

Abstract
Recent studies have implicated a role of the epidermal growth factor receptor (EGFR) pathway in kidney disease. Skin toxicity associated with therapeutics which completely block the EGFR pathway precludes their use in chronic dosing. Therefore, we developed antibodies which specifically neutralize the EGFR ligands TGFα (transforming growth factor‐alpha) and epiregulin but not EGF (epidermal growth factor), amphiregulin, betacellulin, HB‐EGF (heparin‐binding epidermal growth factor), or epigen. The epitope of one such neutralizing antibody, LY3016859, was characterized in detail to elucidate the structural basis for ligand specificity. Here we report a crystal structure of the LY3016859 Fab fragment in complex with soluble human TGFα. Our data demonstrate a conformational epitope located primarily within the C‐terminal subdomain of the ligand. In addition, point mutagenesis experiments were used to highlight specific amino acids which are critical for both antigen binding and neutralization, most notably Ala41, Glu44, and His45. These results illustrate the structural basis for the ligand specificity/selectivity of LY3016859 and could also provide insight into further engineering to alter specificity and/or affinity of LY3016859.
Keywords: transforming growth factor alpha, epiregulin, kidney disease, epitope mapping, X‐ray crystallography
Short abstract
PDB Code(s): 5KN5
Introduction
The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase which signals following ligand‐induced homo‐ or hetero‐dimerization with other ErbB family members.1 EGFR signaling modulates numerous processes including proliferation, survival, differentiation, and migration.2, 3 At least seven ligands bind and activate EGFR: transforming growth factor‐α (TGFα), epiregulin (EREG), epigen (EPGN), amphiregulin (AREG), heparin‐binding epidermal growth factor (HB‐EGF), betacellulin (BTC), and epidermal growth factor (EGF).4 While sequence homology among these seven ligands is low, they all contain a structurally conserved EGF domain.5, 6, 7 The EGF domains contains six conserved cysteine residues which form three intramolecular disulfide bonds and N‐ and C‐terminal subdomains separated by a single residue. Both subdomains contain conserved β‐strand secondary structure.
Dysfunction of EGFR is associated with numerous cancer types making it an attractive and successful therapeutic target.8, 9 The EGFR pathway has also been implicated in numerous other diseases, including experimental chronic kidney disease (CKD)10, 11, 12 with TGFα in particular appearing to play a significant role.13, 14 More recently, we have shown that epiregulin also plays a pathologic role in experimental diabetic kidney nephropathy as specific neutralization of epiregulin reduces disease progression (manuscript in preparation). While EGFR is a potential new target in the treatment of CKD, chronic dosing with pan‐EGFR inhibitors is not feasible due to skin toxicity observed in 50–85% of patients treated with EGFR blockers.15, 16 Rather than targeting the receptor, neutralization of specific receptor ligands such as TGFα and/or epiregulin could be useful for the treatment of CKD while eliminating the skin toxicity associated with full EGFR blockade. Given the limitations of current standard of care17, 18 in preventing or delaying progression to end stage renal disease, there is a significant unmet medical need necessitating novel therapeutics.
To this end, we have developed LY3016859, a humanized monoclonal hIgG4 antibody, which binds and neutralizes only TGFα and epiregulin with high affinity.19 LY3016859 is in clinical investigation for the treatment of diabetic nephropathy. The murine version of LY3016859, mAb41, slowed the progression of kidney disease in both non‐diabetic 129S6 3/4th subtotal nephrectomy and Type 2 diabetic uninephrectomized (uniNx) db/db model (manuscript in preparation). Here we report detailed characterization of the LY3016859 epitope through crystallization and mutagenesis studies. Thorough characterization of this epitope provides a structural basis both for neutralization and selectivity/specificity of LY3016859. Moreover, these results could provide a foundation for engineering to alter specificity of the antibody.
Results
Preliminary characterization of the LY3016859 epitope
We recently reported a humanized monoclonal antibody, LY3016859, which selectively neutralized the EGFR ligands TGFα and epiregulin.19 An epitope region at the C‐terminus of TGFα/epiregulin was proposed based on sequence homology and binding affinities to various human and mouse EGFR ligands (Fig. 1). Previously, Western blot analysis of reduced and nonreduced human TGFα revealed that the epitope was conformational and dependent on the native disulfide bonds. To probe the localization of the epitope to the C‐terminus, a truncated TGFα C‐terminal subdomain peptide (residues 33–50) containing the Cys34‐Cys43 disulfide was characterized by Biacore. The peptide bound LY3016859 with an affinity of 8.9 nM, nearly 100‐fold weaker than the affinity to the full‐length protein. The C‐terminal subdomain is sufficient for binding, but it is not fully binding competent relative to the intact protein.
Figure 1.

