

Abstract
Objectives
Polymyxin B is being increasingly utilized as a last resort against resistant Gram-negative bacteria. We examined the pharmacodynamics of novel dosing strategies for polymyxin B combinations to maximize efficacy and minimize the emergence of resistance and drug exposure against Acinetobacter baumannii.
Methods
The pharmacodynamics of polymyxin B together with doripenem were evaluated in time–kill experiments over 48 h against 108 cfu/mL of two polymyxin-heteroresistant A. baumannii isolates (ATCC 19606 and N16870). Pharmacokinetic/pharmacodynamic relationships were mathematically modelled using S-ADAPT. A hollow-fibre infection model (HFIM) was also used to simulate clinically relevant polymyxin B dosing strategies (traditional, augmented ‘front-loaded’ and ‘burst’ regimens), together with doripenem, against an initial inoculum of 109 cfu/mL of ATCC 19606.
Results
In static time–kill studies, polymyxin B concentrations >4 mg/L in combination with doripenem 25 mg/L resulted in rapid bactericidal activity against both strains with undetectable bacterial counts by 24 h. The mathematical model described the rapid, concentration-dependent killing as subpopulation and mechanistic synergy. In the HFIM, the traditional polymyxin B combination regimen was synergistic, with a >7.5 log10 reduction by 48 h. The polymyxin B ‘front-loaded’ combination resulted in more rapid and extensive initial killing (>8 log10) within 24 h, which was sustained over 10 days. With only 25% of the cumulative drug exposure, the polymyxin B ‘burst’ combination demonstrated antibacterial activity similar to traditional and ‘front-loaded’ combination strategies. The polymyxin B ‘front-loaded’ and ‘burst’ combination regimens suppressed the emergence of resistance.
Conclusions
Early aggressive dosing regimens for polymyxin combinations demonstrate promise for treatment of heteroresistant A. baumannii infections.
Introduction
The emergence of antimicrobial resistance is a major threat to human health and is jeopardizing our ability to treat infections.1 Gram-negative resistance is at the top of the infectious diseases research agenda,2 as new therapeutic options are urgently needed. The CDC has highlighted Acinetobacter baumannii, a Gram-negative ‘superbug’, as a serious threat to human health.3 Gram-negative pathogens account for 70% of the infections among intensive care unit patients and there has been an alarming increase in the incidence of nosocomial infections caused by A. baumannii.4
A. baumannii is an extremely difficult pathogen to treat as it has an ability to adapt through multiple mechanisms of resistance in the face of antibiotic therapy. Clinicians have already been confronted with the reality of infections caused by A. baumannii strains that are resistant to all clinically available agents except polymyxins (polymyxin B and colistin) and tigecycline.5,6 Together with the first report of plasmid-mediated polymyxin resistance in E. coli that could be transferred to other Gram-negative strains,7 the reports send a very strong warning. Colistin is administered as an inactive prodrug, colistin methanesulfonate (CMS), and requires conversion in vivo into the active form of colistin, a process that occurs slowly and incompletely in humans.8 Polymyxin B does not suffer from these limitations as it is administered as the active antibiotic. The pharmacokinetic (PK) and pharmacodynamic (PD) knowledge necessary to optimize dosing for polymyxin B is limited,9,10 and suboptimal dosing at currently recommended doses may have contributed to increased resistance in Gram-negative pathogens.10,11 Dose escalation of daily doses of polymyxin B is impractical as nephrotoxicity is a major dose-limiting adverse effect that occurs in up to 60% of patients even with currently recommended dosage regimens as administered.12–14 Polymyxin B combination therapy has been shown to be more effective against MDR A. baumannii strains as compared with monotherapy with polymyxin B.15,16 Previous in vitro studies have shown that polymyxin combinations with a carbapenem are highly bactericidal against A. baumannii17 although the rapid bactericidal activity of this combination is followed by regrowth with the emergence of resistant subpopulations.18 Therefore, this underscores the need to develop novel strategies to combat A. baumannii focused on polymyxin combinations to maximize bactericidal activity, minimize resistance and decrease drug exposure.
In an attempt to define optimal dosing strategies for polymyxin B combinations with doripenem, we first examined via static time–kill studies the concentration–response relationship across a wide range of polymyxin B and doripenem concentrations alone and in combination against A. baumannii. Next, we employed mechanism-based mathematical models to guide the design of new polymyxin B combination regimens. Finally, we simulated traditional and novel polymyxin B combination regimens in the hollow-fibre infection model (HFIM) and profiled the dynamics of subpopulations over 10 days.
Materials and methods
Antibiotics, medium and bacterial isolates
Analytical grade polymyxin B sulphate was purchased from Sigma-Aldrich (St Louis, MO, USA) and doripenem was provided by Shionogi Inc. (Florham Park, NJ, USA). Fresh stock solutions of polymyxin B and doripenem were prepared immediately prior to each experiment and filter sterilized using a 0.22 μm Millex GP filter (Millipore, Bedford, MA, USA). Cation-adjusted Mueller–Hinton broth (CAMHB) (Difco, Detroit, MI, USA) supplemented with calcium (25 mg/L) and magnesium (12.5 mg/L) was used as the growth medium for all time–kill and HFIM experiments.
