

Abstract
Diabetes and hyperinsulinemia may be risk factors for Alzheimer's disease (AD). We conducted a pilot study of metformin, a medication efficacious in treating and preventing diabetes while reducing hyperinsulinemia, among persons with amnestic mild cognitive impairment (AMCI) with the goal of collecting preliminary data on feasiblity, safety, and efficacy. Participants were 80 men and women aged 55 to 90 years with AMCI, overweight or obese, without treated diabetes. We randomized participants to metformin 1000 mg twice a day or matching placebo for 12 months. The co-primary clinical outcomes were changes from baseline to 12 months in total recall of the Selective Reminding Test (SRT) and the score of the Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-cog). The secondary outcome was change in relative glucose uptake (rCMRgl) in the posterior cingulate-precuneus in brain Fluorodeoxyglucose Positron Emission Tomography. Change in plasma Aβ42 was an exploratory outcome. The mean age of participants was 65 years. Fifty % of participants were women. The only baseline variable that was different between the arms was the ADAS-Cog. Metformin could not be tolerated by 7.5% of participants; 15% tolerated 500 mg/day, 35% tolerated 1000 mg/day, 32.5% tolerated 1500 mg/day, and only 10% tolerated the maximum dose. There were no serious adverse events related to metformin. The 7.5% of persons who did not tolerate metformin reported gastrointestinal symptoms. After adjusting for baseline ADAS-cog, changes in total recall of the SRT favored the metformin group (9.7 ± 8.5 vs. 5.3 ± 8.5; p = 0.02). Differences for other outcomes were not significant. A larger trial seems warranted to evaluate the efficacy and cognitive safety of metformin in prodromal AD.
Keywords: Metformin, insulin, amnestic mild cognitive impairment, memory, ra
Introduction
The most common cause of late onset dementia is Alzheimer's disease (AD)[1], comprising between 60 and 80% of cases. Vascular dementia is the second most common cause, comprising about 10% of cases, but approximately 50% of cases of dementia have a vascular component [1]. Nearly half of persons 85 years and older have late onset AD [2] and the prevalence worldwide will quadruple by mid-century [3]. A key process in AD causation is the deposition of amyloid β (Aβ) in the brain[4], but other factors, such as cerebrovascular disease, may also be important [5]. The natural history of AD starts slowly in late middle age with mild memory deficits that progress to amnestic mild cognitive impairment (AMCI) [6], which in turn progresses to dementia [7]. Thus, AMCI is considered a high risk group for dementia [6] and has become a common target for secondary prevention. Clinical trials in AMCI and AD have thus far been disappointing [8] and an expert panel from NIH concluded in 2010 that there was insufficient evidence to recommend treatment of AMCI or prevention for AD [9]. In this context, the search for strategies for the prevention and treatment of AD continues. Type 2 diabetes (T2D) and its correlates and antecedents, insulin resistance and elevated adiposity (overweight and obesity) have shown strong associations with AD in observational studies [10, 11]. These observations have enormous public health implications because 2/3 of adult Americans are overweight or obese (Body Mass Index (BMI) ≥ 25 m/k2) [12], and are at high risk for insulin resistance and T2D [13]. Among persons 60 years and older, a group with high prevalence of AMCI [14, 15] and AD, 69% were overweight or obese in 2008 [12], and 60% have pre-T2D or T2D[16]. Thus, a majority of Americans in the age group at risk for AD have insulin resistance and T2D [17]. Insulin resistance and T2D are known cerebrovascular risk factors[18]and it seems reasonable to postulate that insulin resistance and T2D could increase AD risk through cerebrovascular disease, a factor increasingly accepted to be important in AD manifestation [5, 19-21]. However, peripheral insulin resistance and hyperinsulinemia may also affect AD directly [22]. Since insulin resistance could increase the risk of AD through both cerebrovascular and Aβ related mechanisms [23] it has been hypothesized that pharmacologic strategies effective in preventing diabetes while decreasing insulin resistance may prevent AD [24]. One of these strategies is the use of the medication metformin. Metformin is a safe, inexpensive medication available in generic form with proven efficacy in treating and preventing T2D though the reduction of peripheral glucose and insulin levels [25, 26]. Thus, we conducted a pilot study of AD prevention among persons with AMCI at risk for T2D to obtain preliminary evidence of feasibilty, safety, and efficacy.
Materials and Methods
Design
This study was a double-blind placebo-controlled randomized pilot trial of metformin 1000 mg twice a day vs. matching placebo for 12 months in 80 subjects with AMCI. Participants were randomized to metformin or placebo in a 1:1 ratio. To balance factors that may influence treatment outcome, randomization was stratified and blocked. The only stratification variable was the presence or absence of APOE-ε4 genotype. The rationale for this stratification was that observational studies have shown that APOE-ε4 genotype modifies the association of hyperinsulinemia and diabetes with AD (risk is much higher in those with APOE-ε4)[27-29] and a pilot randomized trial of the insulin sensitizer rosiglitazone reported that this medication was only efficacious among persons without APOE-ε4 [30]. Allocation concealment was maintained by randomly alternating block sizes. Participants were seen once a week in the first 4 weeks of the study during metformin titration and were then evaluated at months 3, 6, 9, and 12, including the neuropsychological battery. Half the sample (40 participants) were invited to participate in a brain imaging sub-study.
All participants were invited to undergo brain imaging at baseline and after the 12 month visit and were selected if they agreed to undergo brain imaging and if they had no exclusion criteria for brain imaging.
Setting
the study was a single site study based at Columbia University Medical Center in New York City.
Participants
Table 1 summarizes the main inclusion and exclusion criteria. Participants were 80 subjects aged 55 to 90 years with AMCI defined by the Petersen criteria,[31] without treated diabetes, and with a body mass index (BMI) of 25 kg/m2 or higher (overweight or obese by National Heart, Lung, and Blood Institute (NHLBI)) criteria [32]). The rationale for limiting the study to AMCI was that AMCI is a transitional stage from normal cognition to dementia that is considered a target for secondary prevention of dementia [33]. The rationale for the age group was that the 55 to 90 year old group is considered most susceptible to cognitive impairment and thus commonly evaluated in treatment trials of AMCI such as the Alzheimer's Disease Cooperative Study (ADCS) clinical trial of vitamin E and donepezil [34]. The rationale for not including persons with treated T2D is that the hypothesized main mechanism of metformin is the lowering of insulin levels, and persons with diabetes are treated with medications that can both increase insulin levels (exogenous insulin, sulfonylureas) and medications that can decrease insulin levels (metformin, thiazolidinediones)[35]. We screened for T2D by medical history but also by Hemoglobin A1c (HbA1c) in blood. Our exclusion criterion related to T2D changed in the following way during the course of the study. In September of 2008, when one participant had been enrolled, we changed the inclusion criteria from excluding all persons with a history of T2D to accepting persons with T2D with a HbA1c of 6.5% or less who were not on any medication. The justification for this change was that we discovered in our recruitment efforts that there were many persons with a history of T2D that was not treated, leading to a high proportion of exclusion due to untreated T2D. We chose the cutoff of 6.5% in HbA1c because persons with this level of HbA1c seemed unlikely to receive pharmacologic treatment for T2D, including metformin, during the 12 months of the study. In January of 2010 the American Diabetes Association (ADA) adopted diabetes diagnosis guidelines using HbA1c (6.5% or more) for the first time [36]. Thus, we modified our inclusion criteria in March of 2010, when 60 participants had been enrolled. If persons without a history of diabetes were found to have an HbA1c of 6.5%, they were informed that they met criteria for T2D. We allowed participation of subjects who met HbA1c criteria for diabetes, who were not on diabetes treatment, and with a HbA1c of 6.9% or less (The ADA recommends maintaining a HbA1c of less than 7% [36]), and who upon consultation with their physicians were deemed not to need pharmacologic treatment for T2D.
Table 1.