Alignment of the sequences of the seven human EGFR ligands as well as mouse TGFα, epiregulin, and epigen. Measured affinities (K D) of LY3016859 to these ligands are shown on the right side. Based on sequence homology of the binding/non‐binding ligands, the C‐terminal sequence indicated by the box was initially proposed as the epitope region. Further inspection of the alignment showed the conserved (A/V)RCEH sequence for the ligands with sub nM affinity: human TGFα, mouse TGFα, and human epiregulin.
Structure of the LY3016859 Fab/TGFα complex
To understand the structural basis for recognition and selectivity, the crystal structure of the LY30168259 Fab in complex with human TGFα was determined. The structure was solved by molecular replacement and refined to a resolution of 2.8 Å with an R factor of 21.1% and a free R factor of 27.0%. The asymmetric unit contains two Fab‐antigen complexes which form a dimer through tetravalent coordination of a Zn2+ ion by the His18 and His35 sidechains of TGFα (Fig. 2). No direct contacts between the two complexes in the asymmetric unit are observed outside of the metal coordination. The two complexes in the asymmetric unit have virtually identical structures (backbone RMSD of 0.59 Å for 1876 backbone atoms). Density is not observed for the C‐terminus or the 10 N‐terminal amino acids of TGFα. The well‐ordered region of TGFα includes residues 11–49.
Figure 2.
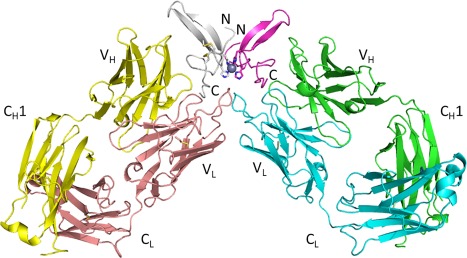
Overview of the LY3016859 Fab/human TGFα complex showing each complex in the asymmetric unit. The N‐ and C‐termini of TGFα are indicated. The coordinated Zn2+ metal ion is represented as a gray sphere. Heavy chain variable domain (V H), heavy chain constant domain 1 (C H1), light chain variable domain (V L), and light chain kappa constant domain (C L) are indicated.
The LY3016859 Fab binds primarily to the structured C‐terminal loop of TGFα (residues 39–49) (Fig. 3). The antigen binding site is a shallow groove formed by the CDR loops L1, L3, H2, and H3. A deeper, positively charged pocket in the groove is also present. The buried surface area upon antigen binding is ∼817 Å2. Interactions are also observed with the β1–β2 loop and the loop preceding the first β1 strand. The surface complementarity of the Fab and TGFα surfaces for both complexes in the asymmetric unit calculated utilizing the Sc program of the ccp4 suite is 0.74 and 0.69. This demonstrates a high degree of shape complementarity relative to average Sc values of 0.64–0.68 for antibody–antigen complexes.20
Figure 3.
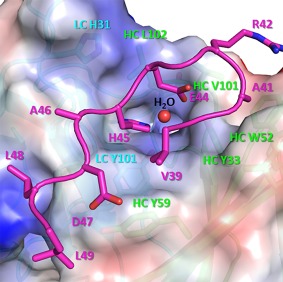
The primary TGFα binding surface of LY3016859 is shown. Only residues 39–49 of TGFα (in magenta) are depicted. The LY3016859 surface is colored by electrostatic potential using Pymol, and several residues which make up the binding interface are annotated with HC amino acids in green and LC amino acids in cyan. Glu44 and His45 sit in a binding pocket, and a coordinated water molecule (red sphere) is observed in this pocket in both complexes in the asymmetric unit. Pockets which accommodate Ala41 and Leu48 are also observed while Val39, Arg42, Ala46, Asp47, and Leu49 all sit on the surface of the Fab.
The crystal structure of TGFα bound to the extracellular domain of EGFR (PDB ID: 1MOX) was reported previously.5 The LY3016859 Fab binds TGFα from a similar orientation as EGFR (Fig. 4). While additional N‐terminal density was observed for TGFα in 1MOX, no significant changes are observed in the core ligand structure in the two complexes (backbone RMSD of 1.23 Å between the 156 atoms of residues 11–49). LY3016859 binds at the C‐terminus of TGFα through the shallow groove between the V H and V L domains, and this region is deeply buried in the EGFR ligand site in a cleft between the L1 and L2 domains. Despite binding the same region of TGFα, these binding sites show little structural similarity. However, this is not unexpected given the increased specificity of LY3016859 compared to EGFR. Nonetheless, it is clear from these structures that LY3016859 and EGFR could not simultaneously bind ligand, thereby providing a structural basis for LY3016859 neutralization of TGFα/epiregulin activity by directly blocking ligand/receptor interaction.
Figure 4.