MIC values were determined in quadruplicate according to CLSI guidelines.19 A. baumannii ATCC 19606 (polymyxin B MIC 0.5 mg/L; doripenem MIC 2 mg/L) and a clinical isolate N16870 (polymyxin B MIC 0.5 mg/L; doripenem MIC 16 mg/L) were studied. Both isolates were polymyxin heteroresistant, demonstrating the presence of subpopulations on agar plates containing >2 mg/L polymyxin B.20
Time–kill experiments
Static time–kill experiments were performed as previously described21 against a starting inoculum (108 cfu/mL) over 48 h to characterize the concentration–effect relationship of polymyxin B and doripenem. In brief, prior to each experiment, bacterial isolates were subcultured on Mueller–Hinton agar and incubated overnight at 37°C. Bacteria from the overnight growth were added to CAMHB to produce a bacterial suspension that was subsequently diluted to provide the starting inoculum mentioned above. Serial samples were collected at 0, 1, 2, 4, 6, 8, 24, 28, 32 and 48 h for the determination of bacterial counts. After appropriate dilutions with saline, samples of bacterial cell suspension (50 μL) were spirally plated on Mueller–Hinton agar plates using a Whitley automatic spiral plater (WASP; Don Whitley Scientific, Shipley West Yorkshire, UK). Colonies were counted by a ProtoCOL automated colony counter (Synbiosis, Cambridge, UK) after an incubation period of 24 h at 37°C. The lower limit of counting was 20 cfu/mL. Time–kill experiments were conducted at polymyxin B concentrations of 0.5, 1, 2, 4, 8, 16, 32 and 64 mg/L, and a doripenem concentration of 25 mg/L, alone and in combination.
HFIM
The HFIM was used to evaluate the effect of selected polymyxin B and doripenem monotherapy and combination regimens on bacterial burden and suppression of resistance against A. baumannii ATCC 19606 over 240 h, as previously described.22 Cellulosic hollow-fibre cartridges (C3008; Fiber Cell System, Inc., Fredrick, MD, USA) were utilized in all experiments. Twenty-three samples (0.5 mL) were serially collected over 240 h to determine bacterial counts for each experiment. Samples were quantified for the total population by depositing appropriately diluted bacterial samples on CAMHA plates, as previously described.23 Aliquots of the diluted sample were plated on CAMHA plates containing 0.5, 1, 2, 3, 4, 6, 8 or 10 mg/L polymyxin B for in-depth analysis of the time-course of resistant subpopulations at pre-dose at 24, 48, 72, 96, 144, 192 and 240 h, as previously described.20
HFIM experiments consisted of clinically achievable monotherapy and combination therapy regimens, which were informed by mechanism-based population modelling of the static time–kill experiments. The rationale for dosing polymyxin B as a continuous infusion was based on the flat PK profile seen in critically ill patients receiving polymyxin B. Patients in this study received polymyxin B doses ranging from 0.45 to 3.38 mg/kg/day every 12 h. Based on this previous study, the unscaled population estimate of the half-life was 11.9 h and fAUCs ranged from 6.2 to 53.5 mg·h/L with a median of 26.5 mg·h/L, average of 29.4 mg·h/L and standard deviation of 10.4 mg·h/L.24 The experimental design consisted of a no treatment control arm and eight monotherapy and combination regimens, as follows:
Polymyxin B traditional monotherapy: free steady-state concentration (fCss) of 2 mg/L administered as a continuous infusion, based on the PK in critically ill patients.24
Polymyxin B traditional monotherapy: as above but with a fCss of 5 mg/L administered as a continuous infusion.
Doripenem monotherapy: fCmax of 25 mg/L every 8 h based on PK in critically ill patients.25
Combination of polymyxin B traditional (fCss of 2 mg/L continuous infusion) and doripenem (fCmax of 25 mg/L every 8 h).
Combination of polymyxin B ‘front-loaded’ (fCss of 5 mg/L continuous infusion for 24 h followed by fCss of 2 mg/L thereafter) and doripenem (fCmax of 25 mg/L every 8 h).
Combination of polymyxin B ‘burst 2’ (fCss of 2 mg/L continuous infusion for 24 h followed by no polymyxin B thereafter) and doripenem (fCmax of 25 mg/L every 8 h).
Combination of polymyxin B ‘burst 5’ (fCss of 5 mg/L continuous infusion for 24 h followed by no polymyxin B thereafter) and doripenem (fCmax of 25 mg/L every 8 h).
Combination of doripenem ‘burst’ (fCmax of 25 mg/L every 8 h × 3 doses followed by no doripenem thereafter) and polymyxin B traditional regimen (fCss of 2 mg/L continuous infusion).
Polymyxin B concentrations were quantified by analysing polymyxin B1 and B2 using a validated high-performance LC-MS/MS approach as previously described by Cheah et al.26 Analysis of independently prepared quality control samples indicated good reproducibility (coefficients of variation ≤8.39%) and accuracy (observed concentrations were within ≤10.0% of target concentrations). The limit of quantification was 0.1 mg/L.
Population mathematical modelling of static time–kill data
Mechanism-based population PD modelling was performed to estimate the exposure–response relationship for time–kill studies for both bacterial isolates to propose front-loaded and other novel regimens for evaluation in the HFIM. Models with differing susceptibilities to polymyxin B and doripenem were considered. The final number of pre-existing subpopulations was based on the objective function, plausibility of parameter estimates, standard diagnostic plots and visual predictive checks. The total bacterial population was modelled as the sum of these pre-existing subpopulations. The final model consisted of four subpopulations for ATCC 19606 and N16870 (Figure S1, available as Supplementary data at JAC Online). Bacterial replication was modelled using the life-cycle growth replication model,21 in which each subpopulation was described by two states: a vegetative state and a replicating state. The two states had the same susceptibility but differed in their growth phase. The action of polymyxin B was described using a target site-binding model, and bacterial killing by polymyxin B was modelled as a second-order process. The killing effect of doripenem was described by a first-order saturable process.
