

Abstract
Diabetes, a risk factor for stroke, leads to elevated blood methylglyoxal (MG) levels. This is due to increased MG generation from the high glucose levels, and because diabetes impairs the glutathione (GSH)-glyoxalase system for MG elimination. MG glycates proteins and causes dicarbonyl stress. We investigated the contribution of MG and GSH to stroke outcome. Cerebral ischemia/reperfusion was performed in chemical-induced (streptozotocin) and genetic Akita mouse models of Type 1 diabetes. Brain infarction and functions of the GSH-dependent MG elimination pathway were determined. Diabetes increased post-ischemia-reperfusion cerebral infarct area in association with elevated MG and diminished GSH levels. Infarct size correlated with brain MG-to-GSH ratio. Expression of glutamate-cysteine ligase catalytic subunit (GCLc) was increased in diabetic brain. GCL activity was unchanged. MG-adducts were elevated in the diabetic brain and, using immunoprecipitation, we identified one of the bands as glycated occludin. This was accompanied by increased blood-brain barrier permeability. Total protein carbonyls were elevated, indicative of oxidative/carbonyl stress. N-acetylcysteine (NAC) corrected MG-to-GSH ratio, and reduced diabetic brain infarct area, occludin glycation and permeability. In addition, protein carbonyls were decreased by NAC. We showed that the diabetic brain exhibited a lower GSH-dependent potential for MG elimination, which contributed to increased protein glycation, and oxidative/carbonyl stress. The consequence of these changes was aggravated post-stroke brain injury. NAC administration protected against the exacerbated brain damage via restored GSH generation and normalization of the MG-to-GSH ratio and possibly by attenuating oxidative/carbonyl stress. This treatment could contribute to the successful management of stroke risk/outcome in diabetes.
Keywords: Brain, carbonyl stress, diabetes, glutamate-cysteine ligase, glutathione, glyoxalase, ischemia-reperfusion, methylglyoxal
Introduction
Diabetes mellitus is an independent risk factor for the development of cardiovascular and cerebrovascular disease. Stroke occurs twice as frequently in hypertensive individuals with diabetes than those with hypertension alone [1]. Moreover, stroke patients with diabetes have a 3-fold higher mortality than their non-diabetic counterparts [2]. The plasma glycemic status is typically used as a predictor of increased cardiovascular disease risk in diabetes, and risk of heart disease and stroke is exaggerated in diabetics with poor glycemic control [3]. However, increased cardiovascular disease risk remains in diabetic individuals with well-controlled blood sugar levels, suggesting involvement of other factors. One candidate is methylglyoxal (MG), a highly reactive dicarbonyl metabolite of α-oxoaldehydes [4] and a potent glycating agent. MG is a precursor of advanced glycation end-products (AGEs), and MG-derived protein modification [5], known as glycation, is a patho-physiologically relevant process.
MG levels are elevated in diabetes [6], and have been implicated in diabetic complications. MG can increase inflammatory cytokine release, upregulate adhesion molecule expression and promote leukocyte adhesion [7]. While all of these responses are implicated in stroke, it is unclear whether MG levels rise sufficiently to contribute to post-stroke injury in non-diabetics. In contrast, these findings do suggest a possible role for MG in the exacerbation of stroke-induced injury by diabetes. We previously demonstrated that MG treatment of human brain microvascular endothelial cells (IHECs) increases barrier permeability [8]. Furthermore, the barrier disruption was worse in the presence of hyperglycemia [9], suggesting that, in addition to being a source of increased MG generation, high glucose levels potentiate the injury caused by MG.
Interestingly, MG-induced IHEC dysfunction was preceded by cellular glutathione (GSH) loss and was prevented by N-acetylcysteine (NAC) [8], a precursor of GSH synthesis, suggesting a role for GSH in endothelial preservation. GSH is decreased during diabetes and after brain ischemia-reperfusion (I/R) [10, 11]. GSH is a rate-limiting co-factor in the elimination of MG via the glyoxalase (Glo) system. Thus it follows that MG-induced disruption of IHEC integrity is potentiated by inhibition of GSH synthesis [8]. Depletion of GSH also exacerbated infarct size and edema following stroke [12], underscoring the importance of endogenous GSH in protection against post-stroke lesion formation. GSH synthesis is controlled by glutamate-cysteine ligase (GCL), the rate-limiting enzyme. GCL is comprised of a catalytic (GCLc) and a modulatory (GCLm) subunit. GCLc and GCLm expressions are attenuated in mesenteric vessels of diabetic (db/db) mice [13] and GCLc is decreased in the retina of streptozotocin-induced diabetic rats [14]. The glyoxalase pathway comprises the glyoxalase I (Glo I) and glyoxalase II (Glo II) enzymes. To date, reports on changes in Glo I and Glo II during diabetes are conflicting [15-22], although Glo I overexpression has been shown to protect against diabetic complications [23, 24].
The glycation reaction of MG with amino acids can generate superoxide radical anions [25]. Hence, protein damage by MG can be mediated by protein glycation, and/or oxidative stress through reactive oxygen species (ROS) generation, which may lead to protein carbonyl formation [26]. GSH also acts as an antioxidant, thus oxidative stress could result from both decreased GSH levels and increased MG. However, we previously showed that MG stimulates only very low levels of ROS generation in IHECs, and even more importantly, MG-induced barrier disruption was not protected by antioxidants. Collectively, these results support our hypothesis that increased MG and diminished GSH during diabetes could worsen post-stroke injury, and that this is more likely to be via glycation than carbonylation. Previous studies have shown diabetes exacerbates cerebral I/R injury in mice [27, 28], however the exact mechanisms remain unclear. The current project seeks to determine the: a) contributions of MG, Glo enzymes, and GSH to I/R brain injury during diabetes; b) mechanism of GSH decrease in diabetic brain.
Materials and Methods
Materials
From Sigma (St. Louis MO, USA): GSH, MG, NAC, STZ, L-buthionine-(S, R)-sulfoximine (BSO), D-lactate, D-lactic dehydrogenase, glutamic-pyruvate transaminase, β-nicotinamide adenine dinucleotide hydrate, S-D-lactoylglutathione, evans blue. Antibodies: Abcam, Cambridge, MA (rabbit-anti-mouse GCLc, HRP-conjugated goat-anti-rabbit IgG); Invitrogen, Camerillo, CA (rabbit-anti-mouse occludin); Genox Corporation, Baltimore, MD (anti-MG); Amersham, Piscataway, NJ (HRP-conjugated sheep-anti-mouse IgG); BD Biosciences, La Jolla, CA (mouse-anti-actin). ECL reagent: BIO-RAD (Berkeley, CA, USA).