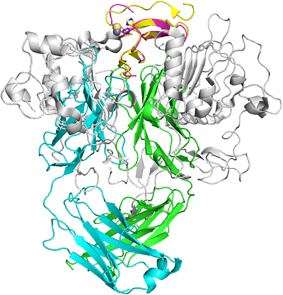
Comparison of the EGFR (1MOX) and LY3016859 binding sites. Structures are aligned by TGFα. TGFα from 1MOX is shown in yellow, and LY3016859 bound TGFα is shown in magenta. EGFR is shown in grey while the LY3016859 Fab is colored cyan (LC) and green (HC). A Cd2+ ion is observed in the 1MOX structure (yellow sphere) and is bound by TGFα similarly to the Zn2+ ion in the LY3016859 bound structure. It is clear from the superposition of the complexes that EGFR and LY3016859 cannot simultaneously bind TGFα, and this provides a structural basis for the neutralization of TGFα by LY3016859.
The primary TGFα epitope residues are the C‐terminal residues 41 and 44‐49. Arg42, which is 100% conserved in the EGFR ligand family sequences, is not involved in binding. Cys43, while not directly involved in binding, is likely critical for maintaining the conformational epitope via the disulfide bond with Cys34. The Glu44 and His45 sidechains are bound in a pocket within the binding groove and form a substantial network of hydrogen bonds which are shown in Figure 5 and listed in Table 1. In addition to these intermolecular hydrogen bonds, the Glu44 sidechain makes two hydrogen bonds to the TGFα backbone, likely stabilizing the binding conformation. Additional hydrogen bonds observed are listed in Table 1. Several hydrophobic interactions are present as well in this region—most importantly the shallow pocket formed by Tyr33, Trp52, Ile57, and Val101 of the HC in which Ala41 sits.
Figure 5.
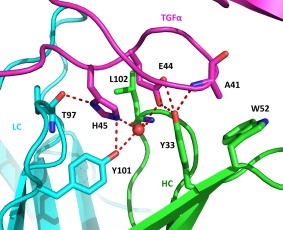
Interactions and key amino acid sidechains in the Glu44/His45 binding pocket are shown. Hydrogen bonds between TGFα (magenta)/Fab (LC in cyan, HC in green) and the bound water molecule (red sphere) are indicated by dashed lines. The water in the binding pocket forms a network of hydrogen bonds with the Glu44 and His45 sidechains, likely stabilizing the epitope conformation. Tyr101 of the LC and Tyr33 of the heavy chain are critical for binding as they form numerous hydrogen bonds to both TGFα and the bound water molecule. The Ala41/HC Trp52 interaction which is important for selectivity is also shown. Substitution with bulkier amino acids such as glutamic acid at this position results in significant loss in affinity.
Table 1.
Intermolecular Hydrogen Bonds Between the LY3016859 Fab and TGFα
| TGFα atom | LY3016859 fab atom | Average distancea (Å) |
|---|---|---|
| His18‐Nε2 | LC Ser32‐Oγ | 2.93 |
| Ala41‐N | HC Tyr33‐OH | 2.85 |
| Glu44‐Oε1 | HC Tyr33‐OH | 2.66 |
| Glu44‐Oε2 | HC Tyr33‐OH | 3.18 |
| Glu44‐O | LC His31‐Nε2 | 2.90 |
| His45‐Nδ1 | LC Thr97‐O | 2.84 |
| His45‐Nε2 | LC Tyr101‐OH | 2.79 |
| Ala46‐O | LC Val99‐N | 2.96 |
| Ala46‐N | LC Val99‐O | 3.32 |
| Asp47‐Oδ1 | HC Tyr59‐OH | 2.74 |
| Leu48‐N | LC Val99‐O | 3.00 |
Average of the distances in the two complexes in the asymmetric unit.
Limited interactions outside of the primary binding region of TGFα are observed. In the loop preceding β1, a hydrogen bond is made between the His18 sidechain and the LC Ser32 sidechains, and Phe17 interacts with a hydrophobic patch formed by the LC Thr33 and HC Leu102 sidechains. Glu27 of the β1–β2 loop makes contacts with Gly54 and Pro55 of the HC, and it also creates a favorable electrostatic interaction with HC Lys74.
Point mutagenesis analysis of the key epitope residues
To further characterize the importance of the core epitope residues identified in the crystal structure, several human TGFα point mutants were made: A41E, R42A, E44A, and H45A. The A41E mutation was chosen based on sequence alignment with human epigen which LY3016859 binds with low affinity. The Glu44 and His45 positions are conserved in epigen, so it was hypothesized that the Ala/Glu difference at position 41 was primarily responsible for the ∼20,000‐fold weaker binding of LY3016859 to epigen relative to TGFα. While not implicated in LY3016859 binding in the crystal structure, the R42A mutation was included as this position is 100% conserved in the EGFR ligand family and therefore could be important for specificity. The point mutants were expressed, purified, and their binding affinities to LY3016859 (Fig. 6) and activities in the MFc‐7 proliferation assay were measured. Results are summarized in Tables 2 and 3.
