

Abstract
The primary cilium is an antenna-like, immotile organelle present on most types of mammalian cells, which interprets extracellular signals that regulate growth and development. Although once considered a vestigial organelle, the primary cilium is now the focus of considerable interest. We now know that ciliary defects lead to a panoply of human diseases, termed ciliopathies, and the loss of this organelle may be an early signature event during oncogenic transformation. Ciliopathies include numerous seemingly unrelated developmental syndromes, with involvement of the retina, kidney, liver, pancreas, skeletal system and brain. Recent studies have begun to clarify the key mechanisms that link cilium assembly and disassembly to the cell cycle, and suggest new possibilities for therapeutic intervention.
Primary cilia emerge from centrioles through a unique mechanism by which one organelle transforms into another with altogether different properties1. Centrosomes are composed of two orthogonally arranged centrioles embedded in proteinaceous material, the pericentriolar matrix, and are present in one or two copies per cell, depending on cell cycle stage (Fig. 1). Centriole duplication occurs in the S phase of the cell cycle, when a new (daughter) centriole assembles perpendicular to the old (mother) centriole. Each centriole is composed of nine microtubule triplets radially distributed with respect to a central lumen2. The centrosome, unlike most organelles, is not encased within a membrane. In contrast, the core microtubule structure of the primary cilium, or axoneme, is encased within a membranous sheath continuous with the plasma membrane. Ultrastructural studies have shown that only the mother centriole can give rise to a primary cilium and that the microtubule triplets in centrioles transition to doublets at the distal end of the basal body, which nucleates the axoneme1.
Figure 1. Linkage of the centrosome–cilium cycle to the cell cycle.

Primary cilia assemble specifically when cells exit the cell cycle and become quiescent or differentiate. Cells are also competent to form cilia in G1. Phases of the cell cycle are indicated, and blue and yellow arrows indicate cilium assembly and disassembly, respectively. Only the mother centriole (light blue) can initiate ciliogenesis. The daughter centrioles are shown in dark blue. During the process of ciliogenesis, an axoneme is assembled. This microtubular structure (indicated with parallel green rods) is disassembled as cells progress towards S phase, concomitant with remodelling of the distal end of the basal body (aqua ring). During S phase, centrosomes commence duplication, at which point cilia have largely disassembled. After mitosis, centrosomes are again competent to assemble primary cilia, either in G0 or in early G1 phase.
The primary cilium is observed primarily in quiescent or differentiated cells (Fig. 1). Major insights into the function of the primary cilium came in the 1990s, when studies of the flagellum in the green alga, Chlamydomonas, revealed the existence of a conserved intraflagellar transport (IFT) system3 required for organelle biogenesis. This led to the discovery that the mouse homologue of the Chlamydomonas IFT88 gene was mutated in a model for polycystic kidney disease (PKD)4,5, in which the cells lining the urinary tract tubules fail to properly assemble primary cilia. This study provided the first evidence that, despite being immotile, primary cilia clearly have a function. Since then, much has been learned about the function of this extraordinary organelle, and a rapidly growing area of interest is its role as a major conduit for key signal transduction pathways. Signalling associated with primary cilia impacts processes as diverse as calcium flux in the kidney, growth and differentiation, and memory and learning6–8. Proteins that contribute to ciliogenesis and human diseases that result from defects in this organelle have been reviewed elsewhere (for example, refs 9–13). In this Review, we focus on exciting new developments in the field, with particular emphasis on the mechanisms that promote the assembly and disassembly of primary cilia and how these processes are subverted in pathological states.
Cilium assembly
Assembly of the primary cilium begins when cells exit the mitotic cycle in response to mitogen deprivation or differentiation cues, although certain differentiated cell lineages (including lymphocytes, hepatocytes, mature adipocytes and skeletal muscle) lack primary cilia14–18. Ciliation can be recapitulated in cell culture through serum withdrawal, and the use of mouse 3T3 fibroblasts and human retinal pigment epithelial (RPE1) cells19–21, in particular, has been instrumental for the analysis of factors required for cilium assembly. Although the nature of proximate cues able to promote ciliogenesis remains largely unknown, multiple initiating events — both intrinsic and extrinsic to the basal body — accompany the rapid remodelling of the distal end of mother centrioles to basal bodies (which then assemble the primary cilium) in an elegant switching mechanism (Fig. 2).
Figure 2. Key players and events in cilium assembly.

Cilium assembly proceeds through a series of orchestrated and well-defined stages (labelled I–IV), resulting in the dramatic remodelling of the maternal centriole. Exit from the cell cycle initiates the recruitment of Rabin8, a guanine nucleotide exchange factor (GEF), to the pericentriolar recycling endosome, whereupon it is activated by Rab11, setting off a cascade that ultimately activates Rab8a vesicles for recruitment and docking to the distal appendages (DA, orange antennae) of mother centrioles (I–II). Shortly after quiescence is induced, small vesicles (distal appendage vesicles, yellow) associated with Rab11 assemble around the DA, docking at the appendages (II). Distal appendage vesicles fuse to form ciliary vesicles through the action of the Ehd1 protein (II–III). After ciliary vesicle (CV) formation, the distal ends of mother centrioles/basal bodies are remodelled (concentric rings) through the action of: the kinase TTBK2, the balance of PtdIns (modulated by INPP5E and PIPKIγ), and CP110 removal, which releases the inhibition of microtubule growth and axonemal extension (III–IV). A second kinase, MARK4, could collaborate with TTBK2 to remove CP110 from distal ends. Vesicular Rab11 probably activates Rab8 for subsequent elongation of the ciliary membrane. After removal of CP110, the transition zone (TZ), which functions as a ciliary gate, is assembled, and IFT helps transport proteins through this gate.
Ciliogenesis can be divided into distinct phases (Fig. 2), encompassing all the events that occur before and after the basal body docks at the plasma membrane. Time-lapse microscopy has revealed a remarkably dynamic process22. Within 10–15 minutes of mitogen withdrawal, small cytoplasmic vesicles thought to originate from the Golgi and the recycling endosome, termed distal appendage vesicles (DAVs), begin to accumulate in the vicinity of distal appendages of the mother centriole, where they seem to dock23–25. This is perhaps the first visible sign of the centriole-to-basal-body transition. Vesicular fusion then produces a membranous cap, or ciliary vesicle, on the distal tip of the mother centriole, which Sorokin designated as the primary ciliary vesicle26. Extension of the microtubules of the centriole at its distal tip proceeds underneath this cap, and subsequent vesicular trafficking enlarges the cap concomitant with extension of the microtubules, ensheathing the growing axoneme in a double membrane. This nascent cilium then docks to the plasma membrane by fusion with the ciliary sheath, establishing continuity of these compartments.
