

Abstract
Glycoproteins are an important class of naturally occurring biomolecules which play a pivotal role in many biological processes. They are biosynthesized as complex mixtures of glycoforms through post-translational protein glycosylation. This fact, together with the challenges associated with producing them in homogeneous form, has hampered detailed structure-function studies of glycoproteins as well as their full exploitation as potential therapeutic agents. By contrast, chemical synthesis offers the unique opportunity to gain access to homogeneous glycoprotein samples for rigorous biological evaluation. Herein, we review recent methods for the assembly of complex glycopeptides and glycoproteins and present several examples from our laboratory towards the total chemical synthesis of clinically relevant glycosylated proteins that have enabled synthetic access to full-length homogeneous glycoproteins.
Keywords: Chemoselective ligation, Glycopeptides, Glycoproteins, Glycosylation, Total synthesis
1 Introduction
The study of protein post-translational modification (PTM) [1] is an active area in protein research. Protein glycosylation is a major form of PTM which expands the functional diversity and structural heterogeneity of proteins. More than 50% of human proteins are reported to be glycosylated [2], a modification known to play an important role in a wide range of biological events, at both cellular (cell–cell interaction, signaling, immune response) and protein levels (molecular recognition, protein folding) [3–5]. Aberrant glycosylation of proteins is also implicated in a number of illnesses, including autoimmune diseases, infectious diseases, and cancer [6, 7]. As a result, glycans and glycoproteins are increasingly being investigated in clinical settings as potential targets for the development of therapeutics, diagnostics, and vaccines [8, 9]. For all these reasons, a great deal of effort has been directed toward elucidating the effects of protein glycosylation on the function, conformation, stability, and bioactivity of glycoproteins [10–13].
Structurally, all native glycoproteins can be classified into two major types: N-linked and O-linked glycoproteins. In N-linked glycoproteins, the glycan (N-acetylglucosamine, GlcNAc) is attached through a β-glycosidic linkage to the amide side chain of an asparagine within an Asn-Xaa-Ser/Thr consensus sequence. In common O-glycoproteins, the sugar (mainly N-acetyl-galactosamine, GalNAc and GlcNAc) is α- or β-linked to the hydroxyl group of serine or threonine residues, respectively (Fig. 1). Other forms of O-glycosylation are also observed with several different glycans such as mannose, xylose, glucose, or fucose [14].
Fig. 1.

Common (β-GlcNAc)-N-linked asparagine and (α-GalNAc)/(β-GlcNAc)-O-linked serine/ threonine in glycosylated proteins
Unlike protein synthesis, protein glycosylation is not regulated genetically, and the biosynthesis of typical N- and O-glycoproteins often comprises multiple steps dictated by the relative activities of a large number of enzymes. As a result, glycoproteins are naturally expressed as complex mixtures of glycoforms possessing the same peptide backbone but differing in both the nature and site of glycosylation [15]. This structural diversity lays the molecular basis for a wide range of biological functions. However, the structural heterogeneity of glycosylation and the difficulty associated with obtaining homogeneous glycoproteins from biological sources has limited progress towards correlating glycoprotein structure with function [16]. The urgent need for chemically pure, synthetic glycoproteins for detailed structure–activity relationship studies and therapeutic applications has been the main driving force toward the development of several strategies for the preparation of these structures [17]. In particular, chemical synthesis has emerged as a practical way to gain access to well-defined glycoproteins, as is demonstrated through several relevant examples described herein.
2 Chemical Synthesis of Glycopeptides
The chemical synthesis of glycoproteins requires the ability to prepare glycosylated peptide fragments efficiently [18, 19]. Peptide synthesis efforts typically rely on solid-phase peptide synthesis (SPPS) [20], a process which allows for the efficient assembly of peptide fragments comprising up to 50 amino acid residues. Of the two conventional SPPS methodologies (Boc method and Fmoc method), Fmoc-based SPPS is usually favored in the synthesis of glycopeptides because the glycosidic linkages are relatively labile in the presence of strong acid.
For the synthesis of a glycopeptide, bearing oligosaccharide side chains, there exist two general strategies: the cassette-based approach and the convergent method. In the more linear cassette approach, the glycan is attached to an amino acid residue and the resulting glycosyl amino acid is then used in SPPS to assemble the complete glycopeptide fragment (Scheme 1). In principle, the pre-formed glycosyl amino acid “cassette” can consist of either the fully elaborated oligosaccharide, or a monosaccharide residue, which, following SPPS, may be elongated further by enzymatic or chemical glycosylation to yield the glycopeptide containing the full glycan. Several research groups have used this stepwise, “cassette-based” approach to generate successfully N-linked [21, 22] and especially O-linked [23, 24] glycopeptide fragments related to natural glycoproteins. A particularly attractive application of this strategy is being implemented in a number of laboratories for the synthesis of mucin-type O-glycopeptides containing tumor carbohydrate antigens (Tn, TF, STn) as promising targets for anticancer vaccines [25–27].
Scheme 1.