Figure 6.
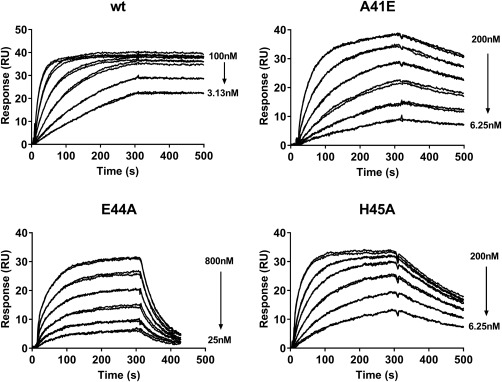
Biacore sensorgrams of LY3016859 binding to wt TGFα and three point mutants within the LY3016859 epitope. Binding kinetics are reported in Table II. The E44A mutation has the most dramatic effect with a 2,500‐fold loss in affinity resulting from an 8‐fold reduction in on‐rate and a 280‐fold increase in off‐rate. While the A41E and H45A mutants have affinity losses of ∼80‐fold, the decrease in H45A affinity is nearly entirely off‐rate driven while A41E suffers from both a decreased on‐rate and an increased off‐rate.
Table 2.
Biacore Binding Kinetics of LY3016859 to the His‐tagged TGFα Mutants
| TGFα mutant | k on (1/Ms × 105) avg ± std dev | k off (1/s × 10−5) avg ± std dev | K D (pM) avg ± std dev |
|---|---|---|---|
| Wt (n = 3) | 4.02 ± 0.86 | 4.28 ± 2.63 | 102 ± 43 |
| A41E (n = 2) | 1.48 ± 0.06 | 110 ± 9 | 7,410 ± 919 |
| R42A (n = 1) | 6.15 | 1.52 | 24.6 |
| E44A (n = 2) | 0.48 ± 0.01 | 1200 ± 212 | 251,500 ± 40,305 |
| H45A (n = 2) | 3.69 ± 0.11 | 304 ± 16 | 8,240 ± 693 |
Table 3.
Activity and LY3016859 Neutralization of the His‐tagged TGFα Mutants in the MFc.7 Proliferation Assay
| TGFα mutant | EC50 (pM) avg ± std dev | IC50 (nM) avg ± std dev |
|---|---|---|
| wt | 11.5 ± 0.7 (n = 2) | 0.46 ± 0.03 (n = 3) |
| A41E (n = 2) | 5.3 ± 1.9 | 328 ± 104 |
| R42A (n = 1) | ∼420 | <0.26 |
| E44A (n = 2) | 6.4 ± 5.4 | >1000 |
| H45A (n = 2) | 39.9 ± 50.0 | 15.6 ± 18.6 |
As expected from the structure, the R42A mutation did not significantly impact LY3016859 binding or neutralization, though it did drastically decrease activity to the receptor. The A41E, E44A, and H45A mutants all had comparable proliferative activity compared to the wild‐type ligand. Given the low sequence homology at these positions across the EGFR ligand family, this result is not surprising. LY3016859 affinity and neutralizing activity, however, were both significantly weakened as anticipated. The E44A mutation had the most significant impact, decreasing affinity approximately 2,500‐fold to ∼250 nM. The decrease in affinity was largely off‐rate driven with a 280‐fold increase in off‐rate compared to an 8‐fold decrease in on‐rate. This decrease in affinity resulted in a >1000‐fold decrease in neutralization by LY3016859, so much so that an accurate IC50 could not be determined. The A41E (7.41 nM) and H45A (8.24 nM) mutations resulted in comparable decreases in affinity of ∼300–400 fold. Again these decreases in affinity were predominantly off‐rate driven. Consistent with the decreased affinities observed, LY3016859 neutralization was decreased for both the A41E and H45A mutants.
Discussion
Complete blockade of the EGFR pathway is associated with skin toxicity which would preclude the use of EGFR inhibitors in a chronic setting such as diabetic nephropathy. To circumvent this toxicity, we developed a monoclonal antibody, LY3016859, to specifically bind and neutralize the EGFR ligands TGFα and epiregulin with high affinity. Indeed, weekly dosing of 100 mg kg−1 for 6 weeks in Cynomolgus monkeys was well tolerated with no observed skin toxicity.19 Understanding the structural basis of the specificity of LY3016859 could provide valuable insights for any future SAR and could potentially allow for rational tuning of the specificity of LY3016859 to selectively neutralize different EGFR ligands.
