat proteins that were loaded onto a Superdex 200 5/150 increase column (GE Healthcare) equilibrated with 20 mM Hepes pH 8 and 150 mM NaCl. The size exclusion column was connected in series with a miniDAWN TREOS light scattering detector (Wyatt Technology, Santa Barbara, CA) and an Optilab T‐rEX refractive index detector (Wyatt) in order to determine the mass of the protein in solution.
SEC‐SAXS analysis
The on‐line size‐exclusion chromatography small angle X‐ray scattering (SEC‐SAXS) experiment was performed at European Molecular Biology Laboratory (EMBL) Hamburg beamline P12 on the storage ring PETRA III.21 50 µL samples of 11.3 mg/mL WT ProCat and of 9.1 mg/mL S386A ProCat were injected onto a Wyatt SEC Protein Column 30 (7.8 x 300) pre‐equilibrated with 50 mM HEPES pH 7.5, 150 mM NaCl, 5% glycerol at a flow rate of 0.5 mL/min. SEC‐SAXS data were measured in continuous flow mode from the SEC column eluates using one‐second exposure periods for a total of 3600 s. Data were recorded using a Pilatus 2M pixel X‐ray detector (Dectris, Switzerland), at a sample to detector distance of 3.1 m and a wavelength of 0.15 nm. The range of momentum transfer 0.05 < s < 4.0 nm−1 was recorded (s = 4π sinθ/λ, where 2θ is the scattering angle). Each frame was radially averaged and normalized to the intensity of the transmitted beam. The frames corresponding to the buffers were averaged and subtracted from each individual frame. The subtracted frames corresponding to the peaks were scaled and subsequently averaged to produce the final scattering profiles. Distance distribution functions were calculated from these scattering patterns using the program GNOM.22 A model for WT ProCat was created by extracting residues 61 − 447 from the full‐length crystal structure of PCSK9 solved at 1.9 Å resolution (PDB 2QTW). The missing 31 residues at the N terminus of the SAXS construct were reconstructed in an extended conformation using the program CORAL.23 The calculated scattering profile of the resulting model closely matches the experimental SAXS data of the ProCat(WT) (Χ 2 = 1.46), but not that of the ProCat(S386A) (X 2 = 3.7). Efforts to generate a monomeric ProCat model matching the experimental mutant data were not successful, owing to the significant shape and apparent mass differences.
Acknowledgments
The authors thank Drs. Alexey Kikhney and Dmitri Svergun (BIOSAXS GmbH/EMBL, Hamburg, Germany) for their assistance with the SAXS characterization of wild‐type and S386A mutant PCSK9.
Contributor Information
Colin W. Garvie, Email: cgarvie@broadinstitute.org
Douglas S. Daniels, Email: doug.daniels@udayton.edu.
References
- 1. Cameron J, Holla OL, Ranheim T, Kulseth MA, Berge KE, Leren TP (2006) Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum Mol Genet 15:1551–1558. [DOI] [PubMed] [Google Scholar]
- 2. Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD (2006) Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest 116:2995–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lambert G, Sjouke B, Choque B, Kastelein JJ, Hovingh GK (2012) The PCSK9 decade. J Lipid Res 53:2515–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M (2003) The secretory proprotein convertase neural apoptosis‐regulated convertase 1 (NARC‐1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA 100:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J, Tumanut C, Gavigan JA, Huang WJ, Hampton EN, Tumanut R, Suen KF, Trauger JW, Spraggon G, Lesley SA, Liau G, Yowe D, Harris JL (2007) Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J 406:203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McNutt MC, Lagace TA, Horton JD (2007) Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem 282:20799–20803. [DOI] [PubMed] [Google Scholar]
- 7. Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derre A, Villeger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 34:154–156. [DOI] [PubMed] [Google Scholar]