Linear “cassette”-based approach for the synthesis of O-linked glycopeptides
In contrast, the convergent approach to glycopeptide synthesis relies on the direct conjugation of an oligosaccharide domain with the full-length peptide to generate the corresponding glycopeptide. Whereas the cassette strategy has been widely used for the synthesis of O-glycopeptides, the convergent method has been mainly applied for the preparation of N-glycopeptide fragments. In this approach, the partially protected peptide is first synthesized by SPPS. Next, the N-glycan, bearing an anomeric amine, is directly introduced through an amide linkage to the free aspartic acid (Asp) side chain of the peptide under Lansbury aspartylation conditions [28]. The Asp residue to be coupled requires an additional orthogonal protecting group which can be selectively removed prior to Lansbury aspartylation. The carboxylic acid is activated using coupling agents (i.e., HATU, HBTU, or PyAOP), and the anomeric amine of the sugar reacts with the activated ester to form the amide bond. When the peptidyl Asp side chain is activated, care should be taken to minimize competitive intramolecular aspartimide formation [29]. To address this problem, we and Unverzagt independently developed a simple solution, involving emplacement of a temporary pseudoproline motif, derived from Ser/Thr, at the n + 2 position (relative to Asp) of the Asn-Xaa-Ser/Thr consensus sequence (Scheme 2) [30, 31]. Adoption of this pseudoproline strategy has enabled the efficient and convergent syntheses of extended glycopeptide fragments, thereby providing enabling progress in the chemical synthesis of homogeneous complex N-glycopeptides [32].
Scheme 2.