Animal Preparation
Four-week-old male C57BL/6J mice (WT), weighing 18-20 g, were purchased from Jackson Laboratory. The genetically diabetic Akita mice (InsAkita+/-), mice deficient in GCLm (GCLm-/-) or their wildtype littermates (InsAkita+/+ and GCLm+/+ respectively) were provided by Drs Harris and Pattillo from their respective breeding colonies at LSUHSC-Shreveport. For the Akita mice, breeding pairs were purchased from Jackson Laboratories, and offspring were produced by crossing InsAkita+/- with InsAkita+/+ mice. Diabetic InsAkita+/- mice were identified by their plasma glucose levels. The Gclm-/- mice were developed by Dr. Terrance Kavanagh, who provided breeding pairs for our colony [29]. Dr. Pattillo generated the experimental mice by crossing heterozygous pairs. The WT and knockout offspring genotypes were determined by PCR of genomic DNA isolated from tail clips, using a two tube reaction system. They shared the same forward primer (5′-GCCCGCTCGCCATCTCTC-3′). The sequence of the reverse primer for the knockout was 5′-CAGTTTGAGGGGACGACGACA-3′, and for the WT was 5′-GTTGAGCAGGTTCCCGGTCT -3′. All animal procedures were approved by the LSUHSC-S Institutional Animal Care and Use Committee and were in accordance with the US NIH guidelines in the Guide for the Care and Use of Laboratory Animals. WT mice were divided randomly into non-diabetic (received citrate buffer vehicle (Veh) IP for 5 consecutive days) or diabetic (received 50 mg/kg of STZ IP for consecutive 5 days) groups. On day 7, mice with non-fasting plasma glucose >16.7 mmol/l were deemed diabetic (“diabetes onset”). Some groups received 2 mmol/l NAC in the drinking water (refreshed every 2 days to avoid autoxidation) starting at 1 wk after diabetes onset for 1 wk or 3 wks. This dose was chosen to give within the mouse equivalent dose range (326-570 mg kg-1 day-1) for the human dose of 20-30 mg kg-1 day-1. Based on body weight and water consumption our diabetic mice received 400±70 mg kg-1. I/R (or sham operation) was performed in mice 5 wks post-STZ (i.e. @ 4 wks diabetes) or post-Veh. In one STZ+NAC group, I/R was performed at 2 wks diabetes (1 wk NAC). Thus the 7 WT groups were:
NT-sham group (n=6): non-treated sham operated group
NT-I/R group (n=5): non-treated I/R group
Veh-I/R group (n=7): vehicle-treated I/R group
STZ-2wk I/R group (n=9): STZ with I/R @2 wks diabetes
STZ-I/R group (n=7): STZ group with I/R @4 wks diabetes
STZ+NAC 1wk-I/R group (n=9): STZ group with I/R @2 wks diabetes; NAC for 1 wk before I/R
STZ+NAC 3wk-I/R group (n=6): STZ group with I/R at @4 wks diabetes; NAC for 3 wks before I/R
Akita non-diabetic (InsAkita+/+) and diabetic (InsAkita+/-) mice, as well as GCLm-/- and wildtype littermates (GCLm+/+) underwent I/R at 12 wks and 10 wks age, respectively.
Middle Cerebral Artery Occlusion/Reperfusion (MCAo/R) and brain infarction
Mice were anesthetized with ketamine (125 mg/kg) and xylazine (6.25 mg/kg) IP. The Koizumi method was employed to induce stroke [30]. The left common, internal, and external carotid arteries were exposed, and the external carotid artery was ligated. A 6-0 silicone-coated nylon filament was introduced through the common carotid artery. The filament was advanced into the internal carotid artery 9.0 to 11.0 mm beyond the carotid bifurcation until an elastic resistance indicated that the tip was properly lodged in the anterior cerebral artery and thus blocked blood flow to the MCA for 45 min (45 min ischemia). Then the filament was pulled out and the blood flow was restored for 24 h (24 h reperfusion). In the sham operation, the filament was removed immediately after proper placement in the anterior cerebral artery.
The mice were euthanized by ketamine/xylazine at 24h after reperfusion, and brains were quickly removed. Six 2-mm-thick coronal sections were cut every 1 mm from posterior of the olfactory bulb. The 1st, 2nd, 5th and 6th slices were rapidly frozen in liquid nitrogen for later GSH, MG, and protein carbonyl content assays (slices 1 and 2) and enzyme activity assay (slice 5) and western blot (slice 6; primarily reflecting the effects of diabetes rather than ischemia). The 3rd and 4th slices were weighed and used to determine infarct size and water content. Brain infarct area was determined using 2,3,5-triphenyltetrazolium chloride (TTC) staining as previously described [31]. Infarct areas were calculated by the formula: 100%×(contralateral hemisphere area – non-infarct ipsilateral hemisphere area)/contralateral hemisphere area, and expressed as the average of the two brain slices. After obtaining the photos of infarct area, the brain slices were dried at 50°C for 72h to determine the water content. The water content in brain was calculated by the formula: 100% × (wet brain weight - dry brain weight)/wet brain weight.
High-performance liquid chromatography (HPLC) quantification of GSH, cysteine, γ-glutamylcysteine (γ-GC) and MG
Determination of GSH, cysteine and γ-GC was performed as previously described [32]. Briefly, TCA-soluble acid supernatants of brain homogenates were derivatized with 6 mmol/l iodoacetic acid and 1% 2,4-dinitrophenyl fluorobenzene (pH adjusted to 7-8 and 7.0, respectively) to yield the S-carboxymethyl and 2,4-dinitrophenyl derivatives, respectively. GSH, cysteine and γ-GC derivatives were separated on a 250×4.6-mm Alltech Lichrosorb NH2 10μm anion-exchange column and detected at 365nm. The tissue contents, expressed as nmol/mg protein, were quantified by comparison to standards derivatized in the same manner.
To confirm that cysteine utilization for GSH synthesis was reflected in its steady-state tissue levels, 2 additional groups of mice were performed: Veh+NAC group (n=7): vehicle-treated WT mice receiving 2 mmol/l NAC in drinking water for 2 wks; Veh+BSO group (n=8): vehicle-treated WT mice receiving 300 μl of BSO (0.5 mol/l in 0.9 % of saline), administered IP every 12 h for 24 h. BSO is a glutathione-synthesis inhibitor that acts by irreversibly inhibiting γ-glutamylcysteine ligase.
Determination of MG was as described [33]. In brief, brain homogenates were treated with 60% perchloric acid (29:1 v/v) and the acid supernatants were derivatized with 0.1 mol/l of o-phenylenediamine for 24h. MG derivatives were separated on a 250×4.6-mm Chromegabond Ultra C-18 reversed phase column, and detected at 315nm. Tissue MG contents, expressed as nmol/mg protein, were quantified by comparison to MG standards derivatized with o-phenylenediamine.
Assay of GCL activity
Brain tissues were homogenized with TES/SE buffer (PH=7.0 - 7.4) containing 20 mmol/l Tris, 1 mmol/l EDTA, 250 mmol/l sucrose, 20 mmol/l sodium borate and 2 mol/l serine. Post-15,000 g supernatants were used in an assay of GCL activity as described with modifications [34]. The GCL reaction cocktail contained 400 mmol/l Tris, 40 mmol/l ATP, 20 mmol/l L-glutamate, 2 mmol/l EDTA, 20 mmol/l sodium borate, 2 mmol/l serine and 40 mmol/l MgCl2. Reaction and baseline samples were prepared. The reaction sample contained the cocktail mixture plus brain supernatant, 20 mmol/l DTT, and 2 mmol/l cysteine to start the reaction. TES/SB buffer replaced cysteine in the baseline sample. The samples were incubated for 45 min at RT, and the reactions were stopped by addition of 50% TCA. The amount of γ-GC formed was quantified by HPLC as above. GCL activity, expressed as nmol (mg protein)-1 min-1, was calculated as the difference in γ-GC concentration in the reaction versus baseline samples, normalized to protein.
Assay of glyoxalase I and II activity
Brain tissues were homogenized (1:20, w/v) in 10 mmol/l Tris-HCl pH 7.4 containing a cocktail of proteinase inhibitors. Post-12,000 g supernatants were used for spectrophotometric assays of GloI and Glo II activities. Glo I activity was determined by S-D- lactoylglutathione (SDL) formation [35]. Glo II activity was determined by GSH regeneration [36]. Glo I and Glo II activities were expressed as nmoles of SDL or GSH formed respectively, per min per mg protein.
Western blot of occludin, GCLc and MG-modified proteins, and Immunoprecipitation
Western immunoblotting was performed on brain tissue homogenates to detect occludin, GCLc and MG-modified proteins as previously described [8]. Brain tissues were homogenized in RIPA lysis buffer containing 50 mmol/l Tris, pH 8.0, 150 mmol/l NaCl, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail. Twenty μg protein per sample was loaded on 10% SDS-polyacrylamide gels. Electrophoresis was performed, followed by transfer, and blocking, with dried milk in the blocking buffer. Although there is some evidence that dried milk contains AGEs and may result in non-specific bands on the western blot, we compared milk, bovine serum albumin and no blocking, and found dried milk gave the least number of, and very distinct, bands, and these were stronger in the diabetic samples, suggesting they were not non-specific. PVDF membranes were probed with the following primary antibodies: rabbit-anti-mouse occludin (1:2000), rabbit-anti-mouse GCLc (1:2000) or mouse anti-MG-modified protein (1:2000), followed by secondary HRP-conjugated goat-anti-rabbit or sheep-anti-mouse (1:10000) antibodies, as appropriate. Protein expression was detected using enhanced chemiluminescence (BIO-RAD) per manufacturer's instructions. Equal protein loading was normalized to actin.