Cilia membrane assembly and trafficking
Recently, significant progress has been made in understanding the trafficking of vesicles to the nascent cilium. Rab GTPases are integral to membrane trafficking, and their activity is under the strict control of a corresponding pair of guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs), which convert Rabs into an active (GTP-bound) or inactive (GDP-bound) state, respectively. Rab proteins regulate distinct steps in membrane trafficking through the control of vesicularization of the donor membrane and the ensuing fusion with the acceptor membrane. Thus, the localization of GEFs and GAPs in distinct vesicular compartments can direct membrane trafficking. Rab8a has been implicated in ciliary membrane assembly22,27 and, in growing RPE1 cells, it is localized to cytoplasmic vesicles and the Golgi/trans-Golgi network — but serum starvation induces its rapid redistribution to the mother centriole22,27. After the formation of DAVs, the Ehd1 protein is recruited, converting these small vesicles to the larger ciliary vesicles, which then elongate through continuous fusion with Rab8-positive vesicles to produce the primary cilium membrane25.
How is Rab8 activated and directed to the distal appendages of the mother centriole in response to a ciliogenic signal? Rab11-positive recycling endosomes may transport the GEF, Rabin8, an activator for Rab8, and the membrane-tethering complex, TrappII, to this location22. Rab8 activation therefore coincides with the fusion of Rab11- and Rab8- positive vesicles and the activation of the ciliary assembly program. Insight into the function of Rab8/Rabin8 at the primary cilium came from an analysis of proteins interacting with the BBSome complex, which includes Bardet–Biedl syndrome protein 4 (BBS4), a protein that localizes to the basal body of the primary cilium28. Defects in some or all of the proteins in the BBSome complex are associated with the human ciliopathy Bardet–Biedl syndrome, although mutations in BBS genes generally perturb ciliary function rather than ciliogenesis per se29. Rabin8 associates with the BBSome at the centrosome and the base of the primary cilium. Thus, ciliogenesis is driven by sustained activation of Rab8 through two different sources of Rabin8: the Rab11-positive recycling endosome and the BBSome. As cilia lack ribosomes, growth and maintenance of the axoneme also requires two macromolecular IFT complexes, IFT-B and IFT-A, which transport hundreds of proteins towards (anterograde) or away (retrograde) from the ciliary tip, respectively29. In addition, the BBSome functions as an adaptor for IFT during ciliary export29–31. Transport within the cilium also requires kinesin and dynein motors that, together with IFT complexes, are necessary for the localization of key ciliary membrane proteins such as G-protein-coupled receptors (GPCRs), one of which (Smoothened, Smo) functions in the Hedgehog (Hh) signalling pathway. Defects in both IFT-A and IFT-B subunits result in multiple ciliopathies, attesting to an essential role for IFT in cilium assembly (Fig. 2).
Basal body docking
Elegant studies have also begun to clarify the succession of events that occur at the distal ends of basal bodies and during distal-appendage-mediated vesicle docking and maturation25,32 (Fig. 2). Components intrinsic to the basal body undergo dramatic remodelling during the early stages of cilium assembly. One element involved in this switch is CP110, a distal-end protein essential for centriole duplication and length regulation33–36. Asymmetric destruction of CP110 and its interacting partner Cep97 on mother centrioles may be an obligate event in the initiation of ciliogenesis37, and the silencing of either gene results in the aberrant assembly of primary cilia in proliferating cells or the formation of extra-long centrioles in non-ciliated cells37.
Extension of the ciliary axoneme occurs after the anchoring (or docking) of basal bodies to intracellular vesicles or the plasma membrane via distal appendages (Fig. 2). The assembly of distal appendages on mother centrioles involves the orchestrated recruitment of five proteins (Cep83, Cep89, Cep164, SCLT1 and FBF1)32. At least one distal appendage protein, Cep164, helps dock vesicles through interactions with Rab8 and Rabin8, and loss of Cep164 or another ciliopathy-associated distal centriolar protein, Talpid3, abrogates the recruitment of ciliary vesicles23,24. All five proteins are required for ciliogenesis, and beyond docking to membranes, the distal appendages are assembly points for the recruitment of IFT proteins38 and other components required to build the cilium and selectively import proteins through the transition zone, which acts as a ciliary gate to the cilium29.
Interestingly, undocked centrioles retain CP110 even after serum withdrawal. Thus, the activation of signalling pathways that induce ciliogenesis is restricted by spatial cues. Certain cell types that do not assemble cilia, such as cytotoxic T lymphocytes (CTLs), can use similar signalling pathways to execute their unique functions17. In these cells, the distal appendages of mother centrioles dock at the plasma membrane, analogous to cilium assembly, forming a protrusion at the immunological synapse necessary for CTL secretion and target killing, without producing an axoneme or primary cilium. Notably, Cep97 and CP110 persist at mother centrioles during synapse formation, suggesting that these cells implement an ‘abortive’ ciliogenesis program.