Lansbury aspartylation reaction facilitated by a pseudoproline motif for the convergent synthesis of N-linked glycopeptides
3 Chemical Synthesis of Glycoproteins
3.1 Advances in Chemical Ligation
Extension of the methodologies described above to the assembly of full-length glycoproteins is hindered by a number of factors, including low coupling efficiency, poor solubility of protected polypeptides, acid-lability of the oligosaccharide chains, increased epimerization, and formation of side-products resulting in low yields and purities of the final target molecules. For these reasons, several chemoselective ligation methods have been developed which permit the assembly of complex glycoproteins from shorter glycosylated peptides [33].
Most notable among these is the powerful native chemical ligation (NCL) method, first described by Kent et al. in 1994 [34]. Over the past 20 years, NCL has proved to be a remarkably robust methodology for the efficient, epimerization-free assembly of proteins. As shown in Scheme 3, the process accomplishes chemoselective ligation, in aqueous solution, of two fully deprotected peptides, one of which presents a C-terminal thioester and the other an N-terminal cysteine residue. The reaction involves transthioesterification of the C-terminal thioester with the N-terminal cysteine to generate a transient thioester intermediate, which then undergoes an irreversible S → N acyl transfer to form the native peptide bond. Interestingly, large peptide fragments may be obtained by recombinant expression and further used as ligation partners through a method termed expressed protein ligation (EPL) [35, 36].
Scheme 3.
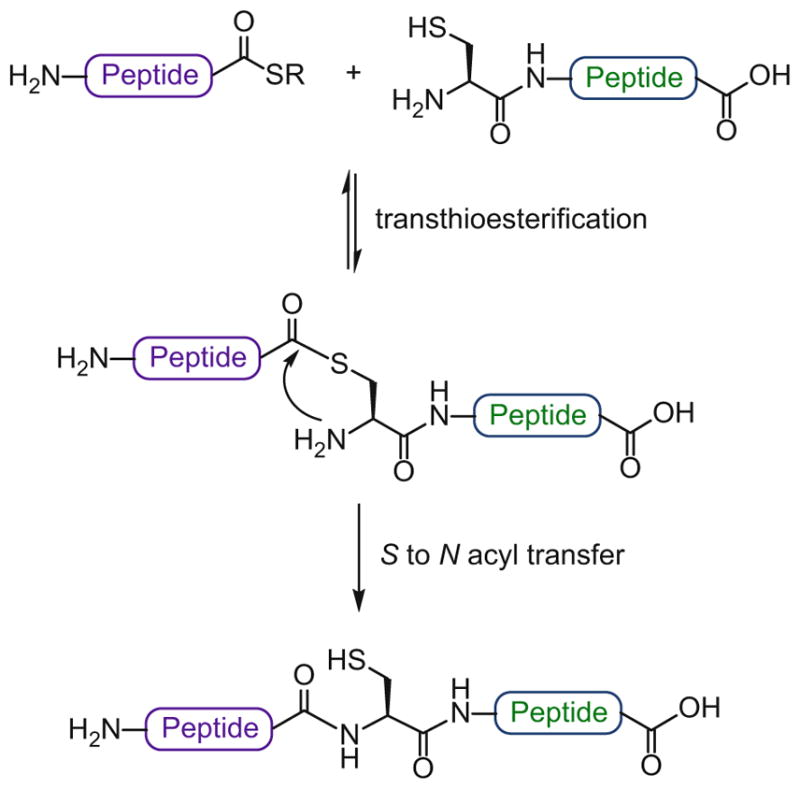
Native chemical ligation (NCL)
To date, NCL-based strategies have been successfully applied to the chemical synthesis of over 200 proteins [37]. This technology has also been adopted for the total synthesis of a number of biologically relevant glycoproteins [38]. The first significant examples were reported by Bertozzi and coworkers, who synthesized several mucin-type glycoproteins bearing GalNAc residues at the relevant O-glycosylation sites [39, 40]. One such target was diptericin, an 82-residue antimicrobial glycoprotein prepared via NCL between two fragments, a 58-mer Cys-glycopeptide and a 24-mer glycopeptide thioester (Scheme 4). Notably, an alkane sulfonamide “safety-catch” linker was used for the Fmoc-based SPPS synthesis of the glycopeptide thioester [39]. Unverzagt et al. also used the “safety-catch” linker strategy followed by NCL to synthesize the first N-linked glycopeptide fragment carrying a complex-type oligosaccharide [41]. The first total chemical synthesis of a full-length glycoprotein bearing a complex type N-glycan was reported by Kajihara and coworkers, who prepared a single glycoform of the 76-amino acid chemokine monocyte chemotactic protein-3 (MCP-3) via two consecutive NCLs of three (glyco)peptide fragments, followed by protein folding (not shown) [42].
Scheme 4.

Total synthesis of antimicrobial O-glycoprotein diptericin by NCL
Despite these impressive examples, efforts to extend directly the NCL method toward the synthesis of complex glycopeptides have been complicated by the difficulties associated with synthesizing the requisite pre-formed glycopeptide thioester fragments, particularly by Fmoc-based SPPS [43]. To address this issue, our group has been engaged in the development of a number of synthetic strategies that may have broad utility for the preparation of homogeneously glycosylated peptides and proteins [44, 45]. In pursuing modified NCL strategies, we and others have sought to replace the labile C-terminal thioester with more stable latent thioester or alternative acyl donor groups.
In a key advance, we synthesized a C-terminal glycopeptide phenolic ester equipped with an o-disulfide moiety which, upon reductive treatment with subsequent unmasking of the free thiol, can rearrange to generate the corresponding aryl thioester via a reversible O → S acyl transfer [46]. The utility of this latent acyl donor in glycopeptide ligation has been widely demonstrated. In the example shown in Scheme 5, two glycopeptide fragments – one bearing the newly crafted o-disulfide phenolic ester at the C-terminus and the other a tBu-thio-protected cysteine at the N-terminus – were subjected to reductive cleavage by the addition of sodium 2-mercaptoethanesulfonate (MESNa). The resulting glycopeptide intermediates underwent ligation in situ to furnish the doubly glycosylated peptide.
Scheme 5.

