

ABSTRACT
Chemotherapy often fails to eradicate cancer stem cells (CSCs) that drive cancer recurrence. In fact, the treated tumors often contain a higher frequency of chemo-resistant CSCs. It is thought that CSC formation is supported by exposure of cancer cells to sub-cytotoxic chemotherapy doses as a result of poor drug penetration in epithelial tumors. We have shown that low-dos cisplatin triggers the transdifferentiation of ovarian cancer cells into CSCs through processes that are also involved in the generation and maintenance of induced pluripotent stem (iPS) cells. Considering similarities between CSCs and iPS cells, we screened a library of 60 synthetic small-molecule compounds, designed to influence EMT/MET signaling in iPS cells on primary ovarian cancer cells. Using a Nanog reporter system we identified a series of compounds capable of blocking the cisplatin triggered formation of CSCs. We then focused on compound GHDM-1515, a drug that acts on pathways that regulate histone demethylases. We demonstrated that co-treatment of primary ovarian cancer cells with GHDM-1515 significantly increased cisplatin induced apoptosis, specifically apoptosis of CSCs. GHDM-1515 inhibited EMT and the cisplatin-induced formation of CSCs. This suggests that GHDM-1515 can sensitize ovarian cancer cells to low-dose cisplatin and potentially enhance the efficacy of cisplatin chemotherapy.
KEYWORDS: Cisplatin, epithelial-to-mesenchymal transition, ovarian cancer, resistance
Introduction
More than 80% of all cancer cases are carcinomas, formed by the malignant transformation of epithelial cells. For most carcinomas, invasion of normal tissues and metastasis is accompanied by a loss of epithelial differentiation and a shift toward a mesenchymal phenotype, i.e. a partial epithelial-to-mesenchymal transition (EMT).1 However, following invasion or metastasis, cells that have undergone EMT can also revert to a well-differentiated epithelial phenotype by a process called mesenchymal to epithelial transition (MET).2-7 Phenotypic transitions involving epigenetic changes such as EMT and MET have been linked to the concept of cancer stem cells (CSC).8,9 CSC are defined as a small sub-population of cancer cells with self-renewing, multi-lineage differentiation, and tumor forming ability. We and others have shown that putative ovarian CSCs are in a transitional phase between epithelial and mesenchymal cell stages and that considerable plasticity exists between non-CSCs and CSCs.7
The epithelial phenotype, specifically, epithelial junctions between tumor cells, is considered to provide protection against innate and adaptive immune attacks. Junctions also represent physical barriers to intratumoral penetration of anti-cancer drugs.10 A series of studies demonstrated that intravenously injected chemotherapy drugs penetrate only a few cell layers from the blood vessel into the tumor.11-13 This implies that more distant tumor cells are exposed to lower drug concentrations that are non-cytotoxic. It is thought that this triggers the formation of CSC that later drive cancer recurrence. Several reports suggest chemotherapy-mediated induction of EMT and CSCs in a number of epithelial cancers, including colon cancer, gastric cancer, head-and-neck cancer, liver cancer, breast cancer and ovarian cancer.14-21
The theoretical basis for our study is the assumption that there are similarities between CSC and induced pluripotent stem cells (iPS cells). The production of iPS cells from somatic cells can be achieved by the overexpression of the transcription factors Oct4, Klf4, Sox2, and c-Myc.22 A key feature of reprogramming fibroblasts into iPSCs entails a dramatic phenotypic change from a spindle mesenchymal-like to a compact epithelial-like morphology, with concomitant upregulation of E-cadherin - a process reminiscent of MET.23,24 Pluripotent iPS cells are therefore in an epithelial/mesenchymal (E/M) hybrid stage, i.e., express both epithelial and mesenchymal markers.23,25 This enables them to differentiate into different lineages upon specific extrinsic stimuli.
Considering the involvement of EMT and MET processes in formation, maintenance and differentiation of both iPS cells and CSCs, we screened a small-molecule library that was developed to study EMT and EMT signaling in iPS cells on ovarian cancer cells. Using a combinatorial scaffold approach, we previously synthesized large diverse chemical libraries consisting of over 100,000 discrete compounds representing over 50 distinct structural classes with drug-like properties. Screening on iPS cells resulted in the selection of drugs that lacked cytotoxicity and induced genome-wide changes in chromatin status and gene expression. Among these drugs, we identified bio-active small-molecules targeting various protein families involved in signaling transduction, transcriptional regulation and epigenetic modifications.26-28 Examples of such drugs are Pluripotin 28 (maintains iPS cell self-renewal), and Pyrintegrin 29 (promotes cell survival).
Here we used 60 small-molecule compounds from this library. In a primary screen, the drugs were tested for their ability to block cisplatin induced CSC formation in patient-derived ovarian cancer cell cultures. Further studies focused on one drug capable of increasing the sensitivity of ovarian cancer cells to cisplatin.
Results and discussion
Previously, we established more than 40 ovarian cancer cultures from biopsies of grade III and IV cancers obtained through the Pacific Ovarian Cancer Research Consortium.30,31 Six of these cultures can be passaged as xenografts in immunodeficient CB17 mice. The vast majority of ovarian cancer cells in patient biopsies and xenograft tumors stained positive for the epithelial marker E-cadherin (Fig. 1A). Tumors in situ also contained a small subset of cancer cells that were positive for E-cadherin and the mesenchymal marker Vimentin, i.e E/M hybrid cells. As outlined above, in our previous studies we showed that E/M hybrid cells are putative CSCs.24,30 Previous studies also showed that during passaging (>5 passages) of ovarian cancer cells isolated from patient biopsies or xenograft tumors, epithelial and E/M cells transdifferentiated into mesenchymal cells. Upon transplantation, mesenchymal cells reverted back to E/M hybrid cells involving MET.24,30 For our studies we used only early-passage ovarian cancer cells derived from xenograft tumors with a predominant epithelial phenotype.
Figure 1.
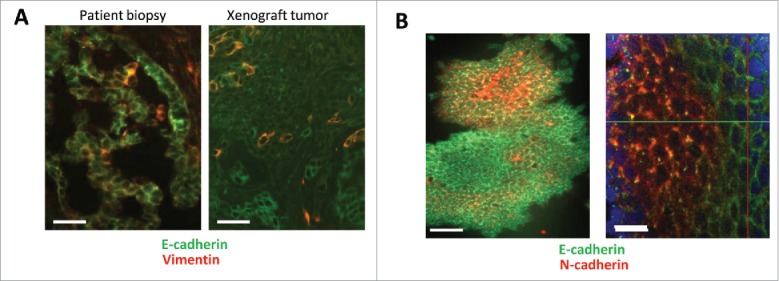
Immunfluorescence analysis of primary ovarian cancer and human iPS cells. (A) Ovarian cancer cells: Epithelial marker E-cadherin and the mesenchymal marker Vimentin on sections from an ovarian cancer patient biopsy and a xenograft tumor established by transplantation of primary ovarian cancer cells into immunodeficient mice. Green: E-cadherin, Red: Vimentin. The scale bar is 20 μm. (B) iPS cells: Cells were grown on glass slides and stained for the indicated markers. Note that culturing iPS cells on glass slides triggers their differentiation at the periphery of colonies. Green: E-cadherin, Red: N-cadherin. The scale bar is 40 μm in the left panel and 20 μm in the right panel.