Inclusion and exclusion criteria.
| Inclusion criteria | Exclusion criteria |
|---|---|
|
|
The rationale for the BMI cutoff is that there is no standard for the diagnosis of hyperinsulinemia in research or clinical practice [17]. Thus, we limited inclusion to persons with AMCI who were overweight or obese (BMI ≥ 25 m/k2), definitions of elevated adiposity in research and clinical practice [37],and a surrogate marker of hyperinsulinemia [38], because they were the most likely to benefit from the insulin lowering action of metformin.
Screening was conducted folllowing a 2-step process. First, participants who were interested in participating were screened by telephone for demographic and medical inclusion and exclusion criteria. The Telephone Interview for Cognitive Status (TICS)[39] was administered if the potential participant agreed to screen out persons who were unlikely to have any memory impairment. A TICS Score > 34 out of 41 was considered normal cognition. Persons with this score were not invited to participate. Persons who passed the telephone screen were invited for an in-person screening that included a physical exam, blood tests, and a neuropsychological battery.
Intervention
Participants were randomized to identical-appearing pills of metformin or matching placebo provided by Merck-Lipha of France. The maximum dose of metformin was 1000 mg twice a day, as is commonly used in clinical practice. Metformin was administered as 500 mg tablets. Metformin was titrated weekly from 500 mg once a day to 1000 mg twice a day over 4 weeks. Subjects were maintained on the highest tolerated dose. Participants who did not tolerate the study drug were invited to continue in the study and were included in the intent to treat (ITT) analyses. Metformin and placebo were supplied every 3 months, when participants were asked for side effects and contraindications to metformin in addition to undergoing safety laboratory tests. The most common side effect of metformin is gastrointestinal intolerance, which can range from bloating to frank diarrhea [40]. In clinical practice patients taking metformin can often break through these symptoms after a few weeks and tolerate the drug well. However, we were conservative and maintained participants on the dose of metformin at which they had no symptoms.
Outcomes
Primary clinical outcomes
The primary outcomes of the study were changes from baseline to month 12 in total recall of the Bushcke Selective Reminding Test [41] and the Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-cog) [42]. The rationale for using the SRT is that it has been extensively used to assess AMCI[14, 43] and AD[44] in the community of Northern Manhattan sorrounding Columbia University Medical Center (CUMC), where the study was based. The rationale for using the ADAS-Cog is that it is commonly used for clinical trials of AMCI[45] and AD. Other clinical measures in the study included the The ADCS Clinical Global Impression of Change for Mild Cognitive Impairment (CGIC-MCI) [46], the logical memory II delayed paragraph recall sub-test of the Wechsler Memory Scale Revised (WMS-R)[47], the Mini-Mental Status Exam(MMSE) [48], the Neuropsychiatric Inventory Questionnaire (NPI-Q) [49], and the digit span backwards [50].
Imaging outcomes
The primary imaging outcome was changes from baseline to month 12 in relative glucose uptake (rCMRgl) in the posterior cingulate-precuneous measured by non-quantitative brain [18]F-labeled 2-deoxy-2-fluoro-D-glucose (FDG) positron emission tomography (PET) with magnetic resonance imaging (MRI) co-registration. PET imaging was performed with a HR+ scanner (Siemens/CTI, Knoxville, TN) at the Columbia Kreitchman PET center. This region of interest was chosen because CMRgl reductions in this region are correlated with dementia severity[51] and progression[52]. FDG PET with MRI coregistration and a Region of Interest (ROI) approach as conducted in this study have been shown to have good six-month test-retest reliability for ROIs affected in AD[7]. Cerebellum rCMRgl is not expected to change with AD progression and was chosen as the reference ROI for standardization[53]. The FDG PET results are shown as changes from baseline to 12 months in ratios of rCMRgl of the ROI weighted by the ROI volume and the the rCMRgl of the cerebellum. We did not expect to have interference of extreme glucose levels with PET readings because we excluded subjects with treated diabetes from the trial and because metformin does not produce hypoglycemia [40]. However, we conducted a washout period of 2 weeks [54] at the end of the trial before performing FDG PET to ensure that changes in rCMRgl were not due to acute changes in glucose due to metformin. It has been estimated that 26% of metabolic effects of metformin are acute and disappear upon withdrawal of the medication [54]. Brain Magnetic Resonance Imaging (MRI) was acquired at the Hatch MRI Research Center at Columbia University Medical Center for co-registration of FDG PET images using a 1.5T Philips Intera scanner with an 8 channel receive coil following a protocol similar to the Alzheimer's Disease Neuroimaging Initiative [55]. The Primary region of interest (ROI) was the posterior cingulate-precunenous [56]. This ROI included the Precuneus & Posterior Cingulate gyrus together, because they share the same pathological features in AD. The cingulate sulcus, the callosal sulcus, the inter-hemispheric fissure and the parieto-occipital fissure were identified in the orthogonal view. The line was then extended between the cingulate and callosal sulci that is the antero-superior limit of the ROI, then posteriorly and inferiorly over the callosal sulcus until reaching the fundus of this sulcus. It was then extended posteriorly and superiorly over the parieto-occipital fissure up to the end. Then, the contour of the precuneus was followed anteriorly and superiorly to the marginal segment of the cingulate sulcus. It was then extended anteriorly over the cingulate sulcus until reaching the other anterior end.
Plasma amyloid β
Plasma Aβ42 levels were measured in plasma using a combination of monoclonal antibody 6E10 (specific to an epitope present on 1 to 16 amino acid residues of Aβ) and rabbit antibodies specific for Aβ42 (R165) in a double-antibody sandwich ELISA [57]. The detection limit for this assay was 10 pg/mL for Aβ42. The test–retest reliability of the measurement of plasma Aβ42 is excellent (Cronbach's α coefficient = 0.91).
Other measures
Genotyping of APOE polymorphisms rs7412 and rs429358 was performed at PreventionGenetics (www.preventiongenetics.com) with an array tape [58] using allele-specific PCR with universal molecular beacons [59, 60]. DNA sequencing of positive control DNA samples were completed to assure correct assignment of alleles. APOE-ε4 genotyping was conducted within one week of acceptance into the trial and was used for randomization purposes. Insulin was measured using a solid-phase chemiluminescent enzyme immunoassay (Immulite, Diagnostic Products Co, Los Angeles, CA). Intra- and inter-assay coefficients of variation are 4.7% and 8.2%, respectively. The normal insulin range for this assay is 6-27microIU/ml. HbA1c and lipids (total cholesterol, high density lipoprotein) were measured at baseline and each follow-up to document the hypothesized metabolic effect which is similar to insulin [61]. High-sensitivity C-Reactive protein (hsCRP) was measured as a marker of systemic inflammation and vascular risk [62, 63] that predicts memory impairment in our population [64] using ELISA (Diagnostic systems laboratories, INC, Webster, Texas). Insulin and hsCRP were measured in all samples at the same time at the end of the study. Thyroid Stimulating Hormone, vitamin B12, and syphillis rapid plasma reagin (RPR) were assessed at screening.
Statistical Analyses
Given that the study was planned as a pilot clinical trial with the goal of collecting preliminary data on safety, feasibility, and efficacy, it was powered to detect a large difference (0.75 standard deviations) in the change in the outcomes at 12 months with a sample size of 30 persons per group (Total 60 groups). We obtained supplementary funding and increased the sample size from 60 to 80 participants. Initial analyses were conducted blinded to true treatment allocation. Treatment unblinding was conducted after analyses were checked for quality. The data coordinating center at Department of Biostatistics at Columbia University Medical Center generated the random allocation sequence. The data coordinating center resided in a building separate from where the study team resided. Only the statistician had access to the randomization sequence. The assistant assigning the participants to interventions and the staff recruiting and evaluating participants did not have access to the randomizaion sequence. All measurements and laboratory assessments were conducted at the Irving institute for Clinical Translational Research at Columbia University Medical Center. The coordinators conducting outcomes assessments were blinded from these data. No interim analyses were planned or conducted during the conduct of the clinical trial.
