

Abstract
Significance: Hydrogen sulfide (H2S) has only recently gained recognition for its physiological effects. It is synthesized widely in the mammalian tissues and regulates several biologic processes ranging from development, angiogenesis, neurotransmission to protein synthesis. Recent Advances: The aim of this review is to critically evaluate the evidence for a role for H2S in kidney function and disease. Critical Issues: H2S regulates fundamental kidney physiologic processes such as glomerular filtration and sodium reabsorption. In kidney disease states H2S appears to play a complex role in a context-dependent manner. In some disease states such as ischemia-reperfusion and diabetic kidney disease it can serve as an agent that ameliorates kidney injury. In other diseases such as cis-platinum-induced kidney disease it may mediate kidney injury although more investigation is needed. Recent studies have revealed that the actions of nitric oxide and H2S may be integrated in kidney cells. Future Directions: Further studies are needed to understand the full impact of H2S on kidney physiology. As it is endowed with the properties of regulating blood flow, oxidative stress, and inflammation, H2S should be investigated for its role in inflammatory and toxic diseases of the kidney. Such in-depth exploration may identify specific kidney diseases in which H2S may constitute a unique target for therapeutic intervention. Antioxid. Redox Signal. 25, 720–731.
Keywords: : renal physiology, acute kidney injury, chronic kidney disease, diabetic kidney disease, hypertension
Introduction
Among the physical forms of matter, gases are gaining attention as important regulators of cell function. Hydrogen sulfide (H2S) has only recently received recognition for its physiological effects in joining gases that are active in mammalian homeostasis, including nitric oxide and carbon monoxide. Although it was known for its toxic effects as an environmental hazard, recent findings have highlighted its regulatory effects on fundamental cellular processes in several tissues. Our review will focus on the role of H2S in kidney physiology and disease.
H2S Synthesis
In mammalian cells, H2S is primarily produced in the cytosol by the enzymes of the trans-sulfuration pathway, cystathionine β-synthase (CBS), and cystathionine γ-lyase (CSE) (13, 70, 83). Homocysteine (HCy) is condensed with serine by CBS to generate cystathionine, which is converted into l-cysteine by CSE. l-cysteine can be used as a substrate by both CBS and CSE to produce H2S (Fig. 1). CSE also catalyzes H2S production by the conversion of HCy into homolanthionine (70). Additionally, cysteine is converted by aspartate aminotransferase in the presence of α-ketogluatarate to form 3-mercaptopyruvate and glutamate. Another enzyme, 3-mercaptopyruvate sulfurtransferase (3-MST) contributes to the endogenous H2S production in the mitochondria in the presence of a reductant using 3-mercaptopyruvate as a substrate (65, 82) (Fig. 1).
FIG. 1.

H2S synthesis. H2S, hydrogen sulfide; CBS, cystathionine β-synthase; CSE, cystathione γ-lyase; 3-MST, 3-mercaptopyruvate sulfurtransferase; AAT, aspartate aminotransferase; DAO, D-amino acid oxidase.
It was recently discovered that H2S can also be produced from d-cysteine involving 3-MST and d-amino acid oxidase (81). In the kidney, more H2S is said to be produced from D-serine than from l-serine (81) (Fig. 1).
H2S Signaling Through Sulfhydration
Recent studies have shown that H2S can regulate several signaling pathways that are involved in cell biological processes; these will be reviewed later. Additionally, H2S can post-translationally modify proteins by forming a covalent link to the SH of cysteine by the process of sulfhydration (Fig. 2). A functional role for sulfhydration of proteins had been demonstrated for xanthine oxidase (60) and aldehyde oxidase (9), both of which have an active site persulfide that is essential for catalysis. In contrast, sulfhydration of rat liver tyrosine aminotransferase, which is CSE dependent, leads to enzyme inactivation (29), and it is enhanced in streptozotocin (STZ)-induced diabetes (28). The physiological importance of sulfhydration has been recently discovered; a proteomic analysis of persulfidation revealed that ∼10–25% of liver proteins harbor this modification (67).
FIG. 2.

Modification of proteins by sulfhydration.
The functional consequences of sulfhydration have been characterized for some proteins. TNFα stimulates Sp1-mediated transcriptional activation of CSE, which increases H2S production and sulfhydration of the p65 subunit of the transcription factor NF-κB, leading to an increase in its DNA binding (76). Sulfhydration of the ATP-sensitive K+ channel (KATP) activates the channel and causes hyperpolarization and vasorelaxation of rodent mesenteric arteries (68). Sulfhydration of protein tyrosine phosphatase, PTP1B, inhibits its activity and potentiates the activation of protein kinase R-like endoplasmic reticulum (ER) kinase (PERK) in response to ER stress (45).
H2S has been proposed to modify cysteine residues by redox-dependent sulfhydration (38). In the reducing intracellular milieu, cysteines are likely to exist predominantly in the thiol form that does not react with H2S. Transient increases in reactive oxygen species (ROS) concentration would increase the proportion of cysteines present in the oxidized form, for example, sulfenic acid and disulfides, both of which are reactive to H2S (Fig. 2). Hydrogen persulfide, formed by two-electron oxidation of H2S, has been proposed as a possible cellular sulfhydration reagent (69).
Renal Effects of H2S
The anatomical and physiological unit of the kidney is the nephron. The mammalian nephron consists of a capillary filtration unit called the glomerulus and a tubular system downstream of the glomerulus. The kidney is a highly vascular organ receiving nearly 25% of the cardiac output. The unique feature of the glomerular capillary is that being interposed between two arterioles it is exposed to much higher pressures than capillaries elsewhere. The hydrostatic pressure in the glomerular capillary is maintained at ∼50 mm Hg, whereas the hydrostatic pressure in other capillaries falls from about 35 mm Hg at the arteriolar end to about 15 mm Hg at the venular end (8, 24).
The glomerulus is connected to a tubule that consists of a series of highly specialized segments that are tasked with elaborate processes for solute and ionic absorption and secretion. The tubules are classified into the proximal tubule, the descending and ascending loops of Henle (the ascending limb has thin and thick segments), the distal convoluted tubule, the connecting duct, and the collecting duct. The proximal tubule performs the bulk of transport functions of the nephron. It reabsorbs nearly two thirds of the filtered sodium facilitated by the actions of the Na-H antiporter. Sodium reabsorption in the ascending limb of the loop of Henle is an active process under the control of Na-K-2Cl cotransporter. Sodium reabsorption in the distal convoluted tubule is under the control of Na-Cl cotransporter, whereas that in the connecting duct and the collecting duct is regulated by the epithelial sodium channel (ENaC). In these segments, the functions of transporters governing sodium reabsorption are integrated with the actions of Na+,K+-ATPase.
The human kidney filters ∼150–200 L of plasma at the glomerulus daily; the tubules extensively reabsorb and modify the composition of the glomerular filtrate such that the volume of urine is about 1–2 L/day. The tubules are surrounded by peritubular blood vessels that are derived from the efferent arteriole; these blood vessels participate in returning to the systemic circulation the vast amount of solutes and water that are reabsorbed by the tubules. It is important to note that the tubules have elaborate secretory pathways that add substances from the peritubular blood vessels to the filtrate, including drugs for elimination in the urine. Since so much of the kidney function is under the control of blood vessels, that is, afferent and efferent arterioles, glomerular capillary, and peritubular blood vessels, it is no wonder that gases such as H2S and nitric oxide (NO), highly vasoactive agents, regulate renal physiology.
H2S production by the kidney
It has been long known that the kidney generates measurable quantities of H2S (86). Both CSE and CBS are present in the kidney. CSE is expressed by the endothelial cells, mesangial cells, and the podocytes that together form the majority of the cells in the glomerulus. It is expressed in the proximal and distal tubular epithelial cells and in the peritubular capillaries; nearly 75% of all renal cells express CSE (7, 49). Yamamoto et al. did not detect either CBS or CSE in the glomerulus or in the renal blood vessels but found their expression in the proximal tubules (98).