A similar E/M hybrid stage was found in iPS cells (Fig. 1B). Confocal microscopy of iPS cell colonies showed the localization of E-cadherin to intercellular junctions in more differentiated epithelial cells (at the periphery of the colony) and cytoplasmic punctuated E-cadherin in undifferentiated iPS cells in the center of the colony. Undifferentiated cells also express membrane N-cadherin, a protein that is expressed upon MET and often considered a marker for a mesenchymal differentiation stage.
Cisplatin treatment triggers transdifferentiation into cells with CSC features. Studies were performed with primary ovarian cell line ovc316.24,30 Ovc316 cells were obtained from biopsy of a patient with high grade serous ovarian cancer in collaboration with the Pacific Ovarian Cancer Research Consortium. Cisplatin (2.5μM) treatment of ovc316 cells increased the Western blot signals for the stem cell markers Oct4, Sox2, and Nanog (Fig. 2A). To better monitor putative CSCs and to develop a high-throughput drug screening assay, we generated primary ovarian cancer cells stably expressing Discosoma red fluorescent protein (DsRed) under the transcriptional control of responsive elements that bind the stem cell factor Nanog (Fig. 2B). To do this ovc316 cells were transduced with a corresponding lentivirus vector and selected with Zeocin. The cells were then passaged as xenograft tumors in CB17 mice. This cell system reports Nanog activity through expression of DsRed. The corresponding cells were called ovc-rNanog/DsRed. The percentage of rNanog/DsRed-positive cells in xenograft derived cultures ranged from 4–10% (Fig. 2C). The vast majority of rNanog/DsRed-positive cells were E/M cells, i.e expressed E-cadherin and Vimentin (Fig. 2D). We also confirmed that rNanog/DsRed-positive cells are CSCs based on functional assays, i.e. formation of tumor spheres in culture (Fig. 2E) and tumors after transplantation into immunodeficient mice. Tumor formation studies showed that 100 rNanog/DsRed-positive cells were sufficient to form tumors in 4 out of 5 animals while 106 rNanog/DsRed-negative cells were required to achieve the same efficacy, indicating that this reporter system marks CSCs (data not shown).
Figure 2.
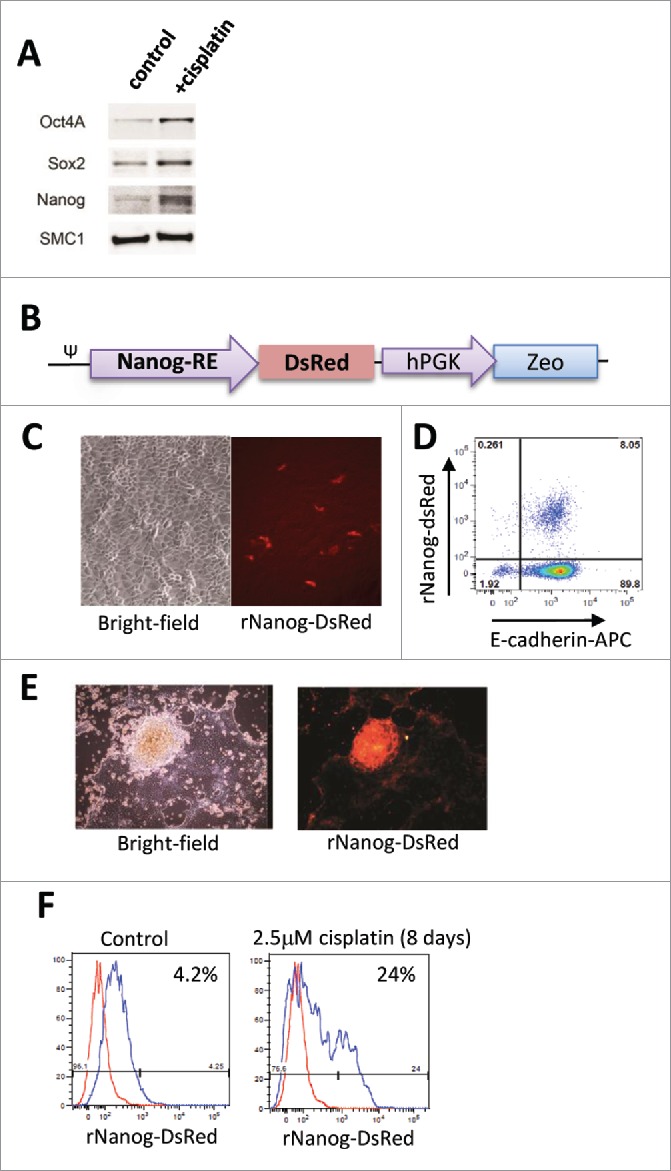
Cisplatin treatment triggers increase in rNanog/DsRed cells in vitro. (A) Induction of stem cell factors by cisplatin. Ovc316 cells were treated with 2.5 µM cisplatin for 8 days and analyzed by Western blot. Cisplatin treatment triggered Oct4, Sox2, Nanog upregulation. SMC1 (Structural maintenance of chromosomes protein 1 served as a loading control. (B) Expression cassette of rNanog/DsRed reporter lentivirus vector. DsRed expression is under the control of a minimal (m) CMV promoter and tandem repeats of the Nanog transcriptional response element. The zeocin resistance gene is under the control of the ubiquitously active phosphoglycerate kinase (PGK) promoter. ψ: lentivirus packaging signal. C-E) Characterization of a representative xenograft-derived cell culture. (C) Bright-field and rNanog/DsRed expression in ovc-rNanog/DsRed cells. (D) Flow cytometry analysis for rNanog/DsRed and E-cadherin. (E) Primary ovarian cancer cells expressing DsRed in tumor spheres. (F) rNanog/DsRed expression before and after treatment with 2.5 μM cisplatin for 8 days (added only once). A representative study is shown. The red peak represents DsRed negative ovc316 cells.
