

Abstract
In an attempt to identify novel nucleoside phosphoramidate analogues for improving the anti-HCV activity of 2’-C-Me-uridine, we have synthesized for the first time a series of L-glutamic acid, L-serine, L-threonine and L-tyrosine containing aryloxyphosphoramidate prodrugs of 2’-C-Me-uridine. Evaluation of their activity against HCV revealed that they displayed very potent anti-HCV activity, with EC50 values that are in the same range as of Sofosbuvir.
Graphical Abstract

A series of L-glutamic acid, L-serine, L-threonine and L-tyrosine containing aryloxyphosphoramidate prodrugs of 2’-C-Me-uridine displayed very potent activity against HCV.
Introduction
Nucleosides and nucleotides have demonstrated wide-spread utility as antiviral agents.1 Antiviral nucleosides are essentially prodrugs since their antiviral activity depends upon their intracellular metabolism within virus-infected cells to form sequentially the mono-, di- and triphosphates. It is these nucleotides, and especially the triphosphates that are the pharmacologically active species, as they are incorporated into a growing DNA or RNA strand by a DNA or RNA polymerase, resulting in chain termination and fraudulent DNA/RNA. The first phosphorylation step leading to the formation of the nucleoside 5’-monophosphate is commonly catalyzed by a nucleoside kinase encoded by the host cell or the virus infecting the host cell. Conversion of the nucleoside monophosphate to the corresponding 5’-diphosphate and triphosphates is carried out by nucleotidyl, and nucleoside diphosphate kinases, respectively. Hence, cellular kinases, as well as virally-encoded kinases play a vital role in the activation of nucleoside drugs.
In most cases, the first step of phosphorylation represents the rate-limiting step of the bioactivation and its inefficiency may limit the therapeutic potential of the nucleoside analogues. Bypassing this rate-limiting activation step may improve the biological activity of the nucleosides. In principle, administration of nucleoside-5’-monophosphates would overcome the drawbacks. However, phosphates are strongly acidic, and thus negatively charged at physiological pH and hence, are not able to penetrate the lipid-rich cell membrane. In addition, phosphohydrolases (acid and alkaline phosphatases, 5’-nucleotidases) rapidly convert the phosphates to the corresponding nucleosides. In order to overcome these limitations, various prodrug or ‘pronucleotide’ approaches have been devised and investigated.2 In general, the goal of these approaches has been to promote stability in the extracellular medium, passive diffusion through the lipophilic cell membranes and to liberate the parent nucleotide, where it can be further phosphorylated to the pharmacologically active species. As a result, several prodrug approaches now exist. The synthetic derivatization has been made by using various protecting groups to shield the phosphate charges. The development of the protecting groups has moved from using simple alkyl groups to more sophisticated structures that may efficiently deliver phosphorylated species into cells. One of the most promising approaches is the “aryloxyphosphoramidate” approach (also known as ProTide approach), pioneered by Jones et al. in the early 1980s, and later developed by McGuigan et al. in the 1990s.3 The cleavage of this class of prodrugs is initiated by an esterase enzyme, then an intramolecular cyclization is believed to take place with displacement of the aryl moiety to form a short-lived five-membered ring intermediate, which is hydrolyzed to phosphoramidic acid. The cleavage of the monoamidate to the active species may be catalyzed by a second enzyme, for example, phosphoramidase or may result from simple hydrolysis in a more acidic subcellular compartment, releasing intracellularly nucleoside-monophosphate. Research on antivirally active ProTides in the past 20 years led to the discovery of a number of clinically efficacious ProTides (Figure 1). Sofosbuvir was the first ProTide that received marketing approval and it has been licensed for the treatment of HCV infected patients.4 Tenofovir alafenamide (TAF) is an example of a phosphono-amidate derivative.5 The first combination pill containing TAF for the treatment of HIV was FDA approved for the treatment of HIV under the trade name Genvoya®. In addition, two other TAF-based HIV treatments are currently under FDA review and in January 2016, Gilead requested FDA approval of stand-alone TAF for hepatitis B. GS-5734 is an example of a phosphoramidate C-nucleoside derivative that exhibits antiviral activity against multiple variants of the Ebola virus in cell-based assays.6 In a rhesus monkey model of Ebola virus infection, treatment with GS-5734 protected the Ebola virus infected animals from the lethal disease. A Phase 1 clinical trial in healthy human volunteers is ongoing to determine its safety, tolerability and pharmacokinetics.
Figure 1.

Known ProTides
In nearly all reports on ProTides, L-alanine is the most commonly used amino acid.7 While keeping the L-alanine motif intact, several structural modifications of the ester functional group have been studied. Small ester moieties, such as methyl, ethyl and isopropyl esters, as well as bulkier groups such as cyclohexyl and benzyl esters have been synthesized and studied for biological activities.8,9 On the other hand, systematic structure-activity relationship (SAR) studies involving the amino acid side chain have not been performed and in most cases only glycine, leucine, valine, leucine, and proline were systematically studied.8,9 From these studies, it was concluded that L-alanine is optimal in terms of antiviral activity of the corresponding nucleoside phosphoramidate.7 The stereochemistry of the amino acid plays also an important role in the antiviral activity of the ProTide. L-alanine is preferred over D-alanine when evaluating the anti-HIV activity of phosphoramidate triester prodrugs of stavudine and zidovudine.10,11 Similarly, the D-alanine analogue of Sofosbuvir completely lacks anti-HCV activity.4 We recently reported the discovery of a novel series of L-aspartic acid based Protides of 2’-C-Me-uridine and 2’-C-Me-cytidine with potent anti-HCV activity. In some cases, compounds exhibited improved activity relative to the corresponding L-alanine derived ProTides, depending on the ester moiety of the two carboxylic acid moieties of L-aspartic acid.12 Given these promising data, we have explored other bifunctional amino acids (L-glutamic acid, L-serine, L-threonine and L-tyrosine) as potential alternatives for L-alanine, using 2’-C-Me-uridine as prototype nucleoside. As in previous reports on ProTides, the L-stereochemistry was found to be superior when compared to the D-enantiomers, we focused on the synthesis of ProTides with amino acids having the natural L-stereochemistry. In this manuscript, the first synthesis of L-glutamic acid, L-serine, L-threonine and L-tyrosine containing aryloxyphosphoramidate prodrugs of 2’-C-Me-uridine is presented, as well as the assessment of their anti-HCV activity.
Chemistry
Synthesis of di-esters of L-glutamic acid
Although we previously used the thionyl chloride/isoamylalcohol system for the preparation of the amino acid esters from the corresponding amino acids,12 we switched to the trimethylchlorosilane reagent for esterification of the carboxylic acid moiety of L-glutamic acid 1.13 The isoamylalcohol /TMSCl is a facile system as compared to the classical methods (using an acid and an appropriate alcohol) used for esterification, because it is more convenient to use and measure but also because it acts as a water scavenger so that the esterification is faster and cleaner.
Synthesis of mono- and di-esters of L-serine
TMSCl is added to a mixture of amylalcohol or isoamylalcohol and O-benzyl-L-serine 3, affording compounds 4 and 5, respectively. Similarly, esterification of the acid group of L-serine 6 affords compounds 7 and 8 (as their hydrochloride salts). Protection of the amino group of compounds 7 and 8 as a Boc furnished the esterified N-Boc-L-serine derivatives 10 and 11. Boc-L-serine methyl ester 9 is commercially available. Conversion of the primary hydroxyl group to an isobutyryl ester by reaction with isobutyryl chloride, is followed by acidic cleavage of the Boc group affording the desired di-esters of L-serine 15–17.
Synthesis of mono- and di-esters of L-threonine
The carboxylic acid moiety of O-benzyl-L-threonine 18 was esterified using isoamyl alcohol and N,N,N′,N′-tetramethyl-O-(6-chloro-1H-benzotriazol-1-yl)uraniumhexafluorophosphate (HCTU) as coupling reagent (Scheme 3). The carboxylic acid moiety of N-Boc-L-threonine 20 was esterified in the same way. The secondary alcohol group was esterified with isobutyryl chloride. Finally, acidic deprotection of the Boc group yielded the desired di-ester derivative of L-threonine 23 as a hydrochloride salt.
Scheme 3.

Synthesis of mono- and di-esters of L-threonine
Synthesis of di-esters of L-tyrosine
Esterification of the carboxylic acid of N-Boc-L-tyrosine 24, is followed by conversion of the phenolic hydroxyl group to an isobutyryl ester moiety yielding intermediate 26. Finally, deprotection affords the desired diester analogue of L-tyrosine 27.
Synthesis of ProTides of 2’-C-uridine
The synthesis of the final nucleoside phenoxy phosphoramidate analogues is shown in Scheme 5.
Scheme 5.

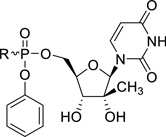
Commercially available phenyl phosphorodichloridate 28 was reacted with the amino acid esters 2, 4–5, 15–17, 19, 23 and 27 in dichloromethane yielding the arylaminoacyl phosphorodichloridates 29. Coupling with 2′-C-methyl-2′,3′-O-isopropylidene-uridine, which was prepared according to a literature protocol,12 furnished the 2’-C-Me-2′,3′-O-isopropylidene-uridine ProTide analogues 30a-k. Finally, acidic deprotection afforded the desired target compounds 31a-k. Each ProTide was generated as a pair of diastereomers, due to the chirality of the phosphorus centre, and compounds were evaluated as isomeric mixtures.
Biological evaluation
The final ProTides 31a-k, as well as the parent 2’-C-Me-uridine, were evaluated for anti-HCV activity as follows. The primary assay uses the genotype 1b (Con1 strain) HCV replicon to evaluate anti-HCV activity. The assay makes use of the cell line ET (luc-ubi-neo/ET), which is a Huh7 human hepatoma cell line harboring a subgenomic HCV replicon that incorporates a stable luciferase (Luc) reporter gene.15 This Luc reporter is used as an indirect measure of HCV replication as its activity is directly proportional to HCV RNA levels. Recombinant human interferon alpha-2b (rIFNα-2b) and Sofosbuvir are included as positive control. EC50, EC90 (compound concentrations reducing HCV replication by 50% and 90% respectively) and CC50 (concentration decreasing cell viability by 50%) values were determined. The results are summarized in Table 1.
Table 1.