Physiological effects of H2S in the kidney
Infusion of H2S in the form of sodium hydrosulfide (NaHS) into the renal artery increases the renal blood flow and augments the glomerular filtration rate, which is an index of the clearance function of the kidney; urinary flow rate was also increased, and these effects were dose dependent (97).
H2S augments the urinary excretion of sodium and potassium, probably by the inhibition of Na-K-2Cl cotransporter in the ascending limb of the loop of Henle and Na K ATPase (97). Patch clamp studies on Xenopus A6 cells have shown that hydrogen peroxide augments phosphatidyl inositol 3 kinase activity to activate the ENaC; this is inhibited by H2S (103). In addition to the Xenopus frog epithelial cells, recent reports have shown that in human lung epithelial cells (H441) H2S inhibits β-adrenergic agonist-induced ENaC-mediated Na transport; H2S also inhibits ENaC-dependent lung fluid clearance in rats (1). Additionally, H2S augments blood flow in the peritubular blood vessels, and NaHS corrects hypoperfusion in diabetic mice (98).
Context-Dependent Role of H2S in Kidney Injury
Recent investigations have revealed a complex role played by H2S in the pathogenesis of kidney injury. Whether it ameliorates kidney injury or whether it participates in bringing it about appears to depend on the disease model being studied.
Acute kidney injury
The kidney is vulnerable to injury from changes in the systemic hemodynamics and after the administration of toxic medications. Thus, acute kidney injury is seen in the form of ischemia/reperfusion injury or when nephrotoxic medications such as anticancer agents and antibiotics are administered.
Renal ischemia/reperfusion injury
Ischemia/reperfusion injury is common during kidney transplantation and may affect the survival and function of the graft. In humans, the levels of renal CSE messenger RNA (mRNA) at the time of organ procurement positively correlate with the glomerular filtration rate measured at 2 weeks after transplantation (7), suggesting that H2S could be protective in the context of renal ischemia. In support of this notion, NaHS has been shown to improve allograft survival and function in rats with bilateral nephrectomy that underwent a subsequent kidney transplant; this was accompanied by a general decrease in the transcription of cellular stress response genes (55).
Ischemia/reperfusion injury that is characterized by oxidative stress has been implicated in delayed function of transplanted kidneys; xanthine oxidoreductase enzyme activation in the form of xanthine oxidase can lead to excessive production of reactive oxygen/nitrogen species and inflammation. Clinically significant changes in the activity of kidney xanthine metabolizing enzymes have been reported in renal transplant patients with delayed graft function when compared with those with early graft function (16). Because H2S improves ischemia/reperfusion injury in the setting of kidney transplantation, these studies suggest a possible link between the reduced H2S expression and the activation of xanthine pathway that generates ROS.
That H2S inhibits ROS generation by the xanthine oxidase pathway was established in the rat model of aspirin-induced gastric ulcers in which oxidative stress plays a major role. Intraperitoneal administration of 2-acetyloxybenzoic acid 4-(3-thioxo-3H-1, 2 dithiol-5-yl) phenyl ester (ACS14, S-aspirin), a form of aspirin that releases H2S, prevented aspirin-induced gastric ulceration; this protective effect of S-aspirin was reproduced by NaHS establishing that H2S was responsible for the ameliorative effect (54). The mechanism of H2S amelioration of aspirin-induced injury involved a decrease in the markers of oxidative stress and a reversal of aspirin-induced increased expression of xanthine oxidase in the gastric mucosa (54).
In animal models of ischemia/reperfusion injury, the renal expression of CBS and CSE and the endogenous production of H2S are decreased (26). Renal ischemia/reperfusion injury is characterized by increased inflammation, oxidative stress, apoptosis of tubular epithelial cells, and vascular dysfunction (6). Administration of H2S donors before or after the ischemia/reperfusion insult restores the levels of H2S-producing enzymes and local H2S production (26), and it accelerates renal recovery (5, 26, 56). This protective effect is accompanied by a reduction in oxidative stress (5, 26) and inflammation (56). A role for H2S deficiency in the pathogenesis of renal ischemia/reperfusion injury was demonstrated by targeting CSE; administration of propargylglycine (PAG), a CSE inhibitor (89), or genetic ablation of CSE (7) significantly worsened both kidney damage and animal mortality.
Recent studies have suggested a protective role for H2S derived from d-cysteine in ischemia/reperfusion injury in the kidney (81). Furthermore, the ameliorative effects of dietary calorie restriction on ischemia/reperfusion injury to the liver and the kidney are dependent on increased production of H2S (30).
Obstructive kidney injury
Renal injury after obstruction of the genitourinary tract is a common cause of both acute and chronic kidney injury, particularly in elderly men. In studies on obstructive kidney injury, investigators commonly employ unilateral obstruction to the ureter, which permits the contralateral kidney to serve as a control. Unilateral obstruction by ureteral ligation resulted in kidney injury characterized by inflammation and renal fibrosis in association with reduction in the generation of H2S in that kidney (85). Reduced H2S generation in this model was due to a decrease in the expression of CBS. Administration of NaHS significantly reduced the aforementioned manifestations of kidney injury, including inflammation (85). In vitro experiments showed that H2S inhibited transforming growth factor β-induced transformation of fibroblasts to myofibroblasts (85), providing a mechanistic basis for reduction in fibrosis. These data and observations in the ischemic/reperfusion model suggest that depending on the nature of acute kidney injury, CSE or CBS can undergo dynamic regulation in the kidney.
Cisplatin and doxorubicin nephrotoxicity
Cisplatin is a chemotherapeutic drug that is used for the treatment of lung, ovarian, and cervical cancers. Platinum complexes bind to and cause crosslinking of DNA, which ultimately triggers apoptosis of cancer cells. However, cisplatin is toxic to the kidneys. The nephrotoxicity of cisplatin seems to be related to oxidative stress, since it can be ameliorated by free radical scavenging agents in animal models. In rats, cisplatin causes inflammation of the renal outer medulla that is characterized by the infiltration of macrophages, neutrophils, and T lymphocytes; pretreatment with PAG, an inhibitor of H2S synthesis, significantly reduces the renal damage induced by cisplatin (14).
In contrast to this report, administration of H2S donors to rats has been reported to prevent cisplatin-induced renal injury by increasing anti-oxidant mechanisms in kidney cells (2). The conflicting data on the role of H2S in cisplatinum kidney injury emphasize the need for further examination of the issue.
Doxorubicin is another chemotherapeutic agent employed in the treatment of malignant tumors. Doxorubicin may cause congestive heart failure and chronic kidney injury in the form of focal segmental sclerosis of the glomerulus that is associated with marked proteinuria and loss of clearance function of the kidney (51). H2S has been proposed as a mediator of kidney injury manifesting as inflammation and proteinuria after the administration of doxorubicin (22); however, H2S donors have been shown to protect the heart from doxorubicin-induced toxicity (102), suggesting the need for clarification of the role of H2S in kidney injury induced by that agent. In addition, studies are also needed on the involvement of H2S in acute kidney injury due to other nephrotoxic agents such as radiocontrast media containing iodine and gadolinium.
Hyperhomocysteinemia
HCy is usually converted into cystathionine by CBS (Fig. 1). Thus, hyperhomocysteinemia (HHCy) occurs either due to decreased expression of CBS or due to inhibition of its activity. HHCy can be caused by deficiencies of vitamins B6, B9, and B12 (62). HHCy has been linked to atherosclerosis and thrombosis (11) and to Alzheimer's disease (64). It is also associated with kidney injury, as indicated by the development of albuminuria (34). Increased levels of HCy repress the expression and activity of the H2S-generating enzymes, CSE, CBS, and 3-MST, leading to a significant decrease in endogenous H2S production (95). It has been shown that HCy increases the methylation of the CSE promoter by DNA methyltransferase, which leads to the repression of CSE transcription (53). Whether the DNA methylation mechanism affects the expression of CBS and 3-MST is not known.