Modelling the combinatorial PD of polymyxin B and doripenem in the HFIM
The interaction between polymyxin B and doripenem was modelled by considering two different synergistic interactions for the combination. Polymyxin B and doripenem are not subject to the same resistance mechanisms.27,28 Hence, the time-course of polymyxin B and doripenem interactions was modelled as ‘subpopulation synergy’, in which polymyxin B killed the doripenem-resistant subpopulations and vice versa. The second synergistic interaction was ‘mechanistic synergy’, in which the killing activity of doripenem was enhanced by polymyxin B and vice versa. The final model included both proposed synergistic interactions of subpopulation and mechanistic synergy (see the Supplementary data available at JAC Online, including Figure S1 and the differential equations).
Model estimation
Candidate models describing the time-course of combination drug effects on bacteria were simultaneously fitted to all viable total population count profiles of the two isolates studied in time–kill experiments. Estimation was performed using parallelized S-ADAPT software (version 1.57) facilitated by SADAPT-TRAN using the importance sampling Monte Carlo parametric expectation maximization method (pmethod = 4).29 An additive residual error model on a log10 scale was used for bacterial counts ≥100 cfu/mL. To account for high sampling error at low concentrations, an error model containing both proportional and Poisson distributions was used for the bacteria.21
Results
The concentration–response profiles for polymyxin B alone and in combination with doripenem against two A. baumannii isolates with a starting inoculum of 108 cfu/mL based on static time–kill studies are shown in Figure 1. The profiles with polymyxin B concentrations of 0.5, 1.0 and 2.0 mg/L against both isolates (Figure 1a and b) were no different from the growth control. High polymyxin B concentrations of 16, 32 and 64 mg/L resulted in early bactericidal activity by 8 h. However, extensive regrowth was observed for all polymyxin B concentrations except at the highest concentration of 64 mg/L. Doripenem monotherapy displayed minimal activity against both isolates with an inoculum of 108 cfu/mL (Figure 1c and d). Interestingly, the combination of polymyxin B (across the concentration range 2–8 mg/L) and doripenem (25 mg/L) was markedly synergistic against both isolates. Against a high bacterial density of ATCC 19606 (Figure 1c) and N16870 (Figure 1d) polymyxin B ≥2 mg/L in combination with doripenem demonstrated rapid bactericidal activity, while polymyxin B ≥4 mg/L in combination with doripenem resulted in undetectable bacterial counts by 28 h.
Figure 1.

Observed and fitted viable counts for the time–kill experiments for mono and combination therapies with polymyxin B and doripenem against ATCC 19606 (a, c) and N16870 (b, d). PMB, polymyxin B; DOR, doripenem. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
The mathematical model described the rapid, concentration-dependent killing of polymyxin B based on known mechanisms of action of polymyxin B and doripenem. The model described the time-course of bacterial killing and regrowth well (Figure 1). Population predictions were reasonably precise and unbiased (Figure 2). The model diagnostics generated based on observed viable counts versus individual (population) fitted shown in Figure 2(a–d) indicates that the individual fits for N16870 (R2 = 0.949) were more precise as compared with ATCC 19606 (R2 = 0.938). On the other hand, the population fits for ATCC 19606 (R2 = 0.856) were better as compared with those for N16870 (R2 = 0.459). Based on the estimated EC50, 55.7% and 70.8% of the binding sites for Mg2+ and Ca2+ (Table S1) are required to be unoccupied by Mg2+ or Ca2+ to achieve an effective polymyxin B concentration that is 50% of its concentration in broth for ATCC 19606 and N16870, respectively. Models with a total of four subpopulations with differing susceptibilities to polymyxin B and doripenem were adequate to describe the antibacterial activity of polymyxin B and doripenem.
Figure 2.

Observed versus individual fitted (population fitted) for ATCC 19606 (a, b) and N16870 (c, d).
Based on the modelling of the time–kill data, polymyxin B achieved 7.4-fold greater killing against the susceptible populations of N16870 as compared with ATCC 19606, and killing of the polymyxin-resistant subpopulations was much slower for both strains (Table S1). In the model for doripenem monotherapy, the maximum killing rate constant (KmaxSS) of the doripenem-susceptible subpopulation was 1.19 h−1 for ATCC 19606 and 1.73 h−1 for N16870. The killing of the doripenem-resistant subpopulation by doripenem was estimated to be very slow. The combination was modelled as polymyxin B effectively killing the doripenem-resistant subpopulations and doripenem killing the polymyxin B intermediate and resistant subpopulations with proposed synergistic interactions between subpopulations and mechanisms (Figure S1).