Luciferase-based HCV 1b replicon activity of ProTides 31a-k
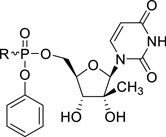 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R | Amino acid | EC50 (µM)a |
EC90 (µM)a |
CC50 (µM)a |
SI50 | clogP |
| 2’-C-Me-uridine | - | - | 6.31 | 55.3 | >100 | >15.8 | −1.69 |
| 31a | 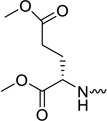 |
L-glutamic acid | 1.52 | 5.6 | >20 | >13 | −1.28 |
| 31b | 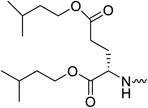 |
L-glutamic acid | 0.08 | 0.21 | >20 | >250 | 2.69 |
| 31c | 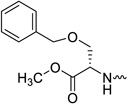 |
L-serine | 0.18 | 0.6 | >20 | >111 | 0.65 |
| 31d | 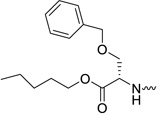 |
L-serine | 0.03 | 0.14 | 5.89 | 196 | 2.77 |
| 31e | 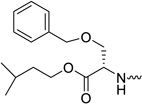 |
L-serine | < 0.06 | 0.13 | 8 | >127 | 2.64 |
| 31f | 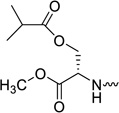 |
L-serine | 0.51 | 1.69 | >20 | >39 | −0.12 |
| 31g | 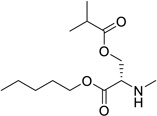 |
L-serine | 0.10 | 0.86 | 6.79 | 68 | 1.99 |
| 31h | 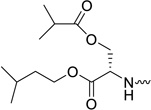 |
L-serine | 0.08 | 0.56 | 7.51 | 94 | 1.87 |
| 31i | 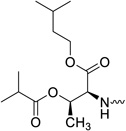 |
L-threonine | 0.29 | 1.39 | >20 | >69 | 2.18 |
| 31j | 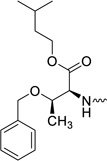 |
L-threonine | 0.47 | 1.83 | 11.46 | 24 | 2.94 |
| 31k | 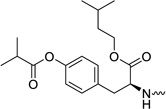 |
L-tyrosine | 0.40 | 1.47 | >20 | >50 | 2.82 |
| IFNα-2b | - | - | 0.15 | 0.48 | >10 | >67 | |
| Sofosbuvir | See Figure 1 | L-alanine | 0.07 | 0.30 | >5.00 | >71 | |
Results are from a single assay generated from quadruplicate assay wells at each concentration
The parent nucleoside 2’-C-Me-uridine shows only marginal anti-HCV activity (EC50 = 6.31 µM) and all ProTides are endowed with a more pronounced antiviral activity. When the dimethyl ester of L-glutamic acid was used as amino acid motif in the ProTide (compound 31a), only weak anti-HCV activity was observed (EC50 = 1.52 µM). Increasing the chain length of the ester moieties from a methyl ester (compound 31a) to an isoamyl group (compound 31b) led to a 20-fold improvement in anti-HCV activity. This might be related to the higher lipophilicity of the compound 31a (clogP = −1.28), when compared to 31b (clogP = 2.69), and hence a better cellular permeability. The choice of the isoamyl ester moiety is dictated by the fact that these ester groups were found to be optimal to confer antiviral activity to L-aspartic acid based aryloxyphosphoramidate nucleoside prodrugs.12
When the methyl ester of O-benzyl-L-serine is used in the aryloxyphosphoramidate prodrugs (compound 31c), less than optimal anti-HCV activity was obtained (EC50 = 0.18 µM). When more lipophilic esters of O-benzyl-L-serine were used (the amylester 31d and the isoamyl ester 31e), potent antiviral activity was observed, displaying EC50 values of 0.03 µM and <0.06 µM, respectively. In analogy with the diesters of L-glutamic acid, a number of diesters of L-serine were synthesized. The most polar derivative (compound 31f, clogP = -0.12) was, as expected, the least active in the HCV replicon assay with a EC50 value of 0.51 µM. Substitution of the methyl ester group by an amyl or isoamyl group afforded compounds 31g and 31h, respectively, both endowed with a minor improvement in antiviral activity. Finally, L-threonine (compounds 31i and 31j) and L-tyrosine (compound 31k) containing ProTides, as other examples of bifunctional amino acids, were prepared. Despite their favorable lipophilicity, these compounds exhibited lower anti-HCV activity in the range of 0.3 – 0.5 µM. These findings suggest that lipophilicity can be used as a predictive tool for HCV activity within a particular amino acid series, but is not useful to compare the biological activities between different series. In general, the prodrugs 31a-k were not cytotoxic towards the Huh7 hepatoma cell line. This is similar as to what is observed for Sofosbuvir. Although in our hands, the highest tested concentration was 5 µM, it is known from literature that Sofosbuvir is completely devoid of cytotoxicity, up to 100 µM.4 For the most potent congeners (compounds 31b, 31e and 31h), a selectivity index of at least 127 was calculated.
To confirm the compounds anti-HCV activity, representative examples were tested in a secondary assay. Briefly, a qRT-PCR (TaqMan) endpoint is used to directly measure HCV RNA copies, rather than the luciferase read-out which is an indirect read-out. Similarly, cellular ribosomal RNA is measured as a cytotoxicity control. The EC50, EC90 and CC50 values for each compound is shown in Table 2. In addition, the potency of the compounds were compared with Sofosbuvir, the only FDA approved nucleoside analogue for the treatment of HCV infected patients. The L-glutamic acid (compound 31b), the L-serine (compounds 31e and 31h), the L-threonine (compound 31i) and L-tyrosine (compound 31k) containing prodrugs are more potent than Sofosbuvir, as evidenced by their very potent EC50 and EC90 values. The strong potency of these ProTides, in combination with low cytotoxicity, gives rise to high selectivity indexes.
Table 2.
qRT-PCR based HCV 1b replicon activity of ProTides 31b and 31e
 | |||||
|---|---|---|---|---|---|
| Cmpd# | R | EC50 (µM)a | EC90 (µM)a | CC50 (µM)a | SI50 |
| 31b | 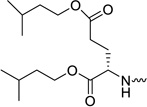 |
0.01 | 0.13 | 9.98 | 998 |
| 31e | 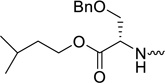 |
0.01 | 0.08 | 4.59 | 459 |
| 31h | 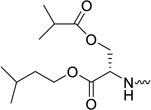 |
0.02 | 0.13 | 5.28 | 264 |
| 31i | 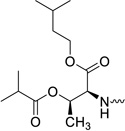 |
0.04 | 0.69 | 7.27 | 182 |
| 31k | 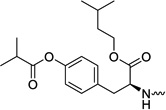 |
0.04 | 0.69 | 18.37 | 459 |
| Sofosbuvir | Structure - Figure 1 | 0.07 | 0.28 | >5.00 | >71 |
Results are from a single assay generated from quadruplicate assay wells at each concentration
To exert their antiviral activity, the ProTides must be metabolized in hepatocytes to release 2’-C-Me-uridine-monophosphate intracellularly, which will then be further phosphorylated yielding the corresponding triphosphate congener, that acts as an inhibitor of the HCV RNA-dependent RNA polymerase NS5B. Human liver microsomes were chosen as a surrogate in vitro model to test for hepatocyte stability. Prodrugs 31b, 31d, 31h, 31i and 31k were incubated at a concentration of 0.1 µM and the parent compound was quantified at different time points (0, 15, 30, 45 and 60 minutes). From these data, the in vitro half-life and the intrinsic clearance values were calculated (Table 3). Extensive metabolism in human liver microsomes leads to very short half lifes and high intrinsic clearance values. Although no metabolite identification has been performed as part of this study, the intracellular activation route for ProTides has extensively been studied in literature,14 and it has been demonstrated that intracellular metabolism of these prodrugs leads to formation of the nucleoside-monophosphate. Taken together, these data indicate that the bifunctional aryloxyphosphoramidate prodrugs of 2’-C-Me-uridine are metabolized to their biologically active species.
Table 3.
Metabolic stability of ProTides 31b, 31d, 31h, 31i and 31k
| Compound | T1/2 (min)a | Clint (µl/min/mg protein)a |
|---|---|---|
| 31b | 8 | 844.60 |
| 31d | 14 | 496.80 |
| 31h | 9 | 763.70 |
| 31i | 10 | 699.00 |
| 31k | 10 | 679.60 |
Results are the mean of two independent experiments
Conclusion
The aryloxyphosphoramidate prodrug technology is a well-known strategy to improve the antiviral activity of nucleoside analogues by bypassing the first and rate-limiting phosphorylation step. Previous research has demonstrated that lipophilic diesters of L-aspartic acid instead of the commonly used L-alanine esters in ProTides, afforded nucleoside analogues with an improved antiviral activity. In this paper, the synthesis of a novel series of ProTides of 2’-C-Me-uridine is described, using L-glutamic acid, L-serine, L-threonine and L-tyrosine as amino acid motif. The most potent congeners display EC50 values in the low nanomolar range against HCV. These data suggest that bifunctional amino acids can be considered as an alternative to L-alanine in the design of antiviral aryloxyphosphoramidate nucleoside prodrugs. The advantage of using a bifunctional amino acid is that there are two sites for structural variation.14 While the α-carboxyl acid group plays an important role in the displacement of the phenoxy group (via neighbouring group assistance), the remaining functional group (the carboxylic acid of L-glutamic acid or the hydroxyl group of L-serine, L-threonine and L-tyrosine) can be used as a chemical handle to tune physicochemical properties, such as lipophilicity and aqueous solubility, by the attachment of suitable functional groups.
Experimental section
General
NMR spectra were recorded on a Bruker Avance II 300 MHz with a 5 mm broad band probe, a 500 MHz spectrometer equipped with a TXI-HCP Z gradient probe or on a Bruker Avance II 600 MHz spectrometer with a 5 mm TCI-HCN Z gradient cryo-probe. The spectra were processed with Bruker Topspin 2.1 software. Chemical shifts (δ) were expressed in parts per million (ppm). The 1H and 13C NMR chemical shifts were referenced relative to the TMS peak (δ = 0.00 ppm). 31P NMR chemical shifts were referenced to an external 85% H3PO4 standard (δ = 0.00 ppm). Mass spectra were acquired on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA). Samples were infused at 3 µL/min and spectra were obtained in positive (or in negative) ionization mode with a resolution of 15 000 (FWHM) using leucine enkephalin as the lock mass. Chemicals of analytical and synthetic grade were obtained from commercial sources and were used as such. Flash silica column chromatography was performed on silica gel 60 A, 0.035–0.070 mm (Acros Organics).
