

Abstract
Glutathione S-transferase P (GSTP) is one member of the GST superfamily that is prevalently expressed in mammals. Known to possess catalytic activity through deprotonating glutathione allowing formation of thioether bonds with electrophilic substrates, more recent discoveries have broadened our understanding of the biological roles of this protein. In addition to catalytic detoxification, other properties so far ascribed to GSTP include chaperone functions, regulation of nitric oxide pathways, regulation of a variety of kinase signaling pathways, and participation in the forward reaction of protein S-glutathionylation. The expression of GSTP has been linked with cancer and other human pathologies and more recently even with drug addiction. With respect to human health, polymorphic variants of GSTP may determine individual susceptibility to oxidative stress and/or be critical in the design and development of drugs that have used redox pathways as a discovery platform.
1. INTRODUCTION
The glutathione S-transferase (GST) family of phase II detoxification enzymes has been critically studied for five decades. The cytosolic GST superfamily is composed of at least 7 classes that share >70% homology and have overlapping substrate specificities (Hayes & Pulford, 1995; McIlwain, Townsend, & Tew, 2006). In humans, a single gene on chromosome 11q13 codes for proteins designated in the human p class (GSTP1). The GSTP1 gene spans 3 kb and encodes 210 amino acids in seven exons (Cowell, Dixon, Pemble, Ketterer, & Taylor, 1988). Polymorphisms at the GSTP1 locus result in four alleles GSTP1*A–D that differ structurally and functionally (Lo & Ali-Osman, 1998). Initially named as a function of its high levels in human placenta, one characteristic of GSTP is that the homodimeric enzyme can undergo heterodimerization with other GST isoenzymes (Pettigrew & Colman, 2001) or other proteins (Ralat, Manevich, Fisher, & Colman, 2006). Distinctively, all mammalian GSTs bind GSH (at the so-called G-site) and activate it to a thiolate anion (GS−) that is used to catalyze subsequent conjugation with various electrophiles (Fig. 4.1) (Graminski, Kubo, & Armstrong, 1989; Graminski, Zhang, Sesay, Ammon, & Armstrong, 1989). High levels of GSTP are found in a number of cancers and in cells resistant to anticancer drugs; factors determinate in targeting GSTP in anticancer drug discovery (Tew, 1994). Moreover, in addition to cancer, there are examples where GSTP has been linked to pathologies such as asthma, neurodegenerative diseases, and inflammatory conditions. The identification of human polymorphisms of GSTP now provides a basis for considering how individual and population differences may be associated with response to oxidative stress (Ali-Osman, Akande, Antoun, Mao, & Buolamwini, 1997; Pal, Hu, Zimniak, & Singh, 2000).
Figure 4.1.
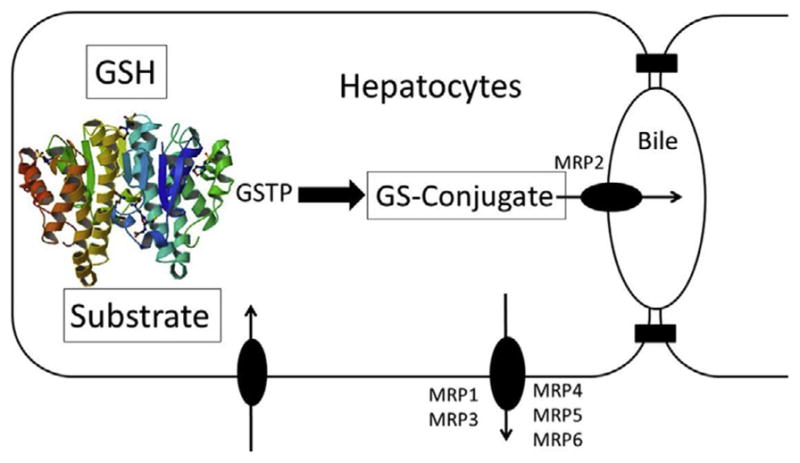
Representative example of phase II detoxification of an electrophilic compound via GSTP, with ATP-binding cassette transporter participation in efflux.
A functional overlap and redundancy invested by the GST isoenzyme family likely explain why the GSTP knockout mouse is viable and healthy, primarily presenting a phenotype with altered sensitivity to certain carcinogens and drugs (Henderson et al., 1998, 2000). Moreover, crosses between this mouse and p53 knockouts only mildly altered susceptibility to the development of spontaneous tumors (Gate, Majumdar, Lunk, & Tew, 2005). Many of the functional attributes of GSTP described in subsequent sections can also be coordinated by other isoenzymes (e.g., GSTA and GSTM), albeit with differing efficiencies. While much of the literature from the 1970s to the 2000s focused upon the detoxification properties of GSTs, more recent evidence has broadened the perspectives on how GSTP contributes to cellular redox homeostasis. The following sections provide information on these advances.
2. SUBCELLULAR DISTRIBUTION OF GSTP
There are many challenges in ascribing enzyme localization to specific organelles. Frequently, the methods used require separation conditions that lead to artifacts or contamination. Immunolocalization techniques also have intrinsic drawbacks. Nevertheless, within the mammalian GST superfamily, three structurally and evolutionarily distinct gene families have tissue- and species-specific distribution and expression patterns, and in some instances, their classifications provide evidence of their subcellular locale and cytosolic (cGST-alpha, cGST-mu, cGST-pi, cGST-omega, cGST-theta, cGST-delta, cGST-sigma, and cGST-zeta), mitochondrial (mGST-alpha, mGST-mu, mGST-pi, and mGST-kappa), and six membrane-bound microsomal (or membrane-associated proteins in eicosanoid and glutathione) transferases (Allocati, Federici, Masulli, & Di Ilio, 2009; Atkinson & Babbitt, 2009; Hayes, Flanagan, & Jowsey, 2005). Recent reports make these designations less rigid, having further characterized GSTs in the plasma membrane, outer mitochondrial membrane, and the nucleus and peroxisomes (Allocati et al., 2009; Gardner & Gallagher, 2001; Goto et al., 2001a; Hayes et al., 2005; Petit et al., 2009; Raza, 2011).
Despite classification as a cytosolic enzyme, GSTP expression has been found in a number of other cellular organelles. GSTP has been detected in human liver mitochondria and the cytoplasm, mitochondria, lysosomes, and nucleus of cancer cells (Ali-Osman, Brunner, Kutluk, & Hess, 1997; Goto et al., 2001b). Mitochondrial GSTP shared the same molecular size as cytoplasmic GSTP, supporting the notion that GSTP mitochondrial targeting does not involve protease processing. Mutation of arginine residues in the N-terminal mitochondrial targeting signal region of full-length GSTP abrogated mitochondrial distribution, suggesting a putative role for positively charged arginine residues in GSTP mitochondrial transport. Mitochondrial GSTP may be involved in protecting the organelle from oxidative stress via the suppression of cardiolipin peroxidation and cytochrome c release from the mitochondrial inner membranes or possibly catalyzing the formation of reactive aldehydes conjugated with GSH.
The presence of nuclear GSTP has been strongly and inversely correlated with patient prognosis in a number of cancers, including ovarian, breast, colon, glioma, lymphomas, and non-small cell lung carcinoma (Ali-Osman, Brunner, et al., 1997; Allen et al., 2007; Huang, Tan, Thiyagarajan, & Bay, 2003; Jankova et al., 2012; Rolland, Raharijaona, Barbarat, Houlgatte, & Thieblemont, 2010; Soh et al., 2005). Nuclear GSTP in Bcl-2-overexpressing breast tumors may gain entry through the nuclear pore and be linked to chemotherapy resistance. Nuclear GSTP immunologic activity is enhanced in cancer cells treated with the chemo-therapeutic agents doxorubicin (DOX) and cis-diamminedichloroplatinum (II) (cisplatin) (Goto et al., 2001a; Rolland et al., 2010). Cancer cells lacking nuclear GSTP expression maintained sensitivity to DOX-mediated apoptosis. The authors suggested that subcellular localization of GSTP in these organelles may be important in metabolizing either DOX or cisplatin, catalyzing the formation of DOX–GSH adducts, thereby enhancing drug efflux, protecting against DNA damage, and contributing to the acquisition and maintenance of chemotherapeutic resistance. However, neither DOX nor cisplatin is a direct substrate of GSTP, and as a consequence, if these correlations are predictive, an alternative function for GSTP would be implicated. Maintenance of nuclear GSTP levels was independent of cytoplasmic levels, further implicating some role of specific nuclear protein transport system. Furthermore, treatment with a nuclear transport inhibitor led to increases in drug accumulation in the nucleus and the inhibition of GSTP nuclear translocation and DOX-stimulated GSTP nuclear activity (Goto et al., 2001a; Rolland et al., 2010).
A recent study has shown that GSTP can be detected in the endoplasmic reticulum (ER) of murine alveolar type II epithelial cells (Anathy et al., 2012). GSTP immunoprecipitation revealed the association of GSTP with the tumor necrosis factor (TNF) receptor family member Fas (CD9/Apo-1) in the ER. Stimulation with its ligand, FasL, enhanced GSTP/Fas interaction in a time-dependent manner, followed by complex translocation to the cytosol and the mediation of apoptotic signaling pathways. This suggests a specific role for GSTP in catalyzing the S-glutathionylation of a latent pool of Fas (Fas-SSG) in the ER. The oxidative processing of ER Fas and the interaction and subsequent translocation of GSTP/Fas-SSG were directly linked to apoptotic pathways involving the formation of death-inducing signaling complexes and increases in caspase activity. Inhibition of GSTP with the specific inhibitor TLK199 diminished the thiolase activity of GSTP and decreased Fas-SSG and caspase activation in epithelial cells. Pharmacological and genetic modulation of GSTP showed that GSTP-mediated S-glutathionylation of Fas plays a role in a bleomycin model of acute lung injury (Anathy et al., 2012). Such studies unveil a mechanism through which GSTP participates in regulation of ER proteins.
In general terms, S-glutathionylated proteins are found in a number of distinct subcellular locations indicating that specific organelle localization would facilitate this process. In this context, additional role for GSTP sub-cellular translocation in mediating protein S-glutathionylation and cell signaling pathways would seem to be likely.