In the HFIM, polymyxin B traditional monotherapy (fCss of 2 mg/L continuous infusion) was bacteriostatic against A. baumannii strain ATCC 19606, with <1 log10 cfu/mL killing over 240 h compared with the growth control (Figure 3 and Figure 4). Emergence of resistance by 24 h in real-time population analysis profiles confirmed the emergence of resistance secondary to monotherapy with polymyxin B, with subpopulations growing on agar plates containing 0–10 mg/L polymyxin B (Figure 4 and Figure 5). Increasing the exposure to polymyxin B in the HFIM with a polymyxin B traditional monotherapy regimen (fCss of 5 mg/L continuous infusion) resulted in initial killing up to 3 log10 in the first 6 h, followed by substantial regrowth (Figure 3). Polymyxin B traditional monotherapy regimens with fCss of 2 and 5 mg/L showed similar activity against the total population beyond 8 h and with respect to the emergence of polymyxin resistance (Figure 5a and b). Doripenem traditional monotherapy (Figure 3) demonstrated bacteriostatic activity over the entire duration of the experiment and did not alter the emergence of resistance to polymyxin B (Figure 5c).
Figure 3.
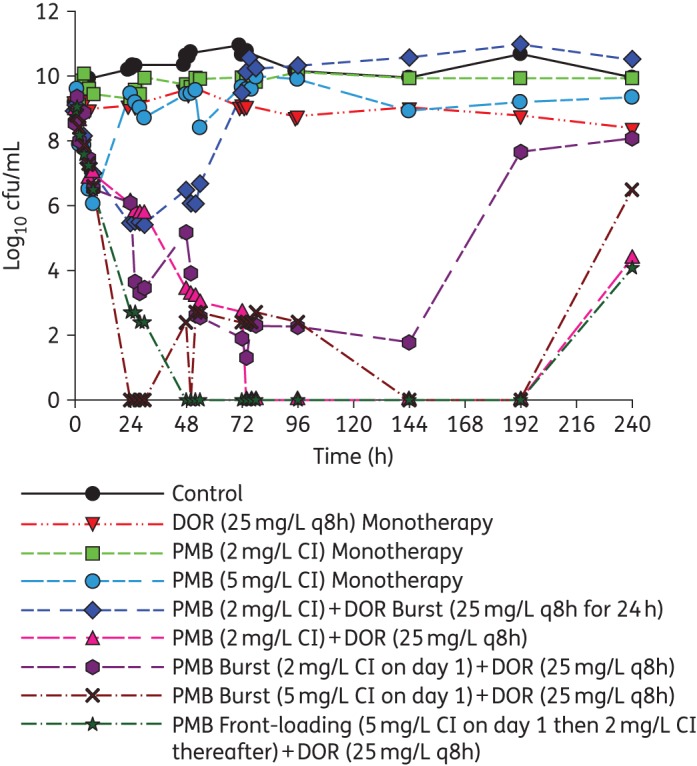
An HFIM was utilized to simulate various mono and combination regimens against a high inoculum of 109 cfu/mL of A. baumannii ATCC 19606. PMB, polymyxin B; DOR, doripenem. The total bacterial counts for the following regimens simulated: Control, growth control with no antibiotic; DOR (25 mg/L q8h) monotherapy, DOR traditional monotherapy (fCmax of 25 mg/L every 8 h); PMB (2 mg/L CI) monotherapy, PMB traditional monotherapy (fCss of 2 mg/L administered as a continuous infusion); PMB (5 mg/L CI) monotherapy, fCss of 5 mg/L administered as a continuous infusion; PMB (2 mg/L CI) + DOR Burst (25 mg/L q8h for 24 h), combination of PMB traditional (fCss of 2 mg/L continuous infusion) and DOR ‘burst’ (fCmax of 25 mg/L every 8 h × 3 doses followed by no DOR thereafter); PMB (2 mg/L CI) + DOR (25 mg/L q8h), combination of PMB traditional (fCss of 2 mg/L continuous infusion) and DOR (fCmax of 25 mg/L every 8 h); PMB Burst (2 mg/L CI on day 1) + DOR (25 mg/L q8h), combination of PMB ‘burst’ (fCss of 5 mg/L continuous infusion for 24 h followed by no PMB thereafter) and DOR (fCmax of 25 mg/L every 8 h); PMB Burst (5 mg/L CI on day 1) + DOR (25 mg/L q8h), combination of PMB ‘burst’ (fCss of 2 mg/L continuous infusion for 24 h followed by no PMB thereafter) and DOR (fCmax of 25 mg/L every 8 h); PMB Front-Loading (5 mg/L CI on day 1 then 2 mg/L CI thereafter) + DOR (25 mg/L q8h), combination of PMB ‘front-loading’ (fCss of 5 mg/L continuous infusion for 24 h followed by fCss of 2 mg/L thereafter) and DOR (fCmax of 25 mg/L every 8 h). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Figure 4.