Diisoamyl ester of glutamic acid (2)
To a suspension of L-glutamic acid 1 (2.0 g, 13.6 mmol) in anhydrous isoamyl alcohol (60 mL) was added trimethylchlorosilane (10.4 mL, 81.6 mmol) dropwise at 0 °C under argon atmosphere. The mixture was allowed to come to room temperature and stirred for 48 h at 35 °C. After evaporation to dryness, hexane was added and the white precipitate was filtered off. Finally, the precipitate was washed several times with hexane to obtain 2 as hydrochloride salt (61%). 1H NMR (300 MHz, DMSO-d6): δ = 8.71 (br s, 3H, -NH3+), 4.19, 4.06 (5H), 2.52 (2H), 2.08 (2H), 1.67, 1.50 (6H), 0.91 (12H). 13C NMR (75 MHz, DMSO-d6): δ = 172.5, 170.0, 65.0, 63.5, 52.0, 37.7, 37.5, 30.0, 26.1, 25.4, 25.2, 23.2, 23.1 ppm. HRMS (ESI+) calcd. for C15H30NO4 [M+H]+ 288.2169, found 288.2166.
Amyl ester of Ser-(OBn)-OH (4)
To a suspension of Ser-(OBn)-OH 3 (1.0 g, 5 mmol) in anhydrous amyl alcohol (30 mL) was added trimethylchlorosilane (4 mL, 30.7 mmol) dropwise at 0 °C under argon atmosphere. The mixture was allowed to come to room temperature and stirred at 35 °C for 72 hours. After evaporation to dryness, hexane was added and the white precipitate was filtered. Finally, the precipitate was washed several times with hexane to obtain 5 as hydrochloride salt (90%). 1H NMR (300 MHz, DMSO-d6): δ = 8.74 (br s, 3H, -NH3+), 7.35-7.34 (m, 5H), 4.54 (dd, 2H), 4.33 (t, 1H), 4.23-4.08 (m, 2H), 3.87 (d, 2H), 1.59-1.52 (m, 2H), 1.30-1.25 (m, 4H), 0.87-0.82 (t, 3H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 168.7, 138.3, 129.1, 128.6, 128.5, 73.3, 68.3, 66.6, 53.3, 28.6, 28.2, 22.6, 14.7 ppm. HRMS (ESI+) calcd for C15H24NO3 [M+H]+ 266.1751, found 266.1750.
Isoamyl ester of Ser-(OBn)-OH (5)
To a suspension of Ser-(OBn)-OH 3 (1.0 g, 5 mmol) in anhydrous isoamyl alcohol (30 mL) was added trimethylchlorosilane (4 mL, 30.7 mmol) dropwise at 0 °C under argon atmosphere. The mixture was allowed to come to room temperature and stirred for 72 h at 35 °C. After evaporation to dryness, hexane was added and the white precipitate was filtered off. The precipitate was washed several times with hexane to obtain 5 as hydrochloride salt (80%). 1H NMR (300 MHz, DMSO-d6): δ = 8.69 (br s, 3H, -NH3+), 7.40-7.29 (m, 5H), 4.54 (dd, 2H), 4.34 (t, 1H), 4.25-4.13 (m, 2H), 3.86 (d, 2H), 1.69-1.58 (m, 1H), 1.52-1.43 (m, 2H), 0.88-0.84 (m, 6H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 168.7, 138.2, 129.1, 128.6, 128.5, 73.4, 68.3, 65.1, 53.3, 37.5, 25.1, 23.1, 23.0 ppm. HRMS (ESI+) calcd for C15H24NO3 [M+H]+ 266.1751, found 266.1748.
Ser-(OH)-O-amyl ester hydrochloride (7)
To a suspension of L-serine 6 (2.0 g, 19 mmol) in anhydrous amyl alcohol (50 mL) was added dropwise trimethylchlorosilane (14.5 mL, 114.2 mmol) at 0 °C under argon atmosphere. The mixture was allowed to come to room temperature and stirred for 24 h at 40 °C. After evaporation to dryness, hexane was added and the white precipitate was filtered off. Finally, the precipitate was washed several times with hexane and ether to obtain 7 as a hydrochloride salt (90%). 1H NMR (300 MHz, DMSO-d6): δ = 9.49 (br s, 3H, -NH3+), 6.52 (s, 1H), 5.06-5.02 (m, 2H), 4.96 (1H), 4.72 (2H), 4.26 (br s, 2H), 2.21 (4H), 1.79, 1.77 (3H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 168.0, 65.5, 59.5, 54.4, 27.6, 27.3, 21.7, 13.8 ppm. HRMS (ESI+) calcd for C8H18NO3 [M+H]+ 176.1281, found 176.1285. HRMS (ESI+) calcd for C8H18NO3 [M+H]+ 176.1281, found 176.1281.
Ser-(OH)-O-isoamyl ester hydrochloride (8)
To a suspension of L-serine 6 (2.0 g, 19 mmol) in anhydrous isoamyl alcohol (50 mL) was added trimethylchlorosilane (14.5 mL, 114.2 mmol) dropwise at 0 °C under argon atmosphere. The mixture was allowed to come to room temperature and stirred for 30 h at 35 °C. After evaporation to dryness, hexane was added and the white precipitate was filtered off. Finally, the precipitate was washed several times with hexane and ether to obtain 8 as hydrochloride salt (90%). 1H NMR (300 MHz, DMSO-d6): δ = 8.60 (br s, 3H, -NH3+), 5.62 (s, 1H), 4.17-4.04 (m, 3H), 3.82 (s, 2H), 4.17-4.64 (m, 1H), 1.53-1.48 (m, 2H), 0.89, 0.87 (6H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 168.0, 64.0, 59.5, 54.4, 36.7, 24.3, 22.3, 22.2 ppm. HRMS (ESI+) calcd for C8H18NO3 [M+H]+ 176.1281, found 176.1285.
Boc-Ser-(OH)-O-isoamyl ester (11)
Compound 8 (3.0 g, 14.8 mmol) was dissolved in anhydrous 1,4-dioxane (60 mL). Di-tert-butyl-bicarbonate (4 g, 17.8 mmol) followed by triethyl amine (3 mL, 22.3 mmol) were added and the reaction mixture was stirred at room temperature for 1.5 h. The reaction mixture was then evaporated to dryness and diluted with ethyl acetate, washed with 0.5 N aq. HCl, followed by aq. sat. sodium bicarbonate and brine. The organic layer was dried over anhydrous MgSO4 and evaporated to dryness. The crude product was purified by flash column chromatography by eluting with 20–30% EtOAc in hexane to obtain compound 11 in 95% yield. Rf = 0.34 (Hexane/EtOAc, 8:2). 1H NMR (500 MHz, CDCl3 ): δ = 5.48 (br s, 1H), 4.36 (br s, 1H), 4.22-4.20 (m, 2H), 3.96-3.91 (m, 2H), 2.54 (m, 1H), 1.74-1.66 (m, 1H), 1.58-1.53 (m, 2H), 1.46 (s, 9H), 0.93, 0.92 (2 s, 6H) ppm. 13C NMR (125 MHz, CDCl3): δ = 171.0, 155.9, 80.4, 64.6, 63.8, 56.0, 37.3, 28.4, 25.1, 22.5, 22.5 ppm. HRMS (ESI+) calcd for C8H18NO3 [M+H]+ 176.1281, found 176.1285. HRMS (ESI+) calcd for C13H25NO5Na [M+Na]+ 298.1625, found 298.1620.
Boc-Ser-(OH)-O-amyl ester (10)
A similar synthetic procedure as for the synthesis of compound 11 is used. Rf = 0.34 (Hexane/EtOAc, 8:2). 1H NMR (500 MHz, CDCl3 ): δ = 5.47 (br s, 1H), 4.37 (br s, 1H), 4.19-4.16 (m, 2H), 3.96-3.92 (m, 2H), 2.52-2.49 (m, 1H), 1.68-1.65 (m, 2H), 1.46 (s, 9H), 1.35-1.32 (m, 4H), 0.92-0.89 (t, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 171.0, 156.0, 80.4, 66.1, 63.9, 56.0, 28.4, 28.3, 28.1, 22.4, 14.0 ppm. HRMS (ESI+) calcd for C13H25NO5Na [M+Na]+ 298.1625, found 298.1627.
Boc-Ser(iBu)-OMe (12)
To a solution of compound 9 (1 g, 4.56 mmol) in dry DCM (50 ml) were added DMAP (56 mg, 0.5 mmol) and TEA (2 mL, 13.7 mmol). Isopropionyl chloride (1.2 mL, 11.4 mmol) was added dropwise. The suspension was then stirred overnight at room temperature. Upon completion, the reaction mixture was diluted with DCM and washed with 0.5N aq. HCl, sat. sodium bicarbonate and with brine. The organic layer was dried over anhydrous MgSO4 and evaporated to dryness. The crude product was then purified in flash silica gel column chromatography eluting with 10–20% ether in hexane to obtain 12 in 95% yield. Rf = 0.38 (Hexane/Et2O, 1:3). 1H NMR (300 MHz, CDCl3): δ = 5.30 (d, 1H), 4.60-4.57 (m, 1H), 4.49-4.44 (m, 1H), 4.33-4.28 (m, 1H), 3.76 (s, 3H), 2.60-2.51 (m, 1H), 1.46 (s, 9H), 1.16 (d, 3H), 1.14 (d, 3H) ppm. 13C NMR (75 MHz, CDCl3): δ = 176.7, 170.5, 155.3, 80.4, 64.1 52.8, 33.9, 28.4, 19.1, 18.9 ppm. HRMS (ESI+) calcd. for C13H23NO6Na [M+Na]+ 312.1418, found 312.1413.