Using the rNanog-reporter system, we showed that cisplatin treatment at a low (non-cytotoxic) dose of 2.5 μM over 8 days resulted in a ∼6-fold increase in the percentage of rNanog/DsRed positive cells (Fig. 2F). This was also observed in vivo in xenograft tumors derived from ovc-rNanog/DsRed cells (Fig. 3). In mice with pre-established tumors, multiple injections of cisplatin at a dose of 0.5 mg/kg over a period of 4 weeks did not control tumor growth (Fig. 3A). The analysis of Nanog protein in tumors by Western blot showed an increase in signal intensity in mice treated with cisplatin (Fig. 3B). Cisplatin also increased the signal for E-cadherin. Increase in Nanog activity upon cisplatin treatment was also observed using the rNanog/DsRed reporter system. After the second cycle of cisplatin injection, in vivo imaging signals for DsRed over the tumor region were significantly higher than in control mice (Fig. 3C, D). Stronger DsRed fluorescence signals were also observed on sections from tumors collected at the end of the experiment (Fig. 3E). CSCs, when cultured in suspension in serum-free medium, form spheres known as tumor spheres. We studied tumorsphere formation upon low-dose cisplatin treatment (Fig. 3F). Spheres with a diameter of ∼200μm formed by day 7 of culture. The percentage of spheres based on the number of plated cells and spheres counted at day 7 was 1.2 (+/−0.3)% for mock-treated cells and 2.8 (+/−0.3)% for cisplatin-treated cells (p < 0.01). This supports our hypothesis that low-dose cisplatin triggers the formation of CSCs in ovc316 cell cultures.
Figure 3.
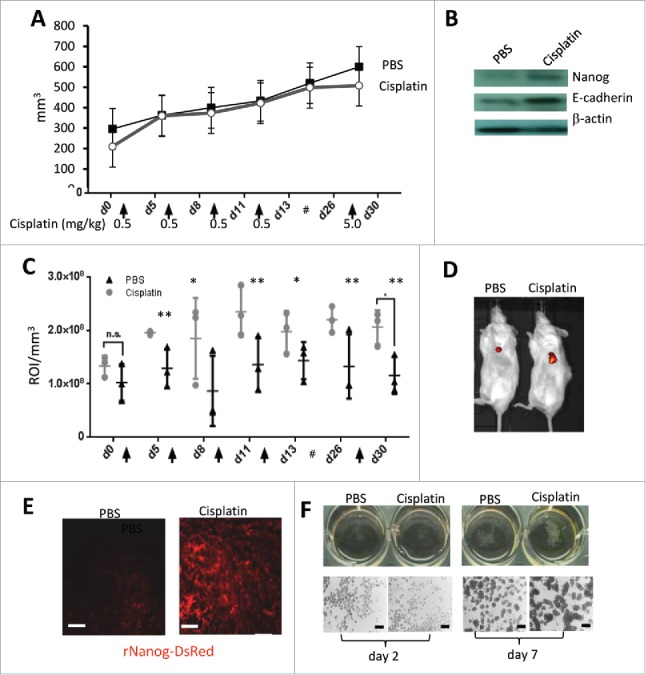
Cisplatin treatment at subtherapeutic doses triggers increase in rNanog/DsRed cells and tumorsphere formation in ovarian cancer xenograft tumors. Ovc-rNanog/DsRed cells were transplanted into the mammary fat pad of CB17 mice. When tumors reached a volume of 200 mm3 mice received intravenous injections of cisplatin at the indicated time points and doses. For the last injection, a dose of 5 mg/kg was used and animals were sacrificed 2 days later. (A) Tumor volume. N = 3. (B) Western blot with Nanog and E-cadherin specific monoclonal antibodies on lysates from tumors that were harvested 2 days after the last cisplatin injection. β-actin serves as a loading control. C and D) DsRed in vivo imaging. (C) Shown is the signal intensity at the region of interest (ROI), i.e., the tumor. The ROI was then normalized to the tumor volume and expressed as signals in ROI/mm3. N = 3; ns-not significant, *p < 0.05, **p < 0.01. The # sign represents a 2 week break before the last treatment. (D) Representative in vivo images taken at day 5 after the first cisplatin injection. (E) DsRed fluorescence on sections from tumors harvested 2 days after the last cisplatin injection. The scale bar is 20 μm. (F) Tumorsphere formation assay. Ovc316 cell were cultured in serum free-medium PRIME-XV Tumorsphere medium with or without cisplatin. Spheres were analyzed at day 2 and day 7. Representative images are shown. The scale bars in the lower panels are 200μm. For day 7 samples, spheres were counted in 3 wells for +/− cisplatin settings.
Importantly, to simulate the clinical situation, where most tumor cells only receive only sub-toxic chemotherapy drug concentrations,11-13 we used, in our subsequent in vitro studies, cisplatin concentrations that would not trigger killing of ovarian cancer cells in vitro and in vivo.
Cisplatin treatment triggers transdifferentiation into E/M hybrid cells that are resistant to low-dose cisplatin. Cisplatin treatment of ovc316 cells triggered an increase in E/M (E-cadherin+/Vimentin+) hybrid cells (Fig. 4A). Based on our previous studies,30 we concluded that the increase in E/M cells involves both partial MET (ME/M) and partial EMT (EE/M). This is also reflected in the flow cytometry analysis which shows a shift of both Vimentin+ and E-cadherin+ cells toward the E/M hybrid phenotype. The involvement of EMT in cisplatin triggered transdifferentation of ovc316 cells is also supported by a functional “wound healing” assay 17 (Fig. 4B). A scratch was made with a sterile tip in a confluent layer of ovc316 cells (marked by a red arrow in PBS/control panel) and wound closure was measured 48h later. While 25.3% wound closure was observed in control cells, cisplatin treatment triggered 83.4% wound closure, indicating the induction of cell proliferation and migration. Cisplatin-triggered wound closure was blocked by the MAPK inhibitor U0126. Notably, MAPK activation is one of the key pathways involved in EMT.32 Along this line, treatment of cells with Epithelial Growth Factor (EGF), a strong activator of MAPK and EMT, resulted in complete wound closure for both the control and the treatment group.
Figure 4.