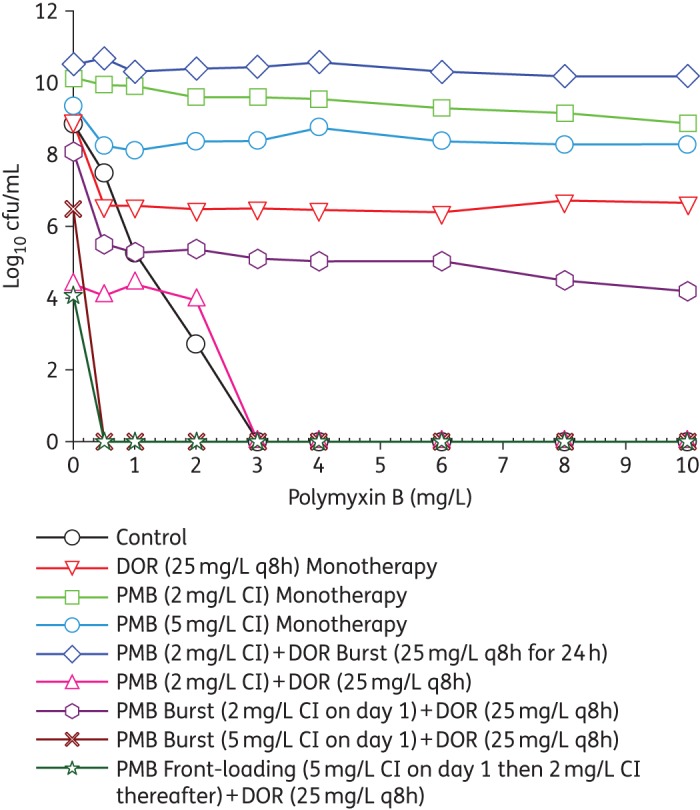
Population analysis profiles at 240 h profiling the emergence of resistant subpopulations grown on polymyxin B-containing agar for the regimens shown in Figure 3. PMB, polymyxin B; DOR, doripenem. Control, bacterial subpopulations present at time 0 h (without any treatment); DOR (25 mg/L q8h) monotherapy, DOR traditional monotherapy (fCmax of 25 mg/L every 8 h); PMB (2 mg/L CI) monotherapy, PMB traditional monotherapy (fCss of 2 mg/L administered as a continuous infusion); PMB (5 mg/L CI) monotherapy, fCss of 5 mg/L administered as a continuous infusion; PMB (2 mg/L CI) + DOR Burst (25 mg/L q8h for 24 h), combination of PMB traditional (fCss of 2 mg/L continuous infusion) and DOR ‘burst’ (fCmax of 25 mg/L every 8 h × 3 doses followed by no DOR thereafter); PMB (2 mg/L CI) + DOR (25 mg/L q8h), combination of PMB traditional (fCss of 2 mg/L continuous infusion) and DOR (fCmax of 25 mg/L every 8 h); PMB Burst (2 mg/L CI on day 1) + DOR (25 mg/L q8h), combination of PMB ‘burst’ (fCss of 5 mg/L continuous infusion for 24 h followed by no PMB thereafter) and DOR (fCmax of 25 mg/L every 8 h); PMB Burst (5 mg/L CI on day 1) + DOR (25 mg/L q8h), combination of PMB ‘burst’ (fCss of 2 mg/L continuous infusion for 24 h followed by no PMB thereafter) and DOR (fCmax of 25 mg/L every 8 h); PMB Front-Loading (5 mg/L CI on day 1 then 2 mg/L CI thereafter) + DOR (25 mg/L q8h), combination of PMB ‘front-loading’ (fCss of 5 mg/L continuous infusion for 24 h followed by fCss of 2 mg/L thereafter) and DOR (fCmax of 25 mg/L every 8 h). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Figure 5.

Time-course of the observed emergence of resistance on polymyxin B-containing agar plate regimens with an initial inoculum of ∼109 cfu/mL in response to the detailed regimens simulated in the HFIM. (a) Polymyxin B monotherapy (2 mg/L CI), fCss of 2 mg/L administered as a continuous infusion. (b) Polymyxin B monotherapy (5 mg/L CI), fCss of 5 mg/L administered as a continuous infusion. (c) Doripenem monotherapy (25 mg/L every 8 h), doripenem traditional monotherapy: fCmax of 25 mg/L every 8 h. (d) Doripenem Burst (25 mg/L every 8 h for 24 h thereafter no doses) + Polymyxin B (2 mg/L CI), combination of doripenem ‘burst’ (fCmax of 25 mg/L every 8 h × 3 doses followed by no doripenem thereafter) and polymyxin B traditional (fCss of 2 mg/L as a continuous infusion), (e) Polymyxin B Traditional (2 mg/L CI) + Doripenem (25 mg/L every 8 h), combination of polymyxin B traditional (fCss of 2 mg/L continuous infusion) and doripenem (fCmax of 25 mg/L every 8 h). (f) Polymyxin B ‘Burst 5’ (5 mg/L CI thereafter no doses) + Doripenem (25 mg/L every 8 h), combination of polymyxin B ‘burst’ (fCss of 5 mg/L continuous infusion for 24 h followed by no polymyxin B thereafter) and doripenem (fCmax of 25 mg/L every 8 h). (g) Polymyxin B ‘Burst 2’ (2 mg/L CI thereafter no doses) + Doripenem (25 mg/L every 8 h), combination of polymyxin B ‘burst’ (fCss of 2 mg/L continuous infusion for 24 h followed by no polymyxin B thereafter) and doripenem (fCmax of 25 mg/L every 8 h). (h) Polymyxin B ‘Front-loading’ (5 mg/L CI for 24 h followed by 2 mg/L CI) + Doripenem (25 mg/L every 8 h), combination of polymyxin B ‘front-loading’ (fCss of 5 mg/L continuous infusion for 24 h followed by fCss of 2 mg/L thereafter) and doripenem (fCmax of 25 mg/L every 8 h). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
The combination of the polymyxin B traditional regimen (fCss of 2 mg/L continuous infusion) and doripenem (fCmax of 25 mg/L every 8 h) was synergistic against A. baumannii ATCC 19606, with a 7.5 log10 cfu/mL reduction by 48 h. This combination regimen resulted in complete eradication at 72 h that was sustained until 192 h (Figure 3) and >3 log10 cfu/mL regrowth at 240 h (Figure 4). The cumulative reduction over time in the log ratio area of bacterial counts is presented for each regimen in Table S2. The combination of polymyxin B traditional regimen and doripenem reduced the time-averaged bacterial load at each timepoint, with a maximal reduction in AUCcfu of 3.36 log10 by 240 h compared with the growth control (Table S2). Population analysis profiles confirmed the emergence of resistance, with polymyxin-resistant subpopulations growing on agar containing 0–2 mg/L polymyxin B up to 24 h and suppression until 192 h (Figure 4).