The core LY3016859/TGFα epitope region is the 41 ARCEH 45 sequence. Glu44 and His45 each form a total of three intermolecular hydrogen bonds: two with the Fab and one each with the bound water molecule in the interface. The water molecule also hydrogen bonds to the Fab providing a network of interactions at this site. Substitution with other amino acids is likely to destabilize this hydrogen bonding network and significantly decrease binding affinity. Furthermore, the pocket is somewhat positively charged providing for favorable electrostatic interactions with Glu44. Based on the requirement of these two amino acids alone, the only EGFR ligands which would be expected to bind LY3016859 are TGFα, epiregulin, and epigen.
Ala41, the other epitope residue characterized by mutagenesis, sits in a shallow hydrophobic pocket with HC Trp52 located at the bottom of the pocket. Substitution of Ala41 to the bulkier hydrophobic amino acid Val is tolerated as evidenced by the high affinity of LY3016859 for mouse TGFα (K D = 70 pM). Further increase in size to Leu, however, appears to be strongly disfavored as this is the only significant difference in the epitope region between the weakly binding mouse epiregulin (KD = 340 nM) and the tightly binding human epiregulin (K D = 854 pM). In addition to increased steric bulk, the A41E mutation adds an unfavorable electrostatic interaction as the Fab interface is somewhat negatively charged in this region. This difference contributes to the significantly weaker affinity of LY3016859 to human epigen (K D > 2 μM).
Though the key epitope residues are found within a five amino‐acid stretch of the C‐terminal loop of TGFα, the epitope of LY3016859 extends beyond this region and is conformational. Previously, we demonstrated that the disulfide bonds in the ligand are required for antigen binding as non‐reduced TGFα was recognized in a Western blot while reduced TGFα was not.19 Subsequent characterization of the binding of the C‐terminal subdomain peptide (residues 33–50) containing the Cys34‐Cys43 disulfide revealed that this C‐terminal subdomain, while encompassing the core epitope sequence and the majority of contacts made with LY3016859, bound with much lower affinity compared to full‐length protein (8.9 nM compared to 102 pM). In addition to the antibody/antigen contacts outside of the C‐terminal subdomain, the high affinity binding conformation of the C‐terminal subdomain is likely stabilized by intramolecular interactions with the N‐terminal subdomain.
While it is difficult to speculate on which subsets of EGFR ligands may prove relevant in various diseases states given that each of the seven EGFR ligands have their own tissue distribution and receptor binding affinity, the ability to tune the selectivity and/or affinities of an EGFR ligand neutralizing antibody could prove useful in disease states in which pan‐EGFR inhibition is unacceptable due to the associated skin toxicity. The structural characterization of the epitope presented in this work could inform SAR to alter the specificity/selectivity of LY3016859 binding for desired EGFR ligands. The unique neutralizing epitope, physiochemical properties, and the favorable pre‐clinical safety assessment of LY3016859 make it an excellent candidate for such specificity engineering as opposed to de novo antibody discovery efforts.
One can hypothesize several ways to alter specificity/affinity of LY3016859 based upon the data presented herein. Betacellulin, for instance, contains the critical Ala41 and Glu44 epitope residues, yet does not bind LY3016859 (Fig. 1). Presumably this is due to the Arg substitution at the His45 position which would cause significant steric hindrance as well as charge repulsion with the HC Arg99 sidechain. Mutation of LC Tyr101 and/or HC Arg99 to Asp/Glu could introduce selectivity to betacellulin by increasing the size of the binding pocket and forming a favorable salt bridge while maintaining the hydrogen bonding network formed through the bound water. The HC Trp52 which forms the bottom of the Ala41 binding pocket is another obvious candidate for engineering to alter specificity. Mutation to a smaller hydrophobic sidechain such as Ala or Val could increase epiregulin binding by better accommodating the bulkier Val/Leu of epiregulin. Alternatively, mutation to hydrophilic amino acids such as Gln, His, Thr, or Ser could make the antibody epigen selective by reducing steric clash and by forming a favorable hydrogen bond to the Glu sidechain of epigen at this position.
In summary, we have characterized the epitope of LY3016859 in detail through determination of the crystal structure in complex with TGFα and subsequent in vitro characterization of TGFα point mutants. These analyses demonstrate a conformational epitope in the C‐terminal loop of TGFα that is anchored by Ala41, Glu44, and His45. The structural basis for recognition of TGFα/epiregulin as well as the selectivity against the other EGFR ligands is elucidated. Finally, these results may provide guidance for potential future rational engineering of LY3016859 to alter specificity/selectivity.