In addition to its repression of H2S synthesis, HHCy has profound effects on the vasculature. It increases inflammation (72), oxidative (72) and nitrosative stress (39), and mitophagy (79) in endothelial cells, leading to their dysfunction. Specifically, in the kidney, there appears to be a vicious cycle with regards to HCy metabolism; a decrease in kidney function leads to an elevation of HCy, which, in turn, leads to kidney injury and loss of function. Chronic HHCy in CBS +/− mice is associated with glomerulosclerosis and proteinuria, which are ameliorated by NaHS (77). Augmented apoptosis of cells in the kidney cortex of mice with HHCy was associated with increased activity of matrix metalloproteinase (MMP)-2 and −9; these changes were inhibited by NaHS, leading the authors to the conclusion that activation of MMPs is involved in remodeling of the renal vascular beds in HHCy due to deficiency of renal H2S content (77).
HHCy in association with decreased H2S in the kidney is also associated with systemic hypertension, increase in the expression of p47 phox subunit of NADPH oxidase (NOX), glomerular infiltration with macrophages, and type IV collagen accumulation; these changes were also ameliorated by NaHS (78).
HHCy also affects cardiomyocytes directly by the inhibition of H2S pathway that is associated with increased oxidative stress, mitochondrial dysfunction, and apoptosis (12, 95). In the brain, HHCy induces the disruption of the blood–brain barrier (39), a reduction of the cerebral blood flow (40), an increase in oxidative and nitrosative stress (39), and an increase in cerebral inflammation (39, 40). The fact that all the consequences of HHCy on the vasculature, the myocardium, and the brain are counteracted by H2S donors (12, 39, 40) suggests that H2S deficiency is the major mechanism leading to tissue injury in HHCy. This is reinforced by the study showing that a triple gene therapy (CBS-CSE-3-MST) blocks the effects of HHCy on vasorelaxation in an ex vivo renal artery culture; this approach also restores the levels of vascular endothelial growth factor (VEGF), CD31 and reduces elevated endostatin levels (79).
Chronic kidney disease of diverse etiology is associated with abnormalities in H2S metabolism. Reduced expression of CBS, CSE, and H2S content in circulating leukocytes has been observed in end-stage kidney disease patients on maintenance hemodialysis (71). Removal of most of the functioning nephrons by 5/6 nephrectomy results in chronic kidney disease. The production rate of H2S and the expression of CBS, CSE, and 3-MST were selectively reduced in the kidney and the liver in rats at 12 weeks after 5/6 nephrectomy (4). These changes were associated with an increase in the markers of oxidative stress, including the expression of NADPH oxidase 4 (NOX4) in the kidney, suggesting that deficiency of H2S could contribute to the pathogenesis of chronic kidney disease by promoting oxidative stress (4).
Hypertension
Hypertension affects ∼20–25% of the adult population in the Western world. In primary hypertension, genetic factors are believed to interact with environmental factors in order elevate the blood pressure. Mice lacking CSE display H2S deficiency and hypertension due to reduced endothelium-mediated vasodilation (100); administration of NaHS lowers the blood pressure in both the hypertensive CSE knockout mice (100) and the normotensive mice (87). Recent studies have identified H2S as the endothelium-derived hyperpolarizing factor (68, 90) that can directly induce vasorelaxation. Further studies have shown that H2S induces vasorelaxation through activation of the cAMP/PKA signaling pathway, activation of calcium signaling (104), and activation of ATP-dependent K+ (KATP) channels and low-threshold voltage-gated K+ (KCNQ) channels (44).
In rats, dexamethasone-induced hypertension is accompanied by a reduction of the H2S-producing enzymes CBS and CSE in the vasculature (mesenteric arterial bed and carotid artery) (15), implying that the reduction of endogenous production of H2S could contribute to the hypertension in this model. In spontaneously hypertensive rats (SHR), a high salt diet causes hypertension that is accompanied by a reduction of renal CBS protein expression and endogenous H2S production; administration of NaHS inhibits salt-induced hypertension and activation of the intra-renal renin-angiotensin system (33).
Systemic infusion of angiotensin II in mice causes hypertension that is accompanied by vascular oxidative stress, reduction in NO bioavailability, and reduced aortic endothelium function; these effects were worsened by a CSE inhibitor and reversed by an H2S donor (3).
H2S/NO crosstalk
In rats, inhibition of endogenous NO production by NO synthase (NOS) inhibitors, such as Nω-nitro-l-arginine methyl ester (L-NAME), causes hypertension that can be rescued by the administration of NaHS, which also restores NO bioavailability (37, 105). These data suggest the existence of an H2S/NO crosstalk in the control of blood pressure. In support of this hypothesis, administration of H2S to normotensive rats lowers blood pressure, an effect that is blocked by an NOS inhibitor, and the hypotensive response to NO donor sodium nitroprusside is abrogated by a CSE inhibitor (91). Interestingly, the effects of H2S donor and NO donor are independent of one another in the SHR rats (91), suggesting that the H2S-NO crosstalk may vary with the model of hypertension.
Pre-eclampsia
Pre-eclampsia is a disorder of pregnancy that is characterized by high blood pressure and proteinuria (19). Circulating H2S levels are reduced in women with pre-eclampsia (94). Studies conducted in rodents have suggested that excess circulating soluble fms-like tyrosine kinase 1 (sFlt1), a truncated VEGF receptor, contributes to the pathogenesis of pre-eclampsia (61). In placental explants, inhibition of CSE activity or genetic ablation of CSE significantly increased the expression and secretion of sFlt1 (94), and in a mouse model of pre-eclampsia, administration of a slow-releasing H2S donor reduced circulating (sFlt1) and restored fetal growth (94).
Adenovirus-mediated sFlt1 overexpression in rats leads to hypertension and proteinuria that are similar to pre-eclampsia; administration of NaHS to these rats reduces circulating sFlt-1, blood pressure, and proteinuria in association with restoration of circulating VEGF levels, in part due to stimulation of VEGF synthesis by the kidney podocytes (31).
These data suggest that endogenous H2S is required for a healthy placental vasculature and that a decrease in endogenous H2S production could contribute to the pathogenesis of pre-eclampsia. In support of this hypothesis, a single nucleotide polymorphism in the CSE gene has been found in association with pre-eclampsia (66).
Diabetes
Diabetic kidney disease
Diabetes is associated with clinically significant kidney injury in about a third of the patients. Kidney injury in diabetes is characterized by kidney growth due to hypertrophy; accumulation of extracellular matrix proteins leading to fibrosis of glomerular and tubulointerstitial compartments; excessive urinary loss of proteins, particularly albumin (proteinuria, albuminuria); and loss of waste clearance function over time. In fact, diabetes is the most common cause of end-stage kidney disease in the United States.
Plasma levels of H2S in patients with type 2 diabetes are lower than in normoglycemic humans (35), and they negatively correlate with markers of adiposity (96). Among chronic dialysis patients, plasma H2S levels have been reported to be lower in diabetic patients compared with those with nondiabetic cause of end-stage kidney disease; plasma H2S levels significantly correlated with indices of uremic atherosclerotic disease, suggesting that reduced H2S may contribute to the latter (52).
Recent investigations have shown that the renal cortical expression of H2S-producing enzymes CBS and CSE is reduced in rodents with type 1 or type 2 diabetes (49, 101, 106). Administration of NaHS inhibits diabetes-induced increase in the expression of transforming growth factor-β, the profibrogenic cytokine, glomerular and tubulointerstitial deposition of extracellular matrix proteins, oxidative stress, and proteinuria, indicating that H2S deficiency contributes to the development of kidney injury in diabetes (101, 106).
