

Bone is a metabolically active tissue regulated by multiple hormones that control formation and resorption to maintain bone mass and mineral homeostasis. But bone is also an endocrine organ that secretes osteocalcin to regulate glucose homeostasis and energy metabolism by targeting G protein–coupled receptor family C group 6 member A (GPRC6A) in beta cells and other tissues1, and fibroblast growth factor-23 (FGF-23), which is a recently discovered phosphate- and vitamin D–regulating hormone2.
FGF-23, which is predominately expressed in osteoblasts and osteocytes, targets FGF receptor–α-klotho complexes in the kidney, leading to inhibition of renal phosphate reabsorption and a decrease in circulating concentrations of 1,25(OH)2D, the active form of vitamin D, owing to decreased production by cytochrome P450, family 27, subfamily B, polypeptide 1 (Cyp27b1) and increased catabolism by Cyp24, enzymes that respectively hydroxylate 25(OH) D to active 1,25(OH)2D and 1,25(OH)2D to inactive 1, 24, 25(OH)2D (ref. 2). This constitutes the FGF-23 bone-kidney axis.
In the pathological setting, FGF-23 has a role in hereditary hypophosphatemic disorders and tumor-induced osteomalacia, where excess FGF-23 results in hypophosphatemia, aberrant vitamin D metabolism and defective bone mineralization that leads to rickets and osteomalacia2. Physiologically, however, FGF-23 participates in a regulatory 1,25(OH)2D feedback loop—1,25(OH)2D stimulates FGF-23 production in bone and FGF-23 suppresses 1,25(OH)2D production by the kidney3—which may protect the organism from vitamin D intoxication (Fig. 1). But how the FGF-23 bone-kidney axis is integrated with the classical parathyroid hormone endocrine axis, which principally functions to maintain serum calcium in a narrow range but also regulates 1,25(OH)2D production and renal phosphate reabsorption, is not fully understood.
Figure 1.
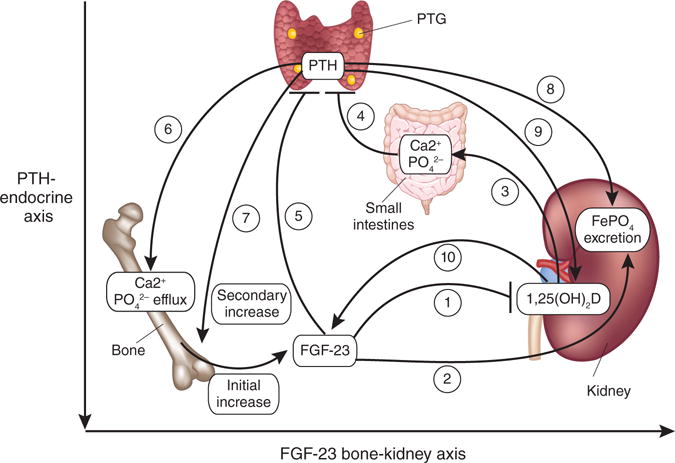
Abnormalities in the FGF-23–parathyroid hormone–vitamin D endocrine network in CKD. Initial increases in FGF-23 secretion from bone triggered by yet-to-be-identified stimuli suppress 1,25(OH)2D production by upregulating its catabolism (1) and inhibit renal phosphate reabsorption (2) to compensate for a reduced glomerular filtration rate in people with CKD. Decreased calcium absorption in the gut in response to lower amounts 1,25(OH)2D (3) stimulates secondary increments in parathyroid hormone (PTH) (4). In another less certain stratum of the axis, FGF-23 suppresses parathyroid hormone in healthy individuals (5), but in people with CKD, FGFR1–α-klotho are downregulated, leading to an increase in parathyroid hormone. This increase in parathyroid hormone leads to enhanced bone remodeling, increased efflux of calcium and phosphate from the bone (6) and further increases in FGF-23 production, thereby amplifying the bone-kidney axis (7). In another level of the axis, the kidney, parathyroid hormone increases excretion of phosphate (8) and stimulates production of 1,25(OH)2D (9), which respectively enhances and offsets the effects of FGF-23, thereby achieving a balance favoring net phosphate losses while attempting to maintain calcium homeostasis. Increased production of 1,25(OH)2D by the kidney or exogenous sources of active vitamin D analogs will increase FGF-23 levels (10). PTG, parathyroid gland.