Boc-Ser(iBu)-O-amyl ester (13)
A similar procedure as for the synthesis of 12 was used. Rf = 0.7 (Hexane/EtOAc, 8:2). 1H NMR (500 MHz, CDCl3 ): δ = 5.29 (br d, 1H), 4.57-4.56 (m, 1H), 4.47-4.44 (m, 1H), 4.33-4.30 (m, 1H), 4.18-4.13 (m, 2H), 2.57-2.52 (m, 1H), 1.66-1.63 (m, 2H), 1.46 (s, 9H), 1.34-1.32 (m, 4H), 1.16-1.14 (m, 6H), 0.90 (t, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 176.7, 170.0, 155.3, 80.3, 66.1, 53.2, 34.0, 28.4, 28.3, 28.0, 22.4, 19.1, 18.9, 14.0 ppm. HRMS (ESI−) calcd for C17H30NO6 [M-H]− 344.2078, found 344.2082.
Boc-Ser(iBu)-O-isoamyl ester (14)
A similar procedure as for the synthesis of 12 was used. Rf = 0.82 (Hexane/EtOAc, 8:2). 1H NMR (500 MHz, CDCl3 ): δ = 5.28 (br d, 1H), 4.57-4.55 (m, 1H), 4.47-4.44 (m, 1H), 4.33-4.30 (m, 1H), 4.23-4.15 (m, 2H), 2.59-2.50 (m, 1H), 1.72-1.64 (m, 1H), 1.58-1.50 (m, 2H), 1.46 (s, 9H), 1.16-1.14 (m, 6H), 0.93-0.91 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3): δ = 176.7, 170.0, 155.3, 80.4, 64.6, 64.3, 53.2, 37.3, 34.0, 28.4, 25.1, 22.5, 22.4, 19.1, 18.9 ppm. HRMS (ESI+) calcd for C17H31NO6Na [M+Na]+ 368.2044, found 368.2036.
Ser(iBu)-OMe hydrochloride (15)
Compound 12 (2.65 g, 9.2 mmol) was dissolved in CH2Cl2. A solution of HCl in isopropanol (5–6 N, 10 mL) was added and the reaction mixture was stirred at 30 °C for 6 h. Upon completion, the reaction mixture was evaporated to dryness to obtain the product as white solid which was then washed with ether and dried overnight to obtain 15 as hydrochloride salt in 95% yield. 1H NMR (300 MHz, DMSO-d6): δ = 8.95 (br s, 3H, -NH3+), 4.45 (3H), 3.75 (s, 3H), 2.64-2.52 (m, 1H), 1.10, 1.07 (2 d, 6H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 175.5, 167.5, 61.3, 53.0, 51.2, 33.0, 18.7, 18.4 ppm. HRMS (ESI+) calcd. for C8H16NO4 [M+H]+ 190.1074, found 190.1076.
Ser(iBu)-O-amyl ester hydrochloride (16)
This compound was made according to the procedure for the synthesis of 15. 1H NMR (500 MHz, DMSO-d6 ): δ = 8.95 (br s, 3H, -NH3+), 4.47-4.43 (m, 3H), 4.23-4.18 (m, 1H), 4.15-4.11 (m, 1H), 2.59-2.53 (m, 1H), 1.61-1.59 (m, 2H), 1.30-1.28 (m, 4H), 1.10-1.07 (m, 6H), 0.87 (t, 3H) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 175.5, 167.1, 63.9, 61.5, 51.3, 33.0, 27.6, 27.3, 21.7, 18.6, 18.4, 13.8 ppm. HRMS (ESI+) calcd for C12H24NO4 [M+H]+ 246.1700, found 246.1702.
Ser(iBu)-O-isoamyl ester hydrochloride (17)
This compound was made according to the procedure for the synthesis of 15. 1H NMR (500 MHz, DMSO-d6): δ = 8.82 (br s, 3H, -NH3+), 4.45-4.42 (m, 3H), 4.26-4.15 (m, 2H), 2.58-2.53 (m, 1H), 1.71-1.62 (m, 1H), 1.52-1.47 (m, 2H), 1.10-1.08 (m, 6H), 0.89-0.87 (m, 6H) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 175.5, 167.1, 64.4, 61.5, 51.3, 36.6, 33.0, 24.2, 22.2, 22.1, 18.6, 18.4 ppm. HRMS (ESI+) calcd for C12H23NO4Na [M+Na]+ 268.1519, found 268.1517.
Isoamyl Boc-L-Thr(Bzl) (17)
To a mixture Boc-Thr(Bzl)-OH (16) (1.55 g, 5.0 mmol) and isoamyl alcohol (3 ml) in anhydrous dichloromethane (30 ml) at 0°C, was added O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (2.30 g, 5.5 mmol) and triethylamine (2 ml) respectively. The mixture was stirred and allowed to warm to room temperature. Stirring was continued till all starting material was consumed according to TLC analysis (about 4 h.). The reaction mixture was then evaporated to dryness under reduced pressure, and the residue was purified by silica gel flash column chromatography (the mobile phase being a mixture of methanol in dichloromethane, in a gradient gradually ranging from 0 to 10% methanol) to yield the title compound 17 as a colorless oil (1.90 g, 99%). 1H NMR (300 MHz, CDCl3): δ = 7.32 (m, 5H), 5.32 (m, 1H), 4.58 (d, J = 11.7 Hz, 1H), 4.38 (d, J = 11.7Hz, 1H), 4.30 (m, 1H), 4.13 (m, 3H), 1.63 (m, 1H), 1.47 (m, 2H), 1.45 (s, 9H), 1.27 (d, J = 6.3 Hz, 3H), 0.90 (d, J = 6.6 Hz, 6H) ppm.
Isoamyl O-benzyl-L-threonine hydrochloride (18)
To a solution of isoamyl Boc-Thr(Bzl) 17 (1.9 g, 5.0 mmol) in dichloromethane (10 ml) was slowly added trifluoroacetic acid (10 ml). The resulting reaction mixture was stirred at room temperature for 1 hour. The reaction mixture was evaporated to dryness under reduced pressure, and the residue was dissolved in dichloromethane (20 ml) and washed with a 5% sodium carbonate solution. To the organic phase, 1.25 N HCl in isopropanol (5 ml) was added. The mixture was concentrated under reduced to yield the title compound 18 as a white solid (1.5 g, 95%). 1H NMR (300 MHz, DMSO-d6): δ = 8.64 (br.s, 3H), 7.32 (m, 5H), 4.60 (d, J = 12.0 Hz, 1H), 4.42 (d, J = 12.0 Hz, 1H), 4.15 (m, 4H), 1.64 (m, 1H), 1.43 (m, 2H), 1.31 (d, J = 6.4 Hz, 3H), 0.84 (d, J = 6.6 Hz, 6H) ppm.
Isoamyl Boc-L-threonine (19)
To a mixture of Boc-Thr-OH 18 (2.19 g, 10 mmol) and isoamyl alcohol (6 ml) in anhydrous dichloromethane (50 ml) at 0°C, was added O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (4.56 g, 11 mmol) and triethylamine (4 ml), respectively. The mixture was stirred and allowed to warm to room temperature. Stirring was continued till all starting material was consumed according to TLC analysis (about 4 hours). The reaction mixture was then evaporated to dryness under reduced pressure, and the residue was purified by silica gel flash column chromatography (the mobile phase being a mixture of methanol in dichloromethane, in a gradient gradually ranging from 0 to 10% methanol) to yield the title compound 19 as a colorless sticky solid (2.60 g, 90%). 1H NMR (300 MHz, CDCl3): δ = 5.31 (s, 1H), 4.24 (m, 1H), 4.22 (t, J = 6.9 Hz, 2H), 1.71 (m, 1H), 1.57 (m, 2H), 1.47 (s, 9H), 1.26 (d, J = 6.4 Hz, 3H), 0.93 (d, J = 6.6 Hz, 6H) ppm.
Isoamyl O-isobutyryl-Boc-L-threonine (20)
To a solution of isoamyl Boc-L-threonine 19 (2.6 g, 9.0 mmol) in anhydrous dichloromethane (20 ml) at 0°C were added isobutyryl chloride (13.5 mmol) and N-methylimidazole (45 mmol) respectively. The resulting mixture was stirred and allowed to room temperature, and stirring was continued till all starting material disappeared according to TLC analysis (about 4 hours). The reaction mixture was then evaporated to dryness under reduced pressure, and the residue was purified by silica gel flash column chromatography (the mobile phase being a mixture of methanol in dichloromethane, in a gradient gradually ranging from 0–20% methanol) to yield the title compound 20 as colorless oil (3.0 g, 93%). 1H NMR (300 MHz, CDCl3): δ = 5.40 (m, 1H), 5.19 (m, 1H), 4.42 (m, 1H), 4.15 (t, J = 7.0 Hz, 2H), 2.48 (m, J = 7.0 Hz, 1H), 1.72 (m, 1H), 1.65 (m, 2H), 1.48 (s, 9H), 1.30 (d, J = 6.4 Hz, 3H), 1.13 (d, J = 7.0 Hz, 6H), 0.93 (d, J = 6.5 Hz, 6H) ppm.
Isoamyl O-Isobutyryl-L-threonine hydrochloride (21)
To a solution of isoamyl-O-isobutyryl-Boc-L-threonine 20 (3.0 g, 8.3 mmol) in dichloromethane (10 ml) was added slowly trifluoroacetic acid (10 ml). The resulting mixture was stirred at room temperature for 1 hour. The reaction mixture was evaporated to dryness under reduced pressure, and the residue was dissolved in dichloromethane (20 ml) and washed with a 5% sodium carbonate solution. To the organic phase, 1.25 N HCl in isopropanol (10 ml) was added. The mixture was concentrated under reduced to yield the title compound 21 as white solid (2.4 g, 98%). 1H NMR (300 MHz, DMSO-d6): δ = 8.85 (br.s, 3H), 5.25 (m, 1H), 4.32 (m, 1H), 4.16 (m, 2H), 2.52 (m, 1H), 1.65 (m, 1H), 1.47 (m, 2H), 1.47 (d, J = 6.5 Hz, 3H), 1.08 (m, 6H), 0.89 (m, 6H) ppm.
Isoamyl Boc-L-tyrosine (23)
To a mixture of Boc-Tyr-OH 22 (1.41 g, 5 mmol) and isoamyl alcohol (6 ml) in anhydrous dichloromethane (30 ml) at 0°C, was added O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (2.28 g, 5.5 mmol) and triethylamine (2 ml) respectively. The mixture was stirred and allowed to warm to room temperature. Stirring was continued till all starting material was consumed according to TLC analysis (about 4 hours). The reaction mixture was then evaporated to dryness under reduced pressure, and the residue was purified by silica gel flash column chromatography (the mobile phase being a mixture of methanol in dichloromethane, in a gradient gradually ranging from 0 to 10% methanol) to yield the title compound 23 as white solid (1.23 g, 70%). 1H NMR (300 MHz, DMSO-d6): δ = 9.21 (s, 1H), 7.20 (d, J = 7.8 Hz, 2H), 7.00 (d, J = 7.8 Hz, 2H), 4.02 (m, 3H), 2.79 (m, 2H), 1.56 (m, 1H), 1.34 (s, 9H), 1.33 (m, 2H), 0.85 (m, 6H) ppm.