An important step towards the identification of proteins that promote ciliogenesis came from a genetic screen in the mouse to identify regulators of the Hh signal transduction pathway, which depends on the integrity of the primary cilium. This screen identified a Tau tubulin kinase 2 (TTBK2)-null mouse mutant, which lacks primary cilia and exhibits defects in Hh signalling39. This was not due to defects in basal body anchoring; rather, loss of TTBK2 abrogated the recruitment of IFT proteins essential for anterograde and retrograde trafficking and axoneme extension and maintenance, and TTBK2-null basal bodies consequently lacked axonemes. Further, since CP110 was retained on basal bodies in null cells, it was inferred that TTBK2 (which is recruited by Cep16440 and localizes at the distal end of the basal body) is necessary for the removal of CP110 from the mother centriole. Interestingly, spinocerebellar ataxia type 11, a neurodegenerative disorder, is caused by dominant, null mutations in the TTBK2 gene41,42. But there are still several gaps in our knowledge of this mechanism. It remains unclear what triggers TTBK2-mediated phosphorylation and how CP110 destruction is restricted to the mother centriole. The identity of proteases that asymmetrically destroy CP110 prior to axoneme growth is still unknown. Whether TTBK2 targets exist beyond the distal appendage protein (Cep164) and Cep97 and how the phosphorylation of these proteins drives ciliogenesis is unknown — although TTBK2 may also play a role in assembly of distal appendages40,43. One priority will be to determine how inductive cues activate this kinase. It is tempting to speculate that a feedback mechanism exists, in which docking of ciliary vesicle triggers the activation of TTBK232: engagement of vesicles by Cep164 might trigger conformational changes in Cep164, TTBK2, or both, stimulating kinase activity and promoting phosphorylation of key substrates, thereby ‘flipping the switch’ that initiates cilium assembly.
Phosphatidylinositol (PtdIns) homeostasis and phosphorylation at the basal body are also linked with TTBK2 regulation and ciliogenesis44. Intriguingly, the balance of two activities — a PtdIns kinase (PIPKIγ) and an opposing phosphatase (INPP5E) — fine-tunes PtdIns(4)P and PtdIns(4,5)P2 levels. PtdIns(4)P negatively regulates recruitment of TTBK2 (thereby promoting CP110 persistence and suppressing ciliogenesis) by modulating its interaction with Cep164 (Fig. 2). Mutations in INPP5E are found in Joubert syndrome, indicating additional roles for phospholipids, which appear to be compartmentalized within the cilium once it is assembled45–47.
In addition to TTBK2, depletion of a second basal-body-associated kinase, MAP/microtubule affinity-regulating kinase 4 (MARK4), results in arrest after vesicle docking but prior to axoneme extension48. It will be interesting to identify both the activators and substrates of MARK4, as this network could collaborate with TTBK2 to promote remodelling of the distal end of the basal body and growth of axonemal microtubules. An exciting possibility is that the tumour suppressor LKB1 (liver kinase B1; also known as serine/threonine kinase 11, STK11) — an activator of mTOR (mammalian target of rapamycin) and AMPK (AMP-activated protein kinase) as well as MARK4, which localizes to centrosomes and cilia49–52 — plays a role here. STK11-knockout cells exhibits abortive cilium assembly that is relieved by inhibition of HDAC652, which plays a key role in cilium resorption, suggesting that cilium loss in these cells is due to excessive organelle disassembly.
Cilium disassembly
In contrast with cilium assembly, much less is known regarding mechanisms underlying disassembly of this organelle in normal or pathological conditions (Fig. 3). Moreover, how cilium disassembly is linked to cell cycle progression remains largely unanswered. Experiments in cultured mammalian cells suggest that cilia disassemble in a biphasic manner19,21, with the first, major ‘wave’ occurring in the G1 phase shortly after mitogen stimulation of quiescent cells (Fig. 1) and a second wave prior to mitosis. These initial studies identified several key regulators of cilium disassembly (Fig. 3): the scaffolding protein HEF1 (also known as NEDD9) and calcium–calmodulin activate Aurora A kinase, which in turn phosphorylates and stimulates the histone deacetylase HDAC6, promoting the de-acetylation of modified, stabilized tubulins within the axoneme21,53. Additional studies will be needed to identify the mechanisms through which HEF1 (NEDD9) is upregulated and recruited to basal bodies following mitogen stimulation. Recent studies have uncovered another critical effector of HDAC6 activity, cortactin, which promotes actin polymerization54. Given that acetylation of cortactin abrogates its interaction with filamentous actin and that actin polymerization counteracts cilium assembly, de-acetylation of cortactin could further explain how HDAC6 activation promotes cilium disassembly by enhancing actin polymerization. Apart from its role as an HDAC6 regulator, Aurora A also activates INPP5E55, which would favour high levels of PtdIns(4)P and suppress ciliogenesis through a second mechanism44.
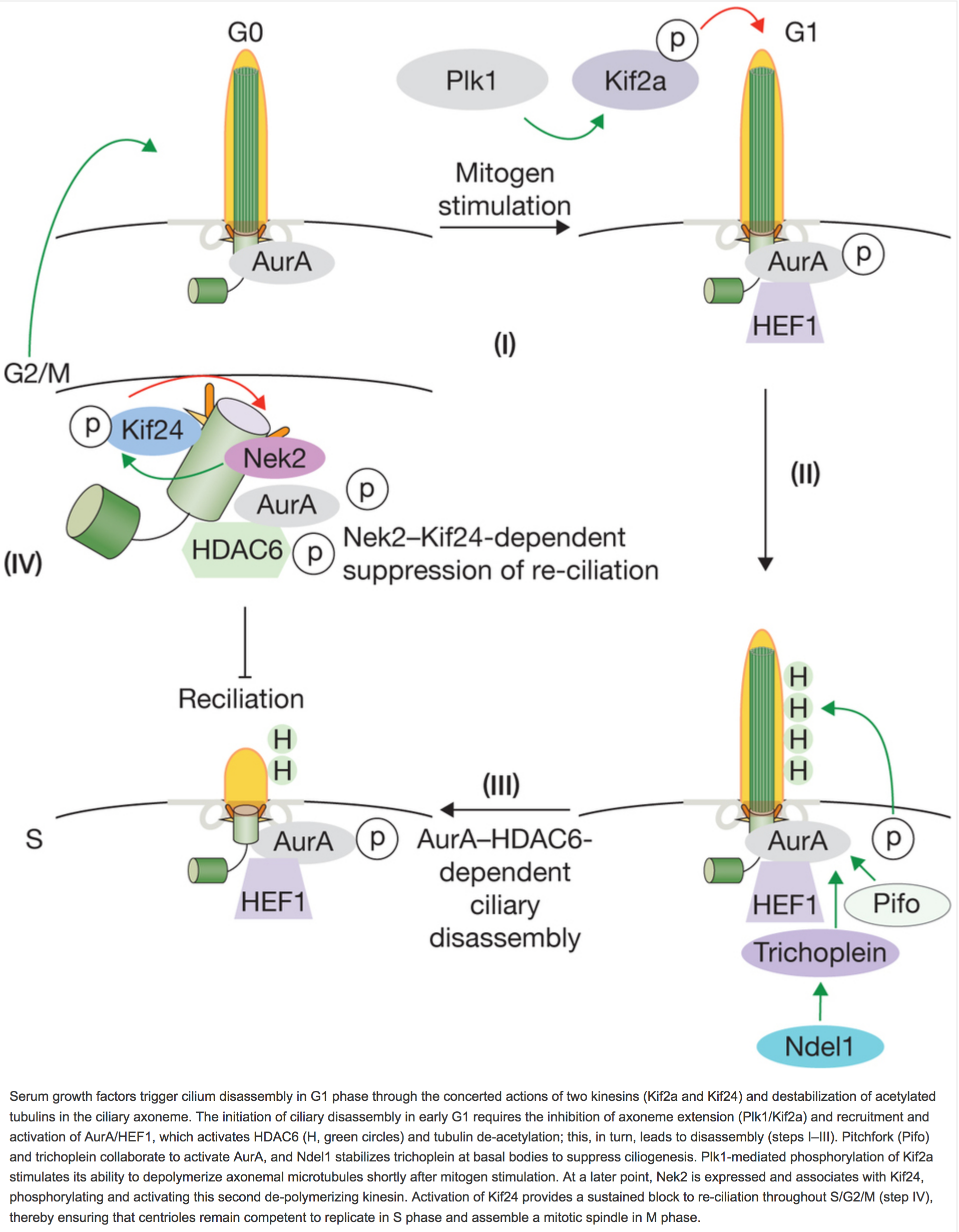
Figure 3. Cilium disassembly.