Initial studies showed that parathyroid hormone does not directly stimulate FGF-23 production or FGF-23 promoter activity in osteoblasts in vitro3 or in calvarial cultures ex vivo4 and that parathyroid hormone administration suppresses circulating FGF-23 levels in normal mice5. FGF-23 levels are also not elevated in people with primary hyperparathyroidism, but there is evidence for indirect upregulation of FGF-23 by parathyroid hormone–mediated increase of 1,25(OH)2D levels3. A role for vitamin D signaling in parathyroid hormone regulation of FGF-23 is supported by the finding that vitamin D receptor–deficient mice have very high levels of parathyroid hormone but low FGF-23 levels.
Studies of the interplay between alterations in FGF-23 expression and disordered mineral metabolism in both animal models and humans with chronic kidney disease (CKD), however, reveal more complex interrelationships between parathyroid hormone, FGF-23 and vitamin D metabolism.
FGF-23 is markedly elevated in end-stage renal disease, and this correlates with the degree of hyperphosphatemia6, predicts refractory hyperparathyroidism7 and is associated with increased mortality. Cross-sectional studies of people with less advanced CKD have shown that FGF-23 is an early marker of renal dysfunction that correlates with increased parathyroid hormone levels and decreased circulating amounts of 1,25(OH)2D (ref. 8). Efforts to establish which comes first—FGF-23 or parathyroid hormone—has led to two competing concepts regarding the pathogenesis of disordered mineral metabolism in CKD.
On the one hand, FGF-23 may be the initial hormonal adaptive response in CKD leading to secondary hyperparathyroidism. In a rat model of anti–glomerular basement membrane nephritis, Hasegawa et al.9 showed that treatment with a neutralizing antibody to FGF-23 increased serum 1,25(OH)2D concentration, along with increased Cyp27b1 and reduced Cyp24 expression, and suppressed parathyroid hormone levels, showing that FGF-23 is the initial and primary factor, leading to secondary reductions in 1,25(OH)2D and elevations in parathyroid hormone.
These findings challenge the traditional view that CKD is a vitamin D–deficient state and raises questions about current treatments for CKD that suppress parathyroid hormone with active vitamin D analogs, which, in fact, can further elevate FGF-23 expression. These results, however, are consistent with the notion that FGF-23–mediated suppression of circulating 1,25(OH)2D levels is an adaptive response that protects against hyperphosphatemia, as decreasing 1,25(OH)2D limits gastrointestinal phosphate absorption and leads to increased parathyroid hormone secretion, which in concert with FGF-23 stimulates phosphaturia to maintain neutral phosphate balance in the setting of declining renal function (Fig. 1).
On the other hand, there is equally compelling evidence supporting that parathyroid hormone is responsible for elevated FGF-23 expression in CKD. There is also a strong association between increased amounts of FGF-23 and the severity of hyperparathyroidism in CKD, and parathyroidectomy reduces FGF-23 levels in people with CKD10. FGF-23 expression is also increased in McCune-Albright syndrome, caused by activating mutations of Gαs, a pathway also activated by parathyroid hormone11.
Moreover, Lavi-Moshayoff et al.12 recently showed that parathyroid hormone increases FGF23 expression and mediates the high FGF-23 abundance in an induced CKD model in rats. In this study, early parathyroidectomy prevented the increase in FGF-23 levels in rats with adenine-induced renal failure, and continuous parathyroid hormone administration at high doses stimulated FGF-23 production in mice and FGF-23 expression in UMR-106 osteoblasts in vitro. Parathyroid hormone–stimulated FGF-23 expression, however, may be an indirect consequence of alterations in bone remodeling, as it was inhibited by disruption of Wnt signaling with sclerostin12.
Given these and other findings, the authors purport the existence of a parathyroid gland-bone axis13: parathyroid hormone stimulates FGF-23 production in the bone, and FGF-23 feeds back on the parathyroid gland to inhibit parathyroid hormone secretion in healthy individuals. FGF-23–mediated suppression of parathyroid hormone is hampered in CKD, owing to the decrease in FGFR1 and α-klotho in the parathyroid glands, leading to an increase in parathyroid hormone in spite of concurrent increments in FGF-23 (ref. 13).
Resolution of these conflicting concepts will require further studies. However, another function of FGF-23, to coordinate renal phosphate handling to balance bone mineralization and remodeling2, may help reconcile the divergent data regarding FGF-23 and parathyroid hormone regulation and function in CKD. Consistent with a modulating effect of bone remodeling on FGF-23 release, intermittent and continuous parathyroid hormone administration in mouse models respectively suppresses and stimulates FGF-23 and correlates with the differential anabolic and catabolic responses to parathyroid hormone5.