Isoamyl O-isobutyryl- Boc-L-tyrosine (24)
To a solution of isoamyl Boc-tyrosine 23 (351 mg, 1.0 mmol) in anhydrous dichloromethane (5 ml) were added isobutyryl chloride (2.0 mmol) and N-methylimidazole(5 mmol), respectively, at 0°C. The resultant mixture was stirred and allowed to room temperature, and the stirring was continued till the starting material disappeared according to TLC analysis (about 4 h.). The reaction mixture was then evaporated to dryness under reduced pressure, and the residue was purified by silica gel flash column chromatography (the mobile phase being a mixture of methanol in dichloromethane, in a gradient gradually ranging from 0–20% methanol) to yield the title compound 24 as colorless oil (400 mg, 95%). 1H NMR (300 MHz, CDCl3): δ = 7.16 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 4.14 (t, J = 7.0 Hz, 2H), 3.09 (m, 2H), 2.80 (m, 1H), 1.63 (m, 1H), 1.51 (m, 2H), 1.44 (s, 9H), 1.32 (d, J = 7.0 Hz, 6H), 0.92 (d, J = 6.5 Hz, 6H) ppm.
Isoamyl O-isobutyryl-L-tyrosine hydrochloride (25)
To a solution of isoamyl O-isobutyryl-Boc-L-tyrosine 24 (400 mg, 0.95 mmol) in dichloromethane (5 ml) was slowly added trifluoroacetic acid (5 ml). The resulting mixture was stirred at room temperature for 1 hour. The reaction mixture was evaporated to dryness under reduced pressure, and the residue was dissolved in dichloromethane (10 ml) and washed with a 5% sodium carbonate solution. To the organic phase, 1.25 N HCl in isopropanol (2 ml) was added. The mixture was concentrated under reduced to yield the title compound 25 as white solid (340 mg, 99%). 1H NMR (300 MHz, DMSO-d6): δ = 8.72 (br.s, 3H), 7.28 (d, J = 8.3 Hz, 2H), 7.08 (d, J = 8.3 Hz, 2H), 4.24 (m, 1H), 4.07 (m, 2H), 3.21 (m, 1H), 3.08 (m, 1H), 2.81 (m, 1H), 1.46 (m, 1H), 1.36 (m, 2H), 1.21 (d, J = 7.0 Hz, 6H), 0.83 (d, J = 6.5 Hz, 6H) ppm. 13C NMR (75 MHz, DMSO-d6): δ = 175.00, 169.17, 149.94, 132.35, 130.55, 121.87, 64.07, 53.28, 36.57, 35.51, 33.45, 24.20, 22.28, 22.24, 18.80 ppm.
General procedure for the synthesis of the 2′-C-methyl-2′,3′-O-isopropyliden-uridine 5’-phosphoramidate analogues (30a-h)
Step 1
A solution/suspension of the appropriate amino acid ester hydrochloride (3.5 equiv.) in anhydrous dichloromethane was cooled to −15 °C. Phenyl dichlorophosphate (2.5 equiv.) was added slowly. After 10 minutes, a solution of N-methylimidazole (10 equiv.) in dry dichloromethane was added dropwise. The mixture was allowed to reach room temperature slowly and left to stir for 10–12 h.
Step 2
In a separate flask, a solution/suspension of an appropriately protected nucleoside (1 equiv.) in anhydrous dichloromethane was cooled to −5 °C. With stirring, the solution prepared in step 1 was added slowly over a period of 1 hour, keeping the temperature around −5 °C. The cooling bath was removed, and the reaction was left to stir at room temperature (for nearly 4–6 h) until TLC indicates a reasonable amount of product formation. The reaction mixture was then evaporated to dryness under reduced pressure, and the residue was purified by flash column chromatography eluting with CH2Cl2–MeOH or EtOAc–hexane in different ratios. The overall yield of the reaction is in the range of 30–90%.
2′-C-Methyl-2′,3′-O-isopropyliden-uridine-5′-[phenylbis(methoxy-L-glutamyl)]phosphate (30a)
Yield: 15%. Rf = 0.39 (Hexane/EtOAc, 2:8). 31P NMR (121 MHz, CDCl3): δ = 3.01, 2.96 ppm. HRMS (ESI−) calcd for C26H33N3O12P [M-H]− 610.1807, found 610.1806.
2′-C-Methyl-2′,3′-O-isopropyliden-uridine-5′-[phenylbis(isoamyl-L-glutamyl)]phosphate (30b)
Yield: 75%. Rf = 0.59 (CH2Cl2/MeOH, 9.5:0.5). 31P NMR (121 MHz, CDCl3): δ = 3.06, 3.04 ppm. HRMS (ESI+) calcd for C34H51N3O12P [M+H]+ 724.3205, found 724.3226.
2′-C-Methyl-2′,3′-O-isopropyliden-uridine-5′-[phenyl(α-methoxy-β-O-benzyl-L-serine)]phosphate (30c)
Yield: 74%. Rf = 0.8 (CH2Cl2/MeOH, 9.5:0.5). 31P NMR (121 MHz, CDCl3): δ = 3.14, 2.77 ppm. HRMS (ESI+) calcd for C30H37N3O11P [M+H]+ 646.2160, found 646.2170.
2′-C-Methyl-2′,3′-O-isopropyliden-uridine-5′-[phenyl(α-amyl-β-O-benzyl-L-serine)]phosphate (30d)
Yield: 62.5%. Rf = 0.58 (Hexane/EtOAc, 1:9). 31P NMR (121 MHz, CDCl3): δ = 3.21, 2.87 ppm. HRMS (ESI−) calcd for C34H43N3O11P [M-H]− 700.2640, found 700.2646.
2′-C-Methyl-2′,3′-O-isopropyliden-uridine-5′-[phenyl(α-isoamyl-β-O-benzyl-L-serine)]phosphate (30e)
Yield: 20%. Rf = 0.53 (Hexane/EtOAc, 1:9). 31P NMR (121 MHz, CDCl3): δ = 3.18, 2.87 ppm. HRMS (ESI+) calcd for C34H45N3O11P [M+H]+ 702.2786, found 702.2770.
2′-C-Methyl-2′,3′-O-isopropyliden-uridine-5′-[phenyl(α-methoxy-β-O-isobutyryl-L-serine)]phosphate (30f)
Yield: 50%. Rf = 0.8 (CH2Cl2/MeOH, 9.5:0.5). 31P NMR (121 MHz, CDCl3): δ = 2.90, 2.71 ppm. HRMS (ESI−) calcd for C27H35N3O12P [M-H]− 624.1964, found 624.1969.
2′-C-Methyl-2′,3′-O-isopropyliden-uridine-5′-[phenyl(α-amyl-β-O-isobutyryl-L-serine)]phosphate (30g)
Yield: 85%. Rf = 0.47 (CH2Cl2/MeOH, 9.5:0.5). 31P NMR (121 MHz, CDCl3): δ = 2.65 ppm. HRMS (ESI−) calcd for C31H43N3O12P [M-H]− 680.2589, found 680.2593.
2′-C-Methyl-2′,3′-O-isopropyliden-uridine-5′-[phenyl(α-isoamyl-β-O-isobutyryl-L-serine)]phosphate (30h)
Yield: 87%. Rf = 0.67 (CH2Cl2/MeOH, 9.5:0.5). 31P NMR (121 MHz, CDCl3): δ = 3.20, 3.00 ppm. HRMS (ESI−) calcd for C31H43N3O12P [M-H]− 680.2589, found 680.2600.
General procedure for the synthesis of the 2′-C-methyl-uridine ProTides (31a-h)
The protected phosphoramidate 30a-h was dissolved in a solution of TFA–H2O (8 : 2, 0.1 M) and was stirred at room temperature until TLC shows no starting material (typically 3–6 h). The reaction mixture was evaporated to dryness and coevaporated three times with toluene. The crude residue was purified by silicagel flash column chromatography eluting with a mixture of methanol and dichloromethane in different ratios (generally 2–5% methanol in dichloromethane) to obtain the title compounds 31a-h as a white solid. The overall yield of the reaction is in the range from 65% to 85%.
2′-C-Methyl-uridine-5′-[phenylbis(methoxy-L-glutamyl)]phosphate (31a)
Yield: 77%. Rf = 0.28 (CH2Cl2/MeOH, 9.5:0.5). 1H NMR (500 MHz, MeOD): δ = 7.68–7.66 (2 d, 1H, H-6), 7.39–7.18 (a series of multiplets, 5H, OPh), 5.97, 5.96 (2 s, 1H, H-1′), 5.65–5.61 (2 d, 1H, H-5), 4.56–4.35 (m, 2H, H-5′ & H-5″), 4.11–4.08 (m, 1H, H-4′), 4.00–3.93 (m, 1H, H-α-Glu), 3.80–3.77 (1H, H-3′), 3.69, 3.66, 3.62 (4 s, 6H, OMe), 2.44–2.26 (m, 2H, H-β-Glu), 2.12–1.81 (m, 2H, H-γ-Glu), 1.16, 1.13 (2 s, 3H, -CH3-2′) ppm. 13C NMR (125 MHz, MeOD): δ = 175.5, 175.2 (-CO-α & -CO-β), 166.7 (C-4), 153.2, 153.1, 153.0 (C-2 & phenyl C), 142.8, 142.7 (C-6), 131.8, 131.7 (phenyl C), 127.2, 127.1 (phenyl C), 122.2–122.1 (phenyl C), 103.7 (C-5), 94.4, 94.3 (C-1′), 82.5, 82.4 (C-4′), 80.5 (C-2′), 74.8, 74.6 (C-3′), 67.3 (d, 2JCP = 5.8 Hz, C-5′), 66.8 (d, 2JCP = 4.9 Hz, C-5′), 56.2, 56.0 (C-α-Glu), 53.7, 53.0 (OMe), 31.5, 31.4 (C-γ-Glu), 30.8–30.6 (C-β-Glu), 21.0 (CH3-2′) ppm. 31P NMR (202 MHz, MeOD): δ = 3.95 ppm. HRMS (ESI−) calcd for C23H29N3O12P [M-H]− 570.1494, found 570.1505.