Serum growth factors trigger cilium disassembly in G1 phase through the concerted actions of two kinesins (Kif2a and Kif24) and destabilization of acetylated tubulins in the ciliary axoneme. The initiation of ciliary disassembly in early G1 requires the inhibition of axoneme extension (Plk1/Kif2a) and recruitment and activation of AurA/HEF1, which activates HDAC6 (H, green circles) and tubulin de-acetylation; this, in turn, leads to disassembly (steps I–III). Pitchfork (Pifo) and trichoplein collaborate to activate AurA, and Ndel1 stabilizes trichoplein at basal bodies to suppress ciliogenesis. Plk1-mediated phosphorylation of Kif2a stimulates its ability to depolymerize axonemal microtubules shortly after mitogen stimulation. At a later point, Nek2 is expressed and associates with Kif24, phosphorylating and activating this second de-polymerizing kinesin. Activation of Kif24 provides a sustained block to re-ciliation throughout S/G2/M (step IV), thereby ensuring that centrioles remain competent to replicate in S phase and assemble a mitotic spindle in M phase.
Two additional activators of Aurora A at the basal body, trichoplein and Pitchfork (Pifo), also play a role in cilium disassembly56,57 (Fig. 3). Similarly to CP110, trichoplein disappears from mother centrioles during ciliogenesis, and its depletion promotes aberrant cilium assembly in proliferating cells57. The process of ubiquitin-mediated protein degradation is required for cilium disassembly58, and trichoplein proteolysis is critical for the initiation of axoneme extension, indicating a similar requirement during cilium assembly59,60. Interestingly, a newly discovered protein, nuclear distribution element (NDE)-like 1 (Ndel1), functions in the trichoplein–Aurora-A pathway, and similarly to trichoplein depletion, loss of Ndel1 triggers aberrant ciliogenesis in growing cells61. Further, Ndel1 blocks ciliogenesis prior to axoneme extension by stabilizing trichoplein at mother centrioles, perhaps by protecting trichoplein from ubiquitin-mediated destruction. The requirement for Pifo in cilium disassembly is illustrated by the observation that Pifo haplo-insufficiency leads to the persistence of cilia in mitosis, abolishes the liberation and duplication of centrioles, and leads to mitotic defects56. Heterozygous mutant Pifo mice and patients exhibit ciliopathy-related manifestations, attesting to the biological consequences of perturbing cilium disassembly.
Cilium disassembly requires the destabilization and de-polymerization of axonemal microtubules, and two members of the Kinesin-13 family of de-polymerizing kinesins, Kif2a and Kif24, are implicated in cilium disassembly62–64 (Fig. 3). Kif2a localizes to proximal ends of both centrioles as well as the sub-distal appendages of the mother centriole, and it is phosphorylated and activated by the G2/M-phase kinase Plk163. Activated Kif2a promotes the de-polymerization of ciliary microtubules, provoking cilium disassembly shortly following a proliferative signal. Kif2a presents an interesting prototype for exploring the role of aberrant cilium disassembly in disease. Premature chromatid separation syndrome (PCS; also known as mosaic variegated aneuploidy syndrome, MVA) is a rare autosomal recessive disorder characterized by a high risk of cancer and symptoms associated with ciliopathies63. Fibroblasts from PCS patients exhibit constitutive activation of Plk1 and Kif2a, as well as reduced ciliogenesis, suggesting that aberrant activation of a disassembly pathway could, in part, underlie the pathology.
A second microtubule de-polymerizing kinesin, Kif24, was identified by virtue of its association with CP110 at the distal end of centrioles62. As for CP110 loss, ablation of Kif24 results in inappropriate assembly of cilia in proliferating cells, although it does not regulate centriole length. The microtubule de-polymerizing activity of Kif24 is enhanced by Nek2- mediated phosphorylation64. Nek2 is expressed during S and G2 phase, ensuring that Kif24 is active in cells that lack cilia. Notably, the ability of Nek2 and Kif24 to suppress cilium assembly can be temporally distinguished from the Aurora-A–HDAC6 pathway, suggesting that Nek2–Kif24 activation ensures the irreversibility of the disassembly process after S phase commences64. These studies suggest that Kif24 acts to safeguard against the extension of de-polymerized microtubules, preventing the aberrant assembly of cilia prior to mitosis. Thus, Kif2a and Kif24 are activated by kinases expressed in S/G2 (Nek2) or G2/M (Plk1) phase, further linking cell cycle progression with the maintenance of a deciliated state necessary for mitosis (Figs 1 and 3). Interestingly, this mode of regulation could represent an ancient, conserved program, as relatives of Kif24 and Nek2 — as well as Aurora A — play a role in axonemal assembly and disassembly in flagellated and ciliated species65–71.