For comparison of baseline characteristics between the metformin and placebo group T-tests were used to compare means, and chi-squared to compare proportions. Analysis of Covariance (ANCOVA) was used to compare outcomes between the treatment groups following an Intent-to-treat (ITT) approach adjusting for variables that were different at baseline if necessary. The primary clinical outcomes were changes from baseline to 12 months in total recall of the SRT and total score of the ADAS-COG. The primary imaging outcome was changes from baseline to 12 months in rCMRgl in the posterior cingulate-precuneous. The exploratory outcome was changes from baseline to 12 months in plasma Aβ-42. Imputation with last-observation-carried-forward (LOCF) was used to account for missing follow-up data. We conducted similar analyses for the secondary outcomes and for changes in pertinent metabolic measures related to the effect of metformin such as insulin and hsCRP. Finally we conducted post-hoc analyses relating the dose of metformin (1, 2, 3, or 4 tablets a day) to the outcomes with linear regression using placebo and the persons who did not tolerate metformin as the reference (completers analysis) and stratified analyses by APOE-ε4, baseline age, BMI, fasting insulin and HbA1c categorized by the median. The level of significance was established at a two-sided alpha of 0.05. This study was approved by the Institutional Review Board of Columbia University Medical Center (AAAC7231) and was registered in ClinicalTrials.gov (ID NCT00620191). All study participants provided written informed consent.
Results
Participant characteristics
Figure 1 shows the CONSORT flowchart. Recruitment was completed in 32 months (2.5 subjects a month) [38]; 1426 subjects were screened by telephone and 331 were screened in person; 65 subjects (81%) completed 12 months, 6 subjects (7.5 %) completed 9 months, and 9 subjects (11.2%) had less than 9 months of follow-up. At 12 months, study completion was similar in the 2 arms (33 persons completed the placebo arm and 32 the metformin arm; p = 0.99), The only statistically significant difference between the groups at baseline (Table 2) was the ADAS-Cog score, which was lower (better) in the metformin group. Thus, the comparison of the primary outcomes is adjusted for this variable.
Figure 1. Flowchart of study participation.
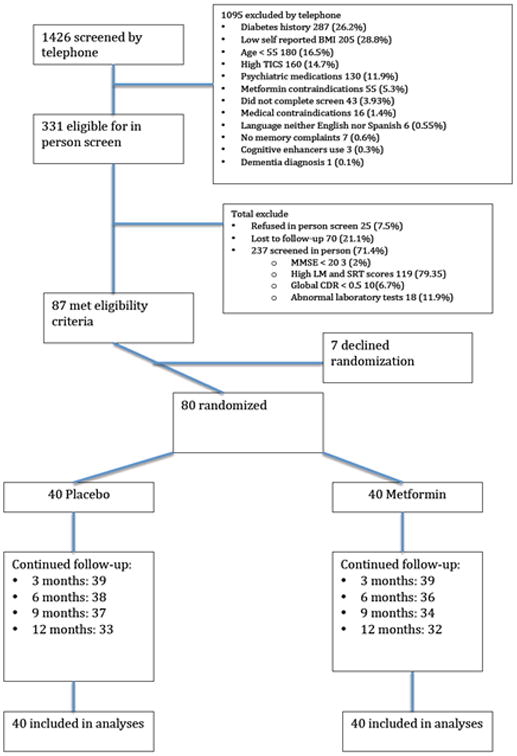
Table 2.
Comparison of characterisitics between the metformin and placebo arms. Continuous variables are presented as means and standards deviations (SD). Categorical variables are presented as counts and percentages. Hypothesis testing was conducted with T tests for continous variables and Chi-Square for categorical variables.
| Metformin (n=40) | Placebo (n=40) | p-value | |
|---|---|---|---|
| Age in years (SD) | 65.3 (7.0) | 64.1 (7.9) | 0.49 |
| Women (%) | 18 (45) | 24 (60) | 0.21 |
| Education in years (SD) | 13.8 (3.4) | 13.1 (4.5) | 0.44 |
| Ethnic group (%) | 0.46 | ||
| Hispanic | 17 (42.5) | 13 (32.5) | |
| Non-Hispanic Black | 11 (27.5) | 15 (37.5) | |
| Non-Hispanic White | 12 (30.0) | 12 (30.0) | |
| Apolipoprotein E ε4 (%) | 10 (25.0) | 11 (27.5) | 0.79 |
| Body Mass Index in kg/m2 (SD) | 30.9 (4.1) | 31.3 (4.7) | 0.65 |
| Systolic Blood Pressure in mmHg (SD) | 130.8 (10.9) | 132.1 (12.4) | 0.62 |
| Total Cholesterol in mg/dl (SD) | 204.2 (43.6) | 208.2 (46.7) | 0.71 |
| High Density lipoprotein in mg/dl (SD) | 51.5 (14.1) | 58.3 (17.7) | 0.06 |
| Hemoglobin A1C in % (SD) | 6.1 (0.8) | 6.1 (0.5) | 0.92 |
| Hemoglobin A1C> 6.5% (%) | 7 (17.5) | 6 (15.0) | 0.76 |
| High Sensitivity C-reactive protein in mg/dl (SD) | 2.9 (3.6) | 3.7 (2.9) | 0.32 |
| Insulin in IU/dl (SD) | 16.3 (9.5) | 13.4 (7.6) | 0.20 |
| ADAS-Cog Score (SD) | 12.0 (4.0) | 14.6 (6.1) | 0.02 |
| Selective Reminding Test Total Recall (SD) | 34.2 (7.9) | 36.1 (9.5) | 0.32 |
ADAS-Cog: Alzheimer's Disease Assessment Scale-cognitive subscale
Compliance with metformin and adverse events
Compliance with metformin was as follows: 7.5% stopped metformin but continued in the study following ITT, 15% remained on 500 mg a day (1 tablet), 35% remained on 1000 mg a day (2 tablets), 32.5% remained on 1500 mg a day (3 tablets), and 10% tolerated the maximum dose of 2,000 mg a day (4 tablets). In terms of safety, there were no serious adverse events related to metformin and the 7.5% of persons who were not able to tolerate metformin reported gastrointestinal symptoms.
Changes of metabolic measures by randomization arms
Fasting insulin increased appreciably more in the placebo group compared with the metformin group as expected, and this difference was close to statistical significance (13.8 vs. 4.7 IU/ml; p = 0.09). hsCRP, a measure of inflammation and vascular risk [63], and a correlate of memory impairment in our center[64] decreased in the metformin group and increased in the placebo group (-0.3 vs. 1.0 mg/dl; p = 0.07). Weight decreased more in the metformin group (-2.7 ± 6.4 Kg) compared with the placebo group (-1.6 ± 4.5 Kg) but this difference was not statistically signficant (p = 0.63). HbA1c decreased modestly more in the metformin group (-0.3 ± 0.7 %) compared with the placebo group (- 0.2 ± 0.5), but this difference was not statistically significant (p=0.64).
Changes in the clinical outcomes
Primary analyses
Table 3 shows the comparison in the primary clinical outcomes of the study, changes from baseline to month 12 in the ADAS-Cog score, and the total recall of the Selective Reminding Test (SRT). Both the SRT and ADAS-COG scores improved in the placebo and metformin groups (increases in SRT total recall, decreases in ADAS-COG scores). Crude analyses showed a greater improvement in the SRT in the metformin group, but the difference for the ADAS-Cog favored the placebo group. However, after adjustment for baseline ADAS-Cog the metformin group showed significantly greater improvement in SRT total recall compared to placebo (difference in changes in total recall of the SRT of metformin vs. placebo= 4.4 ± 8.5 words)and the difference for the ADAS-Cog was attenuated and not significant. The results were similar for delayed recall of the SRT, in which the gain in words was higher in the metformin group (2.3 ± 2.5) compared to the placebo group (1.3 ± 2.3) and was close to statistical significance (p=0.06). There were no differences in delayed recall of the ADAS-cog (0.7±1.8 for metformin vs. 0.0± 2.5 for placebo; p = 0.35).
Table 3.