2′-C-Methyl-uridine-5′-[phenylbis(isoamyl-L-glutamyl)]phosphate (31b)
Yield: 66%. Rf = 0.39 (CH2Cl2/MeOH, 9.5:0.5). 1H NMR (500 MHz, MeOD): δ = 7.68–7.66 (2 d, 1H, H-6), 7.38–7.18 (a series of multiplets, 5H, OPh), 5.97, 5.96 (2 s, 1H, H-1′), 5.66–5.63 (2 d, 1H, H-5), 4.58–4.36 (m, 2H, H-5′ & H-5″), 4.18–4.02 (m, 5H, H-4′ & -OCH2CH2CH(CH3)2), 4.00–3.92 (m, 1H, H-α-Glu), 3.80–3.76 (d, 1H, H-3′), 2.45–2.26 (m, 2H, H-γ-Glu), 2.09–1.81 (m, 2H, H-β-Glu), 1.70–1.61 (m, 2H, -OCH2CH2CH(CH3)2), 1.53–1.45 (m, 4H, -OCH2CH2CH(CH3)2), 1.16, 1.13 (2 s, 3H, -CH3-2′), 0.92–0.90 (m, 12H, -OCH2CH2CH(CH3)2) ppm. 13C NMR (125 MHz, MeOD): δ = 175.1, 175.0, 174.9, 174.8 (-CO-α & -CO-β), 166.6 (C-4), 153.1, 153.0, 152.9 (C-2 & phenyl C), 142.7, 142.6 (C-6), 131.8, 131.7 (phenyl C), 127.2 (phenyl C), 122.2–122.1 (phenyl C), 103.7 (C-5), 94.4, 94.2 (C-1′), 82.4, 82.3 (C-4′), 80.5, 80.4 (C-2′), 74.8, 74.5 (C-3′), 67.3 (d, 2JCP = 5.2 Hz, C-5′), 66.7 (d, 2JCP = 5.2 Hz, C-5′), 65.9, 65.2, 65.1 (-OCH2CH2CH(CH3)2), 56.3, 56.1 (C-α-Glu), 39.3–39.2 (-OCH2CH2CH(CH3)2), 31.8–31.7 (C-γ-Glu), 31.0–30.7 (C-β-Glu), 27.1, 27.0 (-OCH2CH2CH(CH3)2), 23.7– 23.6 (-OCH2CH2CH(CH3)2), 21.1 (CH3-2′) ppm. 31P NMR (202 MHz, MeOD): δ = 3.94 and 3.90 ppm. HRMS (ESI−) calcd for C31H45N3O12P [M-H]− 682.2746, found 682.2753.
2′-C-Methyl-uridine-5′-[phenyl(α-methoxy-β-O-benzyl-L-serine)]phosphate (31c)
Yield: 87%. Rf = 0.38 (CH2Cl2/MeOH, 9.5:0.5). 1H NMR (500 MHz, MeOD): δ = 7.70–7.65 (2 d, 1H, H-6), 7.36–7.17 (a series of multiplets, 10H, OPh & CH2Ph), 5.97, 5.95 (2 s, 1H, H-1′), 5.65–5.59 (2 d, 1H, H-5), 4.60–4.56 (m, 4H, H-5′, H-5″ & CH2Ph), 4.17–4.05 (m, 1H, H-4′ & H-α-Ser), 3.81–3.52 (m, 6H, H-3′, H-β-Ser & OCH3-Ser), 1.13 (s, 3H, -CH3-2′) ppm. 13C NMR (125 MHz, MeOD): δ = 173.9 (d, 3JCP = 4.77 Hz, -CO-α), 173.6 (d, 3JCP = 6.12 Hz, -CO-α), 166.7 (C-4), 153.1–152.9 (C-2 & phenyl C), 142.8, 142.7 (C-6), 140.0, 139.9 (CH2Ph), 131.7–122.2 (phenyl C & CH2Ph), 103.7 (C-5), 94.3, 94.2 (C-1′), 82.4 (C-4′), 80.5, 80.4 (C-2′), 75.0 (CH2Ph), 74.8, 74.6 (C-3′), 73.2 (d, 3JCP = 5.3 Hz, C-β-Ser), 73.0 (d, 3JCP = 6.32 Hz, C-β-Ser), 67.2 (d, 2JCP = 5.21 Hz, C-5′), 66.7 (d, 2JCP = 4.91 Hz, C-5′), 57.3, 57.1 (C-α-Ser), 53.8 (OCH3-Ser), 21.0 (CH3-2′) ppm. 31P NMR (202 MHz, MeOD): δ = 4.14 and 3.91. HRMS (ESI+) calcd for C27H33N3O11P [M+H]+ 606.1847, found 606.1859.
2′-C-Methyl-uridine-5′-[phenyl(α-amyl-β-O-benzyl-L-serine)]phosphate (31d)
Yield: 77%. Rf = 0.53 (CH2Cl2/MeOH, 9.5:0.5). 1H NMR (500 MHz, MeOD): δ = 7.70–7.64 (2 d, 1H, H-6), 7.36–7.17 (a series of multiplets, 10H, OPh & CH2Ph), 5.98, 5.96 (2 s, 1H, H-1′), 5.65–5.60 (2 d, 1H, H-5), 4.60–4.35 (m, 4H, H-5′, H-5″ & CH2Ph), 4.15–4.03 (m, 4H, H-4′, -OCH2CH2CH2CH2CH3 & H-α-Ser), 3.81–3.52 (m, 3H, H-3′ & H-β-Ser), 1.60–1.55 (m, 2H, -OCH2CH2CH2CH2CH3), 1.30–1.27 (m, 4H, -OCH2CH2CH2CH2CH3), 1.13 ( s, 3H, -CH3-2′), 0.88– 0.85 (-OCH2CH2CH2CH2CH3) ppm. 13C NMR (125 MHz, MeOD): δ = 173.5 (d, 3JCP = 4.91 Hz, -CO-α), 173.2 (d, 3JCP = 6.05 Hz, -CO-α), 166.6 (C-4), 153.1–152.9 (C-2 & phenyl C), 142.7, 142.6 (C-6), 139.9 (CH2Ph), 131.8–122.2 (phenyl C & CH2Ph), 103.8, 103.7 (C-5), 94.3, 94.2 (C-1′), 82.4, 82.3 (C-4′), 80.5, 80.4 (C-2′), 75.0 (CH2Ph), 74.8, 74.5 (C-3′), 73.3 (d, 3JCP = 5.13 Hz, C-β-Ser), 73.2 (d, 3JCP = 6.23 Hz, C-β-Ser), 67.6 (-OCH2CH2CH2CH2CH3), 67.2 (d, 2JCP = 5.06 Hz, C-5′), 66.7 (d, 2JCP = 4.70 Hz, C-5′), 57.3, 57.1 (C-α-Ser), 30.2–29.9 (-OCH2CH2CH2CH2CH3), 24.2 (-OCH2CH2CH2CH2CH3), 21.1 (CH3-2′), 15.2 (-OCH2CH2CH2CH2CH3) ppm. 31P NMR (202 MHz, MeOD): δ = 4.17 and 3.89 ppm. HRMS (ESI−) calcd for C31H39N3O11P [M-H]− 660.2327, found 660.2322.
2′-C-Methyl-uridine-5′-[phenyl(α-isoamyl-β-O-benzyl-L-serine)]phosphate (31e)
Yield: 80%; Rf = 0.15 (CH2Cl2/MeOH, 9.7:0.3); 1H NMR (500 MHz, MeOD): δ = 7.70–7.65 (2 d, 1H, H-6), 7.36–7.17 (a series of multiplets, 10H, OPh & CH2Ph), 5.98, 5.95 (2 s, 1H, H-1′), 5.65–5.60 (2 d, 1H, H-5), 4.61–4.34 (m, 4H, H-5′, H-5″ & CH2Ph), 4.21–4.04 (m, 4H, H-4′, -OCH2CH2CH(CH3)2 & H-α-Ser), 3.81–3.52 (m, 3H, H-3′ & H-β-Ser), 1.68–1.59 (m, 1H, -OCH2CH2CH(CH3)2), 1.51–1.44 (m, 2H, -OCH2CH2CH(CH3)2), 1.13 (s, 3H, -CH3-2′), 0.88– 0.86 (m, 6H, -OCH2CH2CH(CH3)2) ppm. 13C NMR (125 MHz, MeOD): δ = 173.5 (d, 3JCP = 4.96 Hz, -CO-α), 173.2 (d, 3JCP = 6.34 Hz, -CO-α), 166.6 (C-4), 153.1–152.9 (C-2 & phenyl C), 142.8, 142.6 (C-6), 140.0, 139.9 (CH2Ph), 131.8–122.2 (phenyl C & CH2Ph), 103.8, 103.7 (C-5), 94.3, 94.2 (C-1′), 82.4 (C-4′), 80.5, 80.4 (C-2′), 75.1 (CH2Ph), 74.8, 74.6 (C-3′), 73.3 (d, 3JCP = 5.40 Hz, C-β-Ser), 73.2 (d, 3JCP = 6.48 Hz, C-β-Ser), 67.3 (d, 2JCP = 4.83 Hz, C-5′), 66.7 (d, 2JCP = 4.83 Hz, C-5′), 66.0 (-OCH2CH2CH(CH3)2), 57.3, 57.1 (C-α-Ser), 39.3, 39.2 (-OCH2CH2CH(CH3)2), 26.9 (-OCH2CH2CH(CH3)2), 23.6 (-OCH2CH2CH(CH3)2), 21.0 (CH3-2′) ppm. 31P NMR (202 MHz, MeOD): δ = 4.15 and 3.88 ppm. HRMS (ESI+) calcd for C31H41N3O11P [M+H]+ 662.2473, found 662.2488.