Cilia can also be removed by severing mechanisms. For example, in Chlamydomonas, deciliation is facilitated by the action of a microtubule-severing enzyme, katanin, which separates basal bodies from axonemes prior to mitosis72,73. In neurons, deciliation can occur through an actomyosin-dependent process of apical abscission, wherein the cilium is pinched off from the centrosome74. As abscission could drastically alter the reception of proliferative and differentiative cues by curtailing Hh signalling74, it will be important to investigate whether this deciliation mechanism is more generally used by other cell types during normal proliferation and differentiation and in pathological states such as cancer.
Whereas these experiments begin to suggest mechanisms for cilium disassembly in mammalian cells, previous studies have mainly examined total populations as opposed to single cells, making it difficult to understand the factors that control equilibria or directionality of the assembly–disassembly process. Thus, it will be important to follow the process in real time in vivo and in single cells. Kinases activated in S/G2/M phase regulate a number of targets linked to cilium disassembly. It will be important to determine whether misregulation of cell cycle kinases causes loss of cilia indirectly due to cell cycle perturbations or directly through defective cilia assembly (or a combination of both). It will also be essential to understand the contributions of each of the disassembly pathways in diverse mammalian tissues using animal knockouts. One surprising result of such an approach is the normal development of HDAC6-null mice despite hyper-acetylation of tubulin75. This suggests that additional, unidentified enzymes could function redundantly with HDAC6 to de-acetylate axonemal microtubules (or bypass mechanisms could be operative), and that HDAC6 inhibition may not be toxic — possibilities that must be considered during drug development. In this regard, the Aurora-A–HDAC6 pathway could be lineage-specific, since inhibitors of either Aurora A or HDAC6 led to inefficient assembly of cilia in only a subset of stem cell lines76.
Two proteins that localize to the centriole or transition zone (TZ), Nde1 and Tctex-1, respectively, also link cilium disassembly with S phase entry. Nde1, a paralogue of Ndel1, is highly expressed in mitotic cells but depleted during quiescence, and silencing of Nde1 in cultured cells and zebrafish leads to marked enhancement of cilium length and a delay in S phase entry77. Nde1 levels are controlled by CDK5 phosphorylation in G0/G1 cells, which targets Nde1 for ubiquitylation (and subsequent destruction) by the F-box protein Fbw7, a tumour suppressor78. Ablation of Nde1 in mice leads to microcephaly, possibly due to proliferative delays provoked by the persistence of cilia in progenitor neurons77,79. Likewise, Tctex-1 ablation led to persistent ciliation and a G1/S phase block80. These studies point to a mechanism wherein cells sense the resorption of the cilium, and cell cycle progression may be constrained by its removal. It will be essential to identify additional regulators of cilium disassembly to support this model and to elucidate how the integrity of this restrictive mechanism is sensed by the cell cycle machinery.
A role for the cilium in cell cycle control and human cancer
As discussed above, ciliogenesis is tightly coordinated with the cell cycle (Fig. 1). With a few exceptions81, the cilium and mitotic spindle assemble in a mutually exclusive manner, and by tethering the centrosome to the plasma membrane, the cell is deprived of a mitotic organizing centre (MTOC), thereby restricting proliferation. This observation has significant implications for mammalian growth and development. For example, one important yet untested implication of the ‘cilium versus centrosome’ model is that the cilium could function in a tumour suppressive capacity. In contrast to defects in cilium assembly, which are linked to ciliopathies, aberrations in cilium disassembly have not been widely linked to disease (with the exceptions described above). However, human cancer may be associated with the loss of primary cilia, and given the parallels with ciliopathies, it may be worthwhile to consider these diverse pathologies simultaneously when devising therapeutic strategies.
The presence or absence of cilia — and therefore integrity of signalling — can be instructive for cell growth. For example, certain ciliary signalling proteins, such as Smo (a key regulator of Hh signalling) or platelet-derived growth factor receptor (PDGFR), are proto-oncogenes that are aberrantly activated at cilia in cancer82,83. Remarkably, whereas some types of cancer (basal cell carcinoma and medulloblastoma) are dependent on cilia84,85, other cancers, including melanoma and breast, pancreatic, renal, and prostate cancer, exhibit loss of cilia, most likely during the early stages of oncogenesis86–90. The mechanisms by which cancer cells lose their cilia are unknown, as is how cilia loss affects signalling and tumour growth. It is notable that proteins associated with cilium disassembly — Aurora A and Nek2 — are widely deregulated in human cancer through overexpression or gene amplification. Inhibitors of these kinases, which have many of the hallmarks of oncoproteins, as well as HDAC6 inhibitors, are currently in clinical trials91. Furthermore, expression of an oncogenic form of PDGFR-α found in gastrointestinal tumours is linked to cilium disassembly through Aurora A activation82. Therefore, inhibitors of these kinases could be useful for the treatment of both tumours and ciliopathies92.
Oncoproteins and tumour suppressors clearly play antagonistic roles in cilium assembly. Indeed, the function of multiple tumour suppressors — including VHL, PTEN, p53, tuberous sclerosis proteins (TSC1 and TSC2) and adenomatous polyposis coli — are linked to normal ciliary assembly and function93–98. Mutations in the von Hippel–Lindau tumour suppressor gene (VHL) predispose patients to renal cysts and clear cell renal cell carcinoma, and loss of pVHL, combined with ablation of a second tumour suppressor protein (p53 or PTEN), provokes ciliary loss, kidney cyst formation and neoplastic growth93,94. PTEN localizes near basal bodies, and its loss leads to severely diminished cilium assembly, most likely as a result of accelerated organelle disassembly95. Moreover, the transition zone protein NPHP4, which is mutated in ciliopathies, modulates the Hippo signalling pathway, which regulates cell proliferation96. Thus, tumour suppressor function is tied to normal cilium biogenesis and function, and drugs that target downstream effectors of these proteins — such as the mTOR signalling pathway, which is aberrantly activated in TSC mutants97 — will be an avenue for future exploration.
