

Abstract
Objectives
There is uncertainty about the optimal teicoplanin regimens for neonates. The study aim was to determine the population pharmacokinetics (PK) of teicoplanin in neonates, evaluate currently recommended regimens and explore the exposure–effect relationships.
Methods
An open-label PK study was conducted. Neonates from 26 to 44 weeks post-menstrual age were recruited (n = 18). The teicoplanin regimen was a 16 mg/kg loading dose, followed by 8 mg/kg once daily. Therapeutic drug monitoring and dose adjustment were not conducted. A standard two-compartment PK model was developed, followed by models that incorporated weight. A PK/pharmacodynamic (PD) model with C-reactive protein serial measurements as the PD input was fitted to the data. Monte Carlo simulations (n = 5000) were performed using Pmetrics. The AUCs at steady state and the proportion of patients achieving the recommended drug exposures (i.e. Cmin >15 mg/L) were determined. The study was registered in the European Clinical Trials Database Registry (EudraCT: 2012-005738-12).
Results
The PK allometric model best accounted for the observed data. The PK parameters medians were: clearance = 0.435 × (weight/70)0.75 (L/h); volume = 0.765 (L); Kcp = 1.3 (h−1); and Kpc = 0.629 (h−1). The individual time-course of C-reactive protein was well described using the Bayesian posterior estimates for each patient. The simulated median AUC96-120 was 302.3 mg·h/L and the median Cmin at 120 h was 12.9 mg/L; 38.8% of patients attained a Cmin >15 mg/L by 120 h.
Conclusions
Teicoplanin population PK is highly variable in neonates, weight being the best descriptor of PK variability. A low percentage of neonates were able to achieve Cmin >15 mg/L. The routine use of therapeutic drug monitoring and improved knowledge on the PD of teicoplanin is required.
Introduction
Gram-positive bacterial pathogens are an important cause of nosocomial infection in neonates1 and risk factors include prematurity and the extensive use of central venous catheters. There is high attributable mortality and the potential for serious longer-term morbidity.1–3 Teicoplanin is a glycopeptide antibiotic with activity against methicillin-resistant staphylococci (MRSA and CoNS) and has several potential advantages over vancomycin, including better tolerability, lower risk of nephrotoxicity and improved ease of administration.4,5 In older children, there is considerable pharmacokinetic (PK) variability in comparison with adults, with a much wider drug exposure (AUC) distribution among children.6 Little is known about the pharmacokinetics (PK) of teicoplanin in neonates.
Serial concentrations of C-reactive protein (CRP) are a useful adjunct to the clinical assessment of neonates with acute infection.7 The diagnosis of neonatal bloodstream infection and its subsequent management remains challenging. Clinical signs are non-specific.2 Blood cultures are notoriously insensitive and often only intermittently positive in this population because of the low yield of small blood volumes collected.8,9 A fall in CRP is reassuring evidence of the response to antimicrobial therapy and CRP is frequently used to guide antimicrobial therapy in neonates with proven or suspected infection.10,11 Much of this decision-making has not been formalized using PK/pharmacodynamic (PD) models or dosing algorithms.
In this study, we developed a population PK/PD mathematical model to describe the serum PK of teicoplanin in neonates with the PD quantified in terms of circulating CRP concentrations. Our objectives were as follows: (i) describe and quantify measures of central tendency and inter-patient neonatal PK variability; (ii) evaluate teicoplanin exposure in the neonatal population with currently recommended regimens; and (iii) investigate the relationship between drug exposure and time-course of CRP. The latter is a first critical step for the development of algorithms to control both serum drug concentrations and clinically relevant biomarkers such as CRP.
Methods
Study design, patient population and sample collection
An open-label, hospital-based PK study using a sparse blood sampling strategy was conducted. Both pre-term and term neonates from 26 to 44 weeks post-menstrual age (PMA) were recruited from Alder Hey Children's NHS Foundation Trust and Liverpool Women's NHS Foundation Trust (Liverpool, UK).
Participants were screened and recruited according to five age categories to ensure a range of sizes was studied. The following categories were used for recruitment: 24–27, 28–31, 32–35, 36–39 and 40–44 weeks PMA. All patients that received teicoplanin for proven or suspected CoNS sepsis and/or central-line associated infection and likely to survive >72 h were eligible for the study. All patients also received ciprofloxacin or gentamicin as part of the combined empirical therapy for central-line associated bloodstream infection. Additional therapeutic interventions were the use of inotropes, diuretics, paracetamol, anticonvulsants, muscle relaxants and assisted mechanical ventilation depending on the individual case.
Teicoplanin was used at the discretion of the treating neonatologist. The regimen for neonates ≤44 weeks PMA was a loading dose of 16 mg/kg, followed 24 h later by 8 mg/kg administered once daily.12 Teicoplanin was infused over 30 min. Therapeutic drug monitoring (TDM) is not routinely performed and dose adjustment was not conducted in this study. The duration of treatment was also at the discretion of the treating neonatologist.
Blood samples (0.2 mL) were obtained throughout the first and last dose intervals (1, 3, 6 and 24 h post-dose). Patients weighing <1000 g at inclusion could have a maximum of two study-specific sampling episodes per dose interval to minimize blood loss. The total sampling period was to a maximum of 168 h for the majority of patients. If the first dose administration occurred before informed consent had been obtained, a pre-dose sample was obtained. Whenever possible, a washout sample was collected 24 h after the last dose. Samples were centrifuged at 1500 g for 10 min and serum was stored at −80°C prior to analysis.