Materials and Methods
Construction of TGFα expression vectors
DNA sequence encoding the mature, soluble human TGFα (amino acids 40–89 of the full length protein) was generated by PCR overlap extension using four primers. All subsequent TGFα residue numbering is given relative to the mature, soluble form. Primer sequences were 5′‐catatggtggtgagccattttaacgattgcccggatagccatacccagttttgcttt‐3′, 5′‐catacccagttttgctttcatggcacctgccgttttctggtgcaggaagat‐3′, 5′‐cacatagccgctatggcacacgcacgccggtttatcttcctgcaccagaaa‐3′, and 5′‐ggtgctcgagctattacgccagcagatccgcatgttcgcaacgcgcgcccacatagccgctatggca‐3′. The PCR fragment was first TOPO TA cloned (Life Technologies) and then subcloned in to pET‐28a (Novagen) between the NdeI and XhoI restriction sites. The resulting construct had a 6x His tag on the N‐terminus of TGFα. Point mutants were produced using the QuikChange II Site‐Directed Mutagenesis Kit (Agilent) according to the manufacturer's protocol. The primers used for the A41E, R42A, E44A, and H45A mutations were 5′‐ggctatgtgggcgaacgttgcgaacat‐3′, 5′‐tatgtgggcgcggcgtgcgaacatgcg‐3′, 5′‐ggcgcgcgttgcgcgcatgcggatctg‐3′, and 5′‐gcgcgttgcgaagcggcggatctgctg‐3′ and their reverse complements, respectively.
Protein expression and purification
The humanized IgG4 antibody LY3016859 was expressed in stably transfected Chinese hamster ovarian (CHO) cells. The antibody was purified from conditioned cell media using MabSelect Protein A Sepharose (GE Healthcare). The Fab fragment was prepared from purified antibody by papain digestion and subsequent purification with MabSelect Protein A Sepharose to remove cleaved Fc fragment along with undigested/partially digested mAb.
Human TGFα point mutants were expressed as inclusion bodies in the BL21‐RIPL E. coli strain (Agilent). Cultures in 2xTY media with 50 μg mL−1 kanamycin were grown at 37°C, induced at an OD600 of ∼0.8 with 1 mM IPTG, and harvested by centrifugation after 4 h of induction. Cell pellets were resuspended in 50 mM Tris pH 8.0 containing ∼0.4 mg mL−1 lysozyme, ∼0.5 μg mL−1 DNAse I (Roche Diagnostics), and 5 mM MgCl2. Resuspended cells were stirred gently at room temperature for 30 min and then sonicated on ice. Inclusion bodies were harvested by centrifugation at 20,000g and then washed with 0.1% Triton X‐100 in 5 mM EDTA pH 8.0. The isolated inclusion bodies were solubilized in IMAC binding buffer (20 mM phosphate pH 7, 0.5 M NaCl, 10 mM imidazole, 7 M urea, 0.025% β‐mercaptoethanol). The reduced, denatured TGFα was purified using Talon IMAC resin (Clontech) equilibrated in binding buffer. Protein was eluted with imidazole (20 mM phosphate pH 7, 0.5 M NaCl, 300 mM imidazole, 7 M urea, 0.025% β‐mercaptoethanol). TGFα was refolded by addition of L‐cysteine to the IMAC pool to a final concentration of 10 mM followed by dialysis overnight at 4°C against 20 mM glycine pH 10. The dialysate was then acidified to pH 3 with 1 N HCl and purified by RP‐HPLC using a linear gradient elution (water/acetonitrile/0.1% TFA buffer system) on a Symmetry C18 semipreparative column (Waters).
Peptide synthesis and refolding
The mature human TGFα (residues 40–89) used for crystallization was chemically synthesized using DIC/HOBt coupling chemistry. Crude peptide was oxidized in dilute solution of 10% DMSO in 0.1 M ammonium bicarbonate. Oxidized peptide was purified by RP‐HPLC as before to >90% purity.
Surface plasmon resonance
Commercially available ligands were purchased from R&D Systems. SPR experiments were conducted on a Biacore 2000/3000 (Biacore/GE Healthcare) at 25°C. Goat anti‐human Fc γ specific antibody (Jackson ImmunoResearch) was coupled to a CM5 chip (GE Healthcare) using standard amine coupling chemistry. Antibody was captured at a density of ∼300–500 RU. Ligand concentration series were injected in duplicate at a flow rate of 30 μL min−1 for 300 s followed by a dissociation period of up to 1800 s. Flow cells were regenerated with 2 x 20 second injections of glycine pH 1.5. Reference subtracted data were fit to a 1:1 binding model using BiaEvaluation Software (Biacore/GE Healthcare). The equilibrium binding constant, K D, was determined as the ratio of the off‐rate to the on‐rate (k off/k on).
Myofibroblast cell proliferation assay
The MFc.7 myofibroblast cell proliferation assay was performed as described previously.19 Briefly, activity of the ligands to the receptor was first determined, and then neutralization by LY3016859 was measured with the ligand concentration fixed at approximately the EC90.