As described above, the proteins that catalyse cilium disassembly may provide an avenue for drug discovery, since restoration of the primary cilium in tumour cells could promote quiescence64, especially if two or more pathways were simultaneously targeted. It will be important to distinguish the contributions of aberrant regulation of cell cycle proteins (such as Nek2, Plk1 and Aurora A) to mitotic defects in chromosome segregation, spindle pole behaviour and other centrosome-associated processes from their effects on cilium assembly and function. Teasing apart these diverse functions will require additional information from proteomics and molecular genetics.
Future directions
Going forward, it will be essential to understand the inductive cues that link ciliogenesis to the cell cycle, mitogen deprivation and autophagic induction99. Super-resolution microscopy100 and proteomic approaches that combine proximity-based labelling101,102 with gene editing will undoubtedly accelerate progress. Given the pivotal role that cilia play in diverse aspects of human biology from development to disease, it will be vital to shed light on unanswered questions regarding the earliest steps in cilium assembly. It will be essential to decipher the signals that mediate the onset of basal body maturation and cilium assembly as well as the remodelling events at the distal ends of basal bodies and vesicles that facilitate capture and growth of early ciliary vesicles and extension of the nascent axoneme. Likewise, it will be important to elucidate the cues that directly promote cilium disassembly, as these mechanisms may be relevant to the early steps of tumorigenesis and may reveal targets for drug therapy.
Supplementary Material
{kind=link}
Acknowledgments
We apologize to the many colleagues whose work could not be cited owing to space constraints. We thank M. Failler, W. Fu, S. Kim, W. Tsang, L. Wang and other colleagues for critical comments and encouragement. Work in B.D.D.’s laboratory was supported by NIH (grant no. 1R01HD069647).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Contributor Information
Irma Sánchez, Email: Irma.sanchez@med.nyu.edu.
Brian David Dynlacht, Email: brian.dynlacht@med.nyu.edu.
References
- 1.Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. J Cell Biol. 2011;193:435–444. doi: 10.1083/jcb.201101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nigg EA, Stearns T. The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13:1154–1160. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci USA. 1993;90:5519–5523. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pazour GJ, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goto H, Inoko A, Inagaki M. Cell cycle progression by the repression of primary cilia formation in proliferating cells. Cell Mol Life Sci. 2013;70:3893–3905. doi: 10.1007/s00018-013-1302-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Einstein EB, et al. Somatostatin signaling in neuronal cilia is critical for object recognition memory. J Neurosci. 2010;30:4306–4314. doi: 10.1523/JNEUROSCI.5295-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membrane Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 9.Chavali PL, Putz M, Gergely F. Small organelle, big responsibility: the role of centrosomes in development and disease. Phil Trans R Soc B. 2014;369:20130468. doi: 10.1098/rstb.2013.0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JE, Gleeson JG. A systems-biology approach to understanding the ciliopathy disorders. Genome Med. 2011;3:59. doi: 10.1186/gm275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaghloul NA, Katsanis N. Mechanistic insights into Bardet–Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma N, Berbari NF, Yoder BK. Ciliary dysfunction in developmental abnormalities and diseases. Curr Top Dev Biol. 2008;85:371–427. doi: 10.1016/S0070-2153(08)00813-2. [DOI] [PubMed] [Google Scholar]
- 13.Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18:1855–1871. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- 14.Wheatley DN, Wang AM, Strugnell GE. Expression of primary cilia in mammalian cells. Cell Biol Int. 1996;20:73–81. doi: 10.1006/cbir.1996.0011. [DOI] [PubMed] [Google Scholar]
- 15.Aughsteen AA. The ultrastructure of primary cilia in the endocrine and excretory duct cells of the pancreas of mice and rats. Eur J Morphol. 2001;39:277–283. doi: 10.1076/ejom.39.5.277.7380. [DOI] [PubMed] [Google Scholar]
- 16.Marion V, et al. Transient ciliogenesis involving Bardet–Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc Natl Acad Sci USA. 2009;106:1820–1825. doi: 10.1073/pnas.0812518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stinchcombe JC, et al. Mother centriole distal appendages mediate centrosome docking at the immunological synapse and reveal mechanistic parallels with ciliogenesis. Curr Biol. 2015;25:3239–44. doi: 10.1016/j.cub.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu W, Asp P, Canter B, Dynlacht BD. Primary cilia control hedgehog signaling during muscle differentiation and are deregulated in rhabdomyosarcoma. Proc Natl Acad Sci USA. 2014;111:9151–9156. doi: 10.1073/pnas.1323265111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tucker RW, Scher CD, Stiles CD. Centriole deciliation associated with the early response of 3T3 cells to growth factors but not to SV40. Cell. 1979;18:1065–1072. doi: 10.1016/0092-8674(79)90219-8. [DOI] [PubMed] [Google Scholar]
- 20.Tucker RW, Pardee AB, Fujiwara K. Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell. 1979;17:527–535. doi: 10.1016/0092-8674(79)90261-7. [DOI] [PubMed] [Google Scholar]
- 21.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–1363. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westlake CJ, et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci USA. 2011;108:2759–2764. doi: 10.1073/pnas.1018823108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt KN, et al. Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J Cell Biol. 2012;199:1083–1101. doi: 10.1083/jcb.201202126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi T, Kim S, Lin YC, Inoue T, Dynlacht BD. The CP110-interacting proteins Talpid3 and Cep290 play overlapping and distinct roles in cilia assembly. J Cell Biol. 2014;204:215–229. doi: 10.1083/jcb.201304153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Q, et al. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat Cell Biol. 2015;17:228–240. doi: 10.1038/ncb3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol. 1962;15:363–377. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshimura S, Egerer J, Fuchs E, Haas AK, Barr FA. Functional dissection of Rab GTPases involved in primary cilium formation. J Cell Biol. 2007;178:363–369. doi: 10.1083/jcb.200703047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nachury MV, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 29.Lechtreck KF. IFT–cargo interactions and protein transport in cilia. Trends Biochem Sci. 2015;40:765–778. doi: 10.1016/j.tibs.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lechtreck KF, et al. The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol. 2009;187:1117–1132. doi: 10.1083/jcb.200909183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eguether T, et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev Cell. 2014;31:279–290. doi: 10.1016/j.devcel.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanos BE, et al. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013;27:163–168. doi: 10.1101/gad.207043.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell. 2002;3:339–350. doi: 10.1016/s1534-5807(02)00258-7. [DOI] [PubMed] [Google Scholar]