The demographic variables with a potential impact on the PK of teicoplanin and/or influence on the determination of teicoplanin (concomitant medications) were collected for each patient (i.e. weight and serum creatinine). Concentrations of CRP before, during and after teicoplanin treatment were measured as part of standard care. Demographic data were analysed with SPSS Statistics version 22 (IBM Corporation, Armonk, NY, USA; http://www-01.ibm.com/software/analytics/spss/downloads.html).
Ethics
The study was approved by the Medicines and Healthcare Products Regulatory Agency (clinical trial authorization reference number: 21362/0003/001-0002) and the National Research Ethics Service and Regional Committee (REC: 13/NW/0023). Written informed consent was obtained from parents and/or legal guardians. The study was registered in the European Clinical Trials Database Registry (EudraCT: 2012-005738-12).
Measurement of teicoplanin concentrations
Teicoplanin concentrations were measured using a commercially available fluorescence polarization immunoassay (Thermo Fisher Scientific, Germany). The limit of quantification was <3.0 mg/L. The dynamic range was 3–100 mg/L and overall precision was <6%.
Measurement of CRP concentrations
A Multigen CRP Vario® (Abbott, Wiesbaden, Germany) latex immunoassay was used for the immunoturbidimetric determination of CRP in the plasma of patients, and was implemented in the Abbott Architect ci4100 system. The limit of quantification was <0.2 mg/L (reported clinically as <4 mg/L) for the standard and wide range methods (analyte concentration at which the CV = 20%). The dynamic range was 0.2–480 mg/L (wide range method) and total precision was ≤6%. A CRP cut-off value >10 mg/L was considered positive.
Microbiological investigation
Microbiological specimens, including blood cultures, were collected as part of routine clinical care. Positive microbiological samples were stored for identification with a Bruker Biotyper MALDI-TOF MS System (Bruker Daltonics, Billerica, MA, USA) and susceptibility testing was performed using Etest® (bioMérieux, Hampshire, UK), following BSAC methodology.13
Population PK models
All data were analysed using Pmetrics.14 The inverse of the estimated assay variance was used as the weighting function for all models. Three structural PK models were explored in this study. The first represented a standard two-compartment PK model with time-delimited zero-order intravenous input and first-order elimination from central compartment. The model is described by the differential Equations (1a) and (1b) below.
| (1a) |
| (1b) |
Where X(1) and X(2) represent the amount of teicoplanin (mg) in the central (c) and peripheral (p) compartments, respectively. R(1) is the rate of infusion of drug into the central compartment (mg/h). The central compartment has volume (Vc) in L, from which there is clearance (SCL) in L/h. The central and peripheral compartments are connected by the first-order rate constants Kcp and Kpc (h−1).
The effect of weight, PMA, postnatal age, serum creatinine and estimated glomerular filtration rate (eGFR) using the Haycock–Schwartz formula (K × height/serum creatinine15; the UK population median height values for age and sex were used for each patient; and K = 0.33 for pre-term neonates and K = 0.45 for term neonates) on the population PK of teicoplanin was explored.16,17 The Bayesian estimates for clearance and volume of distribution from each patient were obtained from the standard model (above) and plotted against weight, PMA, postnatal age, serum creatinine and GFR, using both linear and logarithmic scales. Since both linear and logarithmic relationships between clearance and weight appeared tenable, the linear and allometric models that incorporated weight as a covariate were developed. Ultimately, an allometric power model was used. Such a model has been widely used to determine the effect of size on the PK of various compounds in children and neonates.18,19 The allometric scaling exponent in Equation (2a) was fixed at 0.75. Only clearance and not volume had a relationship with weight, and therefore clearance was normalized to a 70 kg adult, as described elsewhere.19 The differential equations describing the allometric model are as follows:
| (2a) |
| (2b) |
Where SCLstd represents the normalized estimate for clearance in a 70 kg individual, the other parameters are described above.
After establishing the model that best described the PK of teicoplanin, the following PD equation was used to describe the time-course of CRP concentrations:
| (2c) |
Where KCRPprod is the maximum rate of CRP production (mg·h/L), POPmax is the theoretical maximum CRP concentration (mg/L), KCRPinh is the maximum rate of CRP inhibition (mg·h/L), EC50 is the concentration of teicoplanin (mg/L) that produces the half-maximal effect (CRP inhibition) and H is the slope function for the CRP inhibition term.
Given the high PK variability in the population and to avoid biased parameter estimates in the PK/PD model, the Bayesian posterior estimates for each patient's PK parameters (from the final PK model described above) were fixed and the PD parameters were then estimated by fitting the PD component of the model to each patient's CRP data. The Bayesian posterior estimates for each subject were used to estimate the concentration–time profiles for teicoplanin and CRP for each patient. Average AUC and trough (Cmin) for each 24 h of therapy were calculated from the Bayesian posterior estimates.
Monte Carlo simulation
Monte Carlo simulations were performed using a semi-parametric sampling methodology14,20 that generated a simulated population of 5000 neonates receiving a given teicoplanin regimen. For each simulated patient, the weight-based dose of teicoplanin (in mg/kg) was administered to each neonate (30 min infusion) as an absolute dose of teicoplanin (in mg) by multiplying the rate of infusion (in mg/h) by the simulated weight.