Immunoprecipitation was performed as previously shown [8] to determine if occludin was glycated. Brain tissues were lysed with NP-40 buffer (150 mmol/l NaCl, 50 mmol/l Tris-HCl, 1.0% NP-40 and protease inhibitor cocktail) and pre-cleared with protein A/G–agarose beads for 1h at 4 °C. After centrifugation, the supernatant (1.2 mg lysate protein/reaction) was incubated with 5 μg of rabbit anti-occludin or 1.5 μg of rabbit anti-GCLc antibody or rabbit IgG (as a negative control) at 4°C overnight with gentle rotation. Then the samples were added to 30 μl of protein A/G–agarose beads and incubated for another 6h at 4°C. After centrifugation, the agarose beads were washed four times in lysis buffer and the protein was eluted in 40 μl of 2× loading buffer and boiled (100°C, 5min). 20 μl of the supernatant collected by centrifugation was loaded onto a 10% SDS–PAGE gel and blotted with anti-MG as described above.
Assessment of blood-brain barrier (BBB) integrity
BBB breach was assessed by Evans Blue (EB) extravasation. As described previously [31], a 2% EB solution was injected into the femoral vein of mice (4 ml/kg). At 24 h, blood was obtained by cardiac puncture, and the brain is harvested after transcardial perfusion with PBS (100 mmHg, over 5 mins). EB concentrations in plasma and TCA soluble extracts of brain tissues were measured by fluorescence spectrophotometry. Permeability was quantified by: tissue [EB]/plasma [EB].
Assay for protein carbonyls
Total protein carbonyls was determined in brain homogenates as described [37]. Briefly, brain homogenates were incubated with 10 mmol/l 2, 4-dinitrophenylhydrazine (DNPH) in 2 mol/l hydrochloric acid (1:2 v/v) and precipitated with 10% TCA. After washing, protein pellets were redissolved in 6 mol/l guanidine hydrochloride containing 20 mmol/l potassium phosphate (pH 2.3). Absorbance of post-11000 g supernatants was measured at 366nm. Protein carbonyl contents were quantified using the extinction coefficient, 22000 M-1 cm-1.
Statistical analysis
All data are expressed as mean ± SEM. Significance of difference was assessed by Student t test (single comparisons) or by one-way ANOVA with Newman-Keuls post-hoc tests (multiple comparisons). Spearman rank correlation analysis was used to determine the association between glucose, MG, GSH or MG-to-GSH ratio and infarct area. Differences or correlations were considered significant at P < 0.05.
Results
Diabetes potentiates ischemia-reperfusion brain injury
Four weeks of STZ-induced diabetes led to a 2-fold increase in post-stroke infarct versus non-diabetic controls (Fig. 1a&b). Although the experiments were not performed in a blinded manner, the individual data points show the variability in both controls and STZ mice, suggesting bias did not play a role in the findings. Genetically diabetic Akita mice behaved similarly (Fig. 1b&c)
Figure 1. Diabetes exacerbates brain infarct following stroke.
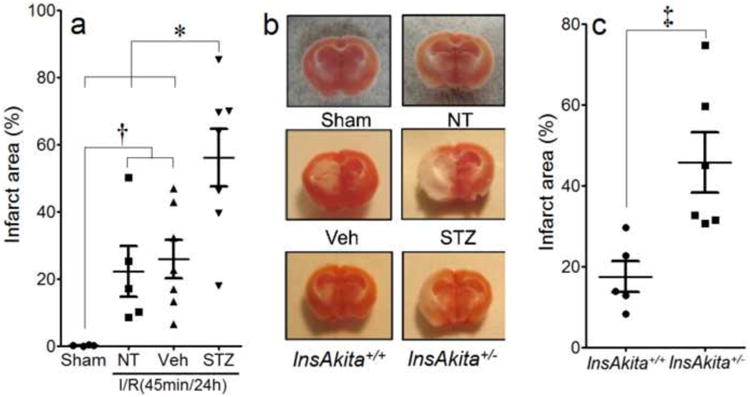
I/R injury in brains of controls and chemically induced diabetic mice (STZ; n=7) (a&b) or genetically diabetic (InsAkita+/-; n=6) mice (b&c). NT=non-treated; n=5; Veh=citrate buffer; n=7; InsAkita+/+=non-diabetic Akita littermates (n=5). The post I/R infarct area is represented by the unstained (white) regions of left brain sections (b). n=6 in Sham. *P<0.05 STZ vs. Sham, NT and Veh, †P<0.05 Sham vs. NT and Veh, ‡P<0.05 InsAkita+/- vs. InsAkita+/+. Data is expressed as mean ± SEM.
Cerebral infarct area correlates with plasma glucose, brain GSH and MG-to-GSH ratio
Diabetes was characterized by a significant elevation in plasma glucose levels (Fig. 2a). The brain injury was statistically positively correlated with plasma glucose. However, the data from the non-diabetic and diabetic mice appeared to divide into two distinct groups. This may have been because we did not have mice with intermediate glucose levels (Fig. 2b), or that glucose does not affect infarct size in a concentration-dependent manner, but rather exerts a threshold effect. Tissue GSH levels were significantly lowered (Fig. 2c) while MG levels were significantly elevated (Fig. 2d) in the diabetic brain. A similar glucose-infarct area relationship, and changes in GSH and MG levels were observed in the Akita diabetic model (Figs. 2e-h).
Figure 2. Diabetes Increases the MG-to-GSH Ratio in the Brain, and This Correlates With Infarct Area.
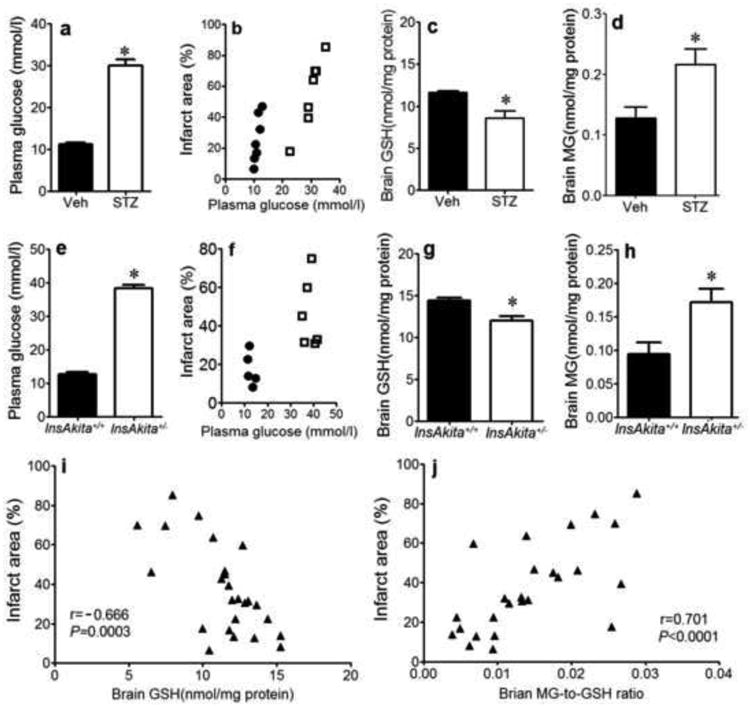
Measurements in chemically-induced (STZ@4wks; n=7: a-d) or genetic (InsAkita+/-; n=6: e-h) models of diabetes compared to their respective controls (Veh; n=7, InsAkita+/+; n=5). a&d: Plasma glucose levels. b&f: correlations of % brain infarct area with plasma glucose levels. c&g: brain GSH levels. d&h: brain MG concentrations. i: correlation of infarct area with GSH for both models combined. j: correlation of infarct area with MG-to-GSH ratio for both models combined. *P<0.05 STZ vs. Veh or InsAkita+/- vs. InsAkita+/+. Correlations: black circles =non-diabetic; white squares =diabetic.