3. GST REGULATION OF KINASE SIGNALING PATHWAYS
GSTP is present at high levels in many solid tumors (particularly ovarian, non-small cell lung, breast, liver, pancreas, and colon cancers and lymphomas) and in a wide range of drug-resistant cell lines and tumors (Tew, 1994). Although the catalytic detoxification function of GSTP is expected to alter drug sensitivity, the physiological significance of increased GSTP in tumor and drug-resistant cells remains unclear because in most instances, the selected drugs were not substrates of GSTP. From this, it can be inferred that the ligand-binding or chaperone properties of GSTs and the capacity of GSTP to regulate the forward reaction of S-glutathionylation (Townsend et al., 2009a) may be of more consequence than its catalytic properties alone. Supporting this conclusion, recent studies have demonstrated the regulatory role of GSTs in the mitogen-activated protein kinase (MAPK) pathways via their noncatalytic, ligand-binding activity. GSTP was the first isoenzyme to emerge as a regulator of redox-mediated kinase signaling (Adler, Yin, Fuchs, et al., 1999; Adler, Yin, Tew, & Ronai, 1999; Gate, Majumdar, Lunk, & Tew, 2004; Wang, Arifoglu, Ronai, & Tew, 2001). Specifically, GSTP binds directly to c-Jun N-terminal kinases (JNKs), which are at the end of the MAPK pathway, and acts as a negative regulator, thereby modulating proliferation and apoptosis (Fig. 4.2). JNKs are encoded by at least three genes (JNK1, JNK2, and JNK3), which are spliced alternatively into 10 JNK isoforms, including both a shorter C-terminus (JNK1α1, JNK1β1, JNK2α1, JNK2β1, and JNK3α1) and longer C-terminus (JNK1α2, JNK1β2, JNK2α2, JNK2β2, and JNK3α2) isoforms. Both JNK1 and JNK2 are expressed ubiquitously, while JNK3 is localized in the brain, testis, and heart (Davis, 2000; Gupta et al., 1996). The basal activity of JNK is necessarily maintained at low levels through its sequestration within a multiprotein complex that includes at least GSTP and JNK. The rise in ROS levels that occurs under conditions of oxidative or chemical stress results in a dissociation of JNK from GSTP and the subsequent activation of JNK as indicated by phosphorylation of c-Jun and/or ATF2. This process further impacts downstream events, which in a cell- or tissue-specific manner can lead to events as divergent as proliferation or apoptosis (Adler, Yin, Fuchs, et al., 1999; Gate et al., 2004; Ruscoe et al., 2001). As such, GSTP serves as a sensor of intracellular changes in either endogenous or exogenous redox potential. It became apparent that this type of function is not restricted to GSTP and that other small redox-active proteins provide a level of redundancy in interacting with specific intracellular signaling molecules and regulate their activity in a redox-dependent fashion. Consistent with this notion, thioredoxin has also been found to interact with, and inhibit, apoptosis signal-regulated kinase 1 (ASK1) activity (Saitoh et al., 1998).
Figure 4.2.

Some examples of how GSTP has been shown to participate in regulating various kinase signaling pathways. GSTP is a negative regulator in TNFα-induced MAPK signaling. It interacts physically with TRAF2, which blocks the interaction of TRAF2 and ASK1, attenuates ASK1 autophosphorylation, and in turn suppresses TNFα–ASK1–JNK/p38 signaling pathways. GSTP is also able to directly sequester JNK in a complex, thus preventing it from acting on downstream targets, c-Jun and ATF2. In contrast, GSTP can amplify Fas-induced MAPK signaling. Stimulation of Fas ligand increased the interaction of GSTP with Fas and ERp57 in the ER leading to Fas S-glutathionylation and subsequent mobilization from ER to cytosol, resulting in enhanced Fas–ASK1–JNK/p38 signaling pathways.
The ligand-binding properties of GSTP seem to be independent of its detoxification activity since mutation at critical sites in GSTP (tyrosine 7 required for catalytic proton transfer and cysteine 47 or 101 required for GSTP dimerization), while abrogating its catalytic activity, has been shown to be as effective as wild-type GSTP in inhibiting JNK activity in mouse embryo fibroblast (MEF) cells from GSTP−/− mice (Adler, Yin, Fuchs, et al., 1999). However, Telintra, a GSH peptidomimetic drug, or TLK199 (γ-glutamyl-S-(benzyl)cysteinyl-R-phenyl glycine diethyl ester), a select GSTP inhibitor, does inhibit the kinase regulatory activity of GSTP in mouse fibroblast 3T3/4A cells (Adler, Yin, Fuchs, et al., 1999) and human myeloid leukemia HL60 cells (Ruscoe et al., 2001). Moreover, NBDHEX (6-(7-nitro-2, 1, 3-benzoxadiazol-4-ylthio) hexanol), another GST inhibitor that forms a σ-complex with GSH in the active site of GSTP, also abolished the interaction between GSTP and JNK1α2 (De Luca, Federici, De Canio, Stella, & Caccuri, 2012). In this context, NBDHEX induces the dissociation of GSTP1-1/JNK1 heterocomplex and the subsequent phosphorylation of JNK1, leading to cell cycle arrest and apoptosis in several tumor cell lines (Turella et al., 2005). Inhibitor binding likely induces changes in the conformation of one or more domains of GSTP and therefore interferes with the GSTP interaction with JNK. Indeed, evidence suggests that the C-terminal domain (residues 194–201) of GSTP, one of the most flexible regions after inhibitor binding, is critical for GSTP and JNK interaction (Monaco et al., 1999). Moreover, the extended C-terminal region (residues 200–424) of JNK was also shown to be critical in this interaction (9), suggesting that the longer JNK isoforms might preferentially interact with GSTs and that the capacity of GSTP to suppress JNK activity could be through an allosteric inhibition mechanism, since the catalytic kinase domain is localized in the N-terminal of JNK. However, similar dissociation constants were reported in a recent study using JNK1Δ or JNK1α2, suggesting that the long C-terminal tail that distinguishes JNK1α2 from the shorter splicing variant JNK1α1 is not implicated in GSTP recognition (De Luca et al., 2012). Evidence has recently been presented to indicate that GSTP binds strongly with the activated phosphorylated form of JNK1 than with JNK2, while the interaction with the unphosphorylated, inactive JNK occurs only in the presence of the substrate ATF2. A direct interaction between GSTP and ATF2 was also demonstrated and explained the ability of GSTP to inhibit JNK catalytic activity as a consequence of the competition between GSTP and the activated JNK for the substrate ATF2 (Thevenin, Zony, Bahnson, & Colman, 2011). Contrary to this, De Luca et al. (2012) found that GSTP is able to form a complex with the unphosphorylated and inactive JNK1α2 isoform, even in the absence of the substrate. The complex strongly reduced the extent of activation of JNK1α2 and preserved GSTP from inactivation. They also showed that GSH exerted a negative effect on the affinity of GSTP for JNK1α2, suggesting that the intracellular levels of this thiol may participate in fine-tuning of the MAPK signaling pathways. Moreover, there are indications that GSTP plays an anti-inflammatory role by preventing lipopolysaccharide-induced production of proinflammatory factors, suggested to occur through the ERK, JNK and p38, and NfkB axes of regulation (Xue et al., 2005). GSTP is also important in the regulation of the transcriptional activity of Stat3 (signal transduction and activator of transcription) and a regulator of the cell cycle via its influence on EGF signaling (Kou, Chen, Feng, Luo, & Yin, 2013).
The stoichiometry of the GSTP–JNK interaction has been difficult to assess by standard procedures. In spite of the catalytic function of GSTP being dependent upon dimerization, in some instances, it was assumed that the monomeric form of GSTP would interact with JNK (Adler, Yin, Fuchs, et al., 1999; Bernardini et al., 2000). A recent study (Gildenhuys et al., 2010) supports the principle that the JNK–GSTP interaction occurs via the dimer of the latter. Using equilibrium folding and unfolding kinetics experiments, as well as molecular modeling, the thermodynamic instability of the GSTP monomer was shown, with the implication that the dimeric GSTP binds to JNK. At this stage, further studies will be required to determine the precise details underlying those mechanisms involved in these reactions. It is worth reflecting that understanding these processes will also be complicated by the fact that different polymorphic variants of GSTP, with differences in residues 105 and 114, triggered different effects. GSTP*C (Val105/Val114) was shown to be the only GSTP haplotype that was able to inhibit JNK apoptotic activity in vivo (Holley et al., 2007). More recently, it was demonstrated that GSTP*C is a more potent inhibitor of JNK activity than the wild-type GSTP*A (Ile105/Ala114) (Thevenin et al., 2011). Such factors when considered in the context of human populations will affect susceptibility traits to stress response through these JNK-mediated pathways.
GSTP also has a negative regulatory role in regulating tumor necrosis factor-alpha (TNFα)-induced MAPK signaling. This occurs through the formation of ligand-binding or chaperone interactions with tumor necrosis factor receptor-associated factor 2 (TRAF2), which blocks the interaction of TRAF2 and ASK1, attenuates ASK1 autophosphorylation, and in turn suppresses ASK1–MEK–JNK/p38 signaling pathways, inhibiting apoptosis. Obverse to this situation, reduced levels of GSTP increased TNFα-dependent TRAF2–ASK1 associations, activating both ASK1 and JNK/p38. Similar to the GSTP–JNK interaction, the GSH conjugation activity of GSTP is not necessary for GSTP–TRAF2binding since mutant GSTP lacking the catalytic tyrosine (Y7F) also represses ASK1 and the interaction between GSTP and TRAF2 is observed only in unstimulated cells. The C-terminal TRAF domain of TRAF2 and the C-terminal TRAF2-binding motif (TWQE amino acids 38–41) in GSTP seem necessary for the binding of TRAF2 to GSTP. A mutant GSTP lacking the TRAF domain-binding motif exhibited a significant decline of capacity to bind TRAF2 and block TRAF2–ASK1 signaling compared with the wild-type GSTP (Wu et al., 2006).
A further indication of functional redundancy is provided by the fact that the direct interaction of JNK with GSTs is not limited to just GSTP. Indeed, in addition to GSTP, GSTA and GSTM were also capable of associating with JNK complex in vitro (Adler, Yin, Fuchs, et al., 1999). GSTP exhibited greater JNK inhibitory activity than did GSTM1-1, which was more potent than GSTA1-1. Furthermore, Romero et al. (2006) had shown that GSTA1-1 interacts physically with JNK to suppress activation of JNK signaling by a proinflammatory cytokine and oxidative stress in Caco-2 cells. They showed that GSTA1-1 levels were higher in postconfluent than in preconfluent cells and that JNK activation was significantly reduced in the former when exposed to stress conditions including interleukin-1β, H2O2, and UV irradiation, implying a protective role for GSTA1-1 in JNK-associated apoptosis. In a different study, Desmots et al. (Desmots, Loyer, Rissel, Guillouzo, & Morel, 2005) demonstrated that GSTA4-4 and JNK coimmunoprecipitate in mouse liver tissue and GSTA4-4 expression was inversely correlated with JNK activities in primary hepatocyte cultures, implying that GSTA4-4 might be an endogenous regulator of JNK activity by direct binding. However, they showed a correlation between increased expression of GSTA4-4 and JNK phosphorylation during hepatocyte isolation and TNFα treatment, suggesting a proapoptotic role for GSTA4-4 in JNK-associated apoptosis. It seems likely that GSTs may be ligand-bound with other upstream receptors, kinases, and/or phosphatases, thus regulating their activities and leading to phosphorylation of JNK. As mentioned earlier (Anathy et al., 2012), GSTP can amplify Fas ligand (FasL)-induced apoptosis in epithelial cells. The stimulation of FasL increased the interaction of GSTP with Fas and ERp57, a member of the TNF receptor superfamily and a protein disulfide isomerase, in the ER leading to Fas S-glutathionylation and subsequent mobilization from the ER to the cytosol, resulting in enhanced apoptosis. Knockdown or inhibition of ERp57 and GSTP enhanced survival. As such, the pleiotropic functions of GSTP are extended to the Fas–ASK1–JNK/p38 signaling pathways as well. These findings indicate complex mechanisms of kinase signaling pathway regulation by GSTs that may serve as checks and balances for commitment to apoptosis versus survival under different stimuli.