All calculations were performed at steady state between day 4 and 5 of treatment. The proportion of patients achieving Cmin >10, >15, 20, 30, 40 and 60 mg/L (recommended safety cut-off) was determined. A comparison of the variability of distribution of drug exposures (AUCs) achieved in the neonatal simulated population was performed with the distribution of drug exposures in older children (1 month–16 years old) and adults receiving currently recommended dosage regimens of teicoplanin. This comparison was based on Monte Carlo simulations conducted on a previous teicoplanin PK study with older children and adults.6
Exposure–response relationships
A newly described PD index (AUC:EC50) was used to link drug exposure with therapeutic response (terminal CRP concentration).21 An Emax sigmoidal model was fitted to the data. The use of a more conventional index (e.g. AUC:MIC ratio) was infeasible because the MIC of the invading microorganism was not available for the majority of patients. EC50 is the estimated drug concentration required to induce half-maximal reduction in the CRP concentrations and it is therefore an in vivo estimate of drug activity.
Results
Demographics
A total of 18 neonates were recruited from two different hospitals in Liverpool (Alder Hey NHS Children's Foundation Trust and Liverpool Women's Hospital) over a 20 month period (between April 2013 and January 2015). Ten patients were recruited from the neonatal intensive care unit at the Liverpool Women's Hospital. The number of recruited patients by sub-category PMA (weeks) was: 24–27 (n = 1); 28–31 (n = 5); 32–35 (n = 2); 36–39 (n = 5); and 40–44 (n = 5). A description of the demographic data is presented in Table 1.
Table 1.
Demographics
| PMA (weeks) | Age (days) | Weight at birth (kg) | Weight at enrolment (kg) | eGFR (mL/min/1.70 m2) | Creatinine (first day of TEC) (μmol/L) | Creatinine (last day of TEC) (μmol/L) | CRP (first day of TEC) (mg/L) | CRP (last day of TEC) (mg/L) | |
|---|---|---|---|---|---|---|---|---|---|
| n | 18 | 18 | 18 | 18 | 18 | 18 | 13 | 18 | 16 |
| Range | 26–44 | 4–69 | 0.69–4.2 | 0.69–5.08 | 5.4–95.2 | 21–265 | 28–114 | 4–172.6 | 4–163.7 |
| Median (IQR) | 37 (29.7–40) | 17 (10.5–26) | 1.42 (0.9–3) | 2.04 (1.16–3.23) | 42.8 (27.8–53.3) | 44.5 (36–57.7) | 38 (34–63.5) | 41.9 (12.1–122.25) | 6.9 (4–13.1) |
Microbiological results
A total of 44.4% of recruited patients (n = 8) had a positive blood culture obtained from either a central or peripheral intravenous line. A total of 33.3% of those recruited (n = 6) were Gram-positive infections [100% CoNS including Staphylococcus haemolyticus (n = 2), Staphylococcus epidermidis (n = 1) and n = 3 unidentified species]. All were susceptible to teicoplanin (MIC ≤4 mg/L using the EUCAST breakpoint).22 The remaining 11.1% (n = 2) were Gram-negative bacterial infections (n = 1 had Pseudomonas aeruginosa and n = 1 had Klebsiella oxytoca). (These two patients only received two doses of teicoplanin each and were excluded from the subsequent exposure–relationship analysis.)
Teicoplanin and CRP concentrations
The concentration–time profile of teicoplanin and corresponding CRP concentrations for each patient is shown in Figure 1(a) and Figure 1(b), respectively. A total of 96 PK samples were available for analysis (mean of 5.3 samples per patient). Fourteen PK concentrations, from four patients were excluded from the analysis because of incorrect or absent sampling times. The mean (SD) from the observed teicoplanin concentrations was 18 (9.11) mg/L and a median of 17.32 mg/L (range 3.1–38.7 mg/L). A total of 104 CRP samples were available for analysis as part of the standard care of the patients.
Figure 1.

(a) Teicoplanin (circles) and (b) CRP (triangles) concomitant concentration–time profiles for the 18 neonates.
Population PK models
Both the population PK linear and allometric models performed similarly with an acceptable fit to the observed data and comparable measures of bias and precision. However, on the basis of the individual Bayesian estimates of the observed versus predicted fit of the data, the allometric PK model better accounted for the observed data and was chosen for further analyses. The model diagnostics are shown in Table 2. For the allometric model the linear regression of observed versus predicted values had a coefficient of determination of r2 = 0.815 with measures of bias and precision of 0.03 and 0.8, respectively. The population PK parameter estimates of the allometric model are shown in Table 2.
Table 2.
Model diagnostics for the PK models
| Model | Log-likelihood | Pop r2a | Post r2a | Slope (95% CI) | Intercept (95% CI) |
|---|---|---|---|---|---|
| Standard PK | −253.8 | 0.159 | 0.814 | 0.925 (−1.3–2.8) | 0.768 (−1.3–2.8) |
| Linear (weight) PK | −254.1 | 0.271 | 0.807 | 0.941 (0.8–1.04) | 0.922 (−1.14–2.98) |
| Allometric (weight) PK | −254.4 | 0.249 | 0.815 | 0.981 (0.9–1.1) | 0.26 (−1.8–2.3) |
Pop, population; Post, individual posteriors.
aRelative to the regression line fitted for the observed versus predicted values before and after the Bayesian step.