The proposed novel dosing strategies of the combination of polymyxin B ‘front-loaded’ and doripenem and the combination of polymyxin B ‘burst’ and doripenem were also evaluated against a dense bacterial inoculum (109 cfu/mL) of A. baumannii ATCC 19606 in the HFIM (Figure 3). The polymyxin B ‘front-loaded’ combination resulted in more rapid and extensive initial killing (>8 log10 cfu/mL) with an improved time to eradication. Front-loaded combination regimens completely eradicated A. baumannii at 48 h compared with traditional combination regimens that resulted in eradication at 72 h. Additionally, there was a modest improvement in bacterial killing with the polymyxin B front-loaded regimen, with AUCcfu reduction of 3.48 log10 by 240 h compared with the growth control. Population analysis profiles for the polymyxin B ‘front-loaded’ combination regimen (Figure 5h) resulted in sustained suppression of resistance beyond 48 h until 192 h, with minimal polymyxin B resistance compared with the polymyxin B traditional combination regimen (Figure 5e).
The combination of doripenem ‘burst’ (fCmax of 25 mg/L every 8 h × 3 doses followed by no doripenem thereafter) and polymyxin B traditional (fCss of 2 mg/L continuous infusion) did not affect the total bacterial population beyond 48 h, given the dramatic regrowth after the initial ∼3 log reduction in cfu/mL between 24 and 48 h (Figure 3). The combination of polymyxin B ‘burst 5’ (fCss of 5 mg/L continuous infusion for 24 h followed by no polymyxin B thereafter) and doripenem (fCmax of 25 mg/L every 8 h) provided rapid initial and sustained killing similar to the combination of polymyxin B ‘front-loaded’ and doripenem regimen. There was a 3.49 log10 reduction in AUCcfu compared with the growth control by 240 h, and cumulative exposure to polymyxin B over 240 h was reduced to only 25% of the exposure resulting from the combination of polymyxin B traditional with doripenem (Table S2). The polymyxin B ‘burst 2’ combination regimen, i.e. combination of polymyxin B ‘burst 2’ (fCss of 2 mg/L continuous infusion for 24 h followed by no polymyxin B thereafter) and doripenem (fCmax of 25 mg/L every 8 h) resulted in >8 log10 cfu/mL reduction by 72 h with ∼6 log10 regrowth beyond 144 h, with an AUCcfu of 2.74 log10 compared with the growth control. The cumulative exposure to polymyxin B was only 10% of the combination of polymyxin B traditional with doripenem regimen (Table S2).
All polymyxin B combination regimens (‘burst’, ‘front-loaded’ and traditional) maintained extremely low bacterial counts near quantifiable limits over 192 h of the 240 h of therapy, even in the complete absence of an immune system. The traditional (Figure 5e), ‘burst 5’ (Figure 5f) and ‘front-loaded’ (Figure 5h) combination regimens demonstrated complete suppression of resistant subpopulations. Further, the ‘burst 5’ polymyxin combination regimen as a 5 mg/L continuous infusion for the first 24 h (Figure 3) was able to achieve similar early antibacterial killing activity as the front-loaded regimen and traditional regimens (Figure 3) with a comparable drug exposure profile. In other words, with only 25% of the cumulative drug exposure, the polymyxin B ‘burst 5’ combination demonstrated antibacterial killing activity similar to traditional and ‘front-loaded’ combination strategies.
Discussion
A. baumannii continues to be a pathogen of great medical significance.30 To combat this difficult-to-treat pathogen, we determined that novel dosing strategies centred on polymyxin B combinations involving ‘front-loading’ and ‘burst’ dosing were highly promising by maximizing initial killing and suppression of resistance while minimizing cumulative drug exposure and potential toxicity. Although the front-loaded regimen demonstrated increased killing, this is balanced against toxicodynamic considerations as the probability of nephrotoxicity increases with increases in cumulative exposure (AUC). Our data suggest that front-loaded regimens achieve an exposure of 552 mg·h/L with similar or greater efficacy over 10 days compared with traditional dosing regimens with an exposure of 480 mg·h/L. The ‘burst 5’ regimen showed very promising efficacy with exposure of 120 mg·h/L over 10 days. Further reduction of over 50% in polymyxin B exposure was achieved by the ‘burst 2’ regimen with an exposure of 48 mg·h/L over 10 days. Importantly, the ‘burst 5’ and ‘burst 2’ polymyxin regimens were able to achieve nearly identical antibacterial activity to traditional and ‘front-loaded’ polymyxin regimens at 25% and 10% of the cumulative polymyxin exposure, respectively. Polymyxin showed rapid bactericidal activity in vitro and the PD of polymyxin B is most closely linked to the AUC/MIC ratio.18 Based on our and previous time–kill studies, monotherapy with polymyxin B leads to rapid initial decline followed by extensive regrowth. Hence, the combination regimens with administration of polymyxin in a high-intensity fashion, based on the mechanism-based modelling, provided an effective way of addressing the subpopulations that are resistant to either polymyxin B or doripenem.