4. GSTP IN REDOX REGULATION AND S-GLUTATHIONYLATION
Posttranslational modification of proteins is an elegant means by which cells have evolved a high degree of functional redundancy through reversible cycling reactions, enhancing protein function in response to varying stimuli in a cell- or tissue-specific manner. In general, kinase signaling pathways have been accepted as one of the cornerstones of signal transduction. The phosphorylation cycle is regulated by kinases and phosphatases that can function as either generalist or specialist enzyme. Redox signaling is gaining traction and is now recognized as physiologically important, and its dys-regulation is attributed to a variety of pathologies (Grek, Zhang, Manevich, Townsend, & Tew, 2013; Tew & Townsend, 2011b; Townsend, 2007; Townsend, Tew, & Tapiero, 2003; Xiong, Uys, Tew, & Townsend, 2011). S-glutathionylation is a posttranslational modification where GSH is conjugated to cysteine residues (P-SSG). Regulation through S-glutathionylation has been ascribed to a large number of proteins that were designated into the following clusters: cytoskeletal, glycolysis/energy metabolism, kinase signaling pathways, calcium homeostasis, antioxidant enzymes, and protein folding (Townsend, 2007) (Fig. 4.3). Here, we will draw comparisons of the S-glutathionylation cycle with examples of thiolases and dethiolases having both generalist and specialist functions.
Figure 4.3.

Representation from the literature of proteins clustered into functional groups that are known to be susceptible to S-glutathionylation.
5. S-GLUTATHIONYLATION REACTIONS
Glutathione is the most prevalent thiol in the cell, and the GSH:GSSG ratio has been used to describe the redox potential. Although recently, Flohe has revisited much of the earlier material that defines GSH:GSSG ratios and posited that small rate constants of related spontaneous reactions when compared with enzyme catalysis define the superiority of kinetic parameters over electrochemical or thermodynamic ones for an in-depth understanding of GSH-dependent biological phenomena. He concludes that at best, the GSH:GSSG potential might be useful analytically to disclose disturbances in redox metabolism, but more importantly, the enzyme-catalyzed reactions are more relevant to the understanding cellular redox homeostasis (Flohe, 2012).
The emerging role of S-glutathionylation underscores the importance of including the protein-associated pool of GSH in evaluating redox homeostasis. The redox potential is decreased following oxidative or nitrosative stress induced by physiological or pathological conditions. It is not surprising that cells have evolved a redox-sensitive subproteome to trigger signaling cascades following exposure to reactive oxygen (ROS) and nitrogen (RNS) species. Both ROS and RNS lead to oxidation of redox-sensitive proteins and subsequent S-glutathionylation (Fig. 4.4). Not all cysteine residues are targets for S-glutathionylation. Rather, cysteines with a low pKa have the nucleophilicity that under oxidative/nitrosative conditions can be oxidized to a thiyl radical (RS) with strong reactivity toward oxygen followed by addition of GSH. S-glutathionylation leads to an increase of 305 MW and a net negative charge the consequences of which impact protein structure, function, and subcellular localization.
Figure 4.4.

S-glutathionylation cycle of redox sensors. PTP1B is a representative protein with a low-pKa residue (redox sensor) that is a target for oxidative or nitrosative stress. Cysteine residues within redox sensors can be oxidized to form protein sulfenic (P-OH) and sulfinic (P-OOH) acids. Protein S-glutathionylation (P-SSG) reactions can be spontaneous or are mediated by GGT, Grx, or GSTP. P-SSG proteins have a wide variety of functions in cellular physiology/pathology.
In vitro studies have shown that S-glutathionylation reactions can occur spontaneously with ROS/RNS generating compounds in the presence of GSH. In cells, several enzymes have been shown to promote the forward reaction of S-glutathionylation, for example, thiolase activity. At present, the details regarding specificity of thiolase enzymes are limited. GSTP is among those enzymes identified to date that have been shown to promote S-glutathionylation reactions.
6. S-GLUTATHIONYLASE ACTIVE PROTEINS
A large proportion of S-glutathionylated proteins that have been identified are compartmentalized intracellularly (Townsend, 2007). However, evidence suggests that extracellular and cell surface proteins can be S-glutathionylated and impact the proliferative/apoptotic balance. Gamma-glutamyl transpeptidase (GGT) is a cell surface enzyme that hydrolyzes GSH to yield glutamate and cysteinylglycine (CG) that can more readily enter cells. As such, GGT impacts intracellular redox homeostasis through enhancing intracellular GSH content. However, GGT plays an active role in the S-glutathionylation cycle (Corti et al., 2005; Pompella, 2005). Both GSH and CG have been shown to form mixed disulfides in proteins in the presence of activated GGT; however, nearly 85% are GSH-bound (Corti et al., 2005; Pompella, 2005). Acivicin, a GGT inhibitor, decreased the formation of protein mixed disulfides. The specificity and targets of GGT-mediated S-glutathionylation remain poorly understood despite the key role GGT plays in the liver and at the blood–brain barrier. With regard to pathophysiology, GGT is overexpressed in a wide variety of cancer cells and is associated with multidrug resistance. Further exploration into the role of GGT in promoting S-glutathionylase reactions of extracellular and cell surface proteins will be insightful to normal physiology and pathophysiology.
Glutaredoxin (Grx) is a member of the thioredoxin superfamily and has thiol disulfide oxidoreductase activity. Grx is widely recognized in the S-glutathionylation cycle as a deglutathionylating enzyme and will be discussed as such in the following section (for review see Mieyal, Starke, Gravina, Dothey, & Chung, 1991; Mieyal, Starke, Gravina, & Hocevar, 1991; Shelton, Chock, & Mieyal, 2005). There are, however, limited conditions in which Grx1 and Grx2 have demonstrated S-glutathionylase activity. Specifically, Grx-mediated S-glutathionylation utilized GS·as the proximal donor with limited redox-sensitive targets, including GAPDH (Gallogly, Starke, Leonberg, Ospina, & Mieyal, 2008). GAPDH is among the few proteins that are S-glutathionylated on cysteine residues not in a basic environment and consequently lacks the characteristic low pKa (Peskin & Winterbourn, 2006). Grx appears to be a “specialist” in terms of glutathionylase activity, and it is plausible that the unique pKa property in GAPDH may be important to the limited substrate specificity of Grx thiolase activity, whereas other characteristic low-pKa cysteines that are modified serve as the generalist/promiscuous substrate for deglutathionylase activity.
GSTP was shown to promote S-glutathionylation reactions and may function as a promiscuous/generalist S-glutathionylase (Anathy et al., 2012; Hutchens, Manevich, He, Tew, & Townsend, 2011; Townsend et al., 2009b). Using MEF from Gstp1p2 wild-type and Gstp1p2-deficient mice, a range of redox-sensitive proteins were subject to S-glutathionylation following treatments with either ROS- or RNS-mediating agents (Townsend et al., 2009b). HEK293 cells transfected with GSTP-WT and a catalytic inactive mutant, Y7F, validated that the catalytic activity is required for S-glutathionylation. Polymorphisms within GSTP impact enzyme activity and may have important implications in an individual’s ability to respond to ROS/RNS, a causative factor in multiple disease states (Townsend et al., 2003). The S-glutathionylation activity of GSTP is autoregulated, much like kinases. Specifically, downregulation of thiolase activity is attributed to S-glutathionylation of GSTP on cys47 and cys101, which acts as an oligomer switch and can cause multimerization of the protein (Townsend et al., 2009b). While GSTP has a wide variety of substrates for S-glutathionylase activity, the cellular consequences of many have not been vetted. However, S-glutathionylation of peroxiredoxin (Prdx) and Fas ligand is attributed to GSTP thiolase activity (Anathy et al., 2012; Manevich, Feinstein, & Fisher, 2004; Manevich, Hutchens, Tew, & Townsend, 2012; Ralat, Misquitta, Manevich, Fisher, & Colman, 2008).
Peroxiredoxins are non-seleno peroxidases with two cysteine-containing active sites that function to reduce hydrogen peroxide through redox reactions with Trx (Rhee, Kang, Chang, Jeong, & Kim, 2001). Peroxiredoxin VI (Prdx6) is a singular catalytic cysteine-containing peroxiredoxin (Choi, Kang, Yang, Rhee, & Ryu, 1998). It is a bifunctional enzyme with phospholipid hydroperoxide peroxidase and acidic Ca2+-independent phospholipase A2 (PLA2) activities (Manevich & Fisher, 2005b). Inactivation of Prdx6 occurs following oxidative stress via formation of sulfinic acid on the catalytic cysteine residue. Catalytic cysteines of multiple enzymes are redox-sensitive and often are buried inside of protein globule to provide a unique environment for catalysis. Commonly, these crucial cysteines are inaccessible to the bulk and polarity of GSH. Recently, it was shown that GSTP could deliver a reducing equivalent (GS−) to the catalytic Cys47 sulfenate (buried inside of protein globule) through its heterodimerization with Prdx6 (Manevich et al., 2004). The evidence of Prdx6–GSTP heterodimerization has been proven by its chromatographic purification and N-terminal sequencing showing equimolar amounts of two proteins in this complex (Ralat et al., 2008). The Prdx6–GSTP heterodimer has been further characterized by determining its molecular mass (52,200 Da, pH 8.0) by dynamic light scattering and by demonstration (using the deletion mutants) that two sites of GSTP, 41–85 and 115–124, are critical for its formation (Ralat et al., 2006, 2008). The delivery of reducing equivalents resulted in transient S-glutathionylation of Prdx6, which affects its folding and brings together disulfide bonds of Prdx6, S-glutathionylated Cys47, and catalytic Cys47 of GSTP (Manevich & Fisher, 2005a; Ralat et al., 2006). As a result of the close proximity of these sites, a disulfide-based heterodimer of Prdx6 and GSTP was formed (Ralat et al., 2006). This influences folding of both proteins and results in exposure of disulfide bond to GSH and, consequently, in sequential reduction of both Prdx6 and GSTP catalytic cysteines. The overall result of this heterodimer-originated and GSTP-catalyzed glutathionylation-driven reduction is activation of both Prdx6 and GSTP. The catalytic cycle of GSTP-mediated activation of Prdx6 is presented in Fig. 4.5.
Figure 4.5.

Catalytic cycle of Prdx6 activation by GSTP1. Reduction of phospholipid hydroperoxide (PLPCOOH) or H2O2 by Prdx6 results in oxidation of its catalytic Cys47 to sulfenic acid (shown in red). This oxidized monomer of Prdx6 forms a heterodimer with thiolate anion-bearing (shown in blue) GSTP. The spontaneous reaction of the thiolate anion with the catalytic Cys47 sulfenate of Prdx6 results in the S-glutathionylation of the latter (shown in box; the critical step of Prdx6 activation). Alignment of the S-glutathionylated catalytic Cys47 of Prdx6 with the catalytic Cys47 of GSTP results in the formation of a disulfide-based heterodimer. GSH access to the disulfide bond results in catalytic cysteine reduction/activation (shown in green) and heterodimer dissociation.