Similarly, parathyroid hormone induction of high bone turnover (osteitis fibrosa) in CKD might lead to increased production of FGF-23, analogous to the catabolic effects of continuous parathyroid hormone administration. If so, elevations of circulating FGF-23, which enhances renal phosphate excretion and reduces 1,25(OH)2D abundance by stimulating Cyp24-mediated catabolism, might be the initial response to the loss of glomerular filtration, which orchestrates a coordinated and progressive adaptation to protect the organism from the adverse effects of excess phosphate retention. As renal failure advances, secondary effects of elevated parathyroid hormone caused by FGF-23–mediated suppression of 1,25(OH)2D protect the organism from hypocalcemia and further stimulate FGF-23 production to work in concert with parathyroid hormone to further increase phosphate excretion, which aims to compensate for progressive reductions in renal function and increased phosphate efflux from bone caused by excessive bone turnover (Fig. 1).
There are several clinical implications for these new mechanistic insights into the FGF-23 bone-kidney axis. FGF-23 may be a useful early marker for abnormal mineral metabolism in CKD. Understanding that treatment with active vitamin D analogs further increases FGF-23 levels, which is independently associated with increased mortality14, may support phosphate restriction in individuals with early-stage CKD, therapeutic efforts to normalize bone remodeling and mineralization and the use of treatment regimens that limit the dose of active vitamin D analogs in late-stage CKD15.
Moreover, if the reductions in 1,25(OH)2D are due to an increase in the catabolic enzyme Cyp24, approaches to selectively inhibit Cyp24 may correct the FGF-23–mediated abnormal vitamin D metabolism without affecting the beneficial phosphaturic actions of FGF-23. This increased catabolism of 25(OH)D into 1,25(OH)2D might explain the unexpectedly high prevalence of low 25(OH)D serum concentrations in people with CKD and the difficulty in normalizing 25(OH)D levels with vitamin D replacement therapy in these patients. Increased amounts of FGF-23 are also observed in unrecognized CKD in elderly individuals, and these increased amounts are associated with adverse outcome and may contribute to the low levels of 25(OH)D.
Finally, as phosphate metabolism is important in energy metabolism, crosstalk between skeletal regulation of energy, phosphate and vitamin D metabolism may occur. For example, leptin stimulates FGF-23 expression, blocks bone remodeling16 and may inhibit the osteocalcin-insulin endocrine loop regulating energy metabolism1.
Footnotes
COMPETING FINANCIAL INTERESTS
The author declares competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturemedicine/.
References
- 1.Katsnelson A. Nature. 2010;466:914–915. doi: 10.1038/466914a. [DOI] [PubMed] [Google Scholar]
- 2.Quarles LD. J Clin Invest. 2008;118:3820–3828. doi: 10.1172/JCI36479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu S, et al. J Am Soc Nephrol. 2006;17:1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- 4.Saji F, et al. Am J Physiol Renal Physiol. 2010;299:F1212–F1217. doi: 10.1152/ajprenal.00169.2010. [DOI] [PubMed] [Google Scholar]
- 5.Samadfam R, et al. Endocrinology. 2009;150:4835–4845. doi: 10.1210/en.2009-0472. [DOI] [PubMed] [Google Scholar]
- 6.Weber TJ, et al. J Bone Miner Res. 2003;18:1227–1234. doi: 10.1359/jbmr.2003.18.7.1227. [DOI] [PubMed] [Google Scholar]
- 7.Komaba H, Fukagawa M. Kidney Int. 2010;77:292–298. doi: 10.1038/ki.2009.466. [DOI] [PubMed] [Google Scholar]
- 8.Gutiérrez O, et al. J Am Soc Nephrol. 2005;16:2205–2215. doi: 10.1681/ASN.2005010052. [DOI] [PubMed] [Google Scholar]
- 9.Hasegawa H, et al. Kidney Int. 2010;78:975–980. doi: 10.1038/ki.2010.313. [DOI] [PubMed] [Google Scholar]
- 10.Sato T, et al. Am J Kidney Dis. 2004;44:481–487. [PubMed] [Google Scholar]
- 11.Riminucci M, et al. J Clin Invest. 2003;112:683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavi-Moshayoff V, et al. Am J Physiol Renal Physiol. 2010;299:F882–F889. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Dov IZ, et al. J Clin Invest. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutierrez OM, et al. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wetmore JB, et al. Clin J Am Soc Nephrol. 2010;5:110–116. doi: 10.2215/CJN.03630509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuji K, et al. J Bone Miner Res. 2010;25:1711–1723. doi: 10.1002/jbmr.65. [DOI] [PubMed] [Google Scholar]