In regards to the mechanistic advantages of polymyxin combinations, we further refined a previously developed mechanism-based mathematical model for colistin monotherapy31 and colistin combinations32 based on the known mechanism of the antibacterial activity of polymyxins and doripenem. The proposed population PD model well characterized the time-course of bacterial growth and killing due to monotherapy and combination regimens against both A. baumannii isolates. This mathematical model described the rapid concentration-based killing of polymyxin B monotherapy well. As polymyxin B is structurally similar to colistin with the exception of one amino acid residue, the d-leucine at position 6,33 we based the combination mathematical model on the known mechanisms of action on the previously constructed colistin and doripenem mathematical models and applied them to polymyxin B. We similarly hypothesized that disruption of the bacterial outer membrane by binding to the lipid A of LPS resulted in increased permeability of the outer membrane that enhanced the penetration of doripenem and binding to its target site in the periplasmic membrane.34 Indeed, the model well characterized the dynamics of bacterial response to polymyxin B combinations. The refinement of this mathematical model that was now applied to polymyxin B may serve as an important framework for the design of future polymyxin-based combination regimens.
By administering polymyxin B in a non-traditional fashion, these new regimens are promising as they decrease the overall exposure while preserving high antibacterial activity. Sandri and colleagues in their PK study in 24 critically ill patients explored the lower limit of the current polymyxin B published dosing guideline of 1.5 mg/kg/day, resulting in an exposure of 228 mg·h/L over 10 days (IQR = 189–292 mg·h/L); at the upper limit of 2.5 mg/kg/day the exposure is 380 mg·h/L over 10 days (IQR = 316–487 mg·h/L).24 Pogue et al.14 evaluated the impact of a colistin loading dose of 5 mg/kg/day on the occurrence of acute kidney injury and concluded that this colistin loading dose in 64 patients out of 162 patients was not associated with an increased risk of acute kidney injury. Therefore, the early, aggressive polymyxin dosing strategies presented here may be of potential benefit for infections in critically ill patients, many of whom are already at significant risk of developing acute renal injury.
We acknowledge potential limitations in the present study. Only two A. baumannii isolates were studied: future investigations should encompass additional strains with differing MIC profiles to confirm these findings. In profiling the detailed bacterial dynamics over 10 days in HFIMs, it is important to appreciate that these results were generated in a broth nutrient-rich environment that was optimal for A. baumannii growth. Although the mathematical model was based on the known mechanisms of action of each drug, we did not identify killing of quiescent versus non-quiescent cells or quantify the relative contributions of each antibiotic to bacterial killing. The PD response of combinations presented in this investigation may also differ in biological fluids, particularly considering the differences in site-specific PK in humans. It is also important to note that interaction of polymyxin B together with doripenem may also differ in patients, as it has been shown that there is a unique relationship between granulocyte-mediated killing and antibiotics.35 Finally, there have been no controlled human studies evaluating these new high-intensity dosing regimens of polymyxin B. Overall, these new findings are promising and systemic evaluation of novel polymyxin combination dosing strategies in vivo is necessary for future translation to the clinical setting.
Funding
This study was supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (R01AI111990). C. B. L. is an Australian National Health and Medical Research Council (NHMRC) Career Development Fellow. J. L. is an NHMRC Senior Research Fellow.
Transparency declarations
None to declare.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Supplementary data
References
- 1.World Health Organisation. Antimicrobial Resistance Global Report on Surveillance 2014. [Google Scholar]
- 2.Infectious Diseases Society of America. The 10 × ‘20 initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50: 1081–3. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control. Antibiotic Resistance Threats in the United States, 2013. 2013. [Google Scholar]
- 4.Gaynes R, Edwards JR. Overview of nosocomial infections caused by gram-negative bacilli. Clin Infect Dis 2005; 41: 848–54. [DOI] [PubMed] [Google Scholar]
- 5.Gordon NC, Wareham DW. A review of clinical and microbiological outcomes following treatment of infections involving multidrug-resistant Acinetobacter baumannii with tigecycline. J Antimicrob Chemother 2009; 63: 775–80. [DOI] [PubMed] [Google Scholar]
- 6.Vasilev K, Reshedko G, Orasan R et al. A Phase 3, open-label, non-comparative study of tigecycline in the treatment of patients with selected serious infections due to resistant Gram-negative organisms including Enterobacter species, Acinetobacter baumannii and Klebsiella pneumoniae. J Antimicrob Chemother 2008; 62 Suppl 1: i29–40. [DOI] [PubMed] [Google Scholar]
- 7.Liu YY, Wang Y, Walsh TR et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 2016; 16: 161–8. [DOI] [PubMed] [Google Scholar]
- 8.Garonzik SM, Li J, Thamlikitkul V et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 2011; 55: 3284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandri AM, Landersdorfer CB, Jacob J et al. Pharmacokinetics of polymyxin B in patients on continuous venovenous haemodialysis. J Antimicrob Chemother 2013; 68: 674–7. [DOI] [PubMed] [Google Scholar]