We combined the data from the chemical and genetic models to test if there was a true correlation between infarct size and GSH, MG or MG:GSH ratio. Regardless of the diabetic model, infarct size inversely correlated with the brain GSH levels (Fig. 2i). Brain infarct was directly correlated with the MG-to-GSH ratio (Fig. 2j), rather than MG contents per se (data not shown), suggesting that post-IR brain injury is dependent on the GSH potential for MG elimination.
To determine if decreased tissue GSH per se would increase brain post-stroke injury, MCAo/R was performed on GCLm-/- mice. Gclm-/- mice exhibited increased infarct size compared with Gclm+/+ littermates (Fig. 3a) in accordance with lower brain GSH levels in these knockout animals (Fig. 3b).
Figure 3. Deficiency in Gclm Increases Post-Stroke Infarct Area and Decreases Brain GSH Levels.
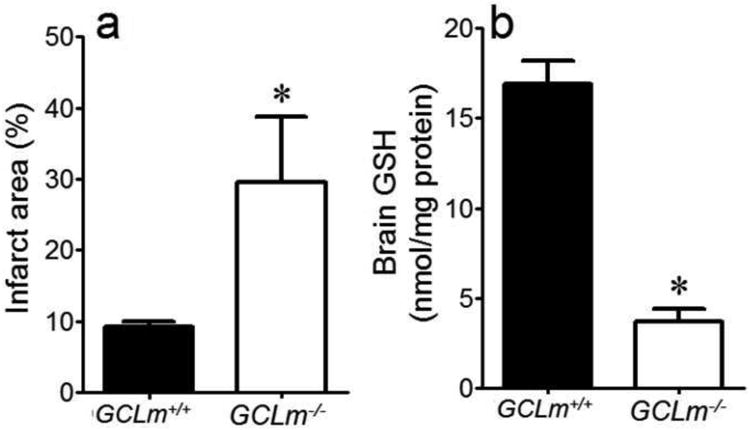
Infarct area (a) and brain GSH (b) in GCLm-/- mice and their littermate controls (GCLm+/+). *P<0.05 vs. GCLm+/+. n=5/grp.
NAC increases brain GSH and attenuates I/R induced diabetic brain infarct
To test for a role for GSH in post-I/R diabetic brain injury, STZ-treated mice were given the GSH precursor, NAC for 1 or 3 wks before MCAo/R. NAC treatment for 3 wks abrogated the exacerbated infarct area at 4wk diabetes (Fig. 4a) that corresponded to significant increases in brain GSH (Fig. 4c). As brief as 2wks diabetes exacerbated infarct size (Fig. 4b), albeit this effect was less than the longer duration of diabetes, indicating a relationship between duration of diabetes and post-stroke injury severity. The infarct in the 2 wks diabetic mice was attenuated by one wk of NAC, which also significantly elevated brain GSH contents (Fig. 4d). Brain infarct area was inversely related to tissue GSH levels (Fig. 4e), consistent with a protective role for GSH.
Figure 4. NAC Corrects Post-Stroke Infarct Area and Brain GSH Levels in Diabetic Mice.
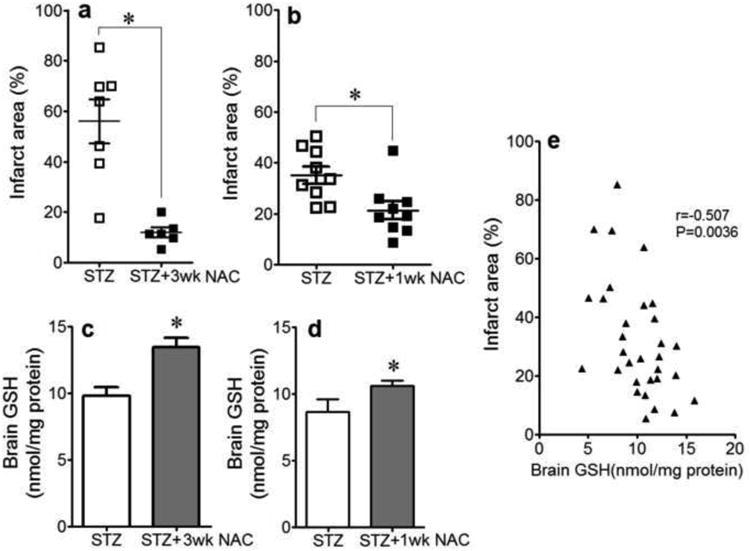
Infarct area (a&b) and brain GSH levels (c&d) in STZ-treated diabetic mice receiving no treatment or 2mmol/l NAC in drinking water for 1 or 3 weeks before MCAoR (a&c: diabetes 4 wks; b&d: diabetes 2 wks). *P<0.05 vs. STZ. (e): correlation between % infarct area and GSH levels. n=7 in STZ (4wk) group; n=6 in STZ (4wk)+3wk NAC group; n=9 in STZ (2wk) group; n=9 in STZ (2wk)+1wk NAC group.
NAC is a cysteine source and increases the GSH potential for MG elimination
GSH is generated in the cytosol in two steps. In the first, and rate-limiting step, GCL catalyzes the formation of the intermediate, γ-glutamylcysteine (γ-GC), from glutamate and cysteine. To further investigate the contribution of NAC to GSH production, we measured brain levels of cysteine as well as γ-GC as an indicator of GCL function. γ-GC contents were not different between veh- or STZ-treated mice at 4 wks (Fig. 5a), suggesting that diabetes did not alter GCL activity per se. The cysteine levels in diabetic brain trended lower than controls, albeit not significant (Fig. 5b). Unexpectedly, although NAC is a cysteine source, the steady-state cysteine levels were further decreased in NAC-treated STZ mice (Fig. 5c). To address if the steady-state cysteine pool within the brain reflects GSH production, veh-treated mice were administered NAC or BSO, an inhibitor of GSH synthesis. BSO significantly increased brain cysteine levels versus NAC-treated animals (Fig. 5c). This is consistent with the notion that enhanced GSH production (e.g., with NAC) would decrease the free cysteine pool; conversely inhibited GSH synthesis (e.g., with BSO) would correlate with tissue accumulation of free cysteine. Brain glutamate levels were unchanged under these conditions (data not shown), further supporting the paradigm that cysteine is the rate-limiting substrate in GSH production.
Figure 5. GCL function and Cysteine Levels in the Brain of Diabetic Mice.
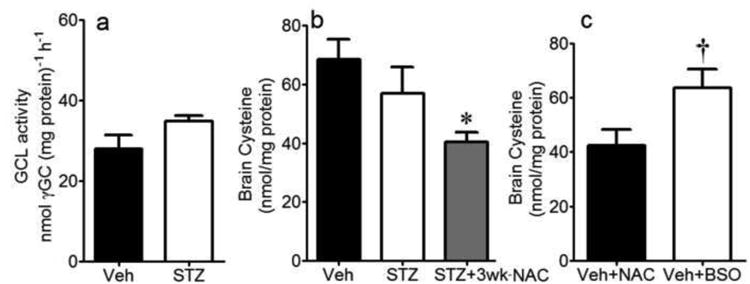
Brain GCL function, determined by γ -glutamylcysteine formation (a), and tissue cysteine levels (b) in Veh- or STZ-treated mice at 4wks diabetes. Cysteine levels were also measured in diabetic mice treated with NAC for 3wks (b), and in NAC-treated non-diabetic mice (n=7) or BSO (n=8) (c). *P<0.05 vs. Veh and STZ; †P<0.05 vs. Veh+NAC.
Three wks of NAC treatment did not alter plasma glucose levels at 4 wk diabetes (Fig. 6a), indicating that NAC did not act by normalizing plasma glucose. While trending lower, NAC did not significantly attenuate MG levels in the diabetic brain (Fig. 6b). However, NAC treatment significantly decreased the MG-to-GSH ratio (Fig. 6c).
Figure 6. Levels of Glucose in Plasma, and MG and MG-to-GSH Ratios in Brain During Diabetes.