Population PK/PD model
The fit of the PK/PD data was acceptable (shown in Figure 2a and b). The linear regression of observed versus predicted values for CRP had a coefficient of determination of r2 = 0.95 with measures of bias and precision of 0.09 and 0.9, respectively (shown in Figure 2b). The time-course of CRP in each individual patient was described with a high degree of precision and minimal bias using the Bayesian posterior median estimates for each patient. The population PK/PD parameter estimates are summarized in Table 3. The Bayesian individual posterior estimates for the linked PK and the PD are shown in Figure 3.
Figure 2.

Individual posterior observed versus predicted plots (after the Bayesian step) from the PK/PD model. (a) PK (teicoplanin concentrations); predicted teicoplanin concentrations = 0.917x − 0.06. (b) PD (CRP concentrations); predicted CRP concentrations = 1.01x + 0.254.
Table 3.
Population PK and PD parameter estimations
| Mean | SD | Median | |
|---|---|---|---|
| Population PK parameter | |||
| CLstd (L/h) | 0.45 | 0.2 | 0.43 |
| Vc (L) | 0.81 | 0.48 | 0.76 |
| Kcp (h−1) | 1.45 | 0.99 | 1.3 |
| Kpc (h−1) | 0.84 | 1.05 | 0.63 |
| Population PD parameter | |||
| KCRPprod (mg·h/L) | 0.05 | 0.03 | 0.05 |
| Popmax (mg/L) | 159.76 | 62.6 | 139.15 |
| H | 18.48 | 3.46 | 19.99 |
| KCRPinh (mg·h/L) | 0.05 | 0.02 | 0.06 |
| EC50 (mg/L) | 7.1 | 6.11 | 5.79 |
| IC3 (mg/L) | 55.32 | 54.24 | 24.99 |
CLstd, clearance standardized [clearance = CLstd × (weight/70)0.75]; Vc, volume of distribution in the central compartment; Kcp and Kpc, first-order rate constants from central compartment to peripheral compartment and from peripheral compartment to central compartment, respectively; KCRPprod, maximum rate of CRP production; Popmax, theoretical maximum CRP concentration; H, Hill slope; KCRPinh, maximum rate of CRP inhibition; EC50, teicoplanin concentration producing half-maximal CRP reduction; IC3, initial condition in CRP concentrations.
Figure 3.

Individual concentration–time plots after the Bayesian step showing teicoplanin (black) and CRP (grey) predicted (continuous line) and observed (crosses) concentrations over time for each of the patients. The y-axis ‘Observation’ refers to both teicoplanin and CRP concentrations. Individuals 1 and 7 were infected with Gram-negative bacteria and only received two doses of teicoplanin. The individual average Cmin and AUC drug exposures are reported for each patient.
Monte Carlo simulation
Based on the simulations, the mean (SD) 24 h steady-state AUC from 96 to 120 h was 365.4 (267.1) with a median of 302.3 mg·h/L. The mean (SD) trough at 96 h was 15.7 (11.7) mg/L with a median of 12.9 mg/L. Only 38.8% of neonates achieved a Cmin at 120 h >15 mg/L. In addition, 69.1%, 22.4%, 8.56%, 3.92% and 1.1% achieved Cmin >10, 20, 30, 40 and 60 mg/L, respectively. Comparative distribution histograms of the achieved AUCs at steady state for the simulated neonates, as well as for simulated older children and adult populations are shown in Figure 4. The neonatal population achieved median AUCs at steady state (302.3 mg·h/L) comparable to the median AUC attained by a population of adults receiving 400 mg/day (291.81 mg·h/L), but with more variability (neonatal AUC IQR = 227.5 versus adult AUC IQR = 101.59 mg·h/L).
Figure 4.

Comparison of the simulated (n = 5000 per population group) teicoplanin AUC (mg·h/L) distribution histograms in different populations receiving the current teicoplanin dosage regimen: (a) neonates (0.7–5 kg); (b), (c) and (d) children >1 month–16 years old with fixed weights of 10, 25 and 50 kg, respectively; and (e) adults.
Exposure–response relationships
The Bayesian posterior estimates for the exposure–response relationships (AUC, Cmin and AUC:EC50) are shown in Figure 5. If patients 1 and 7 (infected with P. aeruginosa and K. oxytoca, respectively) are excluded, 56% of the patients (9 of 16) were able to suppress CRP under the cut-off value of 10 mg/L by 96–120 h. Subject 16 (gastroschisis) was not included in the inhibitory sigmoid Emax model (the patient's data are shown in Figure 5c). An AUC:EC50 of ∼68.3 is predictive of a terminal CRP ≤10 mg/L. The relationship between AUC:EC50 and predicted CRP at the end of therapy is shown in Figure 5(c). Patients with an AUC:EC50 >68.3 tended to have a more consistently lower terminal CRP level than patients with an AUC:EC50 <68.3 (P = 0.002) (Figure 6).
Figure 5.