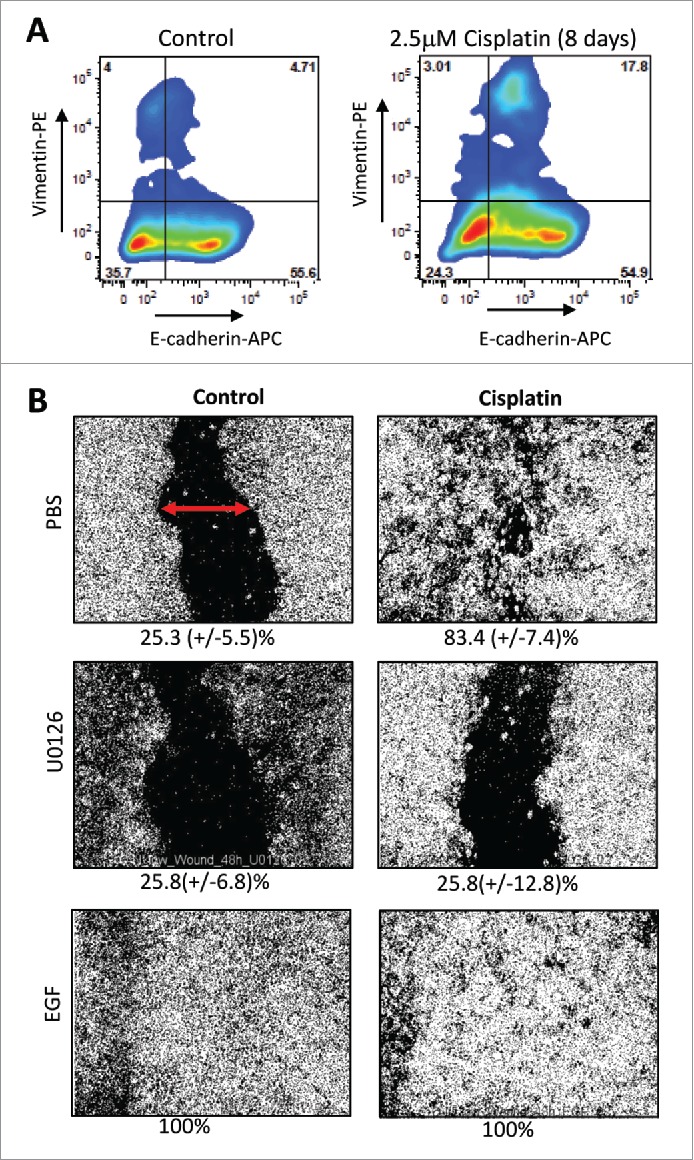
Effect of cisplatin on epithelial-mesenchymal phenotype in ovarian cancer. (A) Flow cytometry analysis for E-cadherin and Vimentin on untreated ovc316 cells and cells treated with 2.5μM cisplatin for 8 days. Representative studies are shown. (B) Wound healing assay. The migratory ability of ovc316 cells was evaluated by a “wound-healing” assay. A scratch was made with a sterile tip in a confluent layer of cells (marked by red arrow) and “wound” closure was observed after 48h of 2.5μM cisplatin treatment. The MAPK inhibitor U0126 (10 μM), was used to block cell migration. As a positive control, EGF (100 ng/ml), a known MAPK pathway activator was used. Wound closure was measured with ImageJ. Representative images are shown. The percentage of wound closure is shown beneath the images. N = 3.
Screening of small-molecule library with EMT/MET inhibitor activity. Our data shown in Figs. 3 and 4 indicate that cisplatin triggers the formation of CSCs with E/M phenotype through EMT/MET. These pathways are also implicated in iPS cells formation and differentiation. We therefore employed a library, consisting of 60 small-molecule compounds, that was generated to influence EMT and/or MET in iPS cells 26-28 and tested their ability to trigger MAPK activation in primary ovarian cancer cells. This would then be a strong indicator for whether the drugs influence EMT/MET processes in ovarian cancer cells. To test this we incorporated a MAPK reporter system into ovc316 cells. This system monitors the activity of Serum Response Factor (SRF)-mediated signal transduction pathways. The ternary complex factors, TCR and Elk-1, form a complex with the SRF over the serum response element (SRE), and activate gene expression. The Elk-1 protein is phosphorylated by MAPK, causing increased DNA binding, ternary complex formation, and transcriptional activation of target genes. The SRE reporter therefore measures the activation of the SRF and the MAPK signal transduction pathway. We generated ovc316 cells that expressed luciferase under SRE/MAPK control (Fig. 5A). Exposure of these cells to cisplatin significantly increased luciferase expression levels compared to DMSO-treated cells (DMSO is used as the drug solvent) (Fig. 5B, first 2 bars). Most of the small-molecule drugs, when combined with cisplatin, triggered lower levels of luciferase expression than cisplatin alone (Fig. 5B, dashed line), indicating that they inhibit cisplatin-induced EMT. The most pronounced effect was seen with the drugs of the GHDM class (see red labels), which are structurally related. The lowest luciferase levels were observed for drug A73 and A148. A number of drugs (e.g. SD41, SD48, A5, A112, A508) increased luciferase expression indicating EMT activation. These data suggest that the drugs are capable of influencing cisplatin-triggered EMT pathways in ovarian cancer cells.
Figure 5.

Induction of MAPK, a pathway involved in EMT of ovc-MAPK cells after treatment with cisplatin and small-molecule compounds. (A) Expression cassette of SRE/MAPK-luc reporter lentivirus vector. Luciferase expression is under control of a minimal CMV promoter and tandem repeats of the SRE transcriptional response element (SRE-TATA). hPGK: human phosphoglycerate kinase promoter, PuroR: puromycin resistance gene, cppt: Central polypurine tract. (B) Luciferase expression in ovc-MAPK cells that were modified with the SRE/MAPK-reporter lentivirus vector. Cells were treated for 4 days with 2.5 µM cisplatin alone (second bar) and with cisplatin in combination with small-molecule compounds at a final concentration of 2 μM. DMSO is the solvent of and was used as a negative control (first bar). EGF was used as a trigger of EMT at a concentration of (100 ng/ml) (third bar). Luciferase activity was normalized to total protein concentration and expressed as RLU/mg protein. N = 3, * p < 0.05.
We then screened the drugs for their ability to block cisplatin-triggered formation of CSCs. To do this, we used the ovc rNanog/DsRed reporter cell line. The goal of this study was to find compounds that would reduce cisplatin-triggered CSC-formation without exerting a significant effect on their own in the absence of cisplatin. Consistent with our earlier observations, cisplatin increased the rNanog/DsRed signal (Fig. 6, upper dotted red line). Combination treatment of cisplatin with drugs resulted in a decrease in the percentage of rNanog/DsRed positive cells with the exception of drug A101, A236 and A195. Members of the GHDM family strongly inhibited the cisplatin-triggered increase of rNanog/DsRed cells (see red labels).
Figure 6.

Screening of small-molecule EMT/MET-regulatory compounds for ability to block cisplatin triggered increase in rNanog/DsRed cells. Ovc-rNanog/DsRed cells were incubated without or with 2.5 μM cisplatin for 4 days in the presence or absence of the indicated compounds at the final concentration of 10 μM. Shown is the percentage of rNanog/DsRed-positive cells. “Control” was set as 100%. DMSO is the solvent for the compounds and is therefore used as a control. The goal was to find compounds that would reduce cisplatin-triggered CSC formation (“w/ Cis”) (upper red line) back to the control level (lower red line) without exerting a significant effect on its own in the absence of cisplatin (“w/o Cis”). N = 3.