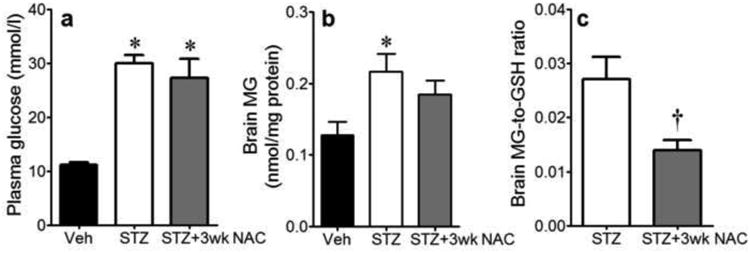
Plasma glucose levels (a), brain MG levels (b) and brain MG-to-GSH ratio (c) in Veh-treated non-diabetic mice and STZ-treated diabetic mice given water (STZ, n=7) or 2mmol/l NAC for 3wks (STZ+3wk NAC, n=6). *P<0.05 vs. Veh; †P<0.05 vs. STZ.
NAC protects against diabetes induced MG-adduct formation, but not GCLc expression
MG-adduct formation in the diabetic mouse brain in vivo was significantly elevated (Fig. 7a), which was decreased by NAC. Taken together with the higher GSH levels, and lower MG:GSH ratio in the NAC group, these data suggest that NAC promotes GSH-catalyzed MG elimination. The two major MG-modified proteins had Mws of 50 kD and 65 kD. Expression of occludin (a 65kD protein) was attenuated in the diabetic brain, although this did not reach statistical significance, and was not altered by NAC (Fig. 7b). However, the 65kD glycated protein was confirmed to be occludin-MG adduct by immunoprecipitating occludin followed by immunoblotting for MG (Fig 7d). Both of these findings are consistent with our previous observations in microvessels from diabetic rat brain [9]. A likely pathophysiological consequence of these changes would be increased blood-brain barrier (BBB) permeability. In agreement with this, Fig. 7e shows that plasma-to-tissue leakage of Evans Blue in diabetic brains was higher than in non-diabetic counterparts, indicating a substantial BBB breach during diabetes. Brain water content was also elevated (data not shown). After treating with NAC for 3 wks, both the occludin glycation as well as the leakage of Evans blue were significantly decreased versus their untreated counterparts.
Figure 7. Western blot analysis of MG-protein adducts, occludin and GCLc in the brain of diabetic mice.
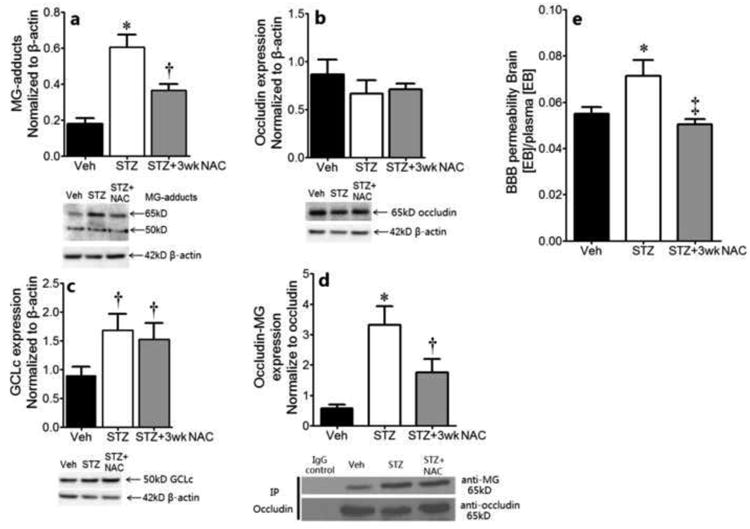
Expression of MG-protein adducts (a), occludin (b) and GCLc (c) in the brain of non-diabetic (Veh) and diabetic (STZ) mice, as measured by Western blot. Representative immunoblots are shown. The bar graphs show the quantitation of the protein band intensities normalized to β-actin; n=5/grp. (d): IP for occludin followed by immunoblot for MG, with quantification of the MG band intensity normalized to occludin. (e) The BBB permeability measured by Evans blue extrusion in control and diabetic mice-/+NAC, n=4/grp. *P<0.05 vs. Veh and STZ+3wk NAC; †P<0.05 vs. Veh; ‡P<0.05 vs. STZ.
Based on the Mw, we hypothesized that the 50kD glycated-protein adduct might be an isoform of mouse GCLc. The expression of GCLc protein was elevated (Fig. 7c). NAC did not reverse this. Unfortunately, we were unable to confirm the glycation of GCLc and if NAC could reverse this using co-immunoprecipitation because the GCLc band overlapped with the IgG heavy chain (which was also obtained in the negative control). Mass spectrometry is being employed in ongoing studies to identify MG-GCLc adducts.
NAC attenuates protein carbonyls in diabetic brain
Given that: i) infarct was correlated with increased MG:GSH; ii) both increased MG and decreased GSH can cause oxidative stress; iii) oxidative stress can promote protein carbonylation, we quantified protein carbonyl levels as a broad indicator of oxidative/carbonyl stress in the diabetic brain, and tested the impact of NAC treatment. Total protein carbonyls were increased in a time-dependent manner after diabetes induction (Fig. 8). To test if NAC could prevent the early rise in protein carbonylation, brain protein carbonyls were determined at 1wk following NAC treatment in 2wks post-STZ diabetic animals. The result shows that NAC abrogated protein carbonyl formation (Fig. 8).
Figure 8. Total Protein Carbonyl Content in the Brain of Diabetic Mice.
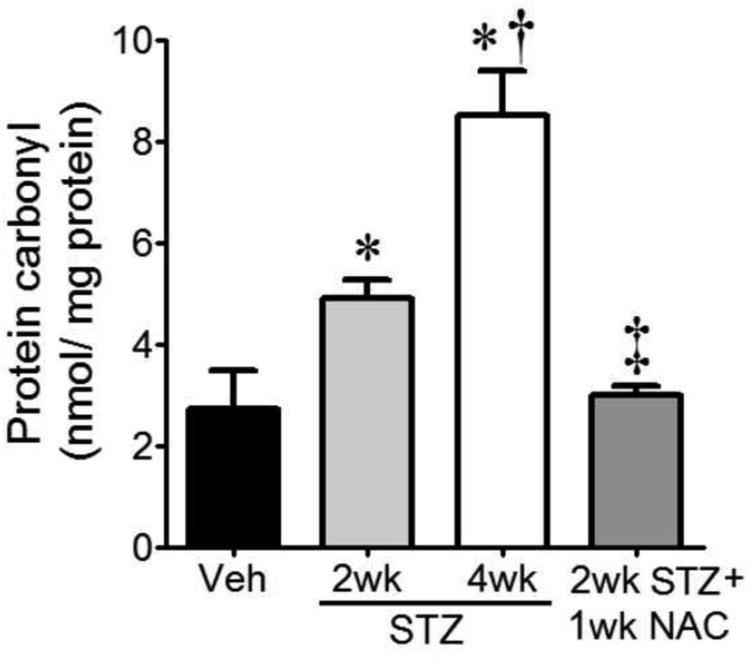
Total protein carbonyl contents, a measure of tissue oxidative/carbonyl stress, were quantified in Veh-treated controls (n=7), mice at 2wk (n=9) and 4wk (n=7) after STZ, and 2wk STZ mice treated with NAC (n=9) for 1wk. *P<0.05 vs. Veh; †P<0.05 vs. STZ 2wk; ‡P<0.05 vs. both STZ groups.
The activities of Glo I and II were determined to assess if differences in enzyme function may contribute to the elevated glycation and oxidative/carbonyl stress in the diabetic brain. Unexpectedly we found no difference in activity of either enzyme in control and diabetic brains (data not shown).