H2S promotes vasodilation and reduces blood pressure in CSE knockout mice with systemic hypertension (100). Changes in renal blood flow and intraglomerular hemodynamics constitute an important contributory mechanism of injury in diabetic kidney disease, In view of its vasoactivity, it is likely that H2S participates in the regulation on renal microcirculation in diabetes. In the initial stages, the renal blood flow and glomerular filtration rate are increased in diabetic kidney disease; whether a transient local increase in glomerular microvascular H2S generation contributes to this phenomenon is not known and needs to be studied.
In established diabetic kidney injury in the Akita mice with type 1 genetic diabetes, Kundu et al. have employed multiple mutually confirmatory approaches, including renal resistive index, to show a decrease in renal cortical blood flow that was associated with reduced vascular density; these changes were improved by the administration of NaHS (46).
Changes in renal microcirculation could include changes in glomerular and/or peritubular capillary systems in diabetic mice. Fibrosis of the tubulo-interstitial compartment in the kidney positively correlates with poor outcome in diabetic kidney disease (63). However, changes in the structure and function of peritubular capillaries in diabetic kidney disease have not been well understood. Renal resistance index appears to correlate inversely with peritubular capillary density in kidney biopsy in patients with diabetic kidney disease (42); this is probably an indirect correlation, since vascular resistance is a function of arterial blood vessels. Peritubular capillary diameter and blood flow are reported to be reduced in type 1 diabetic mice; administration of NaHS reversed these changes.
Matrix remodeling by metalloproteinases may be involved in reduced H2S production in the kidney in diabetes. MMP-9 expression and activity are increased in the renal parenchyma in Akita mice with type 1 diabetes due to oxidative stress. Using MMP9 knockout mice in their studies, Kundu et al. have shown that MMP-9 is involved in the reduced renal expression of CBS and CSE, leading to a decrease in H2S generation in diabetic mice (47). These investigators have proposed a link between MMP-9 activation, reduced H2S generation, and activation of N-methyl-d-aspartate receptor, leading to abnormal regulation of the gap junction proteins, connexins, that may be involved in renovascular remodeling (47). H2S inhibits diabetes-induced kidney injury by several other mechanisms, including inhibition of signaling mechanisms that govern protein synthesis.
Kidney hypertrophy and an increase in matrix protein deposition require stimulation of protein synthesis. mRNA translation is a rate-limiting step in the control of protein synthesis (32), and it is under the direct control of the mechanistic target of rapamycin (mTOR). Studies from our group have shown that the phosphatidylinositol 3 kinase-Akt-mTORC1 signaling pathway is activated, coinciding with renal hypertrophy and onset of matrix protein deposition in rodent models of diabetes (21, 50, 57, 59, 74). In fact, rapamycin, an inhibitor of mTOR, ameliorates kidney injury in early stages of diabetes (74). Recent studies have also shown that reduction in the activity of kidney AMP-activated protein kinase (AMPK) in diabetes permits stimulation of mTOR; thus, stimulation of AMPK ameliorates diabetes-induced kidney injury (18, 50, 80).
The signaling mechanism of NaHS amelioration of high glucose-induced kidney injury involves stimulation of AMPK, which, in turn, inhibits high glucose-induced mTORC1 activity, leading to a reduction in kidney cell hypertrophy and increased matrix protein expression (48, 49). AMPK activity is dependent on Thr172 phosphorylation of the α subunit of the kinase, which is under the control of either LKB1 or calcium calmodulin kinase kinase-β. Employing siRNA against these enzymes, Lee et al. showed that H2S-induced AMPK phosphorylation is dependent on calcium calmodulin kinase kinase-β (49) (Fig. 3).
FIG. 3.

Signaling pathways involved in H2S amelioration of high glucose-induced protein synthesis. NaHS, sodium H2S; CaMKKβ, Ca(2+)/calmodulin-dependent protein kinase kinaseβ; LKB1, liver kinase B1; AMPK, AMP-activated protein kinase; mTOR, mechanistic target of rapamycin; eIF4E, eukaryotic initiation factor 4E; 4E-BP1, eIF4E-binding protein 1; eIF4A, eukaryotic initiation factor 4A; eIF4G, eukaryotic initiation factor 4G; PDCD4, programmed cell death protein 4; p70S6K, p70 S6 kinase; eEF2, eukaryotic elongation factor 2; eEF2K, eEF2 kinase.
mRNA translation, a crucial step in peptide synthesis, consists of initiation, elongation, and termination phases (41). mTORC1 activates mRNA translation, which is required for high glucose induction of matrix protein synthesis (50, 58, 74). In the resting state, the eukaryotic initiation factor 4E (eIF4E) and eIF4A are held inactive by their binding partners, 4E-BP1 and Programmed Cell Death protein 4 (PDCD4), respectively (41). Phosphorylation of 4E-BP1 by mTORC1 releases eIF4E, which binds to eIF4A and eIF4G to form eIF4F complex that promotes mRNA translation (84).
In kidney epithelial cells exposed to high glucose, mTORC1 activity was stimulated as indicated by the increase in phosphorylation of its direct substrates 4E-BP1 and p70S6 kinase that was abolished by NaHS (Fig. 3). High glucose reduced the expression of PDCD4, which was inhibited by NaHS (49). Additionally, high glucose promoted the dissociation of eIF4A from PDCD4 and increased its association with eIF4G; these reactions were also abrogated by NaHS (49).
The elongation phase of translation is facilitated by the eukaryotic elongation factor 2 (eEF2), which is active when dephosphorylated (41). Phosphorylation of eEF2 is under the control of eEF2 kinase; p70S6 kinase, the mTORC1 substrate, phosphorylates eEF2 kinase and inhibits its activity (41). Thus, mTORC1 also regulates the elongation phase of mRNA translation. High glucose promoted the phosphorylation of eEF2 kinase and the dephosphorylation of eEF2; NaHS inhibited these changes (Fig. 3). These observations showed that H2S abrogates high glucose-stimulated initiation and elongation phases of mRNA translation by inhibiting mTORC1 in diverse renal cells.
A fascinating new development in this area is that the ameliorative effect of H2S in kidney cells that are exposed to high glucose involves NO. Diabetes-induced kidney injury is dependent on the production of ROS, mainly by the activation of NOX4 (25). Thus, diabetes-induced kidney hypertrophy, albuminuria, and extracellular matrix protein accumulation are inhibited in NOX4 knockout mice and by the administration of NOX4 inhibitors (25, 36, 75, 92). H2S ameliorates oxidative stress by inhibiting mitochondrial ROS generation (88), increasing GSH production (43), and promoting the transcription of antioxidant genes via the activation of Nrf2 transcription factor (10, 23, 27). H2S has also been reported to inhibit HCy-induced NOX4 expression in the brain endothelial cells (40). Whether H2S regulation of NOX4 involves NO is not known.
We studied high glucose-induced regulation of NOX4 by H2S in the kidney proximal tubular epithelial cells (unpublished). High glucose-induced increase in NOX4 expression, ROS generation, and matrix protein laminin γ1 expression was inhibited by H2S by the activation of AMPK. H2S inhibition of high glucose-induced NOX4 expression was abrogated by L-NAME, an inhibitor of NOS. L-NAME abolished the inhibitory effect of H2S on high glucose-induced laminin γ1 expression. In exploring the type of NOS involved, we found that H2S did not affect the expression of eNOS but increased the expression of inducible nitric oxide synthase (iNOS). siRNA against iNOS abolished the ameliorative effects of H2S on high glucose-induced NOX4 expression. These data suggest that H2S augments iNOS expression to increase NO production, which inhibits high glucose-induced NOX4, ROS, and the downstream consequence of matrix increment (Fig. 4A).
FIG. 4.