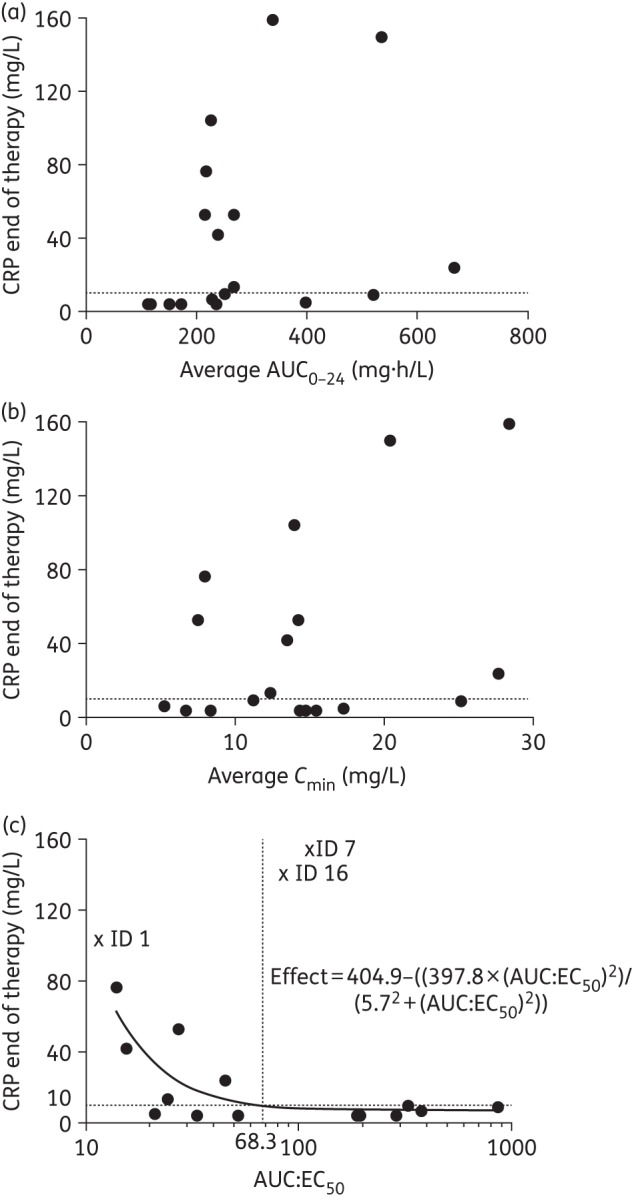
Exposure–response relationships from the Bayesian posteriors from the PK/PD linked model using (a) AUC average, (b) Cmin average and (c) AUC:EC50 ratio (log10 scale) as the PD relevant index versus predicted CRP concentrations at the end of therapy. An Emax model was fitted to the data. Patients with ID 1, 7 (Gram-negative bacterial infection) and 16 (multiple inflammatory comorbidities with persistently high CRP levels >100 mg/L and negative blood culture) were excluded from this analysis, but are shown in (c).
Figure 6.
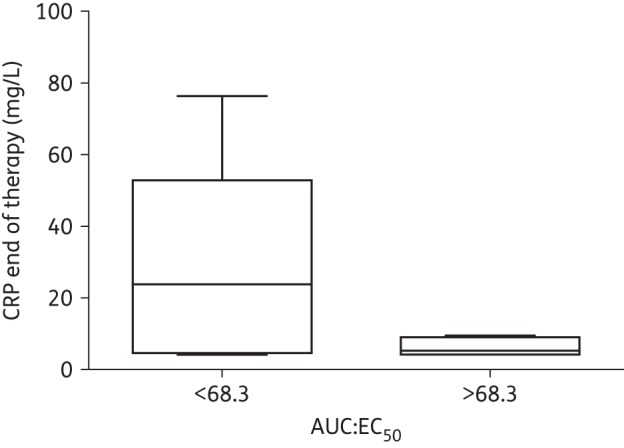
AUC:EC50 box-plot suggesting that patients attaining >68.3 drug exposures (∼>AUC 389.3 mg·h/L) had a more consistently lower CRP at the end of therapy (mean 18.18 versus 5.7 mg/L); P = 0.002 (two sample t-test).
Discussion
Teicoplanin is used for the treatment of serious staphylococcal infections.23–25 Currently, teicoplanin is not licensed in the EU for the treatment of neonates or infants <2 months of age. The PK/PD study provides a rationale to address the appropriate teicoplanin regimen and extent of variability in both drug exposure and response. Furthermore, the study provides the necessary tools to take the next critical steps to provide truly individualized antimicrobial therapy for neonates receiving teicoplanin.
The extent of inter-patient PK variability in this neonatal population was high (Figure 1a). Of the multiple covariates that were studied, only weight accounted for any portion of the observed PK variability. Incorporation of weight into structural PK models resulted in better fits and statistically more likely solutions. Of note, we could have reasonably related weight to clearance using linear or power scaling terms, despite the convention for using a scaling exponent of 0.75.19,26 We could not demonstrate any relationship between teicoplanin clearance and PMA, eGFR or serum creatinine. This is somewhat surprising because teicoplanin is almost completely renally (98%) cleared by glomerular filtration.27 The absence of any relationship probably reflects the small sample size as well as the relatively poor estimates of eGFR in neonates using current nomograms. This finding does call into question whether teicoplanin dosing should be adjusted based on eGFR and further studies are required to address specifically this question. Other factors associated with disease, such as variability in regional blood flow, organ perfusion, changes in acid–base balance or cardiac output might have potentially influenced the drug's disposition characteristics in our patient population; however, this also requires further and targeted study. Using a non-parametric modelling methodology, we took a pragmatic approach by investigating the clinical parameters known to have a significant impact on our patient population PK variability (weight as an estimate of size, age and serum creatinine/eGFR).