Discussion
Despite the availability of anti-glycemic drugs, diabetic individuals remain at elevated risk for cardiovascular disease and stroke, even when blood glucose levels are well-controlled [3], suggesting factors other than high glucose are also important. One possibility is MG, a dicarbonyl metabolite of glucose. In the current study, we showed that diabetic mice experienced larger infarcts following stroke (in agreement with others [27, 28]) and that infarct size inversely correlated with the GSH-dependent capacity of the brain to eliminate MG. Accordingly, the diabetic brain was associated with increased protein glycation, and oxidative/carbonyl stress. NAC provided cysteine for GSH production, and was therefore, effective at decreasing MG stress and MG-adduct formation, and preventing diabetes-induced exacerbation of stroke injury.
Diabetic patients exhibit increased stroke risk and worse stroke outcome [38, 39], likely a result of large and small vessel disease [40]. Here we showed in murine models of chemical-or genetic-induced Type 1 diabetes that diabetes (without the deleterious effects of other metabolic risk factors) increases infarct size following stroke in agreement with others [27, 28]. The positive correlation between plasma glucose levels and infarct area supports a role for hyperglycemia in stroke exacerbation. In fact, hyperglycemia at stroke onset is associated with increased mortality even in non-diabetic individuals. Curiously, diabetic patients with well-controlled blood glucose levels remain at elevated risk for cardiovascular disease, suggesting other factors are involved [3]. Our recent studies demonstrated a role for MG in IHEC apoptosis [41] and barrier dysfunction [8]. The latter was exaggerated under conditions of decreased intracellular GSH, consistent with a relationship between MG stress and GSH in barrier function. This relationship was recapitulated in vivo in the current study in that the brains of both diabetic models exhibited higher MG and lower GSH levels, and the MG-to-GSH ratio linearly correlated with infarct size. Thus, the potential for GSH-driven elimination of MG is a critical determinant of stroke outcome.
Several studies have identified GSH and NAC as antioxidants in protection against ischemic injury, including stroke [10, 42-46]. Our finding that GCLm-/- mice, in the absence of diabetes, had substantially larger infarcts than their wildtype controls underscores the importance of maintaining endogenous GSH homeostasis against I/R brain injury. We found that NAC was highly effective at protecting against the exacerbated infarct in diabetic mice even when administered after diabetes induction. This was associated with restoration of normal GSH levels and attenuation of the MG-to-GSH ratio in the diabetic brain, and is consistent with NAC promoting both GSH synthesis and the capacity for MG elimination. The finding that the steady-state cysteine pool was lower after NAC treatment is in line with enhanced cysteine utilization for GSH production. It is further consistent with cysteine supply being a limiting factor in GSH synthesis. The impact of the NAC effect was a decrease in infarct, dicarbonyl and oxidative/carbonyl stress.
Part of the stress response to oxidative stress, as would occur in diabetes, is an upregulation of the NF-E2-related factor (Nrf2)/antioxidant response element pathway and expression of antioxidant enzymes [47, 48]. Since Nrf2-mediated upregulation of GCL subunits is a primary transcriptional mechanism for increasing GCL activity, it was not surprising that GCLc protein expression was increased 2-fold by diabetes. However, GCL activity in the diabetic brain was proportionally lower than can be accounted for by this increased protein expression, suggesting that the overall function of GCL is diminished during diabetes. This is consistent with decreased brain GSH levels. The presence of a major MG-glycated protein band at 50kD that corresponded to the Mw of mouse GCLc isoform suggests that GCLc may be glycated during diabetes, perhaps leading to compromised function of GCL. We are currently investigating this hypothesis, although we are aware that GCLc function may be disrupted through other mechanisms. We also cannot exclude the alternative possibility that the decrease in GSH levels was due to elevated consumption in the diabetic animals.
A second major MG-modified protein at 65kD was confirmed to be MG-occludin by co-immunoprecipitation, in agreement with our previous findings in IHECs [8] and diabetic rat brain [9]. This was abrogated by NAC. In IHECs, occludin-glycation was implicated in endothelial barrier damage [8]. In this study, the elevated BBB permeability in diabetic mice indicates a breach in endothelial barrier integrity, which was also reversed with NAC. Recent findings of extensive MG-protein glycation, including glycation of occludin, in microvessels in diabetic rat brain [9] support our contention that dicarbonyl stress underlies brain microvascular endothelial dysfunction in vivo during diabetes, and provides further support for a role for NAC in increasing GSH for MG elimination. In previous IHEC studies, we found that ROS played a minimal role in MG-induced endothelial dysfunction, although others have found that NAC may protect against cerebral I/R injury by decreasing oxidative stress. Whether oxidative stress, directly, or indirectly via carbonyl stress, contributes to BBB breach and the exacerbated I/R brain injury in diabetic mice in vivo remain unclear. Alternatively HSP90-HIF-1a interactions [49], or decreased inflammation [10] have been implicated in the protection afforded by NAC in the brain, albeit in non-diabetic models, therefore it is plausible these pathways work in concert with the decrease in dicarbonyl and carbonyl stress we observed in NAC-treated mice to confer protection.
The literature is conflicted regarding diabetes-associated changes in Glo I and Glo II. In a Type 1 diabetes mouse model, Glo I expression was downregulated in the kidney [18]. Glo I, and to a lesser extent Glo II, activity was decreased during diabetes in the Goto-Kakisaki model [20]. The overexpression of Glo I was shown to decrease AGE-adduct formation [23] and protect against progressive diabetic rat retinal lesions. In contrast, other investigators demonstrated that activities of Glo I and II are elevated during diabetes [16, 17, 21]. More recently, Glo I was shown to be upregulated as part of the Nrf2 stress response to carbonyl stress [50] and GSH is central to the redox control of Nrf2 [51]. Our results that GCLc expression was increased in the diabetic brain is consistent with enhanced Nrf2 function. It is somewhat unexpected that we found no alterations in the glyoxalase enzymes, but our results are in agreement with several studies [15, 19, 22]. Whether the conflicting findings are a function of specific models, tissues, or time-points examined is unclear. Studies investigating whether, like GCLc, expressions of Glo I and II proteins are elevated despite their unchanged activities are ongoing.
Conclusion
In summary, our study demonstrated a key role for elevated dicarbonyl stress in the worsening of the stroke-induced infarct during diabetes. The build-up of MG is likely due to both increased generation from the higher glucose levels seen during the diabetic state, and the impaired metabolism of MG to D-lactate. The MG glycates proteins, and our finding that the junctional protein occludin was glycated, and that this was associated with decreased BBB function, indicates the glycation leads to protein dysfunction. GSH-dependent elimination of MG was improved by NAC treatment with associated attenuation of dicarbonyl stress and protection against exacerbated I/R brain injury. This suggests that the disruption of MG elimination is at least in part due to diabetes-associated decreases in GSH available for this pathway. Since Type 2 diabetics also exhibit dicarbonyl stress, increased MG levels and decreased glyoxalase-catalyzed MG elimination [52-54], our current findings could apply to this larger diabetic population, and indicate a prophylactic approach to diminish dicarbonyl stress may offer protection against the worse stroke outcome seen in diabetics.
Highlights.
Diabetes increases dicarbonyl stress and exacerbates stroke-induced brain injury.
The capacity for glutathione to eliminate methylglyoxal is decreased in diabetes.
Methylglyoxal:glutathione correlates with severity of brain injury in diabetic stroke.
Occludin is glycated and blood-brain barrier is disrupted in diabetic brain.
NAC, via GSH synthesis, reverses these changes & protects versus stroke in diabetes.
Acknowledgments
We wish to thank: Ronald E. Maloney (Dept. of Molecular & Cellular Physiology, LSUHSC-S) for his help in the study of the blood-brain barrier with Evans Blue; Chaowei Shang (Dept. of Biochemistry & Molecular Biology, LSUHSC-S) for her help with immunoprecipitation; and Drs Harris and Pattillo (Dept. of Molecular & Cellular Physiology, LSUHSC-S) for kindly providing the genetically diabetic Akita mice (InsAkita+/-), mice deficient in GCLm (GCLm-/-) and their wildtype littermates (InsAkita+/+ and GCLm+/+ respectively).
Funding: This work was supported by NIH grant DK44510 (TYA) and by a Malcolm Feist Cardiovascular Research Fellowship, LSUHSC-CCDS (BW).