2′-C-Methyl-uridine-5′-[phenyl(α-methoxy-β-O-isobutyryl-L-serine)]phosphate (31f)
Yield: 75%. Rf = 0.46 (CH2Cl2/MeOH, 9.5:0.5). 1H NMR (500 MHz, MeOD): δ = 7.67–7.65 (2 d, 1H, H-6), 7.39–7.19 (a series of multiplets, 5H, OPh), 5.97, 5.96 (2 s, 1H, H-1′), 5.64–5.60 (2 d, 1H, H-5), 4.61–4.36 (m, 2H, H-5′, H-5″), 4.31–4.08 (m, 4H, H-4′, H-α-Ser & H-β-Ser ), 3.80–3.78 (1H, H-3′), 3.72, 3.68 (2 s, 3H, -OCH3-Ser), 2.55-2.47 (m, 1H, -CH(CH3)2), 1.15-1.09 ( m, 9H, -CH3-2′ & -CH(CH3)2) ppm. 13C NMR (125 MHz, MeOD): δ = 178.9 (-COCH(CH3)2), 173.1 (d, 3JCP = 5.07 Hz, -CO-α), 172.9 (d, 3JCP = 5.77 Hz, -CO-α), 166.7 (C-4), 153.1–152.9 (C-2 & phenyl C), 142.8, 142.7 (C-6), 131.8–122.1 (phenyl C), 103.7 (C-5), 94.5, 94.3 (C-1′), 82.4, 82.3 (C-4′), 80.5 (C-2′), 74.9, 74.6 (C-3′), 67.4–66.8 (C-β-Ser & C-5′), 56.1, 56.0 (C-α-Ser), 54.0, 53.9 (OCH3-Ser), 35.8 (-CH(CH3)2), 21.1–20.0 (CH3-2′ & -CH(CH3)2) ppm. 31P NMR (202 MHz, MeOD): δ = 3.90 and 3.65 ppm. HRMS (ESI+) calcd for C24H32N3O12P [M+H]+ 586.1796, found 586.1793.
2′-C-Methyl-uridine-5′-[phenyl(α-amyl-β-O-isobutyryl-L-serine)]phosphate (31g)
Yield: 75%. Rf = 0.36 (CH2Cl2/MeOH, 9.5:0.5). 1H NMR (500 MHz, MeOD): δ = 7.67–7.64 (1H, H-6), 7.38–7.18 (a series of multiplets, 5H, OPh), 5.98, 5.97 (2 s, 1H, H-1′), 5.66–5.62 (1H, H-5), 4.62–4.38 (m, 2H, H-5′, H-5″), 4.34–4.05 (m, 6H, H-4′, -OCH2CH2CH2CH2CH3, H-α-Ser & H-β-Ser), 3.81, 3.79 (1H, H-3′), 2.54-2.47 (m, 1H, -CH(CH3)2 of iBu), 1.64–1.59 (m, 2H, -OCH2CH2CH2CH2CH3), 1.33–1.30 (m, 4H, -OCH2CH2CH2CH2CH3), 1.15-1.09 ( m, 9H, -CH3-2′ & -CH(CH3)2 of iBu), 0.90– 0.88 (t, 3H, -OCH2CH2CH2CH2CH3); 13C NMR (125 MHz, MeOD): δ = 178.8, 178.9 (-COCH(CH3)2 of iBu), 172.6 (d, 3JCP = 5.39 Hz, -CO-α), 172.4 (d, 3JCP = 6.18 Hz, -CO-α), 166.6 (C-4), 153.1–152.8 (C-2 & phenyl C), 142.8, 142.6 (C-6), 131.8–122.1 (phenyl C), 103.8, 103.7 (C-5), 94.4, 94.2 (C-1′), 82.4, 82.3 (C-4′), 80.5, 80.4 (C-2′), 74.9, 74.6 (C-3′), 67.8–66.8 (C-β-Ser, C-5′ & -OCH2CH2CH2CH2CH3), 56.2, 56.0 (C-α-Ser), 35.7 (-CH(CH3)2 of iBu), 30.2–29.9 (-OCH2CH2CH2CH2CH3), 24.1 (-OCH2CH2CH2CH2CH3), 21.1–20.0 (CH3-2′ & -CH(CH3)2 of iBu), 15.1 (-OCH2CH2CH2CH2CH3) ppm. 31P NMR (202 MHz, MeOD): δ = 3.92 and 3.64 ppm. HRMS (ESI+) calcd for C28H41N3O12P [M+H]+ 642.2422, found 642.2429.
2′-C-Methyl-uridine-5′-[phenyl(α-isoamyl-β-O-isobutyryl-L-serine)]phosphate (31h)
Yield: 65%. Rf = 0.28 (CH2Cl2/MeOH, 9.5:0.5). 1H NMR (500 MHz, MeOD): δ = 7.67–7.64 (1H, H-6), 7.38–7.18 (a series of multiplets, 5H, OPh), 5.98, 5.96 (2 s, 1H, H-1′), 5.65–5.62 (1H, H-5), 4.62–4.37 (m, 2H, H-5′, H-5″), 4.33–4.09 (m, 6H, H-4′, -OCH2CH2CH(CH3)2, H-α-Ser & H-β-Ser), 3.80, 3.79 (1H, H-3′), 2.55-2.47 (m, 1H, -CH(CH3)2 of iBu), 1.69–1.61 (m, 1H, -OCH2CH2CH(CH3)2), 1.55–1.47 (m, 2H, -OCH2CH2CH(CH3)2), 1.15-1.09 ( m, 9H, -CH3-2′ & -CH(CH3)2 of iBu), 0.91– 0.89 (m, 6H, -OCH2CH2CH(CH3)2) ppm. 13C NMR (125 MHz, MeOD): δ = 178.9, 178.8 (-COCH(CH3)2 of iBu), 172.6 (d, 3JCP = 5.26 Hz, -CO-α), 172.4 (d, 3JCP = 6.07 Hz, -CO-α), 166.6 (C-4), 153.1–152.9 (C-2 & phenyl C), 142.8, 142.6 (C-6), 131.8–122.1 (phenyl C), 103.8, 103.7 (C-5), 94.4, 94.2 (C-1′), 82.4, 82.3 (C-4′), 80.5, 80.4 (C-2′), 74.9, 74.6 (C-3′), 67.5–66.3 (C-β-Ser, C-5′ & -OCH2CH2CH(CH3)2), 56.2, 56.1 (C-α-Ser), 39.2 (-OCH2CH2CH(CH3)2), 35.8, 35.7 (-CH(CH3)2 of iBu), 26.9 (-OCH2CH2CH(CH3)2), 23.6 (-OCH2CH2CH(CH3)2), 21.1–20.0 (CH3-2′ & -CH(CH3)2 of iBu) ppm. 31P NMR (202 MHz, MeOD): δ = 3.91 and 3.64 ppm. HRMS (ESI+) calcd for C28H41N3O12P [M+H]+ 642.2422, found 642.2440.
General procedure for the synthesis of 2′-C-methyl-2′,3′-O-isopropyliden-uridine 5’-phosphoramidate analogues (30i-k)
To a mixture of the appropriate amino acid ester hydrochloride salt (1.0 mmol) in anhydrous dichloromethane (10 ml) was added dichlorophenyl phosphate (165 µl, 1.1 mmol) and N-methylimidazole (420 µl, 5 mmol) at −40°C. The mixture was stirred and allowed to warm to room temperature. The stirring was continued for another 3 hours. The mixture was then cooled to −40°C again, and 2´3´-isopropylidene-2′-C-methyl-uridine (149 mg, 0.5 mmol) was added. The mixture was stirred and warmed to room temperature, and stirring was continued till all starting material disappeared according to TLC analysis. The reaction mixture was then evaporated to dryness under reduced pressure, and the residue was purified by flash column chromatography (using a mixture of methanol in dichloromethane as mobile phase, in a gradient gradually ranging from 0 to 10% methanol) to yield the 2′-C-methyl-2′,3′-O-isopropyliden-uridine-5’-phosphoramidate analogues 30i-k, which were used as such in next step, without any further structural identification.
General procedure for the synthesis of the 2′-C-methyl-uridine ProTides (31i-k)
A solution of aforementioned 2´,3´-isopropylidene-protected product in a mixture of TFA/H2O (4/1, 5 ml) was stirred at room temperature for 2 hours. After concentration under the reduced pressure, the residue was purified by silica gel flash chromatography (the mobile phase being a mixture of methanol in dichloromethane, in a ratio gradually ranging from 0–20% MeOH) to yield the title compounds as white solids.
2′-C-Methyl-uridine-5′-[phenyl(isoamyl-O-benzyl-L-threonine)]phosphate (31i)
This compound was isolated in 46% yield as a white solid. 1H NMR (300 MHz, DMSO-d6): δ = 11.40 (br s, 1H), 7.60 (m, 1H), 7.28 (m, 10H), 5.82 (m, 2H), 5.45 (m, 2H), 5.25 (s, 2H), 4.44 (m, 4H), 3.97 (m, 5H), 3.65 (m, 1H), 1.60 (m, 1H), 1.40 (m, 2H), 1.16 (m, 3H), 1.00 (m, 3H), 0.80 (m, 6H) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 171.36, 171.11, 171.09, 163.00, 162.97, 150.94, 150.90, 150.86, 150.67, 129.72, 129.68, 128.22, 127.59, 127.49, 124.71, 124.64, 120.28, 120.25, 120.21, 101.83, 101.79, 91.54, 79.98, 79.93, 78.02, 77.95, 75.35, 75.30, 75.25, 72.60, 72.44, 70.12, 65.38, 64.72, 63.21, 63.17, 59.07, 59.04, 24.40, 22.34, 22.28, 22.26, 19.96, 15.91, 15.70 ppm. 31P NMR (202 MHz, DMSO-d6): δ = 4.91, 4.83 ppm. HRMS (ES+) calcd for C32H42N3O11P [M+H]+ 676.2630, found 676.2643.