7. DEGLUTATHIONYLASE ACTIVE PROTEINS
Redox signaling events are triggered under physiological levels of oxidative and nitrosative stresses, and the temporal fluxes in homeostasis are restored through deglutathionylation of redox sensors and increased GSH/GSSG levels. In the forward reaction of the S-glutathionylation cycle, GSTP plays a role as a generalist with a multitude of target proteins, while Grx and GGT may have more specialized roles. This paradigm follows through in the reverse reaction, deglutathionylation. Grx mediates the removal of GSH through direct thiol disulfide exchange reactions that lead to the formation of GSSG that is subsequently reduced by glutathione reductase. Grx is a generalist deglutathionylase with a broad spectrum of substrates, dysregulation of some of which can lead to different pathologies (Shelton et al., 2005).
Sulfiredoxin (Srx) is an antioxidant enzyme with oxidoreductase activity. The primary function of Srx is in the metabolism and reactivation of Prdx’s (Baek et al., 2012; Jeong, Bae, Toledano, & Rhee, 2012). Overexpression of Srx in mammalian cells demonstrated that the enzyme has a dual function, including deglutathionylase activity (Bowers, Manevich, Townsend, & Tew, 2012; Findlay, Tapiero, & Townsend, 2005; Findlay et al., 2006; Lei, Townsend, & Tew, 2008; Park, Mieyal, Rhee, & Chock, 2009). Substrates for Srx-mediated deglutathionylation are limited but include Prdx, PTP1B, and actin (Findlay et al., 2006; Park et al., 2009). Srx contains a single cysteine residue in the active site, whereas Grx has a CXXC motif, suggesting distinct molecular mechanism of deglutathionylation. Park et al. evaluated the specificity of the two enzymes toward Prdx1 and determined that Srx has a greater binding affinity than Grx toward Prdx-SSG (Park et al., 2009). Collectively, these data suggest that Grx and Srx play distinct roles in the S-glutathionylation cycle that can be regulated through substrate specificity and/or by tissue and subcellular distribution.
8. GSTP, NITRIC OXIDE SYNTHASES, AND NO HOMEOSTASIS
Under both normal and pathological conditions, nitric oxide (NO) is a critical secondary messenger in controlling cellular signaling events (Isenberg, Martin-Manso, Maxhimer, & Roberts, 2009). NO is generated intracellularly by nitric oxide synthases (NOS) (Stuehr, 1997). It is a reactive molecule (radical) and may either directly or through reaction with molecular oxygen (generating N2O3 with high affinity for thiols; Stamler, Singel, & Loscalzo, 1992) or superoxide anion radicals (generating peroxynitrite; Beckman, Beckman, Chen, Marshall, & Freeman, 1990) lead to nitrosylation or S-glutathionylation of protein thiols (West, Hill, Xuan, & Bhatnagar, 2006). The thiol reaction with peroxynitrite may also cause generation of thiyl radicals or sulfenates (Filipovska & Murphy, 2006), creating a situation where protein glutathionylation status may be independent of the global GSH:GSSG ratio. Dinitrosyl iron complexes (DNIC) ([(RS−)2Fe+(NO+)2 … (−SR)2]−) incorporate NO, increasing its stability and enhancing its target specificity (Martinez & Andriantsitohaina, 2009; Vanin, 2009). Generally, nitrosative stress (RNS) is a composite of nitric oxide (NO) and nitric oxide-derived compounds, including nitroxyl anion (NO− and HNO), nitrosonium cation (NO+), higher oxides of nitrogen (N2O, NO2, and N2O3), peroxynitrite (ONOO−/ONOOH), S-nitrosothiols (RSNO), and DNIC.
In the presence of O2, nitric oxide can react with glutathione sulfhydryls resulting (most likely through intermediate(s) of higher nitrogen oxides) in formation of GSNO, which serves as a physiological scavenger of the NOx species (Wink et al., 1994). GSNO, in turn, can react with GSH yielding GSSG, nitrite, nitrous oxide, and ammonia as end products (Singh, Wishnok, Keshive, Deen, & Tannenbaum, 1996). High intracellular concentrations of GSH can shift the equilibrium of the reaction with NO to GSSG, but site-specifically, some GSNO can still be present. Recent developments with stabilized DNIC–glutathione preparations allow retention of the protein S-nitrosylation for at least 1 year, and these may be precursors for a variety of broad-spectrum NO-containing drug moieties (Vanin, 2009).
9. GSTP BINDING OF NITRIC OXIDE CARRIERS
Using crystallization, isothermal titration, and differential scanning calorimetry techniques, GSNO was shown to bind strongly to the G-site of homodimeric human GSTP. The nitrosyl moiety of bound GSNO is involved in multiple interactions with the protein. The binding of GSNO to wild-type GSTP induces negative cooperativity with the kinetics concomitant with the binding at physiological temperatures. GSNO inhibits the wild-type GSTP enzyme competitively at low temperatures, but covalently at higher temperatures. Because the C47S mutation prevents this covalent modification, S-nitrosylation of the Cys47 sulfhydryl has been implicated. This is compatible with the crucial role of the Cys47 in intersubunit substrate-binding cooperativity and with the physiological role of Cys47 S-nitrosylation by GSNO in certain pathologies (Tellez-Sanz et al., 2006). Both GSNO and DNIC can participate in storage and transport of NO in biological systems. In vitro, without GSH, GSNO induces rapid and stable S-nitrosylation of GSTP Cys47 (101). MS analysis failed to show any S-glutathionylation under those conditions, which presumably indicates an NO (NxOy) release from GSNO. S-nitrosylation of GSTP at Cys47 decreases (~10×) its affinity for GSH. Partial protection of one Cys47 residue of homodimeric GSTP from S-nitrosylation minimizes loss of its enzymatic activity. In living cells, intracellular GSH is generally present at millimolar levels, and another type of NO sequestration—dinitrosyl–diglutathionyl–iron complex (DNDGIC, formed by GSNO decomposition in the presence of traces of ferrous ions) becomes predominant (Lo Bello et al., 2001). In vivo, DNDGIC can serve as an NO donor and iron/GSH transporter (Vasieva, 2011). Through one of its glutathione moieties, DNDGIC binds tightly (Ki <10−12 M) to the G-site of GSTP. The other GSH moiety is displaced by the active site Tyr7, stabilizing the enzyme-inhibitor complex. The NO moieties in this complex are stabilized by interaction with Ile104 and Tyr108 (Fig. 4.6). The half-life of DNDGIC is ~1 min, but in the complex with GSTP, this is substantially increased (~3 h). This binding induces a negative cooperativity in other unoccupied monomer G-sites of the GSTP homodimer, downregulating its basal activity (Lo Bello et al., 2001). In addition, DNDGIC is an irreversible inhibitor of glutathione reductase (GR), although this inhibition was not associated with DNDGIC–GR complexes and/or nitrosation of the active site cysteines 58 and 63. Simultaneously, one NO moiety with acid lability was present per molecule of inhibited monomeric enzyme (Boese, Keese, Becker, Busse, & Mulsch, 1997). Such results indicate that GSTP can serve as a cellular NO donor and/or carrier potentially regulating the activities of other (GR) thiol redox-modifying enzymes.
Figure 4.6.
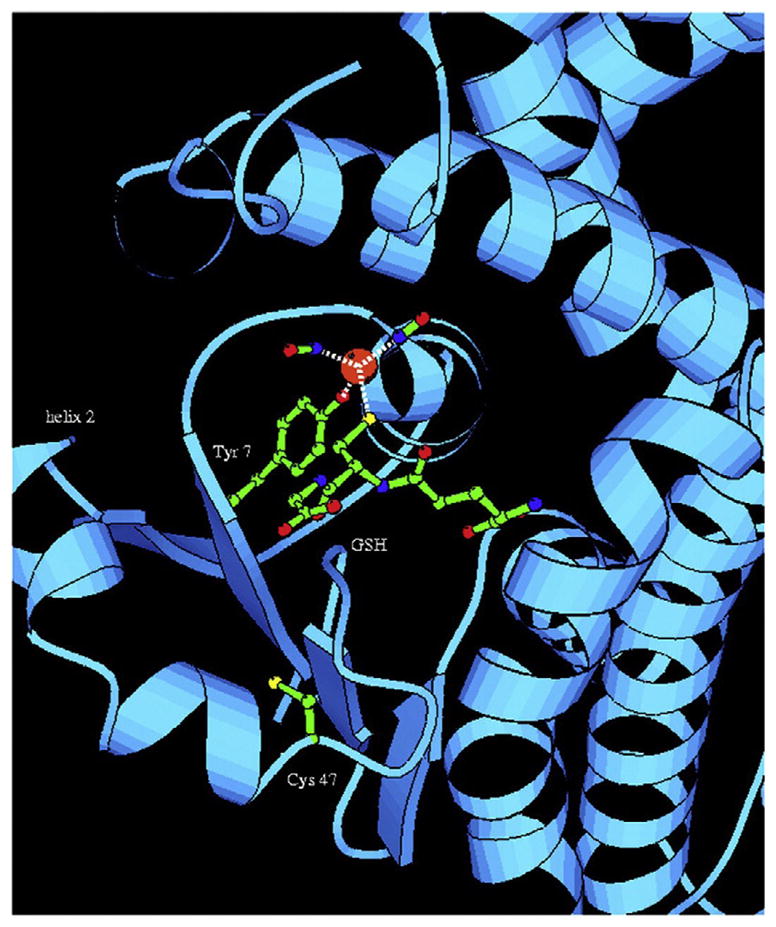
Ribbon depiction of the DNGIC/GSTP1-1 complex. The close-up view of the active site of the covalent DNGIC (as obtained after removal of the excess GSH) is shown. The model shows that (i) one of the GSH ligands of DNDGIC can dock into the G-site and adopt the canonical extended conformation seen in crystal structures of GST–GSH complexes, (ii) Tyr7 is close enough to displace the other GSH ligand to generate a stable enzyme-inhibitor complex, and (iii) the NO moieties of the complex form van der Waal’s interactions with Ile104 and Tyr108. In addition, there are possible polar interactions with Tyr108 and the main chain nitrogen of Gly205. The iron atom is depicted as an orange sphere, oxygen atoms are colored red, nitrogen atoms are blue, sulfur atoms are yellow, and carbon atoms are green. This figure was drawn using MOLSCRIPT. Reproduced from Lo Bello et al. (2001) with permission.
10. GSTP-MEDIATED SITE-SPECIFIC PROTEIN NITROSYLATION/GLUTATHIONYLATION
When GSTP forms heterodimers with target proteins (Prdx6; Ralat et al., 2006, 2008), it can enable the delivery of GS− to cysteines that may otherwise be inaccessible to GSH. GSNO (DNDGIC) bound to GSTP monomers could also modify target protein affinity and impact posttranslational modifications of cysteines. For example, with GSNO bound to the G-site of GSTP, the NO group of the complex is on the surface, in proximity to the Cys47 of GSTP. One could anticipate that NO donor–GSTP complexes may overcome steric constraints and provide specificity for fine-tuning regulation of signal transduction or target enzyme activities. The translational importance of such transnitrosylation reactions has been discussed elsewhere (Marino & Gladyshev, 2010).