- 34.Kohlmaier G, et al. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr Biol. 2009;19:1012–1018. doi: 10.1016/j.cub.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt TI, et al. Control of centriole length by CPAP and CP110. Curr Biol. 2009;19:1005–1011. doi: 10.1016/j.cub.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 36.Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK. CPAP is a cell-cycle regulated protein that controls centriole length. Nat Cell Biol. 2009;11:825–831. doi: 10.1038/ncb1889. [DOI] [PubMed] [Google Scholar]
- 37.Spektor A, Tsang WY, Khoo D, Dynlacht BD. Cep97 and CP110 suppress a cilia assembly program. Cell. 2007;130:678–690. doi: 10.1016/j.cell.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 38.Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr Biol. 2001;11:1586–1590. doi: 10.1016/s0960-9822(01)00484-5. [DOI] [PubMed] [Google Scholar]
- 39.Goetz SC, Liem KF, Jr, Anderson KV. The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell. 2012;151:847–858. doi: 10.1016/j.cell.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cajanek L, Nigg EA. Cep164 triggers ciliogenesis by recruiting Tau tubulin kinase 2 to the mother centriole. Proc Natl Acad Sci USA. 2014;111:2841–2850. doi: 10.1073/pnas.1401777111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Houlden H, et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet. 2007;39:1434–1436. doi: 10.1038/ng.2007.43. [DOI] [PubMed] [Google Scholar]
- 42.Bauer P, et al. Spinocerebellar ataxia type 11 (SCA11) is an uncommon cause of dominant ataxia among French and German kindreds. J Neurol Neurosurg Psychiatry. 2010;81:1229–1232. doi: 10.1136/jnnp.2009.202150. [DOI] [PubMed] [Google Scholar]
- 43.Oda T, Chiba S, Nagai T, Mizuno K. Binding to Cep164, but not EB1, is essential for centriolar localization of TTBK2 and its function in ciliogenesis. Genes Cells. 2014;19:927–940. doi: 10.1111/gtc.12191. [DOI] [PubMed] [Google Scholar]
- 44.Xu Q, et al. Phosphatidylinositol phosphate kinase PIPKIgamma and phosphatase INPP5E coordinate initiation of ciliogenesis. Nat Commun. 2016;7:10777. doi: 10.1038/ncomms10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen VL, et al. Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance. EMBO J. 2015;34:2537–2556. doi: 10.15252/embj.201488044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Gonzalo FR, et al. Phosphoinositides regulate ciliary protein trafficking to modulate Hedgehog signaling. Dev Cell. 2015;34:400–409. doi: 10.1016/j.devcel.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chavez M, et al. Modulation of ciliary phosphoinositide content regulates trafficking and Sonic Hedgehog signaling output. Dev Cell. 2015;34:338–350. doi: 10.1016/j.devcel.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 48.Kuhns S, et al. The microtubule affinity regulating kinase MARK4 promotes axoneme extension during early ciliogenesis. J Cell Biol. 2013;200:505–522. doi: 10.1083/jcb.201206013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lizcano JM, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 51.Boehlke C, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. 2010;12:1115–1122. doi: 10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacob LS, et al. Genome-wide RNAi screen reveals disease-associated genes that are common to Hedgehog and Wnt signaling. Sci Signal. 2011;4:ra4. doi: 10.1126/scisignal.2001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plotnikova OV, et al. Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol Biol Cell. 2012;23:2658–2670. doi: 10.1091/mbc.E11-12-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ran J, Yang Y, Li D, Liu M, Zhou J. Deacetylation of alpha-tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci Rep. 2015;5:12917. doi: 10.1038/srep12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plotnikova OV, et al. INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J Cell Sci. 2015;128:364–372. doi: 10.1242/jcs.161323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinzel D, et al. Pitchfork regulates primary cilia disassembly and left-right asymmetry. Dev Cell. 2010;19:66–77. doi: 10.1016/j.devcel.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inoko A, et al. Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. J Cell Biol. 2012;197:391–405. doi: 10.1083/jcb.201106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang K, Diener DR, Rosenbaum JL. The ubiquitin conjugation system is involved in the disassembly of cilia and flagella. J Cell Biol. 2009;186:601–613. doi: 10.1083/jcb.200903066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kasahara K, et al. Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat Commun. 2014;5:5081. doi: 10.1038/ncomms6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang W, Wu T, Kirschner MW. The master cell cycle regulator APC-Cdc20 regulates ciliary length and disassembly of the primary cilium. eLife. 2014;3:e03083. doi: 10.7554/eLife.03083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inaba H, et al. Ndel1 suppresses ciliogenesis in proliferating cells by regulating the trichoplein–Aurora A pathway. J Cell Biol. 2016;212:409–423. doi: 10.1083/jcb.201507046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kobayashi T, Tsang WY, Li J, Lane W, Dynlacht BD. Centriolar kinesin Kif24 interacts with CP110 to remodel microtubules and regulate ciliogenesis. Cell. 2011;145:914–925. doi: 10.1016/j.cell.2011.04.028. [DOI] [PubMed] [Google Scholar]
- 63.Miyamoto T, et al. The microtubule-depolymerizing activity of a mitotic kinesin protein KIF2A drives primary cilia disassembly coupled with cell proliferation. Cell Rep. 2015;10:664–673. doi: 10.1016/j.celrep.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim S, Lee K, Choi JH, Ringstad N, Dynlacht BD. Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nat Commun. 2015;6:8087. doi: 10.1038/ncomms9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahjoub MR, et al. The FA2 gene of Chlamydomonas encodes a NIMA family kinase with roles in cell cycle progression and microtubule severing during deflagellation. J Cell Sci. 2002;115:1759–1768. doi: 10.1242/jcs.115.8.1759. [DOI] [PubMed] [Google Scholar]