Abbreviations
- BBB
blood-brain barrier
- BSO
L-buthionine-(S, R)-sulfoximine
- EB
evans blue
- GCLc
glutamate-cysteine ligase catalytic subunit
- GCLm
glutamate-cysteine ligase modulatory subunit
- Glo
glyoxalase
- GSH
glutathione
- HPLC
high-performance liquid chromatography
- I/R
ischemia/reperfusion
- MCAo
middle Cerebral Artery Occlusion
- MG
methylglyoxal
- NAC
N-acetylcysteine
- ROS
reactive oxygen species
- SDL
S-D- lactoylglutathione
- STZ
streptozotocin
- TCA
trichloroacetic acid
- TTC
2,3,5-triphenyltetrazolium chloride
- γ-GC
γ-glutamylcysteine
Footnotes
Contribution Statement: BW generated all the data for this study and was involved in data analyses, interpretation and writing the manuscript. TYA was responsible for the overall design and supervision of the study, including data analyses and manuscript preparation. KYS had intellectual input in the study design and part supervision of the study, participated in manuscript preparation and is the guarantor of this work. We acknowledge that no conflicts exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bell DS. Stroke in the diabetic patient. Diabetes Care. 1994;17:213–219. doi: 10.2337/diacare.17.3.213. [DOI] [PubMed] [Google Scholar]
- 2.Singer DE, Moulton AW, Nathan DM. Diabetic myocardial infarction. Interaction of diabetes with other preinfarction risk factors. Diabetes. 1989;38:350–357. doi: 10.2337/diab.38.3.350. [DOI] [PubMed] [Google Scholar]
- 3.Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Liu L, Mancia G, Mogensen CE, Pan C, Poulter N, Rodgers A, Williams B, Bompoint S, de Galan BE, Joshi R, Travert F. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 4.Turk Z. Glycotoxines, carbonyl stress and relevance to diabetes and its complications. Physiol Res. 2010;59:147–156. doi: 10.33549/physiolres.931585. [DOI] [PubMed] [Google Scholar]
- 5.Gomes RA, Vicente Miranda H, Silva MS, Graca G, Coelho AV, Ferreira AE, Cordeiro C, Freire AP. Yeast protein glycation in vivo by methylglyoxal. Molecular modification of glycolytic enzymes and heat shock proteins. Febs J. 2006;273:5273–5287. doi: 10.1111/j.1742-4658.2006.05520.x. [DOI] [PubMed] [Google Scholar]
- 6.Nemet I, Turk Z, Duvnjak L, Car N, Varga-Defterdarovic L. Humoral methylglyoxal level reflects glycemic fluctuation. Clin Biochem. 2005;38:379–383. doi: 10.1016/j.clinbiochem.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Su Y, Lei X, Wu L, Liu L. The role of endothelial cell adhesion molecules P-selectin, E-selectin and intercellular adhesion molecule-1 in leucocyte recruitment induced by exogenous methylglyoxal. Immunology. 2012;137:65–79. doi: 10.1111/j.1365-2567.2012.03608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W, Maloney RE, Circu ML, Alexander JS, Aw TY. Acute carbonyl stress induces occludin glycation and brain microvascular endothelial barrier dysfunction: role for glutathione-dependent metabolism of methylglyoxal. Free Radic Biol Med. 2013;54:51–61. doi: 10.1016/j.freeradbiomed.2012.10.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W, Maloney RE, Aw TY. High glucose, glucose fluctuation and carbonyl stress enhance brain microvascular endothelial barrier dysfunction: Implications for diabetic cerebral microvasculature. Redox Biol. 2015;5:80–90. doi: 10.1016/j.redox.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan M, Sekhon B, Jatana M, Giri S, Gilg AG, Sekhon C, Singh I, Singh AK. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J Neurosci Res. 2004;76:519–527. doi: 10.1002/jnr.20087. [DOI] [PubMed] [Google Scholar]
- 11.Ulusu NN, Sahilli M, Avci A, Canbolat O, Ozansoy G, Ari N, Bali M, Stefek M, Stolc S, Gajdosik A, Karasu C. Pentose phosphate pathway, glutathione-dependent enzymes and antioxidant defense during oxidative stress in diabetic rodent brain and peripheral organs: effects of stobadine and vitamin E. Neurochem Res. 2003;28:815–823. doi: 10.1023/a:1023202805255. [DOI] [PubMed] [Google Scholar]
- 12.Mizui T, Kinouchi H, Chan PH. Depletion of brain glutathione by buthionine sulfoximine enhances cerebral ischemic injury in rats. Am J Physiol. 1992;262:H313–317. doi: 10.1152/ajpheart.1992.262.2.H313. [DOI] [PubMed] [Google Scholar]
- 13.Velmurugan GV, Sundaresan NR, Gupta MP, White C. Defective Nrf2-dependent redox signalling contributes to microvascular dysfunction in type 2 diabetes. Cardiovasc Res. 2013;100:143–150. doi: 10.1093/cvr/cvt125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong Q, Mishra M, Kowluru RA. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2013;54:3941–3948. doi: 10.1167/iovs.13-11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhor VM, Raghuram N, Sivakami S. Oxidative damage and altered antioxidant enzyme activities in the small intestine of streptozotocin-induced diabetic rats. Int J Biochem Cell Biol. 2004;36:89–97. doi: 10.1016/s1357-2725(03)00142-0. [DOI] [PubMed] [Google Scholar]
- 16.Maher P, Dargusch R, Ehren JL, Okada S, Sharma K, Schubert D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One. 2011;6:e21226. doi: 10.1371/journal.pone.0021226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ratliff DM, Vander Jagt DJ, Eaton RP, Vander Jagt DL. Increased levels of methylglyoxal-metabolizing enzymes in mononuclear and polymorphonuclear cells from insulin-dependent diabetic patients with diabetic complications: aldose reductase, glyoxalase I, and glyoxalase II--a clinical research center study. J Clin Endocrinol Metab. 1996;81:488–492. doi: 10.1210/jcem.81.2.8636255. [DOI] [PubMed] [Google Scholar]
- 18.Reiniger N, Lau K, McCalla D, Eby B, Cheng B, Lu Y, Qu W, Quadri N, Ananthakrishnan R, Furmansky M, Rosario R, Song F, Rai V, Weinberg A, Friedman R, Ramasamy R, D'Agati V, Schmidt AM. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes. 2010;59:2043–2054. doi: 10.2337/db09-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakhi AK, Berg JP, Berg TJ. Glyoxalase 1 enzyme activity in erythrocytes and Ala111Glu polymorphism in type 1-diabetes patients. Scand J Clin Lab Invest. 2013;73:175–181. doi: 10.3109/00365513.2013.765028. [DOI] [PubMed] [Google Scholar]
- 20.Skapare E, Konrade I, Liepinsh E, Makrecka M, Zvejniece L, Svalbe B, Vilskersts R, Dambrova M. Glyoxalase 1 and glyoxalase 2 activities in blood and neuronal tissue samples from experimental animal models of obesity and type 2 diabetes mellitus. J Physiol Sci. 2012;62:469–478. doi: 10.1007/s12576-012-0224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skapare E, Riekstina U, Liepinsh E, Konrade I, Makrecka M, Maurina B, Dambrova M. Flow cytometric analysis of glyoxalase-1 expression in human leukocytes. Cell Biochem Funct. 2011;29:171–174. doi: 10.1002/cbf.1723. [DOI] [PubMed] [Google Scholar]
- 22.Thornalley PJ, Hooper NI, Jennings PE, Florkowski CM, Jones AF, Lunec J, Barnett AH. The human red blood cell glyoxalase system in diabetes mellitus. Diabetes Res Clin Pract. 1989;7:115–120. doi: 10.1016/0168-8227(89)90101-0. [DOI] [PubMed] [Google Scholar]
- 23.Berner AK, Brouwers O, Pringle R, Klaassen I, Colhoun L, McVicar C, Brockbank S, Curry JW, Miyata T, Brownlee M, Schlingemann RO, Schalkwijk C, Stitt AW. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia. 2012;55:845–854. doi: 10.1007/s00125-011-2393-0. [DOI] [PubMed] [Google Scholar]