Two cysteines (Cys689 and Cys908) that are highly conserved within the NOS family are susceptible to S-glutathionylation. These are critical for normal eNOS functions, and when modified, the enzyme produces O2•−, a process that cannot be inhibited by either N-nitro-l-arginine methylester (L-NAME) or removal of calcium. S-glutathionylation of eNOS uncouples eNOS in endothelial cells impairing endothelium-dependent vasodilation (Crabtree, Brixey, Batchelor, Hale, & Channon, 2013). Induction of eNOS uncoupling by S-glutathionylation switches eNOS from NO production to O2•− generation, with concomitant loss of endothelial-mediated vasodilation contributing to vascular constriction, which can contribute to hypertensive cardiovascular disease. The involvement of GSTP in mediating this S-glutathionylation seems a viable possibility but is not yet established.
11. GSTP POLYMORPHISMS AND PHARMACOGENETICS
Translational relevance is a desired component of medical research today. One area of GST biology that is likely to provide such an outlet is the identification of human polymorphic variants of GSTP, which arise from nucleotide transitions that change codon 105 from Ile to Val and codon 114 from Ala to Val, generating four GSTP1 alleles: wild-type GSTP1-1A (Ile105/Ala114), GSTP1-1B (Val105/Ala114), GSTP1-1C (Val105/Val114), and GSTP1-1D (Ile105/Val114) (Ali-Osman, Akande, et al., 1997; Watson, Stewart, Smith, Massey, & Bell, 1998). The Ile105→Val105 and Ala114→Val114 substitutions do not alter affinity for GSH, but sterically alter the substrate-binding site of the enzyme (Watson et al., 1998; Zimniak et al., 1994). The allele frequencies for *A–C in Caucasian populations are 0.685, 0.262, and 0.068, respectively (Garte et al., 2001). Altered conformation of the substrate-binding site(s) may contribute to different catalytic rates for the formation of thioether conjugates between GSH and some electrophiles (Ali-Osman, Akande, et al., 1997; Zimniak et al., 1994). The hydrophobicity and size of residue 114 could serve as a determinant of the substrate specificity of each isoenzyme (Hu, Herzog, Zimniak, & Singh, 1999). Since GSTP1-1D has enzyme activity toward CDNB comparable to GSTP1-1A, Val105 may circumvent the influence of Val114 (Watson et al., 1998). There are a few examples where polymorphisms influence response to certain anticancer drugs: GSTP1-1A reportedly enhances the formation of cisplatin–glutathione conjugates (Goto et al., 1999). There are also some early indications of epidemiological correlations of isotype expression with etiological aspects of endometrial (Chan et al., 2005), bladder, and testicular cancers (Harries, Stubbins, Forman, Howard, & Wolf, 1997). Perhaps most directly, these polymorphisms have the potential to influence disease susceptibility and response to ROS/RNS (McIlwain et al., 2006). For example, they have been shown to impact S-glutathionylation and lipid peroxidation (Manevich et al., 2012), altering Prdx6 peroxidase function through their effect on the affinity of GSTP for Prdx6 during heterodimerization (Manevich, Hutchens, Tew, & Townsend, 2013). Specifically, GSTP1*A and GSTP1*C have a lower binding affinity (Kd) for Prdx6 and consequently altered lipid peroxidation. The clinical implication of polymorphisms of GSTP may be attributable to its role in phase II metabolism/detoxification. However, the role of GSTP in the forward reaction of the S-glutathionylation cycle remains a work in progress and clinically could supersede its role in metabolism.
12. GSH PATHWAYS AND GSTP AS DRUG PLATFORMS
Premised upon the elevated levels of GSTP expression in tumors and the induction of expression underlying the drug resistance phenotype, the GSTP platform was used as a target area for anticancer drug discovery. Further, human GST polymorphisms serve to link possible pharmacogenetic variations in catalytic activity and drug metabolism, as well as cancer susceptibility and prognosis (Townsend & Tew, 2003). Early discovery identified GSH analogs, GSH conjugates, small organic molecules, and various natural products as putative GSTP inhibitors (Laborde, 2010; Mahajan & Atkins, 2005; Tew, 1994).
Telintra was designed as a selective inhibitor of GSTP. It is a peptidomimetic of GSH, substituted with aromatic moieties and esterified to enhance membrane permeability. An extensive preclinical and toxicological work established its safety profile and confirmed that inhibition of GSTP led to activation of Jun kinase, a key regulator of cellular growth and differentiation of blood precursor cells. In normal mice, TLK199 treatment increased circulating white blood cells and stimulated bone marrow progenitor proliferation. Early studies using Telintra-treated GSTP-deficient mice indicated that myeloproliferative events were allied with downstream activation of STAT proteins (Gate et al., 2004). Telintra is currently in clinical development (Ezatiostat HCl) for the treatment of low- to intermediate-risk myelodysplastic syndrome (MDS) patients. MDS affects approx. 300,000 people worldwide with etiology as a heterogeneous group of clonal hematopoietic stem cell disorders typically associated with aberrant hematopoiesis of one or more cell lineages (red blood cells, white blood cells, or platelets) and a variable risk of progression to acute myeloid leukemia. Two recent reports (Lyons, Wilks, Young, & Brown, 2011; Raza et al., 2009) have suggested that Telintra has therapeutic benefit in this disease, including durable red blood cell transfusion independence and multilineage responses. Recent trends in drug development have emphasized the value of stratifying patient selection on the basis of biomarkers that might positively influence response. One such study used a gene marker analysis from 30 patients enrolled in a phase II trial (Galili et al., 2012). The Telintra response profile identified two miRNAs that regulate genes implicated in MDS pathology and genes controlling the JNK–c-Jun pathways. This latter correlation is linked with Telintra-mediated activation, and interestingly, JNK–c-Jun were underexpressed in responders and overexpressed in nonresponders, further supporting the mechanism of action of Telintra. Additional attempts to optimize inhibitor allelic specificity, stability, and/or potency have included testing a number of peptidomimetic conjugate analogs with additional modifications of the GSH peptide backbone. Examples of these include compounds that are resistant to cleavage by GGT (Burg, Riepsaame, Pont, Mulder, & van de Water, 2006).
Other concepts have included the design of specific GSH conjugates, where the thiol group is modified and these have met with varying degrees of preclinical success. Allosteric inhibition of GSTP was successfully accomplished by conjugating GSH to DOX. Interestingly, activation of JNK and apoptotic pathways following GSH–DOX–GSTP binding appeared to occur without complex dissociation and be mediated by mitochondrial cytochrome c release (Asakura et al., 2007).
Several small molecule inhibitors and natural products have been reported to successfully target GSTP (see Laborde (2010) for a recent review). However, broad isoenzyme inhibition (i.e., lack of specificity) makes the majority of these less than ideal pharmacological agents. Ethacrynic acid (EA) is a potent, reversible small molecule inhibitor that nonspecifically inhibits GSTP activity by acting both as a GSH conjugate and as a noncompetitive GSH inhibitor for GST binding (Ploemen, Van Schanke, Van Ommen, & Van Bladeren, 1994). While preclinical studies indicated that EA administration effectively sensitized chemotherapy-resistant tumors, EA is also a potent diuretic and this negatively impacted a phase I clinical trial with this agent in combination with thiotepa (O’Dwyer et al., 1991). Similarly, 7-nitro-2, 1, 3-benzoxadiazole derivatives are potent, irreversible suicide GSTP inhibitors that not only induce activation of the c-Jun–JNK-mediated signal transduction pathway but also bind other GSTs and have not found clinical application (Federici et al., 2009a; Ricci et al., 2005).
An alternative approach to targeting GSTP has been tried. Inactive pro-drugs that take advantage of the high GSTP levels in cancer cells have been synthesized and tested. For example, Telcyta (canfosfamide HCl, TLK286) is a conjugate of a GSH analog and an N, N, N′, N′-tetrakis(2-chloroethyl) phosphorodiamidate that is metabolically activated via GSTP cleavage, thus therapeutically targeting GSTP overexpressing cells (Satyam et al., 1996). This drug has been extensively tested preclinically and also in both phase II and phase III clinical trials (Kavanagh et al., 2010; Vergote et al., 2009). Although the phase III results were not positive enough to advance the drug to an NDA application, further clinical trials continue.
GSTP was shown to catalyze NO release from a diazeniumdiolate, O2-{2,4-dinitro-5-[4-(N-methylamino)benzoyloxy]phenyl}1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate (PABA/NO), through addition of GSH and subsequent Meisenheimer complex formation (Saavedra et al., 2006). Antitumor activity and mechanism of action studies confirmed the importance of GSTP in the pharmacology of PABA/NO and related diazenium diolates (Findlay et al., 2004; Townsend et al., 2006). Additionally, it was shown, by crystallization of the complexes, that 6-(7-nitro-2, 1, 3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) is specifically binding to the H-site of GSTM2-2 and resulting in its glutathionylation as a stable σ-complex. Interestingly, NBDHEX binds to the H-site of GSTP, but with lower affinity as compared to GSTM2-2 and without conjugation with GSH in the active center. Apparently, affinity of NBDHEX binding to GSTP was increased ~4× by Ile104Val or Ile104Ala mutation (Federici et al., 2009b). This GSTP property provides a plausible platform for development of novel therapeutics site-specifically releasing NO and other active glutathionylated nitroaromatic compounds (Hutchens et al., 2011; Manevich, Townsend, Hutchens, & Tew, 2010; Tew & Townsend, 2011a).
13. CONCLUSIONS AND PERSPECTIVES
The prevalence and ubiquitous biology of GSTP imply functional properties that extend beyond detoxification. Consistent with this, acting as chaperone-like proteins, GST isoenzymes (and GSTP in particular) have been found to regulate kinase signaling pathways. As a carrier of NO, GSTP also participates in regulating certain aspects of nitrosative stress, important in normal physiology, where imbalance is causally related to specific human disease pathologies (Tew & Townsend, 2011b; Townsend, 2007; Townsend et al., 2003; Xiong et al., 2011). Oxidation/reduction reactions control the activities of a number of proteins that can be functionally clustered. S-glutathionylation of such proteins can occur spontaneously; however, GSTP is documented to facilitate the delivery of GS to acceptor cysteines, catalyzing reactions in parts of the protein that might otherwise be inaccessible. Viability of the GSTP knockout mouse implies functional redundancy within the GST isoenzyme family. Nevertheless, the high level of GSTP in some human cancers and in drug-resistant tumor cells has provided a platform for discovery/development of therapeutic agents and diagnostic tools. Some of these are in clinical trials, and while it remains too early to predict their value, this area of research has translational relevance. Polymorphic expression of GSTP also provides a platform to understand individual differences in response to ROS/RNS (whether through drugs or environmental exposures) and susceptibility of drug addiction. While these developments remain in their early stages of development, they hold great promise for the future.
Acknowledgments
This work was supported by grants from the National Center for Research Resources (5P20RR024485-02) and the National Institute of General Medical Sciences (8 P20 GM103542-02) from the National Institutes of Health and by CA08660, CA117259, and R56 ES017453 and support from the South Carolina Centers of Economic Excellence program. J. Z. was financially supported by the Swedish Research Council (No. 524-2011-6998).