- 66.Pan J, Wang Q, Snell WJ. An aurora kinase is essential for flagellar disassembly in Chlamydomonas. Dev Cell. 2004;6:445–451. doi: 10.1016/s1534-5807(04)00064-4. [DOI] [PubMed] [Google Scholar]
- 67.Quarmby LM, Mahjoub MR. Caught Nek-ing: cilia and centrioles. J Cell Sci. 2005;118:5161–5169. doi: 10.1242/jcs.02681. [DOI] [PubMed] [Google Scholar]
- 68.Bradley BA, Quarmby LM. A NIMA-related kinase, Cnk2p, regulates both flagellar length and cell size in Chlamydomonas. J Cell Sci. 2005;118:3317–3326. doi: 10.1242/jcs.02455. [DOI] [PubMed] [Google Scholar]
- 69.Wloga D, et al. Members of the NIMA-related kinase family promote disassembly of cilia by multiple mechanisms. Mol Biol Cell. 2006;17:2799–2810. doi: 10.1091/mbc.E05-05-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piao T, et al. A microtubule depolymerizing kinesin functions during both flagellar disassembly and flagellar assembly in Chlamydomonas. Proc Natl Acad Sci USA. 2009;106:4713–4718. doi: 10.1073/pnas.0808671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hilton LK, Gunawardane K, Kim JW, Schwarz MC, Quarmby LM. The kinases LF4 and CNK2 control ciliary length by feedback regulation of assembly and disassembly rates. Curr Biol. 2013;23:2208–2214. doi: 10.1016/j.cub.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 72.Lohret TA, McNally FJ, Quarmby LM. A role for katanin-mediated axonemal severing during Chlamydomonas deflagellation. Mol Biol Cell. 1998;9:1195–1207. doi: 10.1091/mbc.9.5.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rasi MQ, Parker JD, Feldman JL, Marshall WF, Quarmby LM. Katanin knockdown supports a role for microtubule severing in release of basal bodies before mitosis in Chlamydomonas. Mol Biol Cell. 2009;20:379–388. doi: 10.1091/mbc.E07-10-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Das RM, Storey KG. Apical abscission alters cell polarity and dismantles the primary cilium during neurogenesis. Science. 2014;343:200–204. doi: 10.1126/science.1247521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bangs FK, Schrode N, Hadjantonakis AK, Anderson KV. Lineage specificity of primary cilia in the mouse embryo. Nat Cell Biol. 2015;17:113–122. doi: 10.1038/ncb3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim S, et al. Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol. 2011;13:351–360. doi: 10.1038/ncb2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maskey D, et al. Cell cycle-dependent ubiquitylation and destruction of NDE1 by CDK5-FBW7 regulates ciliary length. EMBO J. 2015;34:2424–2440. doi: 10.15252/embj.201490831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–293. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 80.Li A, et al. Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol. 2011;13:402–411. doi: 10.1038/ncb2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Riparbelli MG, Callaini G, Megraw TL. Assembly and persistence of primary cilia in dividing Drosophila spermatocytes. Dev Cell. 2012;23:425–432. doi: 10.1016/j.devcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nielsen BS, et al. PDGFRβ and oncogenic mutant PDGFRα D842V promote disassembly of primary cilia through a PLCγ- and AURKA-dependent mechanism. J Cell Sci. 2015;128:3543–3549. doi: 10.1242/jcs.173559. [DOI] [PubMed] [Google Scholar]
- 83.Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013;19:1410–1422. doi: 10.1038/nm.3389. [DOI] [PubMed] [Google Scholar]
- 84.Wong SY, et al. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med. 2009;15:1055–1061. doi: 10.1038/nm.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Han YG, et al. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. 2009;15:1062–1065. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009;69:422–430. doi: 10.1158/0008-5472.CAN-08-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim J, Dabiri S, Seeley ES. Primary cilium depletion typifies cutaneous melanoma in situ and malignant melanoma. PloS ONE. 2011;6:e27410. doi: 10.1371/journal.pone.0027410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Basten SG, et al. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia. 2013;2:2. doi: 10.1186/2046-2530-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hassounah NB, et al. Primary cilia are lost in preinvasive and invasive prostate cancer. PloS ONE. 2013;8:e68521. doi: 10.1371/journal.pone.0068521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Menzl I, et al. Loss of primary cilia occurs early in breast cancer development. Cilia. 2014;3:7. doi: 10.1186/2046-2530-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Frett B, et al. Therapeutic melting pot of never in mitosis gene a related kinase 2 (Nek2): a perspective on Nek2 as an oncology target and recent advancements in Nek2 small molecule inhibition. J Med Chem. 2014;57:5835–5844. doi: 10.1021/jm401719n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang PH, Zhang L, Zhang YJ, Zhang J, Xu WF. HDAC6: physiological function and its selective inhibitors for cancer treatment. Drug Discov Ther. 2013;7:233–242. doi: 10.5582/ddt.2013.v7.6.233. [DOI] [PubMed] [Google Scholar]
- 93.Frew IJ, et al. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J. 2008;27:1747–1757. doi: 10.1038/emboj.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Albers J, et al. Combined mutation of Vhl and Trp53 causes renal cysts and tumours in mice. EMBO Mol Med. 2013;5:949–964. doi: 10.1002/emmm.201202231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shnitsar I, et al. PTEN regulates cilia through Dishevelled. Nat Commun. 2015;6:8388. doi: 10.1038/ncomms9388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Habbig S, et al. NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J Cell Biol. 2011;193:633–642. doi: 10.1083/jcb.201009069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.DiBella LM, Park A, Sun Z. Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Human Mol Genet. 2009;18:595–606. doi: 10.1093/hmg/ddn384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Z, Li W, Song L, Zhu W. Cilia, adenomatous polyposis coli and associated diseases. Oncogene. 2012;31:1475–1483. doi: 10.1038/onc.2011.351. [DOI] [PubMed] [Google Scholar]
- 99.Tang Z, et al. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502:254–257. doi: 10.1038/nature12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang TT, et al. Superresolution pattern recognition reveals the architectural map of the ciliary transition zone. Sci Rep. 2015;5:14096. doi: 10.1038/srep14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gupta GD, et al. A dynamic protein interaction landscape of the human centrosome–cilium interface. Cell. 2015;163:1484–1499. doi: 10.1016/j.cell.2015.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mick DU, et al. Proteomics of primary cilia by proximity labeling. Dev Cell. 2015;35:497–512. doi: 10.1016/j.devcel.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.