Monte Carlo simulations suggest that the median AUCs at steady state in neonates receiving 16 mg/kg as a loading dose, followed by 8 mg/kg every 24 h are comparable to adults receiving 400 mg/day. However, there is a much larger PK variability in the AUCs of neonates (Figure 4). While the matching of measures of central tendency is straightforward, the best way to match two completely different AUC distributions is less clear. The high variability makes identification of a fixed weight-based regimen challenging because of the unacceptably high proportion of neonates with both low and high drug exposure. Any attempt to address this problem results in an unsatisfactory trade between effect and toxicity and eventual acknowledgement that TDM is required to optimize dosing and drug exposure.
While TDM is the only current way teicoplanin dosing can be optimized, there are a number of significant challenges to this process. First and most obviously obtaining repeated blood draws in premature neonates is never trivial, and second, there is persistent uncertainty about drug exposure targets for TDM. A trough concentration of 15 mg/L (measured by fluorescence polarization immunoassay) is proposed in the summary of product characteristics by days 3–5 of therapy for both adults and children, but recently increased to 20 mg/L and 30–40 mg/L for the treatment of deep-seated infections and infective endocarditis, respectively.28 Moreover, concentrations are not recommended to exceed 60 mg/L, despite little evidence for any relationship between serum concentrations and toxicity in neonates.29 Such recommendations are based on scant clinical evidence in adult patients and with only a rudimentary understanding of the PD of teicoplanin.30–32 The use of Bayesian feedback tools for dosage individualization, which requires the availability of robust population PK models and optimally sampled concentrations, may enable the attainment of desired AUC targets (and surrogate trough concentrations) for any individual patient.33
This study is too small to resolve clinical exposure–response relationships. Inadequate power was further compounded by a Gram-positive pathogen being isolated in only 6 of 18 (33.3%) patients. Hence, there was no opportunity to examine the relationship between the magnitude of any traditional PD indices (e.g. AUC:MIC) and outcome. Even in larger datasets, the problem of culture negativity is frequently present. In this situation, most investigators use a population value (e.g. MIC90) to calculate drug exposure for an individual patient. Assuming such patients are infected with the most resistant pathogen is conservative, but necessarily biased. The use of CRP and a novel PD index (the AUC:EC50) circumvents some of these issues. The rationale behind this quotient is that EC50 is an in vivo measure of drug potency and the AUC:EC50 is a measure of the exposure of drug relative to the potency of its effect. A major advantage of this approach is that it allows for drug exposure targets that are more individualized for a specific patient. The EC50 (and therefore AUC:EC50) is influenced by both the patient and characteristics of the infecting organism. The EC50 captures the impact of multiple variables on exposure–response relationships (e.g. in vitro resistance, high bacterial load, a persistent infective focus, biofilms and immune response). In contrast, when the measure of potency is the MIC alone, as for AUC:MIC, it is only the organism's characteristics that are considered, and all the other factors implicit in EC50 are ignored. The Bayesian posterior EC50 estimates ranged widely (0.6–18.7 mg/L), which again reflects highly variable PD and in vivo potency. In this study, the AUC:EC50 predicted the terminal CRP levels after 5 days of therapy (Figure 5c) for the majority of patients.
The use of CRP as a biomarker deserves some comment. CRP is widely used in clinical practice to guide anti-infective therapy, but much of that process is informal and intuitive.11,34,35 In this study, we explicitly link teicoplanin serum concentrations and changes in circulating CRP. The measurement of CRP in an individual patient provides a real-time estimate of the response to drug. There are clearly some advantages to such an approach: CRP is quantitative, widely available, well validated and readily accepted by clinicians. It is the most extensively studied biomarker in neonatal sepsis. In addition, a recent systematic review has showed higher specificity and predictive values at symptom onset and after 24–48 h than procalcitonin in neonatal bacterial sepsis.36 Procalcitonin has been investigated mainly in early onset sepsis and with different cut-off values depending on time after birth. Its value in neonates is limited by a marked physiological increase after birth.37 The ability to link drug concentrations with a biomarker provides the prospect for truly individualized therapy where the dosing of drug is designed to manage a biomarker rather than a serum drug concentration. However, there are some limitations. CRP is a non-specific marker of infection and inflammation, and adjusting a dose solely on the basis of climbing CRP may be dangerous if the CRP elevation is the result of Gram-negative bacteraemia as was the case in patients 1 and 7, or the result of a severe non-infectious inflammation as appears likely for patient 16 (Figure 3). Thus, to guide teicoplanin dosing, there needs to be confidence that the CRP elevation is a result of a teicoplanin-susceptible pathogen. In our study, we had microbiological evidence of a teicoplanin-susceptible organism in a third of patients. However, there was a high clinical suspicion of this being the case for the remaining patients (clinical, laboratory markers of infection and specific risk factors such as a central line inserted). In our setting, teicoplanin and ciprofloxacin constitute the empirical treatment in the context of central-line-associated bloodstream infection. All patients received ciprofloxacin or gentamicin until a blood culture result became available. The other antimicrobial could potentially have had an impact on CRP decline in the case of a Gram-negative microorganism. Nevertheless, CoNS was the most commonly isolated microorganism and teicoplanin was only discontinued in two patients with Gram-negative infection. Interestingly, a recent study has also demonstrated that serial CRP measurements can predict whether an organism is sensitive to the empirical antimicrobial therapy in the first 48 h of treatment of neonatal sepsis.38 These findings now need prospective evaluation.