GHDM-1515 treatment sensitizes ovarian cancer cells to cisplatin. We then focused on compound GHDM-1515. The structure of this compound is shown in Fig. 7A. Because GHDM-1515 appeared to interfere with the EMT pathway and was the least cytotoxic compared to other screened drugs (data not shown). We tested whether this would overcome resistance to treatment with low-dose cisplatin. We therefore measured the percentage of apoptotic cells after treatment with cisplatin in the absence and presence of GHDM-1515. Cells that are in early apoptosis are Annexin-V positive and negative for the vital dye 7-AAD (Fig. 7B). 7-AAD positive cells lost membrane integrity and can therefore not be used for further CSC marker analysis by flow cytometry. Cisplatin treatment (2.5 µM) over 8 days only marginally increased the number of apoptotic cells compared to DMSO-treated control cells (Fig. 7C). Pre-incubation with or simultaneous addition of GHDM-1515 almost doubled the number of apoptotic cells. No significant effect of cisplatin or cisplatin+GHDM-1515 was observed when cells were analyzed at day 4. SD83, a drug that did not block MAPK activation (Fig. 5) and inhibited the formation of rNanog/DsRed+ cells to a lesser degree than GHDM-1515, did not increase cisplatin-mediated apoptosis. Importantly, the apoptosis-enhancing effect of GHDM-1515 was also observed in the cell fraction that was positive for rNanog/DsRed implying that the cisplatin+GHDM-1515 combination kills CSCs (Fig. 7D).
Figure 7.

Studies with compound GHDM-1515 in ovc-rNanog/DsRed cells. (A) Structure of compound GHDM-1515. (B) Flow cytometry-based analysis of apoptosis. Apoptotic cells were defined as Annexin V-positive/7-AAD-negative. (C) Percentage of apoptotic cells after treatment with cisplatin (2.5 μM) and GHDM-1515 (2 μM) for 4 or 8 days. Two settings of combination treatment were tested. Cells were pre-incubated with GHDM-1515 for 24h before adding cisplatin (GHDM-1515cisplatin) or drugs were mixed (GHDM-1515 + cisplatin). SD-83, a compound that did not block cisplatin-triggered MAPK activation (see Fig. 5) was used as a control. N = 3. (D) Percentage of apoptotic cells in the fraction of putative CSCs, i.e. rNanog/DsRed-positive cells.
To further assess the mechanism of action of GHDM-1515 we studied its effect on EMT/MET in ovc316 cells. In the “wound healing” or scratch assay GHDM-1515 significantly inhibited wound closure induced by cisplatin (Fig. 8A). Furthermore, flow cytometry analysis showed that GHDM-1515 reversed cisplatin-mediated upregulation of E-cadherin and Vimentin, rendering cells in primarily Epithelial like phenotype. (Fig. 8B). Activation of p38 MAPK upon cisplatin or GHDM-1515 + cisplatin treatment was analyzed by Western blot with antibodies specific to p38 and the phosporylated (activated) form of p38 (P-p38) (Fig. 8C). No effect on p38 levels was observed. Compared to the P-p38 band intensity in PBS treated cells, cisplatin and cisplatin + DMSO treatment resulted in 3.3-fold and 2.9-fold stronger P-p38 signals, respectively. This effect was reversed by co-treatment with GHDM-1516 (0.8-fold of PBS control). GHDM-1515 alone only slightly (1.3-fold) increased the signal.
Figure 8.

GHDM-1515 counteracts changes triggered by cisplatin in ovc316 cells. (A) Wound healing assay. Assay conditions were as described for Fig. 3. The concentration of GHDM-1515 was 2 μM. Representative image is shown. The percentage of wound closure is shown beneath the images. N = 3. (B) Flow cytometry analysis for E-cadherin and Vimentin was performed as described in Fig. 3. Cells were treated with cisplatin or cisplatin + GHDM-1515 for 4 days. (C) Activation of p38 MAPK. Ovc316 cells in 12-well plates (0.5 ×106 cells per well in 2ml of medium) were treated with PBS, 2.5μM cisplatin, 2μM GHDM-1515 (1000x stock in DMSO), 2.5μM cisplatin +2μl DMSO, and 2.5μM cisplatin +2μM GHDM-1515. Cells were collected 3 days later and lysates were analyzed by Western blot with antibodies specific to the phosphorylated form of p38 (P-p38) (upper panel). Filters were then stripped and incubated with p38-specific antibodies (lower lane).
To corroborate our data obtained in primary ovarian cancer ovc-rNanog/DsRed cells, we performed studies in the established ovarian cancer cell line SKOV3-ip1 (Fig. 9). Treatment with cisplatin at a concentration of 2.5 μM resulted in ∼7% of apoptotic (Annexin V+/7AAD−) cells indicating that SKOV3-ip1 cells are more resistant to cisplatin than ovc-rNanog/DsRed cells. This is not surprising because this cell line was selected to be resistant to chemotherapy drugs.33 The combination of cisplatin with 2 μM GHDM-1515 significantly increased the percentage of apoptotic cells. A lower dose of GHDM-1515 (0.5 μM) did not have an enhancing effect.
Figure 9.
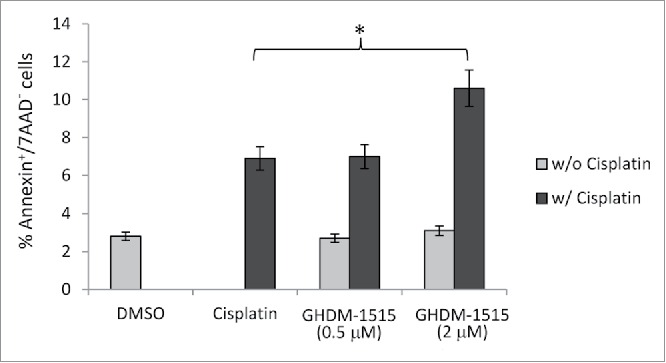
Cytotoxocoty in SKOV3-ip1 cells. Percentage of apoptotic SKOV3-ip1 cells after treatment with cisplatin (2.5 μM) and GHDM-1515 (0.5 and 2 μM) for 4 days. N = 3, * p < 0.05.
In summary, compound GHDM-1515 decreased EMT and the cisplatin-induced formation of E/M hybrid cells and rNanog/DsRed-positive CSCs thereby significantly increasing the percentage of apoptotic cells. This suggests that GHDM-1515 can sensitize ovarian cancer cells to low-dose cisplatin and potentially enhance the efficacy of cisplatin chemotherapy.
The compound GHDM-1515 only dissolved in DMSO or PEG-300-based formulations, which made drug application in mice impossible. Further modifications of GHDM-1515 have to be conducted to increase its solubility in aqueous solutions.
Conclusions
In solid tumors, most cancer cells are exposed to only subtherapeutic drugs doses due to physical barriers to intratumoral drug penetration. We show here in ovarian cancer cells that exposure to non-cytotoxic doses of cisplatin triggers the formation of cancer stem cells that are in an E/M hybrid stage. The small molecular drug GHDM-1515 inhibited this axis and sensitized ovarian cancer cells to cisplatin. If drug solubility problems can be resolved, a combination of GHDM-1515 and cisplatin has therefore the potential to delay or prevent the recurrence of chemoresistant ovarian cancer in patients. Furthermore, our study suggests that similarities between iPS cells and CSCs can be exploited to find new cancer treatments that are targeted toward CSCs.