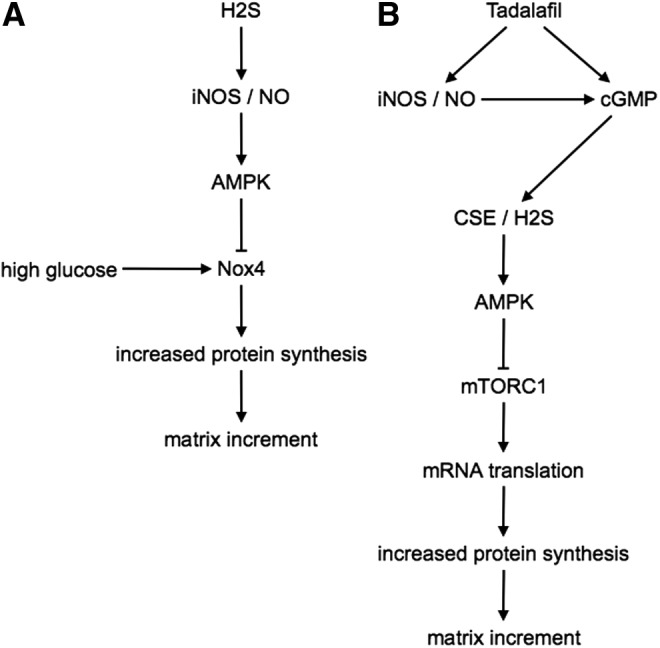
Signaling pathways activated by H2S in renal cells. (A) Signaling pathways involved in H2S amelioration of high glucose-induced renal extracellular matrix increment in kidney cells. (B) Signaling pathways involved in tadalafil (a phosphodiesterase-5 inhibitor)-induced amelioration of high glucose-induced renal extracellular matrix increment in kidney cells. iNOS, inducible nitric oxide synthase; NO, nitric oxide; Nox4, NADPH oxidase 4; mTORC1, mechanistic target of rapamycin complex 1.
Since NaHS is unacceptable to humans because of its foul smell, we investigated whether phosphodiesterase-5 (PDE-5) inhibitors can promote H2S generation in kidney cells, the rationale being that the protective effect of PDE5 inhibitors on cardiac ischemia reperfusion injury is H2S dependent (73). In renal podocytes exposed to high glucose, tadalafil, a long-acting PDE-5 inhibitor, increased the CSE expression, H2S generation, and AMPK stimulation, leading to inhibition of mTOR and a reduction in cell hypertrophy and matrix protein expression. The mechanism of tadalafil regulation of CSE involved not only an increase in cGMP, a well-known effect of that agent due to PDE5 inhibition, but also stimulation of iNOS expression (48) (Fig. 4B).
Together, these observations suggest that H2S recruits NO to ameliorate high glucose-induced kidney cell injury, whereas tadalafil is employed as NO upstream of H2S. These data show a high degree of integration of activities of the two physiological gases, NO and H2S, in the kidney. The surprising development is the ameliorative role played by iNOS in the aforementioned experiments on high glucose-induced kidney cell injury; iNOS is generally believed to mediate tissue injury. These observations emphasize the fact that whether iNOS mediates or ameliorates tissue injury is highly context dependent. PDE-5 inhibitors have been reported to ameliorate diabetic kidney injury in rodent models (20); however, whether H2S mediates the salutary effect of PDE-5 inhibitor was not examined in those studies.
Type 1 diabetes
Type 1 diabetes mellitus results from the autoimmune destruction of the insulin-producing β cells in the islets of Langerhans in the pancreas, resulting in severe insulin deficiency. In mice with STZ-induced type 1 diabetes, STZ directly activates CSE in the pancreatic β cells to increase the local H2S production, leading to cell death. STZ-induced β cell death is attenuated in mice lacking CSE. Furthermore, NaHS significantly reduces insulin secretion and glucose tolerance but does not increase blood glucose in normoglycemic mice (99). Although intra-islet levels of H2S are increased in mice with type 1 diabetes, plasma levels of H2S are lower in mice with type 1 diabetes than in normoglycemic control mice (17). Further studies are needed to understand the role of H2S in the destruction of insulin-secreting cells of the pancreas in spontaneous models of diabetes as distinct from the cytotoxicity induced by STZ.
Conclusion
H2S is a bioactive gas with far-reaching actions that are comparable to those of NO and carbon monoxide. Its regulation of cellular processes is highly cell specific; even in individual cells, its role as a mediator or as an ameliorator of cell injury is highly context specific. In-depth investigations are urgently needed to define its role in individual physiological and pathological states and its evaluation as a potential therapeutic target. For example, inflammatory diseases of the kidney prominently involve glomerular endothelial cells that produce H2S. It is possible that kidney H2S metabolism in dysregulated in inflammatory diseases such as systemic lupus erythematosus.
Several agents have been identified as therapeutic sources of H2S for interventional use, and systematic evaluation is in progress in a variety of nonrenal diseases (93). Following a more complete understanding of the role of H2S in individual acute and chronic kidney diseases due to inflammatory or toxic causes such treatment approaches may prove to be useful in kidney diseases as well.
Abbreviations Used
- 3-MST
3-mercaptopyruvate sulfurtransferase
- AAT
aspartate aminotransferase
- AMPK
AMP-activated protein kinase
- CaMKKβ
calcium calmodulin kinase kinaseβ
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- DAO
d-amino acid oxidase
- eEF2
eukaryotic elongation factor 2
- eEF2K
eEF2 kinase.
- eIF4E
eukaryotic initiation factor 4E
- ENaC
epithelial sodium channel
- ER
endoplasmic reticulum
- H2S
hydrogen sulfide
- HCy
homocysteine
- HHCy
hyperhomocysteinemia
- iNOS
inducible nitric oxide synthase
- LKB1
liver kinase B1
- L-NAME
Nω-nitro-l-argininemethylester
- MMP
matrix metalloproteinase
- mRNA
messenger RNA
- mTOR
mechanistic target of rapamycin
- NaHS
sodium H2S
- NO
nitric oxide
- NOS
NO synthase
- NOX
NADPH oxidase
- Nox4
NADPH oxidase 4
- p70S6K
p70 S6 kinase
- PAG
propargylglycine
- PDCD4
programmed cell death protein 4
- PDE-5
phosphodiesterase-5
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rats
- STZ
streptozotocin
- VEGF
vascular endothelial growth factor
Acknowledgments
The authors thank Dr. Goutam Ghosh Choudhury and Dr. Robert Kunau for their comments on this article. This work was supported by NIH grants DK077295 (to B.S.K.), the Veterans Affairs Research Service (to B.S.K), the Juvenile Diabetes Research Foundation (to D.F.), and the American Heart Association (to D.F.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Agné AM, Baldin J-P, Benjamin AR, Orogo-Wenn MC, Wichmann L, Olson KR, Walters DV, and Althaus M. Hydrogen sulfide decreases β-adrenergic agonist-stimulated lung liquid clearance by inhibiting ENaC-mediated transepithelial sodium absorption. Am J Physiol Regul Integr Comp Physiol 308: R636–R649, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahangarpour A, Abdollahzade Fard A, Gharibnaseri MK, Jalali T, and Rashidi I. Hydrogen sulfide ameliorates the kidney dysfunction and damage in cisplatin-induced nephrotoxicity in rat. Vet Res Forum 5: 121–127, 2014 [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Magableh MR, Kemp-Harper BK, and Hart JL. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens Res 38: 13–20, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Aminzadeh MA. and Vaziri ND. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol Dial Transplant 27: 498–504, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Azizi F, Seifi B, Kadkhodaee M, and Ahghari P. Administration of hydrogen sulfide protects ischemia reperfusion-induced acute kidney injury by reducing the oxidative stress. Ir J Med Sci 2015. Doi: 10.1007/s11845-015-1328-z [DOI] [PubMed] [Google Scholar]
- 6.Bonventre JV. and Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos EM, Wang R, Snijder PM, Boersema M, Damman J, Fu M, Moser J, Hillebrands J-L, Ploeg RJ, Yang G, Leuvenink HGD, and van Goor H. Cystathionine γ-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J Am Soc Nephrol 24: 759–770, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boulpaep EL. The microcirculation. In: Medical Physiology a Cellular and Molecular Approach, edited by Boron WF, Boulpaep EL. Philadelphia: Saunders, 2009, pp. 482–503 [Google Scholar]
- 9.Branzoli U. and Massey V. Evidence for an active site persulfide residue in rabbit liver aldehyde oxidase. J Biol Chem 249: 4346–4349, 1974 [PubMed] [Google Scholar]