Complex formation and crystallization
The Fab/antigen complex was formed by adding ∼10% molar excess of TGFα to 10 mg of Fab followed by purification on a Superdex 75 16/60 column (GE Healthcare) to remove excess antigen. Running buffer was 1X Tris buffered saline (TBS) pH 7.4, and flow rate was 1 mL min−1. Purified Fab/TGFa complex at 8 mg mL−1 (total protein) was crystallized by the vapor diffusion method with a well solution of 100 mM MES pH 6.0, 14% PEG 3350, 200 mM Sodium Sulfate, mixing 0.8 µL of protein with 0.8 µL of well solution. Drops were streak seeded after 45 min with previously grown crystals (well solution: 100 mM MES pH 6.5, 25% PEG MME 550, 10 mM Zinc sulfate monohydrate). Crystals were harvested and frozen in liquid nitrogen after a brief passage in 80% well solution supplemented with 10% (final) glycerol and 10% ethylene glycol.
Structure determination
Synchrotron diffraction data was collected at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL), beamline 31‐ID (LRL CAT) at 100K. Data were integrated and reduced using MOSFLM21 and the CCP4 suite of programs.22, 23, 24 The overall (25–2.8 Å) R‐sym is 10.6% and the highest shell (2.95–2.8 Å) 61.2%. An initial molecular replacement solution was obtained using Phaser (CCP4)22, 25 and subsequently modeled using COOT,26, 27, 28 refined using BUSTER,29 and validated using MolProbity.30 The final 2.8 Å crystal structure is in the P21 spacegroup with cell dimensions a = 63.3, b = 164.4, c = 65.0 Å and a beta angle of 104.2°. The asymmetric unit of the crystal structure contains two Fab and two TGFα molecules. A zinc atom was fit to omit density near Histidines 18 and 35 from both of two adjacent TGFα molecules (along the non‐crystallographic symmetry two‐fold axis). The difference density was too strong for a water molecule and corresponded to a 6σ peak in the anomalous density map. Data collection and refinement statistics are shown in Table 4. Coordinates and structure factors for the TGFα complex with the Fab fragment of the neutralizing antibody LY3016859 have been deposited in the RCSB Protein Data Bank with the accession code 5KN5.
Table 4.
X‐ray Data Collection and Refinement Statistics
| Data collection | |
|---|---|
| Resolution range (outer shell) (Å) | 25.00–2.80 (2.95–2.80) |
| Space group | P21 |
| a (Å) | 63.25 |
| b (Å) | 164.35 |
| c (Å) | 64.97 |
| β (°) | 104.2 |
| Completeness (%) | 98.5 (99.9) |
| Mean redundancy | 3.8 (3.8) |
| R sym (%) | 10.6 (61.2) |
| Mean I/sd(I) | 9.6 (2.0) |
| Refinement | |
| Resolution range (Å) | 25.00–2.80 |
| R (%) | 20.7 |
| R free (%) | 26.3 |
| rmsd bond lengths (Å) | 0.018 |
| rmsd angles (°) | 2.02 |
| Core Ramachandran (%) | 94.05 |
| Disallowed Ramachandran (%) | 1.08 |
| Asymmetric unit | |
| Amino acids | 445 |
| Chains | Heavy chain (A, D), Light chain (B,E), TGFα (C,F) |
| Ordered residues | A: 1–133, 137–217; B: 1–216; C: 10–49; D: 1–132, 135–217; E: 1–216; F: 11–49 |
| Chemical components | 1 SO4, 1 Zn |
| Waters | 150 |
Acknowledgments
The authors thank Robert Cummins for synthesis of the human TGFα used for crystallization, Karen Hill for expression and purification of TGFα point mutants, Kenneth Weichert for crystallization of the complex, and Malgorzata Gonciarz for critical reading of the manuscript. Use of the Lilly Research Laboratories Collaborative Access Team (LRL‐CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
Statement for Broader Audience: We performed epitope mapping studies including X‐ray crystallography to elucidate the specificity of a TGFα/epiregulin antibody which has efficacy in preclinical models of diabetic kidney disease and which does not have the toxicity associated with pan‐EGFR inhibitors. Further, this work could be used for future structure‐based engineering to develop neutralizing antibodies with altered specificity for EGFR ligands.