Despite any potential limitations, this study extends the standard pharmacometric approach whereby the population PK is described, Monte Carlo simulations are performed and post hoc analyses such as the probability of target attainment analyses are performed, often using PD targets of questionable clinical significance. While the current approach has limitations because of the non-specificity of the biomarker, the analyses begin to refocus therapeutic arguments on the individual patient, using real data to deliver a regimen that is both safe and effective for the clinical problem in hand.
Funding
The study and V. R.-M. were funded by the NIHR Alder Hey Clinical Research Facility for Experimental Medicine and Alder Hey NHS Foundation Trust Business Unit.
W. W. H. is supported by a National Institute of Health Research (NIHR) Clinician Scientist Fellowship.
The CRP serial measurements were generated as part of the standard of care of patients in Alder Hey Children's and Liverpool Women's NHS Foundation Trusts and were collected for this study.
Transparency declarations
W. W. H. has received research funding from Pfizer, Gilead, Astellas, AiCuris, Amplyx and F2G, and has acted as a consultant and/or given talks for Pfizer, Basilea, Astellas, F2G, Nordic Pharma, Amplyx, Mayne Pharma and Pulmocide. All other authors: none to declare.
Acknowledgements
We thank the NIHR Alder Hey Clinical Research Facility (CRF) and Alder Hey NHS Foundation Trust Business Unit for supporting the study.
We acknowledge the Neonatal Intensive Care Unit at the Liverpool Women's NHS Foundation Trust and the Paediatric Intensive Care Unit at Alder Hey NHS Foundation Trust.
We recognize the work of the CRF and Liverpool Women's NHS Foundation Trust research nurses Bronagh Howell, Joanne Windrow, Karen Harvey and Gail Wallace for contributing to the recruitment of patients and data collection.
Elaine Scott for supporting the data management and eCRF development.
Richard Drew and Timothy Neal, consultant microbiologists, for their input and support.
We thank all the patients and families that participated in the study.
References
- 1.Stoll BJ, Hansen N, Fanaroff AA et al. Late-onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics 2002; 110: 285–91. [DOI] [PubMed] [Google Scholar]
- 2.Marchant EA, Boyce GK, Sadarangani M et al. Neonatal sepsis due to coagulase-negative staphylococci. Clin Dev Immunol 2013; 2013: 586076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams-Chapman I, Stoll BJ. Neonatal infection and long-term neurodevelopmental outcome in the preterm infant. Curr Opin Infect Dis 2006; 19: 290–7. [DOI] [PubMed] [Google Scholar]
- 4.Svetitsky S, Leibovici L, Paul M. Comparative efficacy and safety of vancomycin versus teicoplanin: Systematic review and meta-analysis. Antimicrob Agents Chemother 2009; 53: 4069–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavalcanti AB, Goncalves AR, Almeida CS et al. Teicoplanin versus vancomycin for proven or suspected infection. Cochrane Database Syst Rev 2010; issue 6: CD007022. [DOI] [PubMed] [Google Scholar]
- 6.Ramos-Martin V, Paulus S, Siner S et al. Population pharmacokinetics of teicoplanin in children. Antimicrob Agents Chemother 2014; 58: 6920–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philip AG, Mills PC. Use of C-reactive protein in minimizing antibiotic exposure: experience with infants initially admitted to a well-baby nursery. Pediatrics 2000; 106: E4. [DOI] [PubMed] [Google Scholar]
- 8.Connell TG, Rele M, Cowley D et al. How reliable is a negative blood culture result? Volume of blood submitted for culture in routine practice in a children's hospital. Pediatrics 2007; 119: 891–6. [DOI] [PubMed] [Google Scholar]
- 9.Lutsar I, Chazallon C, Carducci FIC et al. Current management of late onset neonatal bacterial sepsis in five European countries. Eur J Pediatr 2014; 173: 997–1004. [DOI] [PubMed] [Google Scholar]
- 10.Benitz WE, Han MY, Madan A et al. Serial serum C-reactive protein levels in the diagnosis of neonatal infection. Pediatrics 1998; 102: e41. [DOI] [PubMed] [Google Scholar]
- 11.Ehl S, Gering B, Bartmann P et al. C-reactive protein is a useful marker for guiding duration of antibiotic therapy in suspected neonatal bacterial infection. Pediatrics 1997; 99: 216. [DOI] [PubMed] [Google Scholar]
- 12.BMJ Group, the Royal Pharmaceutical Society of Great Britain and RPL 2014. British National Formulary for Children 2014-2015. 2014; 289–90 http://www.evidence.nhs.uk/formulary/bnfc/current/5-infections/51-antibacterial-drugs/517-some-other-antibacterials/vancomycin-and-teicoplanin/vancomycin.