- 10.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, and Lefer DJ. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res 105: 365–374, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cattaneo M. Hyperhomocysteinemia, atherosclerosis and thrombosis. Thromb Haemost 81: 165–176, 1999 [PubMed] [Google Scholar]
- 12.Chang L, Geng B, Yu F, Zhao J, Jiang H, Du J, and Tang C. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids 34: 573–585, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, and Banerjee R. H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J Biol Chem 284: 11601–11612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coletta Francescato Della H, Cunha FQ, Costa RS, Barbosa Júnior F, Boim MA, Arnoni CP, da Silva CGA, and Coimbra TM. Inhibition of hydrogen sulphide formation reduces cisplatin-induced renal damage. Nephrol Dial Transplant 26: 479–488, 2011 [DOI] [PubMed] [Google Scholar]
- 15.d'Emmanuele di Villa Bianca R, Mitidieri E, Donnarumma E, Tramontano T, Brancaleone V, Cirino G, Bucci M, and Sorrentino R. Hydrogen sulfide is involved in dexamethasone-induced hypertension in rat. Nitric Oxide 46: 80–86, 2015 [DOI] [PubMed] [Google Scholar]
- 16.Dołęgowska B, Błogowski W, and Domański L. Clinical evidence of the association between serum perioperative changes in xanthine metabolizing enzymes activity and early post-transplant kidney allograft function. J Am Coll Surg 211: 587–595, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Dutta M, Biswas UK, Chakraborty R, Banerjee P, Raychaudhuri U, and Kumar A. Evaluation of plasma H2S levels and H2S synthesis in streptozotocin induced Type-2 diabetes-an experimental study based on Swietenia macrophylla seeds. Asian Pac J Trop Biomed 4: S483–S487, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eid AA, Ford BM, Block K, Kasinath BS, Gorin Y, Ghosh Choudhury G, Barnes JL, and Abboud HE. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J Biol Chem 285: 37503–37512, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eiland E, Nzerue C, and Faulkner M. Preeclampsia 2012. J Pregnancy 2012: 586578–7, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang L, Radovits T, Szabó G, Mózes MM, Rosivall L, and Kökény G. Selective phosphodiesterase-5 (PDE-5) inhibitor vardenafil ameliorates renal damage in type 1 diabetic rats by restoring cyclic 3“,5” guanosine monophosphate (cGMP) level in podocytes. Nephrol Dial Transplant 28: 1751–1761, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Feliers D, Duraisamy S, Faulkner JL, Duch J, Lee AV, Abboud HE, Choudhury GG, and Kasinath BS. Activation of renal signaling pathways in db/db mice with type 2 diabetes. Kidney Int 60: 495–504, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Francescato HDC, Marin ECS, Cunha Fde Q, Costa RS, Silva CGAD, and Coimbra TM. Role of endogenous hydrogen sulfide on renal damage induced by adriamycin injection. Arch Toxicol 85: 1597–1606, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Ganster F, Burban M, la Bourdonnaye de M, Fizanne L, Douay O, Loufrani L, Mercat A, Calès P, Radermacher P, Henrion D, Asfar P, and Meziani F. Effects of hydrogen sulfide on hemodynamics, inflammatory response and oxidative stress during resuscitated hemorrhagic shock in rats. Crit Care 14: R165, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giebisch G. and Windhager E. Glomerular filtration and renal blood flow. In: Medical Physiology: A Cellular and Molecular Approach, edited by Boron WF, Boulpaep EL. Philadelphia: Saunders, 2009, pp. 767–781 [Google Scholar]
- 25.Gorin Y, Cavaglieri RC, Khazim K, Lee D-Y, Bruno F, Thakur S, Fanti P, Szyndralewiez C, Barnes JL, Block K, and Abboud HE. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am J Physiol Renal Physiol 308: F1276–F1287, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han SJ, Kim JI, Park J-W, and Park KM. Hydrogen sulfide accelerates the recovery of kidney tubules after renal ischemia/reperfusion injury. Nephrol Dial Transplant 30: 1497–1506, 2015 [DOI] [PubMed] [Google Scholar]
- 27.Han W, Dong Z, Dimitropoulou C, and Su Y. Hydrogen sulfide ameliorates tobacco smoke-induced oxidative stress and emphysema in mice. Antioxid Redox Signal 15: 2121–2134, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hargrove JL, Trotter JF, Ashline HC, and Krishnamurti PV. Experimental diabetes increases the formation of sulfane by transsulfuration and inactivation of tyrosine aminotransferase in cytosols from rat liver. Metab Clin Exp 38: 666–672, 1989 [DOI] [PubMed] [Google Scholar]
- 29.Hargrove JL. Persulfide generated from L-cysteine inactivates tyrosine aminotransferase. Requirement for a protein with cysteine oxidase activity and gamma-cystathionase. J Biol Chem 263: 17262–17269, 1988 [PubMed] [Google Scholar]
- 30.Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L, Longchamp A, Treviño-Villarreal JH, Mejia P, Ozaki CK, Wang R, Gladyshev VN, Madeo F, Mair WB, and Mitchell JR. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160: 132–144, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holwerda KM, Burke SD, Faas MM, Zsengeller Z, Stillman IE, Kang PM, van Goor H, McCurley A, Jaffe IZ, Karumanchi SA, and Lely AT. Hydrogen sulfide attenuates sFlt1-induced hypertension and renal damage by upregulating vascular endothelial growth factor. J Am Soc Nephrol 25: 717–725, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holz MK, Ballif BA, Gygi SP, and Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: 569–580, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Huang P, Chen S, Wang Y, Liu J, Yao Q, Huang Y, Li H, Zhu M, Wang S, Li L, Tang C, Tao Y, Yang G, Du J, and Jin H. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide 46: 192–203, 2015 [DOI] [PubMed] [Google Scholar]
- 34.Jager A, Kostense PJ, Nijpels G, Dekker JM, Heine RJ, Bouter LM, Donker AJ, and Stehouwer CD. Serum homocysteine levels are associated with the development of (micro)albuminuria: the Hoorn study. Arterioscler Thromb Vasc Biol 21: 74–81, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Jain SK, Bull R, Rains JL, Bass PF, Levine SN, Reddy S, McVie R, and Bocchini JA. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid Redox Signal 12: 1333–1337, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HHHW, and Jandeleit-Dahm KA. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25: 1237–1254, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji W, Liu S, Dai J, Yang T, Jiang X, Duan X, and Wu Y. Hydrogen sulfide defends against the cardiovascular risk of Nw-nitro-L-argininemethyl ester-induced hypertension in rats via the nitric oxide/endothelial nitric oxide synthase pathway. Chin Med J 127: 3751–3757, 2014 [PubMed] [Google Scholar]
- 38.Kabil O. and Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal 20: 770–782, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamat PK, Kalani A, Givvimani S, Sathnur PB, Tyagi SC, and Tyagi N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 252: 302–319, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamat PK, Kalani A, Tyagi SC, and Tyagi N. Hydrogen sulfide epigenetically attenuates homocysteine‐induced mitochondrial toxicity mediated through NMDA receptor in mouse brain endothelial (bEnd3) cells. J Cell Physiol 230: 378–394, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasinath BS, Feliers D, Sataranatarajan K, Ghosh Choudhury G, Lee M-J, and Mariappan MM. Regulation of mRNA translation in renal physiology and disease. Am J Physiol Renal Physiol 297: F1153–F1165, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimura N, Kimura H, Takahashi N, Hamada T, Maegawa H, Mori M, Imamura Y, Kusaka Y, Yoshida H, and Iwano M. Renal resistive index correlates with peritubular capillary loss and arteriosclerosis in biopsy tissues from patients with chronic kidney disease. Clin Exp Nephrol 19: 1114–1119, 2015 [DOI] [PubMed] [Google Scholar]