- 10.Zavascki AP, Goldani LZ, Cao G et al. Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin Infect Dis 2008; 47: 1298–304. [DOI] [PubMed] [Google Scholar]
- 11.Gales AC, Jones RN, Sader HS. Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: results from the SENTRY Antimicrobial Surveillance Program (2006-09). J Antimicrob Chemother 2011; 66: 2070–4. [DOI] [PubMed] [Google Scholar]
- 12.Koomanachai P, Tiengrim S, Kiratisin P et al. Efficacy and safety of colistin (colistimethate sodium) for therapy of infections caused by multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii in Siriraj Hospital, Bangkok, Thailand. Int J Infect Dis 2007; 11: 402–6. [DOI] [PubMed] [Google Scholar]
- 13.Kubin CJ, Ellman TM, Phadke V et al. Incidence and predictors of acute kidney injury associated with intravenous polymyxin B therapy. J Infect 2012; 65: 80–7. [DOI] [PubMed] [Google Scholar]
- 14.Pogue JM, Lee J, Marchaim D et al. Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin Infect Dis 2011; 53: 879–84. [DOI] [PubMed] [Google Scholar]
- 15.Falagas ME, Kasiakou SK, Saravolatz LD. Colistin: the revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin Infect Dis 2005; 40: 1333–41. [DOI] [PubMed] [Google Scholar]
- 16.Paul M, Leibovici L. Combination antimicrobial treatment versus monotherapy: the contribution of meta-analyses. Infect Dis Clin North Am 2009; 23: 277–93. [DOI] [PubMed] [Google Scholar]
- 17.Bergen PJ, Tsuji BT, Bulitta JB et al. Synergistic killing of multidrug-resistant Pseudomonas aeruginosa at multiple inocula by colistin combined with doripenem in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 2011; 55: 5685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tam VH, Schilling AN, Vo G et al. Pharmacodynamics of Polymyxin B against Pseudomonas aeruginosa. Antimicrob Agents Chemother 2005; 49: 3624–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: Fifteenth Informational Supplement M100-S15. CLSI, Wayne, PA, USA, 2005. [Google Scholar]
- 20.Bergen PJ, Forrest A, Bulitta JB et al. Clinically relevant plasma concentrations of colistin in combination with imipenem enhance pharmacodynamic activity against multidrug-resistant Pseudomonas aeruginosa at multiple inocula. Antimicrob Agents Chemother 2011; 55: 5134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulitta JB, Ly NS, Yang JC et al. Development and qualification of a pharmacodynamic model for the pronounced inoculum effect of ceftazidime against Pseudomonas aeruginosa. Antimicrob Agents Chemother 2009; 53: 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuji BT, Rybak MJ, Lau KL et al. Evaluation of accessory gene regulator (agr) group and function in the proclivity towards vancomycin intermediate resistance in Staphylococcus aureus. Antimicrob Agents Chemother 2007; 51: 1089–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergen PJ, Bulitta JB, Forrest A et al. Pharmacokinetic/pharmacodynamic investigation of colistin against Pseudomonas aeruginosa using an in vitro model. Antimicrob Agents Chemother 2010; 54: 3783–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandri AM, Landersdorfer CB, Jacob J et al. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: Implications for selection of dosage regimens. Clin Infect Dis 2013; 57: 524–31. [DOI] [PubMed] [Google Scholar]
- 25.Matthews SJ, Lancaster JW. Doripenem monohydrate, a broad-spectrum carbapenem antibiotic. Clinical Therapeutics 2009; 31: 42–63. [DOI] [PubMed] [Google Scholar]
- 26.Cheah S-E, Bulitta JB, Li J et al. Development and validation of a liquid chromatography–mass spectrometry assay for polymyxin B in bacterial growth media. J Pharm Biomed Anal 2014; 92: 177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paterson DL, Depestel DD. Doripenem. Clin Infect Dis 2009; 49: 291–8. [DOI] [PubMed] [Google Scholar]
- 28.Davies TA, Shang W, Bush K et al. Affinity of doripenem and comparators to penicillin-binding proteins in Escherichia coli and Pseudomonas aeruginosa. Antimicrob Agents Chemother 2008; 52: 1510–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bulitta JB, Landersdorfer CB. Performance and robustness of the Monte Carlo importance sampling algorithm using parallelized S-ADAPT for basic and complex mechanistic models. AAPS J 2011; 13: 212–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peleg AY, Seifert H, Paterson DL. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 2008; 21: 538–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulitta JB, Yang JC, Yohonn L et al. Attenuation of colistin bactericidal activity by high inoculum of Pseudomonas aeruginosa characterized by a new mechanism-based population pharmacodynamic model. Antimicrob Agents Chemother 2010; 54: 2051–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ly NS, Bulitta JB, Rao GG et al. Colistin and doripenem combinations against Pseudomonas aeruginosa: profiling the time course of synergistic killing and prevention of resistance. J Antimicrob Chemother 2015; 70: 1434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Velkov T, Roberts KD, Nation RL et al. Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol 2013; 8: 711–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenthal KS, Storm DR. Disruption of the Escherichia coli outer membrane permeability barrier by immobilized polymyxin B. J Antibiot (Tokyo) 1977; 30: 1087–92. [DOI] [PubMed] [Google Scholar]
- 35.Drusano GL, Liu W, Kulawy R et al. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother 2011; 55: 5300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.