Comparison of changes from baseline to 12 months in the Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-cog) and total recall of the Buschke Selective Reminding Test (SRT) between metformin and placebo. Crude (unadjusted) analyses are from T-tests. Adjusted analyses are from Analyses of Covariance. The only variable that was adjusted for was the baseline score of the ADAS-cog. Table 1 shows that this was the only baseline variable that was different between the randomization arms.
| Metformin | Placebo | p-value | |
|---|---|---|---|
| ADAS-Cog | |||
| Baseline | 12.0 ± 4.0 | 14.6 ± 6.1 | 0.02 |
| Last visit | 12.1 ± 3.8 | 12.8 ± 6.2 | 0.52 |
| Crude difference | 0.0 ± 3.3 | -1.98 ± 5.5 | 0.06 |
| Adjusted difference | -0.5 ± 4.1 | -1.4 ± 4.1 | 0.34 |
| Total recall SRT | |||
| Baseline | 34.2 ± 7.9 | 36.1 ± 9.5 | 0.32 |
| Last visit | 43.6 ± 9.1 | 41.5 ± 8.4 | 0.31 |
| Crude difference | 9.4 ± 8.5 | 5.7 ± 8.7 | 0.05 |
| Adjusted difference | 9.5 ± 6.1 | 5.4 ± 6.1 | 0.05 |
There were no differences between metformin and placebo in changes in digit span backwards, the neuropsychiatric inventory, the MMSE, paragraph recall, or CGIC-MCI (Supplemental table 1). One person in the placebo group and none in the metformin group converted to dementia.
Post hoc analyses of primary outcomes
We conducted linear regression models examining the relation of the metformin dose with the primary outcomes, changes in total recall of the SRT and the total score of the ADAS-cog. The highest metformin dose (1000 mg twice a day) was associated with a statistically significant increase of 5.3±10.0 more words in total recall of the SRT (p = 0.03) compared to those in the placebo group and those who could not tolerate metformin. There was no association between the highest metformin dose and changes in the ADAS-Cog (0.7 ± 4.8; p = 0.56).
Table 4 shows the results of stratified analyses by age, APOE-ε4 status, BMI category, baseline HbA1c, and fasting insulin levels. Metformin showed better performance compared with placebo in younger persons, those without APOE-ε4, those with lower HbA1c, and those with higher insulin levels. There were no differences for the ADAS-Cog in any of the strata.
Table 4.
Stratified analyses of intent to treat comparisons of the primary outcomes, Changes in total recall of the selective reminding test (SRT) and Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-Cog). Analyses are stratified by age median, APOE-ε4, Body Mass Index (obese vs. overweight), HbA1c median, and fasting insulin median. Group comparisons were conducted with analyses of covariance adjusting for baseline ADAS-Cog.
| Metformin | Placebo | ||||
|---|---|---|---|---|---|
| Total Recall Selective Reminding Test | |||||
| N | Mean + SD | N | Mean + SD | P | |
| Age group | |||||
| ≤ 63.7 years | 18 | 12.2 ± 7.5 | 22 | 6.3 ± 7.5 | 0.02 |
| > 63.7 years | 22 | 6.9 ± 9.8 | 18 | 5.4 ± 6.7 | 0.67 |
| APOE-ε4 | |||||
| Negative | 30 | 10.7 ± 8.2 | 29 | 5.9 ± 8.6 | 0.04 |
| Positive | 10 | 5.7 ± 9.1 | 11 | 5.2 ± 9.2 | 0.89 |
| Body Mass Index | |||||
| < 30 kg/m2 | 21 | 9.0 ± 10.0 | 19 | 3.9 ± 10.0 | 0.13 |
| ≥ 30 kg/m2 | 19 | 10.3 ± 6.9 | 21 | 6.9 ± 6.8 | 0.14 |
| HbA1c | |||||
| ≤ 6.0 % | 23 | 12.2 ± 7.1 | 20 | 6.6 ± 7.1 | 0.01 |
| > 6.0 % | 17 | 6.3 ± 9.0 | 20 | 4.0 ± 9.8 | 0.49 |
| Insulin | |||||
| ≤ 9 IU/dl | 21 | 6.7 ± 8.7 | 19 | 4.4 ± 8.7 | 0.42 |
| > 9 IU/dl | 19 | 12.4 ± 7.8 | 21 | 6.8 ± 8.0 | 0.03 |
| ADAS-Cog | |||||
| Age group | |||||
| ≤ 63.7 years | 18 | - 0.2 ± 3.5 | 22 | -1.2 ± 3.5 | 0.38 |
| > 63.7 years | 22 | -0.5 ± 5.2 | 18 | - 1.8 ± 6.3 | 0.50 |
| APOE-ε4 | |||||
| Negative | 30 | 0.1 ± 3.8 | 29 | -1.2 ± 4.1 | 0.24 |
| Positive | 10 | -2.3 ± 4.4 | 11 | -2.2 ± 4.4 | 0.97 |
| BMI | |||||
| < 30 kg/m2 | 21 | -0.8 ± 6.1 | 19 | -2.2 ± 6.5 | 0.56 |
| ≥ 30 kg/m2 | 19 | -0.2 ± 3.7 | 21 | -0.7 ± 3.6 | 0.72 |
| HbA1c > 6.0% | |||||
| ≤ 6.0 % | 23 | 0.3 ± 3.9 | 20 | -0.1 ± 3.9 | 0.66 |
| > 6.0 % | 17 | -1.8 ± 4.4 | 20 | -2.8 ± 4.3 | 0.51 |
| Insulin | |||||
| ≤ 9 IU/dl | 21 | -0.51 ± 4.5 | 19 | -0.4 ± 4.4 | 0.93 |
| > 9 IU/dl | 19 | -0.5 ± 3.9 | 21 | -2.4 ± 3.9 | 0.15 |
Changes in Biomarker outcomes
Follow-up MRI and PET were completed in 33 out of 40 participants (15 in the metformin group, 18 in the placebo group, 82.5% completion overall). Changes from baseline to 12 month in the posterior cingulate-precuneus rCMRgl, adjusted for cerebellar CMRgl uptake, showed a difference favoring metformin that was not statistically significant (2.0 ± 6.3% vs. 0.0 ± 6.0%; p=0.36). Secondary ROIs including hippocampus (2.4 ± 4.7% vs. 1.0 ± 5.1%; p =0.73), para-hippocampus (3.3 ± 5.5% vs. 2.0 ± 5.1%; p =0.76), and entorhinal cortex (5.3± 9.0% vs. 1.3 ± 6.0; p =0.16) favored metformin but were not statistically significant. None of the differences in changes in plasma Aβ-42 were statistically significant. Plasma Aβ-42 increased in the metformin group (0.69 ± 18.5 pg/ml) and decreased in the placebo group (-4.40 ± 23.51 pg/ml; p=0.3).
Discussion
We found in a pilot study that conducting a randomized trial of metformin in persons with AMCI was feasible and safe. We also found preliminary evidence of efficacy for one of the primary outcomes, total recall in the Selective Reminding Test, but not the other, the ADAS-Cog, after adjusting for the baseline difference in the ADAS-Cog. Post hoc analyses suggest that the benefit on the SRT total recall is higher with the highest metformin dose, among younger subjects, among those without APOE-ε4, among those with the lower HbA1c, and those with the highest baseline fasting insulin. Differences between metformin and placebo in changes in the primary and exploratory biomarker outcomes were not significantly different but the direction of the difference modestly favored metformin and supported the finding for the SRT. However, given the pilot nature of our study with a relatively small sample size and the potential for chance findings, our preliminary evidence of efficacy should be taken with caution.