Material and methods
Characterization of compound
GHDM-1515 is a derivative of pyrimidine. This small molecule was obtained through custom synthesis. Its structure was rigorously established by 1H, 13C and 19F nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS). NMR spectra of GHDM-1515 were recorded on a Varain Inova 400 spectrometer. The chemical shifts are given in parts per million (ppm) on the delta (δ) scale. Deuterated dimethyl sulfoxide (DMSO-d6) was used as the solvent for NMR analysis. The chemical shifts of the solvent residual peak were used as reference values (for 1H NMR, 2.50 ppm; for 13C NMR, 39.5 ppm).34 For 1H NMR, the following abbreviations were used to designate multiplicities: s = singlet, d = doublet, t = triplet. Coupling constant (J) were expressed in Hz. MS data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an electrospray ionization source. The mass-to-charge ratio (m/z) of the quasi-molecular ion (M+H) of GHDM-1515 was recorded.
1H NMR (400 MHz, DMSO-d6): δ 10.05 (s, 1H), 8.61 (d, J = 5.0 Hz, 1H), 8.57 (s, 1H), 8.22 (s, 1H), 8.17 (d, J = 8.4 Hz, 1H), 7.55 (t, J = 8.2 Hz, 1H), 7.46 (d, J = 5.0 Hz, 1H), 7.29 (d, J = 7.3 Hz, 1H), 4.17 (s, 3H).
13C NMR (100 MHz, DMSO-d6): δ 159.8, 159.2, 157.4, 145.4, 141.3, 129.7, 129.3, 125.5, 124.4, 122.1, 117.4, 114.7, 108.0, 36.7.
19F NMR (376 MHz, DMSO-d6): δ −61.15.
m/z (M+H): 321.11 (theoretical), 321.10 (observed).
Cells
Ovc316 cells are primary ovarian cancer cells derived from a patient biopsy.31 Work with patient derived tumors cells was approved by the Fred Hutchinson Cancer Research Center Institutional Review Board (protocol: 6289 “Secondary use of human cells”). Primary ovarian cancer cells were cultured in MEGM (MEBM containing 3μg/L hEGF, 5μg/L insulin, 5 mg/L hydrocortisone, 26 mg/L bovine pituitary extract, 25 mg/L amphotericin B) (Lonza, Basel, Switzerland), supplemented with 10% FBS (Gibco, Waltham, MA), 100 I.U. penicillin, 100μg/L streptomycin. The ovarian cancer cell line SKOV3-ip1 (obtained from Dr. David Curiel, UAB) was maintained in RPMI-1640 supplemented with 10% FBS and 100 I.U. penicillin, 100μg/L streptomycin.
MAPK reporter system
Ovc316 cells were transduced with the MAPK/Erk-luc reporter lentivirus (Cignal Lenti, Hilden, Germany). Cells were selected with 1μg/ml Puromycin for 4 weeks to obtain stable clones. Luciferase expression was measured using the Promega Luciferase Assay System (Promega, Madison, WI). The relative light units (RLU) were normalized to the total protein concentration in the sample. The data are expressed in RLU/mg.
rNanog/DsRed reporter system
Nanog reporter (rNanog) cells express DsRed under the control of a minimal (m) CMV promoter and tandem repeats of the Nanog transcriptional response element. Zeocin resistance gene is under the ubiquitous PGK promoter. Clones were generated using the pRedZeo-hNanog lentivirus (System Biosciences, Mountain View, CA). Low passage ovc316 cells were infected for 2 days and subsequently treated with 100 µg/mL Zeocin for 4 weeks. Zeocin resistant cells were then passaged in CB17 mice as xenographt tumors.
iPS cells
iPS cells were grown in TeSR™2 (StemCell Technologies) on matrigel (BD Biosciences) coated dishes as described previously.35 The following antibodies were used for analysis of iPS cells: FITC-conjugated anti-E-cadherin (Abcam, Cambridge, MA) and mouse monoclonal anti-N-cadherin (Gene Tex, Inc., Irvine, CA).
Flow cytometry
Adherent cells were detached from tissue culture plates by treatment with Versene (Gibco, Waltham, MA) for 15–30 min. Cells were washed with RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco). Cell pellets were resuspended in ice-cold PBS with 1% FBS and blocked with anti-mouse CD16/CD32 Fc block (eBioscience, San Diego, CA). Cells were incubated with antibodies in a total of 100 μl for 45 min on ice. All subsequent incubation steps were carried out in the dark. Antibody dilutions were as follows, anti E-cadherin-APC (Biolegend, San Diego CA) diluted 1:50, anti Vimentin-PE (Abcam., Eugene, CA) diluted 1:50. Cells were washed with 3 ml PBS/1% FBS and centrifuged at 400g for 5 min at 4°C. Following the PBS/1% FBS wash, cells were subjected to analysis. Stained samples were analyzed on a BD-LSRII (BD Bioscience, San Jose, CA). FITC Annexin V Apoptosis Detection Kit with 7-AAD (Biolegend) was used for Annexin V staining. Cells were stained according to manufacturer's protocol. Unspecific backgrounds of individual channels were determined with matching isotype controls and color compensation was done on single color-stained samples. Dead cells were excluded with DAPI (Sigma). Flow data were analyzed with FlowJo9.1.
Wound healing assay
Ovc316 cells with epithelial phenotype, maintained by culturing in the presence of Pluriton™ Reprogramming Medium (Stemgent, Cambridge, MA) (in the absence of FBS), were plated in 24 well plates until fully confluent. Scratches were made with a 250 μl pipette tip through the center of the well. Compounds were added after the scratch was made. As a negative control we chose MAPK inhibitor U0126 (10 μM), due to its known inhibition of cell migration. Wound closure was observed under the microscope at 0, 24 and 48 hours. Images were then processed with ImageJ software where the percentage of wound closure was calculated as wound area at a given time compared to the initial wound surface. Images were taken with a Leica DMLB Microscope (Wetzlar, Germany), using Leica DFC300FX Digital camera and Leica Application Suite Version 2.4.1 R1 software (Heerbrugg, Germany).
Tumorsphere formation assay
1×106 ovc316 cells were plated in 10ml of PRIME-XV Tumorsphere Serum-Free Medium (IrvineScientific, Santa Anna, CA) supplemented with 2U/ml Heparin and 0.5μg/ml hydrocortisone in low-adherence 10 cm plastic Petri dishes. Cells were collected at day 2 and day 7, allowed to settle to the bottom of a 15ml falcon tube, and spheres were counted.
In vivo imaging
In vivo rNanog/DsRed imaging was performed on an IVIS Xenogen (PerkinElmer Inc., Waltham, MA). For analysis, regions of interest (ROIs) were put over the tumor site and the total flux (photons per second summed over the area of the ROI) was measured using Living Image 4.0 Software (PerkinElmer Inc.).