- 43.Kimura Y. and Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J 18: 1165–1167, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Köhn C, Dubrovska G, Huang Y, and Gollasch M. Hydrogen sulfide: potent regulator of vascular tone and stimulator of angiogenesis. Int J Biomed Sci 8: 81–86, 2012 [PMC free article] [PubMed] [Google Scholar]
- 45.Krishnan N, Fu C, Pappin DJ, and Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal 4: ra86, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kundu S, Pushpakumar S, and Sen U. MMP-9- and NMDA receptor-mediated mechanism of diabetic renovascular remodeling and kidney dysfunction: hydrogen sulfide is a key modulator. Nitric Oxide 46: 172–185, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kundu S, Pushpakumar SB, Tyagi A, Coley D, and Sen U. Hydrogen sulfide deficiency and diabetic renal remodeling: role of matrix metalloproteinase-9. Am J Physiol Endocrinol Metab 304: E1365–E1378, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HJ, Feliers D, Mariappan MM, Sataranatarajan K, Choudhury GG, Gorin Y, and Kasinath BS. Tadalafil integrates nitric oxide-hydrogen sulfide signaling to inhibit high glucose-induced matrix protein synthesis in podocytes. J Biol Chem 290: 12014–12026, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee HJ, Mariappan MM, Feliers D, Cavaglieri RC, Sataranatarajan K, Abboud HE, Choudhury GG, and Kasinath BS. Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J Biol Chem 287: 4451–4461, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee M-J, Feliers D, Mariappan MM, Sataranatarajan K, Mahimainathan L, Musi N, Foretz M, Viollet B, Weinberg JM, Choudhury GG, and Kasinath BS. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am J Physiol Renal Physiol 292: F617–F627, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Lee VWS. and Harris DCH. Adriamycin nephropathy: a model of focal segmental glomerulosclerosis. Nephrology (Carlton) 16: 30–38, 2011 [DOI] [PubMed] [Google Scholar]
- 52.Li H, Feng S-J, Zhang G-Z, and Wang S-X. Correlation of lower concentrations of hydrogen sulfide with atherosclerosis in chronic hemodialysis patients with diabetic nephropathy. Blood Purif 38: 188–194, 2014 [DOI] [PubMed] [Google Scholar]
- 53.Li J-J, Li Q, Du H-P, Wang Y-L, You S-J, Wang F, Xu X-S, Cheng J, Cao Y-J, Liu C-F, and Hu L-F. Homocysteine triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int J Mol Sci 16: 12560–12577, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu L, Cui J, Song C-J, Bian J-S, Sparatore A, Soldato PD, Wang X-Y, and Yan C-D. H2S-releasing aspirin protects against aspirin-induced gastric injury via reducing oxidative stress. PLoS One 7: e46301, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lobb I, Davison M, Carter D, Liu W, Haig A, Gunaratnam L, and Sener A. Hydrogen sulfide treatment mitigates renal allograft ischemia-reperfusion injury during cold storage and improves early transplant kidney function and survival following allogeneic renal transplantation. J Urol 2015. Doi: 10.1016/j.juro.2015.07.096 [DOI] [PubMed] [Google Scholar]
- 56.Lobb I, Zhu J, Liu W, Haig A, Lan Z, and Sener A. Hydrogen sulfide treatment ameliorates long-term renal dysfunction resulting from prolonged warm renal ischemia-reperfusion injury. Can Urol Assoc J 8: E413–E418, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mariappan MM, D'Silva K, Lee M-J, Sataranatarajan K, Barnes JL, Choudhury GG, and Kasinath BS. Ribosomal biogenesis induction by high glucose requires activation of upstream binding factor in kidney glomerular epithelial cells. Am J Physiol Renal Physiol 300: F219–F230, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mariappan MM, Feliers D, Mummidi S, Choudhury GG, and Kasinath BS. High glucose, high insulin, and their combination rapidly induce laminin-beta1 synthesis by regulation of mRNA translation in renal epithelial cells. Diabetes 56: 476–485, 2007 [DOI] [PubMed] [Google Scholar]
- 59.Mariappan MM, Shetty M, Sataranatarajan K, Choudhury GG, and Kasinath BS. Glycogen synthase kinase 3beta is a novel regulator of high glucose- and high insulin-induced extracellular matrix protein synthesis in renal proximal tubular epithelial cells. J Biol Chem 283: 30566–30575, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Massey V. and Edmondson D. On the mechanism of inactivation of xanthine oxidase by cyanide. J Biol Chem 245: 6595–6598, 1970 [PubMed] [Google Scholar]
- 61.Maynard SE, Min J-Y, Merchan J, Lim K-H, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, and Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111: 649–658, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller JW, Nadeau MR, Smith D, and Selhub J. Vitamin B-6 deficiency vs folate deficiency: comparison of responses to methionine loading in rats. Am J Clin Nutr 59: 1033–1039, 1994 [DOI] [PubMed] [Google Scholar]
- 63.Mise K, Hoshino J, Ueno T, Hazue R, Hasegawa J, Sekine A, Sumida K, Hiramatsu R, Hasegawa E, Yamanouchi M, Hayami N, Suwabe T, Sawa N, Fujii T, Hara S, Ohashi K, Takaichi K, and Ubara Y. Prognostic value of tubulointerstitial lesions, urinary N-acetyl-β-d-glucosaminidase, and urinary β2-microglobulin in patients with type 2 diabetes and biopsy-proven diabetic nephropathy. Clin J Am Soc Nephrol 2016. Doi: 10.2215/CJN.04980515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morris MS. Homocysteine and Alzheimer's disease. Lancet Neurol 2: 425–428, 2003 [DOI] [PubMed] [Google Scholar]
- 65.Módis K, Coletta C, Erdélyi K, Papapetropoulos A, and Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J 27: 601–611, 2013 [DOI] [PubMed] [Google Scholar]
- 66.Mrozikiewicz PM, Bogacz A, Omielańczyk M, Wolski H, Bartkowiak-Wieczorek J, Grześkowiak E, Czerny B, Drews K, and Seremak-Mrozikiewicz A. The importance of rs1021737 and rs482843 polymorphisms of cystathionine gamma-lyase in the etiology of preeclampsia in the Caucasian population. Ginekol Pol 86: 119–125, 2015 [DOI] [PubMed] [Google Scholar]
- 67.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, and Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal 2: ra72, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, and Snyder SH. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109: 1259–1268, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nagy P. and Winterbourn CC. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem Res Toxicol 23: 1541–1543, 2010 [DOI] [PubMed] [Google Scholar]
- 70.Paul BD. and Snyder SH. H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol 13: 499–507, 2012 [DOI] [PubMed] [Google Scholar]
- 71.Perna AF, Luciano MG, Ingrosso D, Pulzella P, Sepe I, Lanza D, Violetti E, Capasso R, Lombardi C, and De Santo NG. Hydrogen sulphide-generating pathways in haemodialysis patients: a study on relevant metabolites and transcriptional regulation of genes encoding for key enzymes. Nephrol Dial Transplant 24: 3756–3763, 2009 [DOI] [PubMed] [Google Scholar]