The finding that diabetes and insulin resistance are related to an increased risk of Alzheimer's dementia has prompted the exploration of related strategies for treatment and prevention of AD [11]. This is supported by basic and experimental data showing that diabetes and insulin resistance may affect amyloid clearance in the brain [22]. Pursuing these strategies is feasible because of the existence of known effective pharmacologic and non-pharmacologic strategies for the prevention of diabetes and the improvement of insulin resistance. The best and safest strategy for preventing diabetes and decreasing insulin resistance is lifestyle interventions. Lifestyle interventions (exercise and diet) are the best insulin sensitizers, but require substantial support and resources in clinical trials that are difficult to sustain in the long term and replicate in “real world” settings [65]. Among the pharmacologic strategies, the thiazolidenidiones, also known as PPAR-gamma agonists, are powerful insulin sensitizers effective in lowering insulin resistance [66] and preventing T2D [67], with efficacy similar to lifestyle strategies and greater than metformin [68]. However, they have concerning side effects including edema, congestive heart failure (CHF), and in the case of rosiglitazone, myocardial infarction (MI) [69], which led to a black box warning from the Food and Drug Administration (FDA) [70]. The thiazolidinedione rosiglitazone was tested for secondary prevention of cognitive decline in mild AD among persons without T2D and was found to be non-efficacious in a phase III trial[71] after promising results in a pilot study [72]. However, it seems reasonable to speculate that the negative vascular effects of rosiglitazone [70], including cerebrovascular effects, may have eclipsed the beneficial effects on AD [11].
Metformin is a medication belonging to the biguanide class[40, 73]. It treats and prevents diabetes by suppression of hepatic glucose output, increasing insulin mediated glucose disposal, by increased intestinal glucose use, and by decreasing fatty acid oxidation [74]; this is accompanied by reduced pancreatic insulin secretion and lower insulin levels in blood in response to glucose loads. While the mechanisms for the action of metformin are not completely understood [26], it clearly reduces insulin levels[25], inflammation and thrombosis[75], and the risk of the metabolic syndrome[76] and T2D[77] in persons at risk. Metformin is usually the first step in pharmacological treatment of T2D [78], but it is increasingly used for T2D prevention based on the findings of the landmark Diabetes Prevention Program (DPP)[77], the largest trial of metformin among persons without T2D. In the DPP, metformin was more effective than lifestyle intervention in preventing T2D after 10 years [65]. The only common side effect of metformin in clinical trials has been gastrointestinal intolerance, occurring in a minority of subjects. In the DPP, the rate of serious adverse events for metformin was the same as for placebo. The side effects and safety for metformin in our trial were similar to published research and clinical experience. Metformin is not directly active in the brain [40], and we postulate that it acts on the AD process through reduction of peripheral insulin levels that affect brain clearance of Aβ [79-81]. In addition, metformin also decreases Advanced Glycation End Products (AGE)[82, 83], inflammation [75], coagulation [75], and prevents the metabolic syndrome (diabetes, hypertension, obesity, dyslipidemia) [76], factors that may also influence AD risk through cerebrovascular or neurodegenerative mechanisms [11]. However, metformin has been reported to increase production of Aβ in a cell culture model [84], and two retrospective studies reported that among persons with T2D, a diagnosis of dementia was associated with metformin use[85, 86]. These reports conflict with animal studies showing that metformin decreases AD neuropathology [87, 88] and an epidemiologic study that showed that metformin is related to lower AD risk among persons with T2D [89].
Several of our observations merit further discussion. First, our study showed improvements in both clinical outcomes, contrary to the idea that cognition declines with time. However, we interpret this finding as evidence of practice effects, which are increasingly recognized in AMCI[90], are inversely related with conversion to AD, and can be examined as an outcome in clinical trials [91]. Second, we found a result benefiting metformin for the the SRT but not for the ADAS-Cog. Moreover, the difference for the ADAS-cog seemed to benefit placebo in unadjusted analyses. The difference for the ADAS-cog favoring placebo dissapeared after adjusting for the significant differences in baseline ADAS-cog, while the differences in the SRT persisted. The opposite directions of effect for the ADAS-cog and the SRT seems to have been explained by a baseline difference in the ADAS-cog. While the ADAS-Cog has been the standard for measurement of cognitive change in clinical trials, it is increasingly recognized that it may not be sensitive enough to detect change in AMCI, and more sensitive instruments such as measures of recall are increasingly being considered in clinical trials individually or or to strenghen the ADAS-Cog [92]. In post hoc analyses we found that younger persons, those without the APOE-ε4 allele, those with lower HbA1c, and those with higher insulin, seemed to benefit from metformin in terms of SRT performance. Although these results could be due to chance, we can speculate about their plausiblity. In terms of age group, the benefits of metformin on diabetes prevention have been shown to be stronger in younger persons[65], consistent with our findings. In terms of the APOE-ε4 allele, a previous study of the insulin sensitizer rosiglitazone had similar findings[72] that led to a phase III trial restricted to persons without a APOE-ε4 allele[71]. This makes sense if one considers that persons with both insulin resistance and an APOE-ε4 allele have a higher risk of AD than with either one or none of these risk factors[27]. In terms of HbA1c, those with higher levels may have more advanced hyperglycemia that may be less amenable to prevention-hyperglycemia ensues when pancreatic insulin production can no longer overcome insulin resistance, reflecting a more advanced stage in the natural history leading to T2D. In terms of insulin levels, it seems reasonable that those with the higher insulin have more opportunity for a decrease in insulin with medications and a better opportunity for benefit. Our main imaging outcome did not show statistically significant differences. It has been estimated that sample sizes of several hundred persons with prodromal AD are needed to detect appreciable differences in rCMRgl at longer follow-up periods than our trial [93]. Thus, we were not powered to detect significant differences, but our results were not inconsistent with those showing benefit for the SRT. The decrease in plasma Aβ-42 observed in the placebo group was similar to that in a pilot study of rosiglitazone in mild AD [72] and decreases in plasma Aβ-42 predict future AD[94]. Although the results for plasma Aβ-42 were not significant and this biomarker was exploratory, the findings show a direction of benefit as with the SRT.
It seems safe and feasible to conduct a randomized trial of metformin among persons with MCI although we encountered important challenges. These challenges included a long recruitment period due in part to a large number of subjects needed to be screened, the inability to achieve the maximum metformin dose in the majority of participants, and the inability of some participants to tolerate metformin at all. Potential ways to overcome recruitment challenges include conducting a clinical trial at multiple sites and extending the inclusion criteria to include earlier forms of prodromal AD, such early MCI and subjective memory impairment [95] instead of restricting the sample to the original MCI criteria. A potential way to improve tolerance and achievement of the maximum dose is to use long-acting forms of metformin [96] instead of the short acting form that we used. Despite these challenges, we cautiously believe that there are preliminary findings justify the conduct of larger clinical trial. We acknowledge that our results must be interpreted with caution because of the inconsistency of the results for the SRT and ADAS-cog, because the results of post-hoc analyses are are subject to chance. This study needs to be followed by a larger efficacy trial with suffficient sample size to examine results in subgroups of age, APOE-ε4, and levels of glycemia and insullin. The difference of approximately 4 words in total SRT recall (out of a possible 72) observed between metformin and placebo represents a small but potentially clinically significant difference of approximately 0.5 standard deviations [97]. Our biomarker analyses did not provide insight into the potential impact of metformin on neurodegeneration. Future studies should ideally include AD biomarkers in CSF or imaging markers such as PET with an amyloid ligand. Future studies should also include systematic measures of cerebrovascular disease, which were not part of our protocol. Among the strategies related to insulin resistance that could potentially be used for AD prevention, metformin seems to be the safest, cheapest, and most sustainable. In addition, since metformin is so widely used, it is important to establish its cognitive safety in light of conflicting evidence about its effect on AD.
Supplementary Material
Acknowledgments
This project was funded by the National Institute on Aging (R01 AG026413), the Alzheimer's Disease drug discovery foundation (Grant # 270901), and a Florence and Herbert Irving Scholar Award from the Irving Institute for Clinical Translatiional Research at Columbia University Medical Center. Metformin and matching placebo were provided free of cost by Merck Santé s.a.s. (Lyon, France). Support for laboratory assays and outpatient evaluations was provided by the Irving Institute for Clinical Translational research, funded by a Clinical Transalational Science Award (UL1 RR024156 and UL1 TR000040). We would like to thank all the persons who approached us to enroll in the study, and in particular, the 80 persons who generously participated in the trial.