- 13.Andrews JM. Determination of Minimum Inhibitory Concentrations. 2006; 1–19. http://www.bsac.org.uk/wp-content/uploads/2012/02/Chapter-2-Determination-of-MICs-2006updated.pdf. [DOI] [PubMed]
- 14.Neely MN, van Guilder MG, Yamada WM et al. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 2012; 34: 467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martini S, Prévot A, Mosig D et al. Glomerular filtration rate: measure creatinine and height rather than cystatin C! Acta Paediatr 2003; 92: 1052–7. [DOI] [PubMed] [Google Scholar]
- 16.Brion LP, Fleischman AR, McCarton C et al. A simple estimate of glomerular filtration rate in low birth weight infants during the first year of life: noninvasive assessment of body composition and growth. J Pediatr 1986; 109: 698–707. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz GJ, Feld LG, Langford DJ. A simple estimate of glomerular filtration rate in full-term infants during the first year of life. J Pediatr 1984; 104: 849–54. [DOI] [PubMed] [Google Scholar]
- 18.Wurthwein G, Groll AH, Hempel G et al. Population pharmacokinetics of amphotericin B lipid complex in neonates. Antimicrob Agents Chemother 2005; 49: 5092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hope WW, Seibel NL, Schwartz CL et al. Population pharmacokinetics of micafungin in pediatric patients and implications for antifungal dosing. Antimicrob Agents Chemother 2007; 51: 3714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goutelle S, Bourguignon L, Maire PH et al. Population modeling and Monte Carlo simulation study of the pharmacokinetics and antituberculosis pharmacodynamics of rifampin in lungs. Antimicrob Agents Chemother 2009; 53: 2974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huurneman LJ, Neely M, Veringa A et al. Pharmacodynamics of voriconazole in children: further steps along the path to true individualized therapy. Antimicrob Agents Chemother 2016; 60: 2336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.EUCAST. Breakpoint Tables for Interpretation of MICs and Zone Diameters. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_5.0_Breakpoint_Table_01.pdf.
- 23.Kacet N, Dubos J, Roussel-Delvallez M. Teicoplanin and amikacin in neonates with staphylococcal infection. Pediatr Infect Dis J 1993; 12: 10. [Google Scholar]
- 24.Yalaz M, Cetin H, Akisu M et al. Experience with teicoplanin in the treatment of neonatal staphylococcal sepsis. J Int Med Res 32: 540–8. [DOI] [PubMed] [Google Scholar]
- 25.Fanos V, Kacet N, Mosconi G. A review of teicoplanin in the treatment of serious neonatal infections. Eur J Pediatr 1997; 156: 423–7. [DOI] [PubMed] [Google Scholar]
- 26.Wade KC, Wu D, Kaufman DA et al. Population pharmacokinetics of fluconazole in young infants. Antimicrob Agents Chemother 2008; 52: 4043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson AP. Clinical pharmacokinetics of teicoplanin. Clin Pharmacokinet 2000; 39: 167–83. [DOI] [PubMed] [Google Scholar]
- 28.The electronic Medicines Compendium. Targocid 200 mg—Summary of Product Characteristics (SPC). 2014. http://www.medicines.org.uk/emc/medicine/27319/SPC.
- 29.Yamada T, Kubota T, Nakamura M et al. Evaluation of teicoplanin concentrations and safety analysis in neonates. Int J Antimicrob Agents 2014; 44: 458–62. [DOI] [PubMed] [Google Scholar]
- 30.Matthews PC, Taylor A, Byren I et al. Teicoplanin levels in bone and joint infections: are standard doses subtherapeutic? J Infect 2007; 55: 408–13. [DOI] [PubMed] [Google Scholar]
- 31.Sato M, Chida K, Suda T et al. Recommended initial loading dose of teicoplanin, established by therapeutic drug monitoring, and outcome in terms of optimal trough level. J Infect Chemother 2006; 12: 185–9. [DOI] [PubMed] [Google Scholar]
- 32.Ueda T, Takesue Y, Nakajima K et al. Evaluation of teicoplanin dosing designs to achieve a new target trough concentration. J Infect Chemother 2012; 18: 296–302. [DOI] [PubMed] [Google Scholar]
- 33.Neely MN, Youn G, Jones B et al. Are vancomycin trough concentrations adequate for optimal dosing? Antimicrob Agents Chemother 2014; 58: 309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bomela HN, Ballot DE, Cory BJ et al. Use of C-reactive protein to guide duration of empiric antibiotic therapy in suspected early neonatal sepsis. Pediatr Infect Dis J 2000; 19: 531–5. [DOI] [PubMed] [Google Scholar]
- 35.Pourcyrous M, Bada HS, Korones SB et al. Significance of serial C-reactive protein responses in neonatal infection and other disorders. Pediatrics 1993; 92: 431–5. [PubMed] [Google Scholar]
- 36.Hedegaard SS, Wisborg K, Hvas A-M. Diagnostic utility of biomarkers for neonatal sepsis—a systematic review. Infect Dis (Lond) 2015; 47: 117–24. [DOI] [PubMed] [Google Scholar]
- 37.Turner D, Hammerman C, Rudensky B et al. Procalcitonin in preterm infants during the first few days of life: introducing an age related nomogram. Arch Dis Child Fetal Neonatal Ed 2006; 91: F283–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patil S, Dutta S, Attri SV et al. Serial C reactive protein values predict sensitivity of organisms to empirical antibiotics in neonates: a nested case–control study. Arch Dis Child Fetal Neonatal Ed 2016; doi:10.1136/archdischild-2015-309158. [DOI] [PubMed] [Google Scholar]