Animal studies
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Washington, Seattle, WA (Protocol: 3108–01). Mice were housed in specific-pathogen-free facilities.
Immunodeficient NOD.CB17-Prkdcscid/J (CB17) mice were obtained from the Jackson Laboratory. OVC-Nanog/DsRed tumors were established by injection of the corresponding tumor cells into the mammary fat pad of CB17 mice. When tumors reached a volume of ∼200mm3, mice were treated with cisplatin twice a week over the course of 30 days. Mice were treated with 0.5 mg/kg cisplatin i.v. the first 2 weeks, then with 5 mg/kg i.v. on the last week of the experiment. The mice were imaged with Xenogen IVIS-200 at 7 time points a day after each injection. The DsRed signal was detected at fixed exposure time (10 sec.) and 535nm excitation. The ROI (region of interest) of the dsRed signal was derived by selecting the tumor area with the ROI tool and total ROI was taken from 3 images. The average ROIs were normalized to the tumor volume for each mouse.
Western blot
Xenograft tumor tissue was dissected, homogenized and incubated for 30 min in protein lysis buffer (20 mM Hepes (pH 7.5), 2 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM PMSF, 200 μM Na3VO4 (Sigma, St. Louis, MO), and protease inhibitors on ice. After 30 seconds of sonication (Fisher Scientific Model 120 Dismembrator) on ice, samples were pelleted, and protein containing supernatant stored at −80°C. A total of 20 μg of total protein was used for Western blotting. Protein samples were separated by polyacrylamide gel electrophoresis using 4–15% gradient gels (BioRad, Hercules, CA) followed by transfer onto nitrocellulose membranes according to the supplier's protocol (MiniPROTEAN 3, BioRad). Membranes were blocked in PBS+0.1% Tween20 (PBS-T, Fisher, #BP337) and 5% nonfat dry milk (BioRad, #170–6404)). Incubation times for primary and secondary antibodies were 2h and 1h at room temperature, respectively. Antibodies were diluted in PBS-T and 5% dry-milk powder. Membranes were washed 5 times in PBS-T between antibody incubations, and films were developed using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, Little Chalfont, UK). Mouse anti-Nanog antibodies (eBioscience) were diluted 1:500, goat anti-mouse HRP 1:3000 (BD Bioscience), goat anti-Ecadherin 1:500, mouse anti-ActinB (Sigma) rabbit anti-goat HRP 1:5000 (BioRad). For p38 the following antibodies were used: rabbit mAb anti-p38 (clone D13E1, Cell Signaling Technology, Danvers, MA), rabbit mAb anti-pp38 (clone D3F9, Cell Signaling Technology, Danvers, MA).
Immunofluorescence
Tumor sections of xenografts were embedded in OCT compound (Tissue-Tek, Sakura Fineteck, Torrence, CA) and frozen on dry ice. OCT embedded tissues were then stored at −80°C and equilibrated to −20°C for at least 1h prior to sectioning. Tumor tissue was sliced (8 μm) using a Leica CM 1850 cryostat (Leica Microsystems) and then transferred onto Superfrost Plus microscope slides (Fisher Scientific, Hampton, NJ). Slides were fixed in 4% PFA (Fisher Scientific) for 10 min at −20°C. After two rinses with PBS slides were blocked with 2% nonfat dry milk in PBS for 20 min at room temperature. Immunofluorescence analyses were performed with goat anti E-cadherin (R&D) diluted 1:250, anti Vimentin-FITC (eBiosciences), donkey anti-goat AF488 1:500 (LifeSciences). All immunofluorescence images were taken with Leica DM1000 microscope featuring a Leica DFC FX camera (Leica Microsystems).
Statistical analysis
All results are expressed as mean +/− SD. Student's t-test or 2-Way ANOVA for multiple testing, were applied when applicable. A p-value < 0.05 was considered significant.
Disclosure of potential conflicts of interest
The authors have no competing financial interest.
Funding
This work was supported by the Pacific Ovarian Cancer Research Consortium/Specialized Program of Research Excellence in Ovarian Cancer Grant P50 CA83636, NIH grants R01 CA080192, R01 HLA078836, and the Danish Cancer Society.
Author Contribution
KS, SD, and AL conceived and designed the experiments. KS, RS, MX, MR, RvR conducted the experiments. KS, SD, AL analyzed the results. CD provided patient tumor biopsy. KS, AL wrote the paper. JB, CD, AE, and SD provided crucial comments and revised the manuscript. All authors approved the final submitted version.
References
- 1.Turley EA, Veiseh M, Radisky DC, Bissell MJ. Mechanisms of disease: epithelial-mesenchymal transition-does cellular plasticity fuel neoplastic progression? Nat Clin Practice 2008; 5(5):280-90; PMID:18349857; http://dx.doi.org/ 10.1038/ncponc1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem 2013; 59:168-79; PMID:23220226; http://dx.doi.org/ 10.1373/clinchem.2012.184655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res 2006; 66:8319-26; PMID:16951136; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-0410 [DOI] [PubMed] [Google Scholar]
- 4.Huang RY, Chung VY, Thiery JP. Targeting pathways contributing to epithelial-mesenchymal transition (EMT) in epithelial ovarian cancer. Curr Drug Targets 2012; 13:1649-53; PMID:23061545; http://dx.doi.org/ 10.2174/138945012803530044 [DOI] [PubMed] [Google Scholar]
- 5.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139:871-90; PMID:19945376; http://dx.doi.org/ 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 6.Thiery JP, Lim CT. Tumor dissemination: an EMT affair. Cancer Cell 2013; 23:272-3; PMID:23518345; http://dx.doi.org/ 10.1016/j.ccr.2013.03.004 [DOI] [PubMed] [Google Scholar]
- 7.Ye X, Weinberg RA. Epithelial-Mesenchymal Plasticity: A central regulator of cancer progression. Trends Cell Biol 2015; 25:675-86; PMID:26437589; http://dx.doi.org/ 10.1016/j.tcb.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br J Cancer 2015; 112:1725-32; PMID:25965164; http://dx.doi.org/ 10.1038/bjc.2015.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nomura A, Banerjee S, Chugh R, Dudeja V, Yamamoto M, Vickers SM, Saluja AK. CD133 initiates tumors, induces epithelial-mesenchymal transition and increases metastasis in pancreatic cancer. Oncotarget 2015; 6:8313-22; PMID:25829252; http://dx.doi.org/ 10.18632/oncotarget.3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beyer I, van Rensburg R, Lieber A. Overcoming physical barriers in cancer therapy. Tissue Barriers 2013; 1:e23647; PMID:24665377; http://dx.doi.org/ 10.4161/tisb.23647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beyer I, Cao H, Persson J, Song H, Richter M, Feng Q, Yumul R, van Rensburg R, Li Z, Berenson R, et al.. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin Cancer Res 2012; 18:3340-51; PMID:22535153; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kyle AH, Baker JH, Gandolfo MJ, Reinsberg SA, Minchinton AI. Tissue penetration and activity of camptothecins in solid tumor xenografts. Mol Cancer Ther 2014; 13:2727-37; PMID:25143448; http://dx.doi.org/ 10.1158/1535-7163.MCT-14-0475 [DOI] [PubMed] [Google Scholar]