Footnotes
Trial registration: Clinical trials.gov ID NCT00620191.
Contributor Information
José A. Luchsinger, Departments of Medicine and Epidemiology, Columbia University Medical Center, 630 West 168th street, New York, NY 10032. USA
Thania Perez, Email: tp82@cumc.columbia.edu, Deparment of Medicine, Columbia University Medical Center, 630 West 168th street, New York, NY 10032. USA.
Helena Chang, Email: Helena.Chang@mountsinai.org, Department of Statistics, Mt. Sinai Medical Center, 1425 Madison Avenue, New York, NY, 10029, USA.
Pankaj Mehta, Email: pdmehtaphd@gmail.com, New York Institute for Basic Research, 1050 Forest Hill Road, Staten Island, NY 10314, USA.
Jason Steffener, Email: js2746@columbia.edu, Gertrude H. Sergievsky Center, Columbia University, 630 West 168th Street, New York, NY 10032, USA.
Gnanavalli Pradabhan, Email: ggp14@columbia.edu, Department of Psychiatry, Columbia University Medical Center, and Division of Geriatric Psychiatry, New York State Psychiatric Institute, 1051 Riverside Drive, New York, NY 10032, USA.
Masanori Ichise, Email: mi2193@columbia.edu, Department of Radiology, Columbia University Medical Center, 622 West 168th street, New York, NY 10032, USA.
Jennifer Manly, Email: jjm71@columbia.edu, Gertrude H. Sergievsky Center, Columbia University, 630 West 168th Street, New York, NY 10032, USA.
Devangere P. Devanand, Email: dpd3@cumc.columbia.edu, Department of Psychiatry, Columbia University Medical Center, and Division of Geriatric Psychiatry, New York State Psychiatric Institute, 1051 Riverside Drive, New York, NY 10032, USA.
Emilia Bagiella, Email: emila.bagiella@mountsinai.org, Department of Statistics, Mt. Sinai Medical Center, 1425 Madison Avenue, New York, NY, 10029, USA.
References
- 1.Association As. 2014 Alzheimer's Disease Facts and Figures. Alzheimer's & dementia. 2014;10 doi: 10.1016/j.jalz.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, Chown MJ, Hebert LE, Hennekens CH, Taylor JO. Prevalence of Alzheimer's disease in a community population of older persons. Higher than previously reported. Jama. 1989;262:2551–2556. [PubMed] [Google Scholar]
- 3.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selkoe DJ. The origins of Alzheimer disease: a is for amyloid. Jama. 2000;283:1615–1617. doi: 10.1001/jama.283.12.1615. [DOI] [PubMed] [Google Scholar]
- 5.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. Jama. 1997;277:813–817. [PubMed] [Google Scholar]
- 6.Petersen RC, Roberts RO, Knopman DS, Boeve BF, Geda YE, Ivnik RJ, Smith GE, Jack CR., Jr Mild Cognitive Impairment: Ten Years Later. Arch Neurol. 2009;66:1447–1455. doi: 10.1001/archneurol.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummings JL. Alzheimer's Disease. N Engl J Med. 2004;351:56–67. doi: 10.1056/NEJMra040223. [DOI] [PubMed] [Google Scholar]
- 8.Karran E, Hardy J. Antiamyloid therapy for Alzheimer's disease--are we on the right road? N Engl J Med. 2014;370:377–378. doi: 10.1056/NEJMe1313943. [DOI] [PubMed] [Google Scholar]
- 9.NIH Consensus Development Conference Statement on Preventing Alzheimer's Disease and Cognitive Decline. NIH Consensus and State-of-the-Art Statements. 2010;27 [PubMed] [Google Scholar]
- 10.Luchsinger JA. The relationship between the continuum of elevated adiposity, hyperinsulinemia, and type 2 diabetes and late onset Alzheimer's disease: an epidemiologic perspective. In: Craft S, Christen Y, editors. Diabetes, Insulin, and Alzheimer's Disease. Springer; Berlin Heidelberg: 2010. pp. 89–107. [Google Scholar]
- 11.Luchsinger JA. J Alzheimers Dis [Google Scholar]
- 12.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and Trends in Obesity Among US Adults, 1999-2008. JAMA. 303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 13.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999-2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 14.Luchsinger JA, Reitz C, Patel B, Tang MX, Manly JJ, Mayeux R. Relation of Diabetes to Mild Cognitive Impairment. Arch Neurol. 2007;64:570–575. doi: 10.1001/archneur.64.4.570. [DOI] [PubMed] [Google Scholar]
- 15.Reitz C, Tang MX, Manly J, Mayeux R, Luchsinger JA. Hypertension and the risk of mild cognitive impairment. Arch Neurol. 2007;64:1734–1740. doi: 10.1001/archneur.64.12.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(2007) Centers For Disease Control and Prevention.
- 17.Luchsinger JA, Small S, Biessels GJ. Should we target insulin resistance to prevent dementia due to Alzheimer disease? Arch Neurol. 68:17–18. doi: 10.1001/archneurol.2010.339. [DOI] [PubMed] [Google Scholar]
- 18.Boden-Albala B, Cammack S, Chong J, Wang C, Wright C, Rundek T, Elkind MS, Paik MC, Sacco RL. Diabetes, fasting glucose levels, and risk of ischemic stroke and vascular events: findings from the Northern Manhattan Study (NOMAS) Diabetes Care. 2008;31:1132–1137. doi: 10.2337/dc07-0797. [DOI] [PubMed] [Google Scholar]
- 19.Schneider JA, Wilson RS, Cochran EJ, Bienias JL, Arnold SE, Evans DA, Bennett DA. Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology. 2003;60:1082–1088. doi: 10.1212/01.wnl.0000055863.87435.b2. [DOI] [PubMed] [Google Scholar]
- 20.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62:1148–1155. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 21.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 22.Craft S. Insulin resistance and Alzheimer's disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–152. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- 23.Luchsinger JA. Type 2 diabetes, related conditions, in relation and dementia: an opportunity for prevention? J Alzheimers Dis. 20:723–736. doi: 10.3233/JAD-2010-091687. [DOI] [PubMed] [Google Scholar]
- 24.Luchsinger JA. Type 2 diabetes, related conditions, in relation and dementia: an opportunity for prevention? J Alzheimers Dis. 2010;20:723–736. doi: 10.3233/JAD-2010-091687. [DOI] [PubMed] [Google Scholar]
- 25.The Diabetes Prevention Program Research G. Role of Insulin Secretion and Sensitivity in the Evolution of Type 2 Diabetes in the Diabetes Prevention Program: Effects of Lifestyle Intervention and Metformin. Diabetes. 2005;54:2404–2414. doi: 10.2337/diabetes.54.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrannini E. The Target of Metformin in Type 2 Diabetes. New England Journal of Medicine. 2014;371:1547–1548. doi: 10.1056/NEJMcibr1409796. [DOI] [PubMed] [Google Scholar]
- 27.Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63:1187–1192. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 28.Peila R, Rodriguez BL, White LR, Launer LJ. Fasting insulin and incident dementia in an elderly population of Japanese-American men. Neurology. 2004;63:228–233. doi: 10.1212/01.wnl.0000129989.28404.9b. [DOI] [PubMed] [Google Scholar]
- 29.Peila R, Rodriguez BL, Launer LJ. Type 2 Diabetes, APOE Gene, and the Risk for Dementia and Related Pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 30.Hsueh W. Genetic discoveries as the basis of personalized therapy: rosiglitazone treatment of Alzheimer's disease. Pharmacogenomics J. 2006;6:222–224. doi: 10.1038/sj.tpj.6500383. [DOI] [PubMed] [Google Scholar]
- 31.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 32.Expert Panel on the Identification EaToOaOiA. Executive Summary of the Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults. Arch Intern Med. 1998;158:1855–1867. doi: 10.1001/archinte.158.17.1855. [DOI] [PubMed] [Google Scholar]