- 24.Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, Sauer SK, Eberhardt M, Schnolzer M, Lasitschka F, Neuhuber WL, Kichko TI, Konrade I, Elvert R, Mier W, Pirags V, Lukic IK, Morcos M, Dehmer T, Rabbani N, Thornalley PJ, Edelstein D, Nau C, Forbes J, Humpert PM, Schwaninger M, Ziegler D, Stern DM, Cooper ME, Haberkorn U, Brownlee M, Reeh PW, Nawroth PP. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med. 2012;18:926–933. doi: 10.1038/nm.2750. [DOI] [PubMed] [Google Scholar]
- 25.Yim HS, Kang SO, Hah YC, Chock PB, Yim MB. Free radicals generated during the glycation reaction of amino acids by methylglyoxal. A model study of protein-cross-linked free radicals. J Biol Chem. 1995;270:28228–28233. doi: 10.1074/jbc.270.47.28228. [DOI] [PubMed] [Google Scholar]
- 26.Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein carbonylation in human diseases. Trends Mol Med. 2003;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 27.Jouihan SA, Zuloaga KL, Zhang W, Shangraw RE, Krasnow SM, Marks DL, Alkayed NJ. Role of soluble epoxide hydrolase in exacerbation of stroke by streptozotocin-induced type 1 diabetes mellitus. J Cereb Blood Flow Metab. 2013;33:1650–1656. doi: 10.1038/jcbfm.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalani A, Kamat PK, Tyagi N. Diabetic Stroke Severity: Epigenetic Remodeling and Neuronal, Glial, and Vascular Dysfunction. Diabetes. 2015;64:4260–4271. doi: 10.2337/db15-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McConnachie LA, Mohar I, Hudson FN, Ware CB, Ladiges WC, Fernandez C, Chatterton-Kirchmeier S, White CC, Pierce RH, Kavanagh TJ. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol Sci. 2007;99:628–636. doi: 10.1093/toxsci/kfm165. [DOI] [PubMed] [Google Scholar]
- 30.Engel O, Kolodziej S, Dirnagl U, Prinz V. Modeling stroke in mice - middle cerebral artery occlusion with the filament model. J Vis Exp. 2011 doi: 10.3791/2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terao S, Yilmaz G, Stokes KY, Russell J, Ishikawa M, Kawase T, Granger DN. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke. 2008;39:2560–2570. doi: 10.1161/STROKEAHA.107.513150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reed DJ, Babson JR, Beatty PW, Brodie AE, Ellis WW, Potter DW. High-performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides. Anal Biochem. 1980;106:55–62. doi: 10.1016/0003-2697(80)90118-9. [DOI] [PubMed] [Google Scholar]
- 33.Dhar A, Desai K, Liu J, Wu L. Methylglyoxal, protein binding and biological samples: are we getting the true measure? J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1093–1100. doi: 10.1016/j.jchromb.2009.02.055. [DOI] [PubMed] [Google Scholar]
- 34.White CC, Viernes H, Krejsa CM, Botta D, Kavanagh TJ. Fluorescence-based microtiter plate assay for glutamate-cysteine ligase activity. Anal Biochem. 2003;318:175–180. doi: 10.1016/s0003-2697(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 35.Shinohara M, Thornalley PJ, Giardino I, Beisswenger P, Thorpe SR, Onorato J, Brownlee M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. 1998;101:1142–1147. doi: 10.1172/JCI119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talesa V, Uotila L, Koivusalo M, Principato G, Giovannini E, Rosi G. Demonstration of glyoxalase II in rat liver mitochondria. Partial purification and occurrence in multiple forms. Biochim Biophys Acta. 1988;955:103–110. doi: 10.1016/0167-4838(88)90183-5. [DOI] [PubMed] [Google Scholar]
- 37.Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- 38.Kameyama M, Fushimi H, Udaka F. Diabetes mellitus and cerebral vascular disease. Diabetes Res Clin Pract. 1994;24 Suppl:S205–208. doi: 10.1016/0168-8227(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 39.Mankovsky BN, Metzger BE, Molitch ME, Biller J. Cerebrovascular disorders in patients with diabetes mellitus. J Diabetes Complications. 1996;10:228–242. doi: 10.1016/s1056-8727(96)90006-9. [DOI] [PubMed] [Google Scholar]
- 40.Arboix A, Rivas A, Garcia-Eroles L, de Marcos L, Massons J, Oliveres M. Cerebral infarction in diabetes: clinical pattern, stroke subtypes, and predictors of in-hospital mortality. BMC Neurol. 2005;5:9. doi: 10.1186/1471-2377-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okouchi M, Okayama N, Aw TY. Preservation of cellular glutathione status and mitochondrial membrane potential by N-acetylcysteine and insulin sensitizers prevent carbonyl stress-induced human brain endothelial cell apoptosis. Curr Neurovasc Res. 2009;6:267–278. doi: 10.2174/156720209789630348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuzzocrea S, Mazzon E, Costantino G, Serraino I, Dugo L, Calabro G, Cucinotta G, De Sarro A, Caputi AP. Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130:1219–1226. doi: 10.1038/sj.bjp.0703421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knuckey NW, Palm D, Primiano M, Epstein MH, Johanson CE. N-acetylcysteine enhances hippocampal neuronal survival after transient forebrain ischemia in rats. Stroke. 1995;26:305–310. doi: 10.1161/01.str.26.2.305. discussion 311. [DOI] [PubMed] [Google Scholar]
- 44.Sekhon B, Sekhon C, Khan M, Patel SJ, Singh I, Singh AK. N-Acetyl cysteine protects against injury in a rat model of focal cerebral ischemia. Brain Res. 2003;971:1–8. doi: 10.1016/s0006-8993(03)02244-3. [DOI] [PubMed] [Google Scholar]
- 45.Yabuki Y, Fukunaga K. Oral administration of glutathione improves memory deficits following transient brain ischemia by reducing brain oxidative stress. Neuroscience. 2013;250:394–407. doi: 10.1016/j.neuroscience.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Zhu Z, Liu J, Hu Z. Protective effect of N-acetylcysteine (NAC) on renal ischemia/reperfusion injury through Nrf2 signaling pathway. J Recept Signal Transduct Res. 2014;34:396–400. doi: 10.3109/10799893.2014.908916. [DOI] [PubMed] [Google Scholar]
- 47.Ha KN, Chen Y, Cai J, Sternberg P., Jr Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: implication for protection against oxidative stress. Invest Ophthalmol Vis Sci. 2006;47:2709–2715. doi: 10.1167/iovs.05-1322. [DOI] [PubMed] [Google Scholar]
- 48.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Yan J, Taheri S, Liu KJ, Shi H. Hypoxia-inducible factor 1 contributes to N-acetylcysteine's protection in stroke. Free Radic Biol Med. 2014;68:8–21. doi: 10.1016/j.freeradbiomed.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham N, Park BK, Souma T, Moriguchi T, Yamamoto M, Thornalley PJ. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem J. 2012;443:213–222. doi: 10.1042/BJ20111648. [DOI] [PubMed] [Google Scholar]
- 51.Hansen JM, Watson WH, Jones DP. Compartmentation of Nrf-2 redox control: regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol Sci. 2004;82:308–317. doi: 10.1093/toxsci/kfh231. [DOI] [PubMed] [Google Scholar]
- 52.McLellan AC, Thornalley PJ. Glyoxalase activity in human red blood cells fractioned by age. Mech Ageing Dev. 1989;48:63–71. doi: 10.1016/0047-6374(89)90026-2. [DOI] [PubMed] [Google Scholar]
- 53.Sarkar P, Kar K, Mondal MC, Chakraborty I, Kar M. Elevated level of carbonyl compounds correlates with insulin resistance in type 2 diabetes. Ann Acad Med Singapore. 2010;39:909–904. [PubMed] [Google Scholar]
- 54.Thornalley PJ. The glyoxalase system in health and disease. Mol Aspects Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]