References
- Abdelsaid MA, El-Remessy AB. S-Glutathionylation of LMW-PTP regulates VEGF-mediated FAK activation and endothelial cell migration. Journal of Cell Science. 2012;125(Pt 20):4751–4760. doi: 10.1242/jcs.103481. [DOI] [PubMed] [Google Scholar]
- Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, et al. Regulation of JNK signaling by GSTp. The EMBO Journal. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase Pi gene variants. Evidence for differential catalytic activity of the encoded proteins. The Journal of Biological Chemistry. 1997;272:10004–10012. doi: 10.1074/jbc.272.15.10004. [DOI] [PubMed] [Google Scholar]
- Ali-Osman F, Brunner JM, Kutluk TM, Hess K. Prognostic significance of glutathione S-transferase pi expression and subcellular localization in human gliomas. Clinical Cancer Research. 1997;3:2253–2261. [PubMed] [Google Scholar]
- Allen TC, Granville LA, Cagle PT, Haque A, Zander DS, Barrios R. Expression of glutathione S-transferase pi and glutathione synthase correlates with survival in early stage non-small cell carcinomas of the lung. Human Pathology. 2007;38:220–227. doi: 10.1016/j.humpath.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Allocati N, Federici L, Masulli M, Di Ilio C. Glutathione transferases in bacteria. The FEBS Journal. 2009;276:58–75. doi: 10.1111/j.1742-4658.2008.06743.x. [DOI] [PubMed] [Google Scholar]
- Anathy V, Roberson E, Cunniff B, Nolin JD, Hoffman S, Spiess P, et al. Oxidative processing of latent Fas in the endoplasmic reticulum controls the strength of apoptosis. Molecular and Cellular Biology. 2012;32:3464–3478. doi: 10.1128/MCB.00125-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura T, Sasagawa A, Takeuchi H, Shibata S, Marushima H, Mamori S, et al. Conformational change in the active center region of GST P1-1, due to binding of a synthetic conjugate of DXR with GSH, enhanced JNK-mediated apoptosis. Apoptosis. 2007;12:1269–1280. doi: 10.1007/s10495-007-0053-0. [DOI] [PubMed] [Google Scholar]
- Atkinson HJ, Babbitt PC. Glutathione transferases are structural and functional outliers in the thioredoxin fold. Biochemistry. 2009;48:11108–11116. doi: 10.1021/bi901180v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek JY, Han SH, Sung SH, Lee HE, Kim YM, Noh YH, et al. Sulfiredoxin protein is critical for redox balance and survival of cells exposed to low steady-state levels of H2O2. The Journal of Biological Chemistry. 2012;287:81–89. doi: 10.1074/jbc.M111.316711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini S, Bernassola F, Cortese C, Ballerini S, Melino G, Motti C, et al. Modulation of GST P1-1 activity by polymerization during apoptosis. Journal of Cellular Biochemistry. 2000;77:645–653. [PubMed] [Google Scholar]
- Boese M, Keese MA, Becker K, Busse R, Mulsch A. Inhibition of glutathione reductase by dinitrosyl-iron-dithiolate complex. The Journal of Biological Chemistry. 1997;272:21767–21773. doi: 10.1074/jbc.272.35.21767. [DOI] [PubMed] [Google Scholar]
- Bowers RR, Manevich Y, Townsend DM, Tew KD. Sulfiredoxin redox-sensitive interaction with S100A4 and non-muscle myosin IIA regulates cancer cell motility. Biochemistry. 2012;51:7740–7754. doi: 10.1021/bi301006w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg D, Riepsaame J, Pont C, Mulder G, van de Water B. Peptide-bond modified glutathione conjugate analogs modulate GSTpi function in GSH-conjugation, drug sensitivity and JNK signaling. Biochemical Pharmacology. 2006;71:268–277. doi: 10.1016/j.bcp.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Chan QK, Khoo US, Ngan HY, Yang CQ, Xue WC, Chan KY, et al. Single nucleotide polymorphism of pi-class glutathione s-transferase and susceptibility to endometrial carcinoma. Clinical Cancer Research. 2005;11:2981–2985. doi: 10.1158/1078-0432.CCR-04-2038. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Kang SW, Yang CH, Rhee SG, Ryu SE. Crystal structure of a novel human peroxidase enzyme at 2.0 A resolution. Nature Structural Biology. 1998;5:400–406. doi: 10.1038/nsb0598-400. [DOI] [PubMed] [Google Scholar]
- Corti A, Paolicchi A, Franzini M, Dominici S, Casini AF, Pompella A. The S-thiolating activity of membrane gamma-glutamyltransferase: Formation of cysteinylglycine mixed disulfides with cellular proteins and in the cell microenvironment. Antioxidants & Redox Signaling. 2005;7:911–918. doi: 10.1089/ars.2005.7.911. [DOI] [PubMed] [Google Scholar]
- Cowell IG, Dixon KH, Pemble SE, Ketterer B, Taylor JB. The structure of the human glutathione S-transferase pi gene. The Biochemical Journal. 1988;255:79–83. doi: 10.1042/bj2550079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree MJ, Brixey R, Batchelor H, Hale AB, Channon KM. Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. The Journal of Biological Chemistry. 2013;288:561–569. doi: 10.1074/jbc.M112.415992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- De Luca A, Federici L, De Canio M, Stella L, Caccuri AM. New insights into the mechanism of JNK1 inhibition by glutathione transferase p1-1. Biochemistry. 2012;51:7304–7312. doi: 10.1021/bi300559m. [DOI] [PubMed] [Google Scholar]
- Desmots F, Loyer P, Rissel M, Guillouzo A, Morel F. Activation of C-Jun N-terminal kinase is required for glutathione transferase A4 induction during oxidative stress, not during cell proliferation, in mouse hepatocytes. FEBS Letters. 2005;579:5691–5696. doi: 10.1016/j.febslet.2005.08.088. [DOI] [PubMed] [Google Scholar]
- Federici L, Lo Sterzo C, Pezzola S, Di Matteo A, Scaloni F, Federici G, et al. Structural basis for the binding of the anticancer compound 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol to human glutathione s-transferases. Cancer Research. 2009a;69:8025–8034. doi: 10.1158/0008-5472.CAN-09-1314. [DOI] [PubMed] [Google Scholar]
- Federici L, Lo Sterzo C, Pezzola S, Di Matteo A, Scaloni F, Federici G, et al. Structural basis for the binding of the anticancer compound 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol to human glutathione s-transferases. Cancer Research. 2009b;69:8025–8034. doi: 10.1158/0008-5472.CAN-09-1314. [DOI] [PubMed] [Google Scholar]
- Filipovska A, Murphy MP. Overview of protein glutathionylation. Current Protocols in Toxico. 2006;Chapter 6(Unit 6):10. doi: 10.1002/0471140856.tx0610s28. [DOI] [PubMed] [Google Scholar]
- Findlay VJ, Tapiero H, Townsend DM. Sulfiredoxin: A potential therapeutic agent? Biomedicine & Pharmacotherapy. 2005;59:374–379. doi: 10.1016/j.biopha.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Research. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay VJ, Townsend DM, Saavedra JE, Buzard GS, Citro ML, Keefer LK, et al. Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Molecular Pharmacology. 2004;65:1070–1079. doi: 10.1124/mol.65.5.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flohe L. The fairytale of the GSSG/GSH redox potential. Biochimica et Biophysica Acta. 2012;1830(5):3139–3142. doi: 10.1016/j.bbagen.2012.10.020. [DOI] [PubMed] [Google Scholar]
- Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. The Journal of Biological Chemistry. 2010;285:8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili N, Tamayo P, Botvinnik OB, Mesirov JP, Brooks MR, Brown G, et al. Prediction of response to therapy with ezatiostat in lower risk myelodysplastic syndrome. Journal of Hematology & Oncology. 2012;5:20. doi: 10.1186/1756-8722-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallogly MM, Starke DW, Leonberg AK, Ospina SM, Mieyal JJ. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: Implications for intracellular roles. Biochemistry. 2008;47:11144–11157. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. The Journal of Biological Chemistry. 2010;285:39646–39654. doi: 10.1074/jbc.M110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner JL, Gallagher EP. Development of a peptide antibody specific to human glutathione S-transferase alpha 4-4 (hGSTA4-4) reveals preferential localization in human liver mitochondria. Archives of Biochemistry and Biophysics. 2001;390:19–27. doi: 10.1006/abbi.2001.2352. [DOI] [PubMed] [Google Scholar]
- Garte S, Gaspari L, Alexandrie AK, Ambrosone C, Autrup H, Autrup JL, et al. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiology, Biomarkers & Prevention. 2001;10:1239–1248. [PubMed] [Google Scholar]
- Gate L, Majumdar RS, Lunk A, Tew KD. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. The Journal of Biological Chemistry. 2004;279:8608–8616. doi: 10.1074/jbc.M308613200. [DOI] [PubMed] [Google Scholar]
- Gate L, Majumdar RS, Lunk A, Tew KD. Influence of glutathione S-transferase pi and p53 expression on tumor frequency and spectrum in mice. International Journal of Cancer. 2005;113:29–35. doi: 10.1002/ijc.20540. [DOI] [PubMed] [Google Scholar]
- Gildenhuys S, Wallace LA, Burke JP, Balchin D, Sayed Y, Dirr HW. Class Pi glutathione transferase unfolds via a dimeric and not monomeric intermediate: Functional implications for an unstable monomer. Biochemistry. 2010;49:5074–5081. doi: 10.1021/bi100552d. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Dosal R, Horan KA, Rahbek SH, Ichijo H, Chen ZJ, Mieyal JJ, et al. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: Role for S-glutathionylation of TRAF3 and 6. PLoS Pathogens. 2011;7:e1002250. doi: 10.1371/journal.ppat.1002250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Ihara Y, Urata Y, Izumi S, Abe K, Koji T, et al. Doxorubicin-induced DNA intercalation and scavenging by nuclear glutathione S-transferase pi. The FASEB Journal. 2001a;15:2702–2714. doi: 10.1096/fj.01-0376com. [DOI] [PubMed] [Google Scholar]
- Goto S, Ihara Y, Urata Y, Izumi S, Abe K, Koji T, et al. Doxorubicin-induced DNA intercalation and scavenging by nuclear glutathione S-transferase pi. The FASEB Journal. 2001b;15:2702–2714. doi: 10.1096/fj.01-0376com. [DOI] [PubMed] [Google Scholar]
- Goto S, Iida T, Cho S, Oka M, Kohno S, Kondo T. Overexpression of glutathione S-transferase pi enhances the adduct formation of cisplatin with glutathione in human cancer cells. Free Radical Research. 1999;31:549–558. doi: 10.1080/10715769900301121. [DOI] [PubMed] [Google Scholar]
- Graminski GF, Kubo Y, Armstrong RN. Spectroscopic and kinetic evidence for the thiolate anion of glutathione at the active site of glutathione S-transferase. Biochemistry. 1989;28:3562–3568. doi: 10.1021/bi00434a062. [DOI] [PubMed] [Google Scholar]
- Graminski GF, Zhang PH, Sesay MA, Ammon HL, Armstrong RN. Formation of the 1-(S-glutathionyl)-2,4,6-trinitrocyclohexadienate anion at the active site of glutathione S-transferase: Evidence for enzymic stabilization of sigma-complex intermediates in nucleophilic aromatic substitution reactions. Biochemistry. 1989;28:6252–6258. doi: 10.1021/bi00441a017. [DOI] [PubMed] [Google Scholar]