- 33.Grundman M, Petersen RC, Ferris SH, Thomas RG, Aisen PS, Bennett DA, Foster NL, Jack CR, Jr, Galasko DR, Doody R, Kaye J, Sano M, Mohs R, Gauthier S, Kim HT, Jin S, Schultz AN, Schafer K, Mulnard R, van Dyck CH, Mintzer J, Zamrini EY, Cahn-Weiner D, Thal LJ. Mild Cognitive Impairment Can Be Distinguished From Alzheimer Disease and Normal Aging for Clinical Trials. Arch Neurol. 2004;61:59–66. doi: 10.1001/archneur.61.1.59. [DOI] [PubMed] [Google Scholar]
- 34.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ the Alzheimer's Disease Cooperative Study Group. Vitamin E and Donepezil for the Treatment of Mild Cognitive Impairment. N Engl J Med. 2005 doi: 10.1056/NEJMoa050151. NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 35.DeFronzo RA. Pharmacologic therapy for type 2 diabetes mellitus. Annals of Internal Medicine. 2000;133:73–74. doi: 10.7326/0003-4819-133-1-200007040-00016. [DOI] [PubMed] [Google Scholar]
- 36.Standards of medical care in diabetes--2010. Diabetes Care. 33 Suppl 1:S11–61. doi: 10.2337/dc10-S011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strawbridge WJ, Wallhagen MI, Shema SJ. New NHLBI clinical guidelines for obesity and overweight: will they promote health? 2000:340–343. doi: 10.2105/ajph.90.3.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez T, Thareja S, Arana E, Chang H, Bagiela E, Luchsinger JA. II Clinical Trial of Metformin in Amnestic MCI: Rationale, Methods, and Barriers and Enablers of Successful Recruitment. J Nutr Health Aging. 2011;15:S14. [Google Scholar]
- 39.Lipton RB, Katz MJ, Kuslansky G, Sliwinski MJ, Stewart WF, Verghese J, Crystal HA, Buschke H. Screening for Dementia by Telephone Using the Memory Impairment Screen. J Am Geriatr Soc. 2003;51:1382–1390. doi: 10.1046/j.1532-5415.2003.51455.x. [DOI] [PubMed] [Google Scholar]
- 40.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 41.Buschke H, Fuld PA. Evaluating storage, retention, and retrieval in disordered memory and learning. Neurology. 1974;24:1019–1025. doi: 10.1212/wnl.24.11.1019. [DOI] [PubMed] [Google Scholar]
- 42.Raghavan N, Samtani MN, Farnum M, Yang E, Novak G, Grundman M, Narayan V, Dibernardo A. The ADAS-Cog revisited: Novel composite scales based on ADAS-Cog to improve efficiency in MCI and early AD trials. Alzheimers Dement. 2012 doi: 10.1016/j.jalz.2012.05.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manly JJ, Bell-McGinty S, Tang MX, Schupf N, Stern Y, Mayeux R. Implementing diagnostic criteria and estimating frequency of mild cognitive impairment in an urban community. Arch Neurol. 2005;62:1739–1746. doi: 10.1001/archneur.62.11.1739. [DOI] [PubMed] [Google Scholar]
- 44.Stern Y, Andrews H, Pittman J, et al. Diagnosis of dementia in a heterogeneous population. Development of a neuropsychological paradigm-based diagnosis of dementia and quantified correction for the effects of education. Arch Neurol. 1992;49:453–460. doi: 10.1001/archneur.1992.00530290035009. [DOI] [PubMed] [Google Scholar]
- 45.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 46.Schneider LS, Olin JT, Doody RS, Clark CM, Morris JC, Reisberg B, Schmitt FA, Grundman M, Thomas RG, Ferris SH. Validity and reliability of the Alzheimer's Disease Cooperative Study-Clinical Global Impression of Change. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11 Suppl 2:S22–32. doi: 10.1097/00002093-199700112-00004. [DOI] [PubMed] [Google Scholar]
- 47.Weschler D. Wechsler Memory Scale-revised manual. Psychological corporation; New York: 1987. [Google Scholar]
- 48.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 49.Kaufer DI, Cummings JL, Ketchel P, Smith V, MacMillan A, Shelley T, Lopez OL, DeKosky ST. Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci. 2000;12:233–239. doi: 10.1176/jnp.12.2.233. [DOI] [PubMed] [Google Scholar]
- 50.Weschler D. Manual for the adult Weschler intelligence scale-Revised. Psychological corporation, Harcourt Brace Jovanovich, Inc; New York: 1981. [Google Scholar]
- 51.Minoshima S, Frey KA, Koeppe RA, Foster NL, Kuhl DE. A diagnostic approach in Alzheimer's disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J Nucl Med. 1995;36:1238–1248. [PubMed] [Google Scholar]
- 52.Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer's Disease Treatment Studies. Am J Psychiatry. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- 53.Signorini M, Paulesu E, Friston K, Perani D, Colleluori A, Lucignani G, Grassi F, Bettinardi V, Frackowiak RSJ, Fazio F. Rapid Assessment of Regional Cerebral Metabolic Abnormalities in Single Subjects with Quantitative and Nonquantitative [18F]FDG PET: A Clinical Validation of Statistical Parametric Mapping. NeuroImage. 1999;9:63–80. doi: 10.1006/nimg.1998.0381. [DOI] [PubMed] [Google Scholar]
- 54.The Diabetes Prevention Program Research G. Effects of Withdrawal From Metformin on the Development of Diabetes in the Diabetes Prevention Program. Diabetes Care. 2003;26:977–980. doi: 10.2337/diacare.26.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Y, Rinne JO, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, Kemppainen N, Nagren K, Kim BC, Tsui W, de Leon MJ. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2008;35:2169–2181. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luchsinger JA, Tang MX, Miller J, Green R, Mehta PD, Mayeux R. Relation of plasma homocysteine to plasma amyloid beta levels. Neurochem Res. 2007;32:775–781. doi: 10.1007/s11064-006-9207-7. [DOI] [PubMed] [Google Scholar]
- 58.Chudyk A, Masiuk M, Myslak M, Domanski L, Sienko J, Sulikowski T, Machalinski B, Giedrys-Kalemba S. Soluble HLA class I molecules exert differentiated influence on renal graft condition. Transplant Proc. 2006;38:90–93. doi: 10.1016/j.transproceed.2005.11.090. [DOI] [PubMed] [Google Scholar]
- 59.Hawkins JR, Khripin Y, Valdes AM, Weaver TA. Miniaturized sealed-tube allele-specific PCR. Hum Mutat. 2002;19:543–553. doi: 10.1002/humu.10060. [DOI] [PubMed] [Google Scholar]
- 60.Myakishev MV, Khripin Y, Hu S, Hamer DH. High-throughput SNP genotyping by allele-specific PCR with universal energy-transfer-labeled primers. Genome Res. 2001;11:163–169. doi: 10.1101/gr.157901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tuan CY, Abbasi F, Lamendola C, McLaughlin T, Reaven G. Usefulness of plasma glucose and insulin concentrations in identifying patients with insulin resistance. The American Journal of Cardiology. 2003;92:606–610. doi: 10.1016/s0002-9149(03)00735-5. [DOI] [PubMed] [Google Scholar]
- 62.Ridker PM. High-sensitivity C-reactive protein, inflammation, and cardiovascular risk: from concept to clinical practice to clinical benefit. American Heart Journal. 2004;148:S19–S26. doi: 10.1016/j.ahj.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 63.Ridker PM. Inflammatory biomarkers and risks of myocardial infarction, stroke, diabetes, and total mortality: implications for longevity. Nutr Rev. 2007;65:S253–259. doi: 10.1111/j.1753-4887.2007.tb00372.x. [DOI] [PubMed] [Google Scholar]
- 64.Noble JM, Manly JJ, Schupf N, Tang MX, Mayeux R, Luchsinger JA. Association of C-reactive protein with cognitive impairment. Arch Neurol. 67:87–92. doi: 10.1001/archneurol.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knowler WC, Fowler SE, Hamman RF, Christophi CA, Hoffman HJ, Brenneman AT, Brown-Friday JO, Goldberg R, Venditti E, Nathan DM. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet. 2009;374:1677–1686. doi: 10.1016/S0140-6736(09)61457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 67.investigators TDDrawrarm. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. The Lancet. 2006;368:1096–1105. doi: 10.1016/S0140-6736(06)69420-8. [DOI] [PubMed] [Google Scholar]