- 13.Mugabe C, Matsui Y, So AI, Gleave ME, Baker JH, Minchinton AI, Manisali I, Liggins R, Brooks DE, Burt HM. In vivo evaluation of mucoadhesive nanoparticulate docetaxel for intravesical treatment of non-muscle-invasive bladder cancer. Clin Cancer Res 2011; 17:2788-98; PMID:21357680; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2981 [DOI] [PubMed] [Google Scholar]
- 14.Baribeau S, Chaudhry P, Parent S, Asselin E. Resveratrol inhibits cisplatin-induced epithelial-to-mesenchymal transition in ovarian cancer cell lines. PLoS One 2014; 9:e86987; PMID:24466305; http://dx.doi.org/ 10.1371/journal.pone.0086987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G 2nd, Samuel S, Kim MP, Lim SJ, Ellis LM. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res 2009; 69:1951-7; PMID:19244128; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011; 146:633-44; PMID:21854987; http://dx.doi.org/ 10.1016/j.cell.2011.07.026 [DOI] [PubMed] [Google Scholar]
- 17.Latifi A, Abubaker K, Castrechini N, Ward AC, Liongue C, Dobill F, Kumar J, Thompson EW, Quinn MA, Findlay JK, et al.. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J Cell Biochem 2011; 112:2850-64; PMID:21618587; http://dx.doi.org/ 10.1002/jcb.23199 [DOI] [PubMed] [Google Scholar]
- 18.Nor C, Zhang Z, Warner KA, Bernardi L, Visioli F, Helman JI, Roesler R, Nör JE. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia 2014; 16:137-46; PMID:24709421; http://dx.doi.org/ 10.1593/neo.131744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pirozzi G, Tirino V, Camerlingo R, Franco R, La Rocca A, Liguori E, Martucci N, Paino F, Normanno N, Rocco G. Epithelial to mesenchymal transition by TGFbeta-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS One 2011; 6:e21548; PMID:21738704; http://dx.doi.org/ 10.1371/journal.pone.0021548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009; 9:265-73; PMID:19262571; http://dx.doi.org/ 10.1038/nrc2620 [DOI] [PubMed] [Google Scholar]
- 21.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29:4741-51; PMID:20531305; http://dx.doi.org/ 10.1038/onc.2010.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663-76; PMID:16904174; http://dx.doi.org/ 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 23.Polo JM, Hochedlinger K. When fibroblasts MET iPSCs. Cell Stem Cell 2010; 7:5-6; PMID:20621040; http://dx.doi.org/ 10.1016/j.stem.2010.05.018 [DOI] [PubMed] [Google Scholar]
- 24.Strauss R, Hamerlik P, Lieber A, Bartek J. Regulation of stem cell plasticity: mechanisms and relevance to tissue biology and cancer. Mol Ther 2012; 20:887-97; PMID:22314288; http://dx.doi.org/ 10.1038/mt.2012.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Mah N, Prigione A, Wolfrum K, Andrade-Navarro MA, Adjaye J. A transcriptional roadmap to the induction of pluripotency in somatic cells. Stem Cell Rev 2010; 6:282-96; PMID:20336394; http://dx.doi.org/ 10.1007/s12015-010-9137-2 [DOI] [PubMed] [Google Scholar]
- 26.Li W, Li K, Wei W, Ding S. Chemical approaches to stem cell biology and therapeutics. Cell Stem Cell 2013; 13:270-83; PMID:24012368; http://dx.doi.org/ 10.1016/j.stem.2013.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin T, Ambasudhan R, Yuan X, Li W, Hilcove S, Abujarour R, Lin X, Hahm HS, Hao E, Hayek A, et al.. A chemical platform for improved induction of human iPSCs. Nat Methods 2009; 6:805-8; PMID:19838168; http://dx.doi.org/ 10.1038/nmeth.1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen S, Do JT, Zhang Q, Yao S, Yan F, Peters EC, Schöler HR, Schultz PG, Ding S. Self-renewal of embryonic stem cells by a small molecule. Proc Natl Acad Sci U S A 2006; 103:17266-71; PMID:17088537; http://dx.doi.org/ 10.1073/pnas.0608156103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y, Zhu X, Hahm HS, Wei W, Hao E, Hayek A, Ding S. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc Natl Acad Sci U S A 2010; 107:8129-34; PMID:20406903; http://dx.doi.org/ 10.1073/pnas.1002024107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strauss R, Li ZY, Liu Y, Beyer I, Persson J, Sova P, Möller T, Pesonen S, Hemminki A, Hamerlik P, et al.. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS One 2011; 6:e16186; PMID:21264259; http://dx.doi.org/ 10.1371/journal.pone.0016186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strauss R, Sova P, Liu Y, Li ZY, Tuve S, Pritchard D, Brinkkoetter P, Möller T, Wildner O, Pesonen S, et al.. Epithelial phenotype confers resistance of ovarian cancer cells to oncolytic adenoviruses. Cancer Res 2009; 69:5115-25; PMID:19491256; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tania M, Khan MA, Fu J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol 2014; 35:7335-42; PMID:24880591; http://dx.doi.org/ 10.1007/s13277-014-2163-y [DOI] [PubMed] [Google Scholar]
- 33.Ueno NT, Bartholomeusz C, Herrmann JL, Estrov Z, Shao R, Andreeff M, Price J, Paul RW, Anklesaria P, Yu D, et al.. E1A-mediated paclitaxel sensitization in HER-2/neu-overexpressing ovarian cancer SKOV3.ip1 through apoptosis involving the caspase-3 pathway. Clin Cancer Res 2000; 6:250-9; PMID:10656456 [PubMed] [Google Scholar]
- 34.Gottlieb HE, Kotlyar V, Nudelman A. NMR chemical shifts of common laboratory solvents as trace impurities. J Org Chem 1997; 62:7512-5; PMID:11671879 [DOI] [PubMed] [Google Scholar]
- 35.van Rensburg R, Beyer I, Yao XY, Wang H, Denisenko O, Li ZY, Russell DW, Miller DG, Gregory P, Holmes M, et al.. Chromatin structure of two genomic sites for targeted transgene integration in induced pluripotent stem cells and hematopoietic stem cells. Gene Ther 2013; 20:201-14; PMID:22436965; http://dx.doi.org/ 10.1038/gt.2012.25 [DOI] [PMC free article] [PubMed] [Google Scholar]