- Grek CL, Townsend DM, Uys JD, Manevich Y, Coker WJ, 3rd, Pazoles CJ, et al. S-glutathionylated serine proteinase inhibitors as plasma biomarkers in assessing response to redox-modulating drugs. Cancer Research. 2012;72:2383–2393. doi: 10.1158/0008-5472.CAN-11-4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and consequences of cysteine S-glutathionylation. The Journal of Biological Chemistry. 2013;288:26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. The EMBO Journal. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Harries LW, Stubbins MJ, Forman D, Howard GC, Wolf CR. Identification of genetic polymorphisms at the glutathione S-transferase Pi locus and association with susceptibility to bladder, testicular and prostate cancer. Carcinogenesis. 1997;18:641–644. doi: 10.1093/carcin/18.4.641. [DOI] [PubMed] [Google Scholar]
- Hartmanova T, Tambor V, Lenco J, Staab-Weijnitz CA, Maser E, Wsol V. S-Nitrosoglutathione covalently modifies cysteine residues of human carbonyl reductase 1 and affects its activity. Chemico-Biological Interactions. 2013;202:136–145. doi: 10.1016/j.cbi.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annual Review of Pharmacology and Toxicology. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Critical Reviews in Biochemistry and Molecular Biology. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Smith AG, Ure J, Brown K, Bacon EJ, Wolf CR. Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5275–5280. doi: 10.1073/pnas.95.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson CJ, Wolf CR, Kitteringham N, Powell H, Otto D, Park BK. Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12741–12745. doi: 10.1073/pnas.220176997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley SL, Fryer AA, Haycock JW, Grubb SE, Strange RC, Hoban PR. Differential effects of glutathione S-transferase pi (GSTP1) haplotypes on cell proliferation and apoptosis. Carcinogenesis. 2007;28:2268–2273. doi: 10.1093/carcin/bgm135. [DOI] [PubMed] [Google Scholar]
- Hu X, Herzog C, Zimniak P, Singh SV. Differential protection against benzo [a]pyrene-7,8-dihydrodiol-9,10-epoxide-induced DNA damage in HepG2 cells stably transfected with allelic variants of pi class human glutathione S-transferase. Cancer Research. 1999;59:2358–2362. [PubMed] [Google Scholar]
- Huang J, Tan PH, Thiyagarajan J, Bay BH. Prognostic significance of glutathione S-transferase-pi in invasive breast cancer. Modern Pathology. 2003;16:558–565. doi: 10.1097/01.MP.0000071842.83169.5A. [DOI] [PubMed] [Google Scholar]
- Hutchens S, Manevich Y, He L, Tew KD, Townsend DM. Cellular resistance to a nitric oxide releasing glutathione S-transferase P-activated prodrug, PABA/NO. Investigational New Drugs. 2011;29:719–729. doi: 10.1007/s10637-010-9407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg JS, Martin-Manso G, Maxhimer JB, Roberts DD. Regulation of nitric oxide signalling by thrombospondin 1: Implications for anti-angiogenic therapies. Nature Reviews Cancer. 2009;9:182–194. doi: 10.1038/nrc2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankova L, Robertson G, Chan C, Tan KL, Kohonen-Corish M, Fung CL, et al. Glutathione S-transferase Pi expression predicts response to adjuvant chemotherapy for stage C colon cancer: A matched historical control study. BMC Cancer. 2012;12:196. doi: 10.1186/1471-2407-12-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong W, Bae SH, Toledano MB, Rhee SG. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radical Biology & Medicine. 2012;53:447–456. doi: 10.1016/j.freeradbiomed.2012.05.020. [DOI] [PubMed] [Google Scholar]
- Jin X, Yu L, Wu Y, Zhang S, Shi Z, Chen X, et al. S-Glutathionylation underscores the modulation of the heteromeric Kir4.1-Kir5.1 channel in oxidative stress. The Journal of Physiology. 2012;590:5335–5348. doi: 10.1113/jphysiol.2012.236885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh JJ, Levenback CF, Ramirez PT, Wolf JL, Moore CL, Jones MR, et al. Phase 2 study of canfosfamide in combination with pegylated liposomal doxorubicin in platinum and paclitaxel refractory or resistant epithelial ovarian cancer. Journal of Hematology & Oncology. 2010;3:9. doi: 10.1186/1756-8722-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kil IS, Shin SW, Park JW. S-glutathionylation regulates GTP-binding of Rac2. Biochemical and Biophysical Research Communications. 2012;425:892–896. doi: 10.1016/j.bbrc.2012.07.169. [DOI] [PubMed] [Google Scholar]
- Kim HS, Ullevig SL, Zamora D, Lee CF, Asmis R. Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2803–E2812. doi: 10.1073/pnas.1212596109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou X, Chen N, Feng Z, Luo L, Yin Z. GSTP1 negatively regulates Stat3 activation in epidermal growth factor signaling. Oncology Letters. 2013;5:1053–1057. doi: 10.3892/ol.2012.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laborde E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death and Differentiation. 2010;17:1373–1380. doi: 10.1038/cdd.2010.80. [DOI] [PubMed] [Google Scholar]
- Lei K, Townsend DM, Tew KD. Protein cysteine sulfinic acid reductase (sulfiredoxin) as a regulator of cell proliferation and drug response. Oncogene. 2008;27:4877–4887. doi: 10.1038/onc.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bello M, Nuccetelli M, Caccuri AM, Stella L, Parker MW, Rossjohn J, et al. Human glutathione transferase P1-1 and nitric oxide carriers; a new role for an old enzyme. The Journal of Biological Chemistry. 2001;276:42138–42145. doi: 10.1074/jbc.M102344200. [DOI] [PubMed] [Google Scholar]
- Lo HW, Ali-Osman F. Structure of the human allelic glutathione S-transferase-pi gene variant, hGSTP1 C, cloned from a glioblastoma multiforme cell line. Chemico-Biological Interactions. 1998;111–112:91–102. doi: 10.1016/s0009-2797(97)00153-1. [DOI] [PubMed] [Google Scholar]
- Lock JT, Sinkins WG, Schilling WP. Protein S-glutathionylation enhances Ca2+–induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. The Journal of Physiology. 2012;590:3431–3447. doi: 10.1113/jphysiol.2012.230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons RM, Wilks ST, Young S, Brown GL. Oral ezatiostat HCl (Telintra(R), TLK199) and Idiopathic Chronic Neutropenia (ICN): A case report of complete response of a patient with G-CSF resistant ICN following treatment with ezatiostat, a glutathione S-transferase P1-1 (GSTP1-1) inhibitor. Journal of Hematology & Oncology. 2011;4:43. doi: 10.1186/1756-8722-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan S, Atkins WM. The chemistry and biology of inhibitors and pro-drugs targeted to glutathione S-transferases. Cellular and Molecular Life Sciences. 2005;62:1221–1233. doi: 10.1007/s00018-005-4524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux RJ, Fu A, Robson-Doucette C, Allister EM, Wheeler MB, Screaton R, et al. Glutathionylation state of uncoupling protein-2 and the control of glucose-stimulated insulin secretion. The Journal of Biological Chemistry. 2012;287:39673–39685. doi: 10.1074/jbc.M112.393538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevich Y, Feinstein SI, Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3780–3785. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radical Biology & Medicine. 2005a;38:1422–1432. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radical Biology & Medicine. 2005b;38:1422–1432. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Manevich Y, Hutchens S, Tew KD, Townsend DM. Allelic variants of glutathione S-transferase P1-1 differentially mediate the peroxidase function of per-oxiredoxin VI and alter membrane lipid peroxidation. Free Radical Biology & Medicine. 2012;54C:62–70. doi: 10.1016/j.freeradbiomed.2012.10.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevich Y, Hutchens S, Tew KD, Townsend DM. Allelic variants of glutathione S-transferase P1-1 differentially mediate the peroxidase function of peroxiredoxin VI and alter membrane lipid peroxidation. Free Radical Biology & Medicine. 2013;54:62–70. doi: 10.1016/j.freeradbiomed.2012.10.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevich Y, Townsend DM, Hutchens S, Tew KD. Diazeniumdiolate mediated nitrosative stress alters nitric oxide homeostasis through intracellular calcium and S-glutathionylation of nitric oxide synthetase. PloS One. 2010;5:e14151. doi: 10.1371/journal.pone.0014151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino SM, Gladyshev VN. Structural analysis of cysteine S-nitrosylation: A modified acid-based motif and the emerging role of trans-nitrosylation. Journal of Molecular Biology. 2010;395:844–859. doi: 10.1016/j.jmb.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MC, Andriantsitohaina R. Reactive nitrogen species: Molecular mechanisms and potential significance in health and disease. Antioxidants & Redox Signaling. 2009;11:669–702. doi: 10.1089/ars.2007.1993. [DOI] [PubMed] [Google Scholar]
- McIlwain CC, Townsend DM, Tew KD. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene. 2006;25:1639–1648. doi: 10.1038/sj.onc.1209373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieyal JJ, Starke DW, Gravina SA, Dothey C, Chung JS. Thioltransferase in human red blood cells: Purification and properties. Biochemistry. 1991;30:6088–6097. doi: 10.1021/bi00239a002. [DOI] [PubMed] [Google Scholar]
- Mieyal JJ, Starke DW, Gravina SA, Hocevar BA. Thioltransferase in human red blood cells: Kinetics and equilibrium. Biochemistry. 1991;30:8883–8891. doi: 10.1021/bi00100a023. [DOI] [PubMed] [Google Scholar]
- Mollica JP, Dutka TL, Merry TL, Lamboley CR, McConell GK, McKenna MJ, et al. S-glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. The Journal of Physiology. 2012;590:1443–1463. doi: 10.1113/jphysiol.2011.224535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco R, Friedman FK, Hyde MJ, Chen JM, Manolatus S, Adler V, et al. Identification of a glutathione-S-transferase effector domain for inhibition of jun kinase, by molecular dynamics. Journal of Protein Chemistry. 1999;18:859–866. doi: 10.1023/a:1020679229110. [DOI] [PubMed] [Google Scholar]
- Niwa T, Naito C, Mawjood AH, Imai K. Increased glutathionyl hemoglobin in diabetes mellitus and hyperlipidemia demonstrated by liquid chromatography/electrospray ionization-mass spectrometry. Clinical Chemistry. 2000;46:82–88. [PubMed] [Google Scholar]
- O’Dwyer PJ, LaCreta F, Nash S, Tinsley PW, Schilder R, Clapper ML, et al. Phase I study of thiotepa in combination with the glutathione transferase inhibitor ethacrynic acid. Cancer Research. 1991;51:6059–6065. [PubMed] [Google Scholar]
- Pal A, Hu X, Zimniak P, Singh SV. Catalytic efficiencies of allelic variants of human glutathione S-transferase Pi in the glutathione conjugation of alpha, beta-unsaturated aldehydes. Cancer Letters. 2000;154:39–43. doi: 10.1016/s0304-3835(00)00390-6. [DOI] [PubMed] [Google Scholar]
- Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. The Journal of Biological Chemistry. 2009;284:23364–23374. doi: 10.1074/jbc.M109.021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskin AV, Winterbourn CC. Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phosphate dehydrogenase. Free Radical Biology & Medicine. 2006;40:45–53. doi: 10.1016/j.freeradbiomed.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Petit E, Michelet X, Rauch C, Bertrand-Michel J, Terce F, Legouis R, et al. Glutathione transferases kappa 1 and kappa 2 localize in peroxisomes and mitochondria, respectively, and are involved in lipid metabolism and respiration in Caenorhabditis elegans. The FEBS Journal. 2009;276:5030–5040. doi: 10.1111/j.1742-4658.2009.07200.x. [DOI] [PubMed] [Google Scholar]
- Petrushanko IY, Yakushev S, Mitkevich VA, Kamanina YV, Ziganshin RH, Meng X, et al. S-glutathionylation of the Na, K-ATPase catalytic alpha subunit is a determinant of the enzyme redox sensitivity. The Journal of Biological Chemistry. 2012;287:32195–32205. doi: 10.1074/jbc.M112.391094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew NE, Colman RF. Heterodimers of glutathione S-transferase can form between isoenzyme classes pi and mu. Archives of Biochemistry and Biophysics. 2001;396:225–230. doi: 10.1006/abbi.2001.2629. [DOI] [PubMed] [Google Scholar]
- Ploemen JH, Van Schanke A, Van Ommen B, Van Bladeren PJ. Reversible conjugation of ethacrynic acid with glutathione and human glutathione S-transferase p11. Cancer Research. 1994;54:915–919. [PubMed] [Google Scholar]
- Pompella A. Redox events and cell membrane proteins. Archives of Biochemistry and Biophysics. 2005;434:1–2. doi: 10.1016/j.abb.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Ralat LA, Manevich Y, Fisher AB, Colman RF. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry. 2006;45:360–372. doi: 10.1021/bi0520737. [DOI] [PubMed] [Google Scholar]
- Ralat LA, Misquitta SA, Manevich Y, Fisher AB, Colman RF. Characterization of the complex of glutathione S-transferase pi and 1-cysteine peroxiredoxin. Archives of Biochemistry and Biophysics. 2008;474:109–118. doi: 10.1016/j.abb.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: Implications in oxidative stress, toxicity and disease. The FEBS Journal. 2011;278:4243–4251. doi: 10.1111/j.1742-4658.2011.08358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza A, Galili N, Callander N, Ochoa L, Piro L, Emanuel P, et al. Phase 1-2a multicenter dose-escalation study of ezatiostat hydrochloride liposomes for injection (Telintra, TLK199), a novel glutathione analog prodrug in patients with myelodysplastic syndrome. Journal of Hematology & Oncology. 2009;2:20. doi: 10.1186/1756-8722-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG, Kang SW, Chang TS, Jeong W, Kim K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life. 2001;52:35–41. doi: 10.1080/15216540252774748. [DOI] [PubMed] [Google Scholar]
- Ricci G, De Maria F, Antonini G, Turella P, Bullo A, Stella L, et al. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. The Journal of Biological Chemistry. 2005;280:26397–26405. doi: 10.1074/jbc.M503295200. [DOI] [PubMed] [Google Scholar]
- Rolland D, Raharijaona M, Barbarat A, Houlgatte R, Thieblemont C. Inhibition of GST-pi nuclear transfer increases mantle cell lymphoma sensitivity to cisplatin, cytarabine, gemcitabine, bortezomib and doxorubicin. Anticancer Research. 2010;30:3951–3957. [PubMed] [Google Scholar]
- Romero L, Andrews K, Ng L, O’Rourke K, Maslen A, Kirby G. Human GSTA1-1 reduces c-Jun N-terminal kinase signalling and apoptosis in Caco-2 cells. The Biochemical Journal. 2006;400:135–141. doi: 10.1042/BJ20060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscoe JE, Rosario LA, Wang T, Gate L, Arifoglu P, Wolf CR, et al. Pharmacologic or genetic manipulation of glutathione S-transferase P1-1 (GSTpi) influences cell proliferation pathways. The Journal of Pharmacology and Experimental Therapeutics. 2001;298:339–345. [PubMed] [Google Scholar]
- Saavedra JE, Srinivasan A, Buzard GS, Davies KM, Waterhouse DJ, Inami K, et al. PABA/NO as an anticancer lead: Analogue synthesis, structure revision, solution chemistry, reactivity toward glutathione, and in vitro activity. Journal of Medicinal Chemistry. 2006;49:1157–1164. doi: 10.1021/jm050700k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. The EMBO Journal. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez G, Escobar M, Pedrozo Z, Macho P, Domenech R, Hartel S, et al. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: Possible role in cardioprotection. Cardiovascular Research. 2008;77:380–386. doi: 10.1093/cvr/cvm011. [DOI] [PubMed] [Google Scholar]
- Satyam A, Hocker MD, Kane-Maguire KA, Morgan AS, Villar HO, Lyttle MH. Design, synthesis, and evaluation of latent alkylating agents activated by glutathione S-transferase. Journal of Medicinal Chemistry. 1996;39:1736–1747. doi: 10.1021/jm950005k. [DOI] [PubMed] [Google Scholar]
- Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: Role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxidants & Redox Signaling. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- Singh SP, Wishnok JS, Keshive M, Deen WM, Tannenbaum SR. The chemistry of the S-nitrosoglutathione/glutathione system. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:14428–14433. doi: 10.1073/pnas.93.25.14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh Y, Goto S, Kitajima M, Moriyama S, Kotera K, Nakayama T, et al. Nuclear localisation of glutathione S-transferase pi is an evaluation factor for drug resistance in gynaecological cancers. Clinical Oncology (Royal College of Radiologists) 2005;17:264–270. doi: 10.1016/j.clon.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ. Structure-function aspects in the nitric oxide synthases. Annual Review of Pharmacology and Toxicology. 1997;37:339–359. doi: 10.1146/annurev.pharmtox.37.1.339. [DOI] [PubMed] [Google Scholar]
- Sun R, Eriksson S, Wang L. Oxidative stress induced S-glutathionylation and proteolytic degradation of mitochondrial thymidine kinase 2. The Journal of Biological Chemistry. 2012;287:24304–24312. doi: 10.1074/jbc.M112.381996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellez-Sanz R, Cesareo E, Nuccetelli M, Aguilera AM, Baron C, Parker LJ, et al. Calorimetric and structural studies of the nitric oxide carrier S-nitrosoglutathione bound to human glutathione transferase p1-1. Protein Science. 2006;15:1093–1105. doi: 10.1110/ps.052055206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Research. 1994;54:4313–4320. [PubMed] [Google Scholar]
- Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, Townsend DM. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radical Biology & Medicine. 2011;51:299–313. doi: 10.1016/j.freeradbiomed.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew KD, Townsend DM. Redox platforms in cancer drug discovery and development. Current Opinion in Chemical Biology. 2011a;15:156–161. doi: 10.1016/j.cbpa.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew KD, Townsend DM. Regulatory functions of glutathione S-transferase P1-1 unrelated to detoxification. Drug Metabolism Reviews. 2011b;43:179–193. doi: 10.3109/03602532.2011.552912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenin AF, Zony CL, Bahnson BJ, Colman RF. GST pi modulates JNK activity through a direct interaction with JNK substrate, ATF2. Protein Science. 2011;20:834–848. doi: 10.1002/pro.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM. S-glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Molecular Interventions. 2007;7:313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Findlay VJ, Fazilev F, Ogle M, Fraser J, Saavedra JE, et al. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Molecular Pharmacology. 2006;69:501–508. doi: 10.1124/mol.105.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. The Journal of Biological Chemistry. 2009a;284:436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. The Journal of Biological Chemistry. 2009b;284:436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend D, Tew K. Cancer drugs, genetic variation and the glutathione-S-transferase gene family. American Journal of Pharmacogenomics. 2003;3:157–172. doi: 10.2165/00129785-200303030-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomedicine & Pharmacotherapy. 2003;57:145–155. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turella P, Cerella C, Filomeni G, Bullo A, De Maria F, Ghibelli L, et al. Proapoptotic activity of new glutathione S-transferase inhibitors. Cancer Research. 2005;65:3751–3761. doi: 10.1158/0008-5472.CAN-04-3903. [DOI] [PubMed] [Google Scholar]
- Vanin AF. Dinitrosyl iron complexes with thiolate ligands: Physico-chemistry, biochemistry and physiology. Nitric Oxide. 2009;21:1–13. doi: 10.1016/j.niox.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Vasieva O. The many faces of glutathione transferase pi. Current Molecular Medicine. 2011;11:129–139. doi: 10.2174/156652411794859278. [DOI] [PubMed] [Google Scholar]
- Vergote I, Finkler N, del Campo J, Lohr A, Hunter J, Matei D, et al. Phase 3 randomised study of canfosfamide (Telcyta, TLK286) versus pegylated liposomal doxorubicin or topotecan as third-line therapy in patients with platinum-refractory or -resistant ovarian cancer. European Journal of Cancer. 2009;45:2324–2332. doi: 10.1016/j.ejca.2009.05.016. [DOI] [PubMed] [Google Scholar]
- Wang T, Arifoglu P, Ronai Z, Tew KD. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. The Journal of Biological Chemistry. 2001;276:20999–21003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: Relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19:275–280. doi: 10.1093/carcin/19.2.275. [DOI] [PubMed] [Google Scholar]
- West MB, Hill BG, Xuan YT, Bhatnagar A. Protein glutathiolation by nitric oxide: An intracellular mechanism regulating redox protein modification. The FASEB Journal. 2006;20:1715–1717. doi: 10.1096/fj.06-5843fje. [DOI] [PubMed] [Google Scholar]
- Wink DA, Nims RW, Darbyshire JF, Christodoulou D, Hanbauer I, Cox GW, et al. Reaction kinetics for nitrosation of cysteine and glutathione in aerobic nitric oxide solutions at neutral pH. Insights into the fate and physiological effects of intermediates generated in the NO/O2 reaction. Chemical Research in Toxicology. 1994;7:519–525. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- Wu Y, Fan Y, Xue B, Luo L, Shen J, Zhang S, et al. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene. 2006;25:5787–5800. doi: 10.1038/sj.onc.1209576. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxidants & Redox Signaling. 2011;15:233–270. doi: 10.1089/ars.2010.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Wu Y, Yin Z, Zhang H, Sun S, Yi T, et al. Regulation of lipopolysaccharide-induced inflammatory response by glutathione S-transferase P1 in RAW264.7 cells. FEBS Letters. 2005;579:4081–4087. doi: 10.1016/j.febslet.2005.06.034. [DOI] [PubMed] [Google Scholar]
- Zimniak P, Nanduri B, Pikula S, Bandorowicz-Pikula J, Singhal SS, Srivastava SK, et al. Naturally occurring human glutathione S-transferase GSTP1-1 isoforms with isoleucine and valine in position 104 differ in enzymic properties. European Journal of Biochemistry. 1994;224:893–899. doi: 10.1111/j.1432-1033.1994.00893.x. [DOI] [PubMed] [Google Scholar]