- 72.Pushpakumar S, Kundu S, and Sen U. Endothelial dysfunction: the link between homocysteine and hydrogen sulfide. Curr Med Chem 21: 3662–3672, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salloum FN, Chau VQ, Hoke NN, Abbate A, Varma A, Ockaili RA, Toldo S, and Kukreja RC. Phosphodiesterase-5 inhibitor, tadalafil, protects against myocardial ischemia/reperfusion through protein-kinase G-dependent generation of hydrogen sulfide. Circulation 120: S31–S36, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sataranatarajan K, Mariappan MM, Lee M-J, Feliers D, Choudhury GG, Barnes JL, and Kasinath BS. Regulation of elongation phase of mRNA translation in diabetic nephropathy: amelioration by rapamycin. Am J Pathol 171: 1733–1742, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CRJ, Burns KD, Cooper ME, Jandeleit-Dahm K, Page P, Szyndralewiez C, Heitz F, Hebert RL, and Touyz RM. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of type 2 diabetes. Clin Sci 124: 191–202, 2013 [DOI] [PubMed] [Google Scholar]
- 76.Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, and Snyder SH. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol Cell 45: 13–24, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sen U, Basu P, Abe OA, Givvimani S, Tyagi N, Metreveli N, Shah KS, Passmore JC, and Tyagi SC. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am J Physiol Renal Physiol 297: F410–F419, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sen U, Munjal C, Qipshidze N, Abe O, Gargoum R, and Tyagi SC. Hydrogen sulfide regulates homocysteine-mediated glomerulosclerosis. Am J Nephrol 31: 442–455, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sen U, Sathnur PB, Kundu S, Givvimani S, Coley DM, Mishra PK, Qipshidze N, Tyagi N, Metreveli N, and Tyagi SC. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am J Physiol Cell Physiol 303: C41–C51, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, and Goldstein BJ. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118: 1645–1656, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, Fukui K, Nagahara N, and Kimura H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun 4: 1366, 2013 [DOI] [PubMed] [Google Scholar]
- 82.Shibuya N, Mikami Y, Kimura Y, Nagahara N, and Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem 146: 623–626, 2009 [DOI] [PubMed] [Google Scholar]
- 83.Singh S, Padovani D, Leslie RA, Chiku T, and Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem 284: 22457–22466, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sonenberg N. and Hinnebusch AG. New modes of translational control in development, behavior, and disease. Mol Cell 28: 721–729, 2007 [DOI] [PubMed] [Google Scholar]
- 85.Song K, Wang F, Li Q, Shi Y-B, Zheng H-F, Peng H, Shen H-Y, Liu C-F, and Hu L-F. Hydrogen sulfide inhibits the renal fibrosis of obstructive nephropathy. Kidney Int 85: 1318–1329, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stipanuk MH. and Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J 206: 267–277, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stubbert D, Prysyazhna O, Rudyk O, Scotcher J, Burgoyne JR, and Eaton P. Protein kinase G Iα oxidation paradoxically underlies blood pressure lowering by the reductant hydrogen sulfide. Hypertension 64: 1344–1351, 2014 [DOI] [PubMed] [Google Scholar]
- 88.Suzuki K, Olah G, Módis K, Coletta C, Kulp G, Gerö D, Szoleczky P, Chang T, Zhou Z, Wu L, Wang R, Papapetropoulos A, and Szabo C. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A 108: 13829–13834, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tan Z, Shi Y, Yan Y, Liu W, Li G, and Li R. Impact of endogenous hydrogen sulfide on toll-like receptor pathway in renal ischemia/reperfusion injury in rats. Ren Fail 37: 727–733, 2015 [DOI] [PubMed] [Google Scholar]
- 90.Tang G, Yang G, Jiang B, Ju Y, Wu L, and Wang R. H2S is an endothelium-derived hyperpolarizing factor. Antioxid Redox Signal 19: 1634–1646, 2013 [DOI] [PubMed] [Google Scholar]
- 91.Testai L, D'Antongiovanni V, Piano I, Martelli A, Citi V, Duranti E, Virdis A, Blandizzi C, Gargini C, Breschi MC, and Calderone V. Different patterns of H2S/NO activity and cross-talk in the control of the coronary vascular bed under normotensive or hypertensive conditions. Nitric Oxide 47: 25–33, 2015 [DOI] [PubMed] [Google Scholar]
- 92.Thallas-Bonke V, Jha JC, Gray SP, Barit D, Haller H, Schmidt HHHW, Coughlan MT, Cooper ME, Forbes JM, and Jandeleit-Dahm KAM. Nox-4 deletion reduces oxidative stress and injury by PKC-α-associated mechanisms in diabetic nephropathy. Physiol Rep 2: e12192, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wallace JL. and Wang R. Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat Rev Drug Discov 14: 329–345, 2015 [DOI] [PubMed] [Google Scholar]
- 94.Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PWF, Wang R, Gratacós E, Buhimschi IA, Buhimschi CS, and Ahmed A. Dysregulation of hydrogen sulfide producing enzyme cystathionine γ-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 127: 2514–2522, 2013 [DOI] [PubMed] [Google Scholar]
- 95.Wang Y, Shi S, Dong S, Wu J, Song M, Zhong X, and Liu Y. Sodium hydrosulfide attenuates hyperhomocysteinemia rat myocardial injury through cardiac mitochondrial protection. Mol Cell Biochem 399: 189–200, 2015 [DOI] [PubMed] [Google Scholar]
- 96.Whiteman M, Gooding KM, Whatmore JL, Ball CI, Mawson D, Skinner K, Tooke JE, and Shore AC. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia 53: 1722–1726, 2010 [DOI] [PubMed] [Google Scholar]
- 97.Xia M, Chen L, Muh RW, Li P-L, and Li N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J Pharmacol Exp Ther 329: 1056–1062, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamamoto J, Sato W, Kosugi T, Yamamoto T, Kimura T, Taniguchi S, Kojima H, Maruyama S, Imai E, Matsuo S, Yuzawa Y, and Niki I. Distribution of hydrogen sulfide (H2S)-producing enzymes and the roles of the H2S donor sodium hydrosulfide in diabetic nephropathy. Clin Exp Nephrol 17: 32–40, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang G, Tang G, Zhang L, Wu L, and Wang R. The pathogenic role of cystathionine γ-lyase/hydrogen sulfide in streptozotocin-induced diabetes in mice. Am J Pathol 179: 869–879, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, and Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322: 587–590, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yuan P, Xue H, Zhou L, Qu L, Li C, Wang Z, Ni J, Yu C, Yao T, Huang Y, Wang R, and Lu L. Rescue of mesangial cells from high glucose-induced over-proliferation and extracellular matrix secretion by hydrogen sulfide. Nephrol Dial Transplant 26: 2119–2126, 2011 [DOI] [PubMed] [Google Scholar]
- 102.Zhang H, Zhang A, Guo C, Shi C, Zhang Y, Liu Q, Sparatore A, and Wang C. S-diclofenac protects against doxorubicin-induced cardiomyopathy in mice via ameliorating cardiac gap junction remodeling. PLoS One 6: e26441, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang J, Chen S, Liu H, Zhang B, Zhao Y, Ma K, Zhao D, Wang Q, Ma H, and Zhang Z. Hydrogen sulfide prevents hydrogen peroxide-induced activation of epithelial sodium channel through a PTEN/PI(3,4,5)P3 dependent pathway. PLoS One 8: e64304, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao W. and Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol 283: H474–H480, 2002 [DOI] [PubMed] [Google Scholar]
- 105.Zhong G, Chen F, Cheng Y, Tang C, and Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens 21: 1879–1885, 2003 [DOI] [PubMed] [Google Scholar]
- 106.Zhou X, Feng Y, Zhan Z, and Chen J. Hydrogen sulfide alleviates diabetic nephropathy in a streptozotocin-induced diabetic rat model. J Biol Chem 289: 28827–28834, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]