- 68.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- 69.Nathan DM, Berkwits M. Trials that matter: rosiglitazone, ramipril, and the prevention of type 2 diabetes. Ann Intern Med. 2007;146:461–463. doi: 10.7326/0003-4819-146-6-200703200-00015. [DOI] [PubMed] [Google Scholar]
- 70.Nathan DM. Rosiglitazone and Cardiotoxicity -- Weighing the Evidence. N Engl J Med. 2007 doi: 10.1056/NEJMe078117. NEJMe078117. [DOI] [PubMed] [Google Scholar]
- 71.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006;6:246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 72.Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S. Preserved Cognition in Patients With Early Alzheimer Disease and Amnestic Mild Cognitive Impairment During Treatment With Rosiglitazone: A Preliminary Study. Am J Geriatr Psychiatry. 2005;13:950–958. doi: 10.1176/appi.ajgp.13.11.950. [DOI] [PubMed] [Google Scholar]
- 73.Dunn CJ, Peters DH. Metformin. A review of its pharmacological properties and therapeutic use in non-insulin-dependent diabetes mellitus. 1995 May;:721–749. doi: 10.2165/00003495-199549050-00007. [DOI] [PubMed] [Google Scholar]
- 74.Tian J, Shi J, Bailey K, Lendon CL, Pickering-Brown SM, Mann DMA. Association between apolipoprotein E e4 allele and arteriosclerosis, cerebral amyloid angiopathy, and cerebral white matter damage in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:696–699. doi: 10.1136/jnnp.2003.012096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.The Diabetes Prevention Program Research Group. Intensive Lifestyle Intervention or Metformin on Inflammation and Coagulation in Participants With Impaired Glucose Tolerance. Diabetes. 2005;54:1566–1572. doi: 10.2337/diabetes.54.5.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Orchard TJ, Temprosa M, Goldberg R, Haffner S, Ratner R, Marcovina S, Fowler S for the Diabetes Prevention Program Research Group. The Effect of Metformin and Intensive Lifestyle Intervention on the Metabolic Syndrome: The Diabetes Prevention Program Randomized Trial. Ann Intern Med. 2005;142:611–619. doi: 10.7326/0003-4819-142-8-200504190-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Diabetes Prevention Program Research Group. Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nathan DM. Initial Management of Glycemia in Type 2 Diabetes Mellitus. N Engl J Med. 2002;347:1342–1349. doi: 10.1056/NEJMcp021106. [DOI] [PubMed] [Google Scholar]
- 79.Park CR. Cognitive effects of insulin in the central nervous system. Neurosci Biobehav Rev. 2001;25:311–323. doi: 10.1016/s0149-7634(01)00016-1. [DOI] [PubMed] [Google Scholar]
- 80.Watson GS, Craft S. Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer's disease. European Journal of Pharmacology. 2004;490:97–113. doi: 10.1016/j.ejphar.2004.02.048. [DOI] [PubMed] [Google Scholar]
- 81.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Beneficial effects of metformin and irbesartan on advanced glycation end products (AGEs)-RAGE-induced proximal tubular cell injury. Pharmacol Res. 2012;65:297–302. doi: 10.1016/j.phrs.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 83.Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Metformin Inhibits Advanced Glycation End Products (AGEs)-induced Renal Tubular Cell Injury by Suppressing Reactive Oxygen Species Generation via Reducing Receptor for AGEs (RAGE) Expression. Horm Metab Res. 2012;44:891–895. doi: 10.1055/s-0032-1321878. [DOI] [PubMed] [Google Scholar]
- 84.Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao FF. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci U S A. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Imfeld P, Bodmer M, Jick SS, Meier CR. Metformin, other antidiabetic drugs, and risk of Alzheimer's disease: a population-based case-control study. J Am Geriatr Soc. 2012;60:916–921. doi: 10.1111/j.1532-5415.2012.03916.x. [DOI] [PubMed] [Google Scholar]
- 86.Moore EM, Mander AG, Ames D, Kotowicz MA, Carne RP, Brodaty H, Woodward M, Boundy K, Ellis KA, Bush AI, Faux NG, Martins R, Szoeke C, Rowe C, Watters DA. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36:2981–2987. doi: 10.2337/dc13-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li J, Deng J, Sheng W, Zuo Z. Metformin attenuates Alzheimer's disease-like neuropathology in obese, leptin-resistant mice. Pharmacol Biochem Behav. 2012;101:564–574. doi: 10.1016/j.pbb.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gupta A, Bisht B, Dey CS. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer's-like changes. Neuropharmacology. 2011;60:910–920. doi: 10.1016/j.neuropharm.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 89.Hsu CC, Wahlqvist ML, Lee MS, Tsai HN. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimers Dis. 24:485–493. doi: 10.3233/JAD-2011-101524. [DOI] [PubMed] [Google Scholar]
- 90.Mura T, Proust-Lima C, Jacqmin-Gadda H, Akbaraly TN, Dubois B, Berr C. Measuring Cognitive Change from Mild Cogntive Impairment to Prodromal Alzheimer's Disease. J Nutr Health Aging. 2012;4 doi: 10.1136/jnnp-2013-305078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Duff K, Lyketsos CG, Beglinger LJ, Chelune G, Moser DJ, Arndt S, Schultz SK, Paulsen JS, Petersen RC, McCaffrey RJ. Practice effects predict cognitive outcome in amnestic mild cognitive impairment. Am J Geriatr Psychiatry. 2011;19:932–939. doi: 10.1097/JGP.0b013e318209dd3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Skinner J, Carvalho JO, Potter GG, Thames A, Zelinski E, Crane PK, Gibbons LE. The Alzheimer's Disease Assessment Scale-Cognitive-Plus (ADAS-Cog-Plus): an expansion of the ADAS-Cog to improve responsiveness in MCI. Brain Imaging Behav. 2012;6:489–501. doi: 10.1007/s11682-012-9166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grill JD, Di L, Lu PH, Lee C, Ringman J, Apostolova LG, Chow N, Kohannim O, Cummings JL, Thompson PM, Elashoff D. Estimating sample sizes for pre-dementia Alzheimer's trials based on the Alzheimer's Disease Neuroimaging Initiative. Neurobiology of Aging. 2013;34:62–72. doi: 10.1016/j.neurobiolaging.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, Mehta PD. Plasma A{beta}40 and A{beta}42 and Alzheimer's disease: Relation to age, mortality, and risk. Neurology. 2003;61:1185–1190. doi: 10.1212/01.wnl.0000091890.32140.8f. [DOI] [PubMed] [Google Scholar]
- 95.Jessen F, Wolfsgruber S, Wiese B, Bickel H, Mosch E, Kaduszkiewicz H, Pentzek M, Riedel-Heller SG, Luck T, Fuchs A, Weyerer S, Werle J, van den Bussche H, Scherer M, Maier W, Wagner M. AD dementia risk in late MCI, in early MCI, and in subjective memory impairment. Alzheimers Dement. 2014;10:76–83. doi: 10.1016/j.jalz.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 96.Ali S, Fonseca V. Overview of metformin: special focus on metformin extended release. Expert Opin Pharmacother. 2012;13:1797–1805. doi: 10.1517/14656566.2012.705829. [DOI] [PubMed] [Google Scholar]
- 97.Cohen J. Statistical Power Analysis for the Behavioral Sciences. Lawrence Erlbaum Associates; Hillsdale, NJ: 1988. [Google Scholar]
- 98.Berg L. Clinical Dementia Rating (CDR) Psychopharmacol Bull. 1988;24:637–639. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.