References
- 1. Schlessinger J (2002) Ligand‐induced, receptor‐mediated dimerization and activation of EGF receptor. Cell 110:669–672. [DOI] [PubMed] [Google Scholar]
- 2. Carpenter G (1983) The biochemistry and physiology of the receptor‐kinase for epidermal growth factor. Mol Cell Endocrinol 31:1–19. [DOI] [PubMed] [Google Scholar]
- 3. Schlessinger J, Schreiber AB, Levi A, Lax I, Libermann T, Yarden Y (1983) Regulation of cell proliferation by epidermal growth factor. CRC Crit Rev Biochem 14:93–111. [DOI] [PubMed] [Google Scholar]
- 4. Harris RC, Chung E, Coffey RJ (2003) EGF receptor ligands. Exp Cell Res 284:2–13. [DOI] [PubMed] [Google Scholar]
- 5. Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, Jorissen RN, Nice EC, Burgess AW, Ward CW (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 110:763–773. [DOI] [PubMed] [Google Scholar]
- 6. Puddicombe SM, Chamberlin SG, MacGarvie J, Richter A, Drummond DR, Collins J, Wood L, Davies DE (1996) The significance of valine 33 as a ligand‐specific epitope of transforming growth factor alpha. J Biol Chem 271:15367–15372. [DOI] [PubMed] [Google Scholar]
- 7. Davis CG (1990) The many faces of epidermal growth factor repeats. New Biol 2:410–419. [PubMed] [Google Scholar]
- 8. Yarden Y (2001) The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. Eur J Cancer 37:S3–S8. [DOI] [PubMed] [Google Scholar]
- 9. Yewale C, Baradia D, Vhora I, Patil S, Misra A (2013) Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials 34:8690–8707. [DOI] [PubMed] [Google Scholar]
- 10. Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, Friedlander G, Terzi F (2005) Angiotensin II and EGF receptor cross‐talk in chronic kidney diseases: a new therapeutic approach. Nat Med 11:867–874. [DOI] [PubMed] [Google Scholar]
- 11. Liu N, Guo JK, Pang M, Tolbert E, Ponnusamy M, Gong R, Bayliss G, Dworkin LD, Yan H, Zhuang S (2012) Genetic or pharmacologic blockade of EGFR inhibits renal fibrosis. J Am Soc Nephrol 23:854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Terzi F, Burtin M, Hekmati M, Federici P, Grimber G, Briand P, Friedlander G (2000) Targeted expression of a dominant‐negative EGF‐R in the kidney reduces tubulo‐interstitial lesions after renal injury. J Clin Invest 106:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Laouari D, Burtin M, Phelep A, Bienaime F, Noel LH, Lee DC, Legendre C, Friedlander G, Pontoglio M, Terzi F (2012) A transcriptional network underlies susceptibility to kidney disease progression. EMBO Mol Med 4:825–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laouari D, Burtin M, Phelep A, Martino C, Pillebout E, Montagutelli X, Friedlander G, Terzi F (2011) TGF‐alpha mediates genetic susceptibility to chronic kidney disease. J Am Soc Nephrol 22:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li T, Perez‐Soler R (2009) Skin toxicities associated with epidermal growth factor receptor inhibitors. Target Oncol 4:107–119. [DOI] [PubMed] [Google Scholar]
- 16. Robert C, Soria JC, Spatz A, Le Cesne A, Malka D, Pautier P, Wechsler J, Lhomme C, Escudier B, Boige V, Armand JP, Le Chevalier T (2005) Cutaneous side‐effects of kinase inhibitors and blocking antibodies. Lancet Oncol 6:491–500. [DOI] [PubMed] [Google Scholar]
- 17. Lewis EJ (1996) Influence of ACE inhibition on the kidney function of diabetics. Br J Clin Pract Suppl 84:23–24. [PubMed] [Google Scholar]
- 18. Lewis EJ (2003) Angiotensin II receptor blockade in diabetic nephropathy. Am J Hypertens 16:100–101. [DOI] [PubMed] [Google Scholar]
- 19. Beidler CB, Petrovan RJ, Conner EM, Boyles JS, Yang DD, Harlan SM, Chu S, Ellis B, Datta‐Mannan A, Johnson RL, Stauber A, Witcher DR, Breyer MD, Heuer JG (2014) Generation and activity of a humanized monoclonal antibody that selectively neutralizes the epidermal growth factor receptor ligands transforming growth factor‐alpha and epiregulin. J Pharmacol Exp Ther 349:330–343. [DOI] [PubMed] [Google Scholar]
- 20. Lawrence MC, Colman PM (1993) Shape complementarity at protein/protein interfaces. J Mol Biol 234:946–950. [DOI] [PubMed] [Google Scholar]
- 21. Leslie AGW, Powell HR. Processing diffraction data with mosflm In: Read RJ, Sussman JL, Ed. (2007) Evolving methods for macromolecular crystallography: the structural path to the understanding of the mechanism of action of CBRN Agents. Dordrecht: Springer Netherlands, pp 41–51. [Google Scholar]
- 22. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Howell PL, Smith GD (1992) Identification of heavy‐atom derivatives by normal probability methods. J Appl Cryst 25:81–86. [Google Scholar]
- 24. French S, Wilson K (1978) On the treatment of negative intensity observations. Acta Cryst A 34:517–525. [Google Scholar]
- 25. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 27. Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Cryst D60:2184–2195. [DOI] [PubMed] [Google Scholar]
- 28. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst D60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 29. Bricogne GBE, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO (2011) BUSTER version 2.11.5. Cambridge, United Kingdom: Global Phasing Ltd. [Google Scholar]
- 30. Chen VB, Arendall WB, III , Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]