- 8. Seidah NG, Awan Z, Chretien M, Mbikay M (2014) PCSK9: a key modulator of cardiovascular health. Circ Res 114:1022–1036. [DOI] [PubMed] [Google Scholar]
- 9. Mayne J, Dewpura T, Raymond A, Bernier L, Cousins M, Ooi TC, Davignon J, Seidah NG, Mbikay M, Chretien M (2011) Novel loss‐of‐function PCSK9 variant is associated with low plasma LDL cholesterol in a French‐Canadian family and with impaired processing and secretion in cell culture. Clin Chem 57:1415–1423. [DOI] [PubMed] [Google Scholar]
- 10. Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, Subashi TA, Varghese AH, Ammirati MJ, Culp JS, Hoth LR, Mansour MN, McGrath KM, Seddon AP, Shenolikar S, Stutzman‐Engwall KJ, Warren LC, Xia D, Qiu X (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol 14:413–419. [DOI] [PubMed] [Google Scholar]
- 11. Piper DE, Jackson S, Liu Q, Romanow WG, Shetterly S, Thibault ST, Shan B, Walker NP (2007) The crystal structure of PCSK9: a regulator of plasma LDL‐cholesterol. Structure 15:545–552. [DOI] [PubMed] [Google Scholar]
- 12. Benjannet S, Hamelin J, Chretien M, Seidah NG (2012) Loss‐ and gain‐of‐function PCSK9 variants: cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J Biol Chem 287:33745–33755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chorba JS, Shokat KM (2014) The proprotein convertase subtilisin/kexin type 9 (PCSK9) active site and cleavage sequence differentially regulate protein secretion from proteolysis. J Biol Chem 289:29030–29043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bryan PN (2002) Prodomains and protein folding catalysis. Chem Rev 102:4805–4816. [DOI] [PubMed] [Google Scholar]
- 15. Naureckiene S, Ma L, Sreekumar K, Purandare U, Lo CF, Huang Y, Chiang LW, Grenier JM, Ozenberger BA, Jacobsen JS, Kennedy JD, DiStefano PS, Wood A, Bingham B (2003) Functional characterization of Narc 1, a novel proteinase related to proteinase K. Arch Biochem Biophys 420:55–67. [DOI] [PubMed] [Google Scholar]
- 16. Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J (2008) Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA 105:1820–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mitchell T, Chao G, Sitkoff D, Lo F, Monshizadegan H, Meyers D, Low S, Russo K, DiBella R, Denhez F, Gao M, Myers J, Duke G, Witmer M, Miao B, Ho SP, Khan J, Parker RA (2014) Pharmacologic profile of the Adnectin BMS‐962476, a small protein biologic alternative to PCSK9 antibodies for low‐density lipoprotein lowering. J Pharmacol Exp Ther 350:412–424. [DOI] [PubMed] [Google Scholar]
- 18. Blanchet CE, Svergun DI (2013) Small‐angle X‐ray scattering on biological macromolecules and nanocomposites in solution. Annu Rev Phys Chem 64:37–54. [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y, Eigenbrot C, Zhou L, Shia S, Li W, Quan C, Tom J, Moran P, Di Lello P, Skelton NJ, Kong‐Beltran M, Peterson A, Kirchhofer D (2014) Identification of a small peptide that inhibits PCSK9 protein binding to the low density lipoprotein receptor. J Biol Chem 289:942–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nassoury N, Blasiole DA, Tebon Oler A, Benjannet S, Hamelin J, Poupon V, McPherson PS, Attie AD, Prat A, Seidah NG (2007) The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 8:718–732. [DOI] [PubMed] [Google Scholar]
- 21. Blanchet CE, Spilotros A, Schwemmer F, Graewert MA, Kikhney A, Jeffries CM, Franke D, Mark D, Zengerle R, Cipriani F, Fiedler S, Roessle M, Svergun DI (2015) Versatile sample environments and automation for biological solution X‐ray scattering experiments at the P12 beamline (PETRA III, DESY). J Appl Cryst 48:431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Svergun DI (1992) Determination of the regularization parameter in indirect‐transform methods using perceptual criteria. J Appl Cryst 25:495–503. [Google Scholar]
- 23. Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI (2012) New developments in the ATSAS program package for small‐angle scattering data analysis. J Appl Cryst 45:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]