2′-C-Methyl-uridine-5′-[phenyl(isoamyl-O-isobutyryl-L-threonine)]phosphate (31j)
This compound was isolated in 49% yield as a white solid. 1H NMR (300 MHz, CDCl3): δ = 9.78 (br s, 1H), 7.55 & 7.48 (d, J = 8.1 Hz, 1H), 7.20-7.35 (m, 5H), 6.00 (d, J = 7.8 Hz, 1H), 5.67 (d, J = 8.1 Hz, 1H), 5.32 (m, 1H), 4.54 (m, 2H), 4.12 (m, 5H), 3.78 (m, 1H), 2.48 (m, 1H), 1.65 (m, 1H), 1.49 (m, 2H), 1.13 (m, 12H), 0.91 (m, 6H) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 175.34, 175.25, 170.54, 170.26, 162.93, 162.90, 150.87, 150.84, 150.63, 140.12, 129.71, 129.28, 129.01, 124.73, 124.66, 120.25, 120.23, 120.16, 120.13, 101.78, 101.73, 91.52, 79.92, 79.86, 77.94, 77.89, 72.65, 72.51, 70.28, 70.24, 70.16, 70.12, 65.52, 64.90, 63.40, 63.35, 58.12, 57.95, 36.82, 33.28, 30.78, 24.29, 22.32, 22.19, 19.94, 18.71, 18.66, 16.20, 16.04 ppm. 31P NMR (202 MHz, DMSO-d6): δ = 4.21, 3.64 ppm. HRMS (ES+) calcd for C29H42N3O12P [M+H]+ 656.2579, found 656.2584.
2′-C-Methyl-uridine-5′-[phenyl(isoamyl-O-isobutyryl-L-tyrosine)]phosphate (31k)
This compound was isolated in 50% yield as a white solid. 1H NMR (300 MHz, DMSO-d6): δ = 11.42 (br s, 1H), 7.50 (m, 1H), 6.90-7.33 (m, 9H), 6.30 (m, 1H), 5.87 (m, 1H), 5.43 (m, 2H), 5.24 (m, 1H), 4.00 (m, 6H), 3.57 (m, 1H), 2.93 (m, 1H), 2.80 (m, 2H), 1.6 (m, 1H), 1.46 (m, 2H), 1.22 (d, J = 7.0 Hz, 6H), 0.98 (d, J = 6.8 Hz, 3H), 0.80 (m, 6H) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 175.07, 172.36, 162.94, 162.88, 150.78, 150.73, 150.65, 149.44, 140.75, 139.83, 134.39, 134.37, 130.34, 129.74, 129.72, 124.63, 124.56, 121.53, 120.07, 120.04, 119.78, 119.75, 101.78, 101.72, 91.58, 79.90, 79.84, 79.78, 77.97, 77.86, 72.70, 72.35, 65.25, 64.25, 63.06, 56.27, 56.17, 24.37, 24.33, 22.32, 22.28, 22.25, 19.94, 18.80 ppm. 31P NMR (202 MHz, DMSO-d6): δ = 3.57, 3.05 ppm. HRMS (ES+) calcd for C34H44N3O12P [M+H]+ 718.2735, found 718.2748.
Biological evaluation
Primary HCV antiviral assay
For the primary HCV antiviral screening assay, compounds were evaluated for anti-HCV activity using a luciferase (Luc) reporter gene endpoint incorporated within a subgenomic HCV replicon. The Luc reporter is used as an indirect measure of HCV replication as its activity is directly proportional to HCV RNA levels. Assessment of cytotoxicity is conducted in parallel. Drug stocks are prepared in DMSO and are diluted with tissue culture medium to the desired high-test concentration. For each assay, the compounds are then further diluted in tissue culture medium as required. After incubation, the cells are processed to derive EC50 and EC90 (compound concentrations reducing HCV replication by 50% and 90% respectively) values. In addition, CC50 (concentration decreasing cell viability by 50%) and SI50 (CC50/EC50) values are determined and reported.
HCV replicon antiviral evaluation protocol
The genotype 1b (Con1 strain) HCV replicon was used for primary screening and uses the cell line ET (luc-ubi-neo/ET), which is an Huh7 human hepatoma cell line that contains an HCV replicon with a stable luciferase (Luc) reporter and three cell culture-adaptive mutations.15 Recombinant human interferon alpha-2b (rIFNα-2b) and the NS5B inhibitor Sofosbuvir is included in each run as a positive control. Briefly, sub-confluent cultures of the ET cell line are plated into 96-well plates that are dedicated for the analysis of cytotoxicity or antiviral activity and the next day compounds are added to the appropriate wells. Cells are processed 72 hrs later when the cells are still sub-confluent. Compounds are evaluated using 6 half-log serial dilutions to derive where applicable EC50, EC90, CC50 and SI50 values. HCV replicon levels are assessed as HCV replicon-derived Luc activity (Steady-Glo® Luciferase Assay System, Promega) and compound cytotoxicity is assessed on replicate plates using a fluorometric method for determining cell viability (CytoTox-ONE™ Homogeneous Membrane Integrity Assay, Promega).
The antiviral and cytotoxicity data are the result of one assay in which the compounds were tested in dose-response with each concentration evaluated in quadruplicate. The average values at each concentration were then graphed and used to generate the reported EC50 and EC90 values. The %CV for quadruplicate values are generally below 15%, and the assay is robust and highly reproducible.
Secondary HCV antiviral assay
A qRT-PCR (TaqMan) endpoint is used to directly measure HCV RNA copies to confirm compound activity. In the secondary assay, instead of assessing luciferase activity at the assay endpoint, the cells are processed for intracellular RNA extraction with an RNeasy 96 kit (Qiagen). The level of HCV RNA is determined by qRT-PCR/TaqMan assay using TaqMan® One-Step RT-PCR Master Mix Reagents (Applied Biosystems, Foster City, CA) and an ABI Prism 7900 sequence detection system (Applied Biosystems). Compound cytotoxic effects are measured from the same plate/samples with TaqMan® Ribosomal RNA Control Reagents (Applied Biosystems) as an indication of viable cell numbers.
Metabolic stability assay (human liver microsomes)
The stability assay was contracted and carried out by Eurofins (Eurofins ref. 0607). Pooled liver microsomes are preincubated with NADPH-regenerating system (1 mM NADP, 5 mM G6P, and 1 U/mL G6PDHase) in phosphate buffer (pH 7.4) containing 3 mM MgCl2 and 1 mM EDTA in a 2 mL-block 96-well plate for 10 min in a 37 °C shaking water bath. The reaction was initiated by adding the test compound (final concentration 0.1 µM) and incubated in a final volume of 700 µL in the 37 °C shaking water-bath. At 0, 15, 30, 45, and 60 min after reaction initiation, 100 µL of the incubation mixture is transferred to 100 µL of acetonitrile/methanol (50/50, v/v) in a 0.8 mL V-bottom 96-well plate. Samples are then mixed on a plate shaker for 5 minutes and centrifuged at 2550 × g for 15 min at room temperature. Each supernatant is transferred to a clean cluster tube, followed by HPLC MS/MS analysis on a Thermo Electron triplequadrupole system.
Supplementary Material
Scheme 1.

Synthesis of L-glutamic acid di-ester
Scheme 2.

Synthesis of mono- and di-esters of L-serine
Scheme 4.

Synthesis of a di-ester of L-tyrosine
Acknowledgments
This project has been funded in part with Federal funds from the Division of Microbiology and Infectious Diseases, National Institute of Allergy and infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272201100013I.
Footnotes
Electronic supplementary information (ESI) available: NMR spectra of final compounds.
References
- 1.Jordheim LP, Durantel D, Zoulim F, Dumontet C. Nat. Rev. Drug Discov. 2013;12:447. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 2.Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. Chem. Rev. 2014;114:9154. doi: 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehellou Y, Balzarini J, McGuigan C. ChemMedChem. 2009;4:1779. doi: 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- 4.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy G, Ross BS, Wang P, Zhang H, Bansal S, Espiritu C, Keilman M, Lam AM, Micolochick Steuer HM, Niu C, Otto MJ, Furman PA. J. Med. Chem. 2010;53:7202. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 5.Ray AS, Fordyce MW, Hitchcock MJ. Antiviral Res. 2016;125:63. doi: 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Warren R, Jordan TK, Lo MK, Ray AS, Mackman RL, Soloveva V, Siegel D, Perron M, Bannister R, Hui HC, Larson N, Strickley R, Wells J, Stuthman KS, Van Tongeren SA, Garza NL, Donnelly G, Shurtleff AC, Retterer CJ, Gharaibeh D, Zamani R, Kenny T, Eaton BP, Grimes E, Welch LS, Gomba L, Wilhelmsen CL, Nichols DK, Nuss JE, Nagle ER, Kugelman JR, Palacios G, Doerffler E, Neville S, Carra E, Clarke MO, Zhang L, Lew W, Ross B, Wang Q, Chun K, Wolfe L, Babusis D, Park Y, Stray KM, Trancheva I, Feng JY, Barauskas O, Xu Y, Wong P, Braun MR, Flint M, McMullan LK, Chen S, Fearns R, Swaminathan S, Mayers DL, Spiropoulou CF, Lee WA, Nichol ST, Cihlar T, Bavari S. Nature. 2016;531:381. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahard D, McGuigan C, Balzarini J. Mini-Rev. Med. Chem. 2004;4:371. doi: 10.2174/1389557043403936. [DOI] [PubMed] [Google Scholar]
- 8.Slusarczyk M, Lopez MH, Balzarini J, Mason M, Jiang WG, Blagden S, Thompson E, Ghazaly E, McGuigan C. J. Med. Chem. 2014;57:1531. doi: 10.1021/jm401853a. [DOI] [PubMed] [Google Scholar]
- 9.McGuigan C, Murziani P, Slusarczyk M, Gonczy B, Vande Voorde J, Liekens S, Balzarini J. J. Med. Chem. 2011;54:7247. doi: 10.1021/jm200815w. [DOI] [PubMed] [Google Scholar]
- 10.McGuigan C, Salgado A, Yarnold C, Harries TY, De Clercq E, Balzarini J. Antiviral. Chem. Chemother. 1996;7:184–188. [Google Scholar]
- 11.Saboulard D, Naesens L, Cahard D, Salgado A, Pathirana R, Velazquez S, Mcguigan C, De Clercq E, Balzarini J. Mol. Pharmacol. 1999;56:693. [PubMed] [Google Scholar]
- 12.Maiti M, Maiti M, Rozenski J, De Jonghe S, Herdewijn P. Org. Biomol. Chem. 2015;13:5158. doi: 10.1039/c5ob00427f. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Sha Y. Molecules. 2008;13:1111. doi: 10.3390/molecules13051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groaz E, Herdewijn P. Curr. Med. Chem. 2015;22:3980. doi: 10.2174/092986732234151119155207. [DOI] [PubMed] [Google Scholar]
- 15.Pietschmann T, Lohmann V, Kaul A, Krieger N, Rinck G, Rutter G, Strand D, Bartenschlager R. J. Virol. 2002;76:4008. doi: 10.1128/JVI.76.8.4008-4021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.