NCL-based assembly of a complex model N-glycopeptide using an o-disulfide phenolic ester as latent thioester
Although this method proved highly useful for a wide range of substrates, we next sought to increase the acyl transfer capability of the o-thiophenolic ester in order to improve ligation efficiency with peptides bearing hindered C-terminal amino acids. Along these lines, we found that, when activated p-nitrophenyl or p-cyanophenyl oxo-ester variants were employed as thioester surrogates [47], efficient ligation was attainable at sterically demanding C-terminal sites. Indeed, this strategy was ultimately adopted for the synthesis of a challenging glycopeptide fragment en route to erythropoietin (EPO) [48].
Another important limitation for the general applicability of the NCL is the requirement that an N-terminal cysteine be present on the target peptide or glycopeptide fragment. Given the relative scarcity of cysteine residues in naturally occurring proteins and glycoproteins, it is often difficult to locate a feasible ligation site containing this amino acid, which makes direct ligation at cysteine itself of somewhat limited practical value. As a consequence, a number of alternative strategies have emerged for the cysteine-free, ligation-based assembly of glycopeptides and glycoproteins [49]. Early studies led to the development of thiol-based auxiliaries, wherein a mercapto group present in a provisional N-terminal moiety serves to mimic the function of the cysteine thiol. A pioneering example by Macmillan and Anderson demonstrated the utility of this ligation approach at an accessible glycine–glycine junction in the synthesis of an O-glycopeptide fragment bearing a single GalNAc residue [50]. We have reported several examples of the use of such auxiliary-based ligations for the construction of complex glycopeptides bearing multiple O- and N-glycans [51]. In a representative case, a glycopeptide acceptor containing a cleavable thiol-containing trimethoxybenzyl auxiliary on the N-terminus was ligated with a masked glycopeptide thioester at a glycine –glutamine junction to afford the desired glycopeptide bearing two N-linked glycans (Scheme 6).
Scheme 6.

Synthesis of a complex model N-glycopeptide using an auxiliary-based, cysteine-free ligation strategy
As shown, under the reducing ligation conditions, O → S acyl transfer occurred at the o-disulfide phenolic ester as previously described (see above), to provide the thioester, which underwent transthioesterification with the thiol-containing glycopeptide (upon in situ reduction of the auxiliary disulfide bond); the transient intermediate underwent an S → N acyl transfer to generate the thermodynamically favored amide bond of the doubly glycosylated peptide adduct. The auxiliary was subsequently removed through sequential methylation of the free thiol to prevent the reverse acid-mediated N → S acyl shift, followed by TFA treatment.
While useful as a tool for peptide or glycopeptide ligation, the general application of these cysteine-free ligation strategies presents some drawbacks. First, steric hindrance at the ligation site renders the key S → N acyl transfer step difficult and, as a result, this method can only be effectively applied to junctions where at least one of the amino acids is glycine or alanine. Another potential problem lies in the harsh acidic conditions required for cleavage of the thiol-auxiliary, which could compromise the labile glycosidic linkages within the glycopeptide. An alternative approach to the development of non-cysteine-based ligation strategies is to utilize a thiol-containing amino acid surrogate located at the N-terminus of the peptide fragment. In this strategy, the thiol moiety acts as a temporary “handle” to promote ligation with the corresponding peptide coupling partner. After the ligation reaction, the thiol functionality is removed through desulfurization to afford the natural amino acid at the site of ligation (Scheme 7).
Scheme 7.
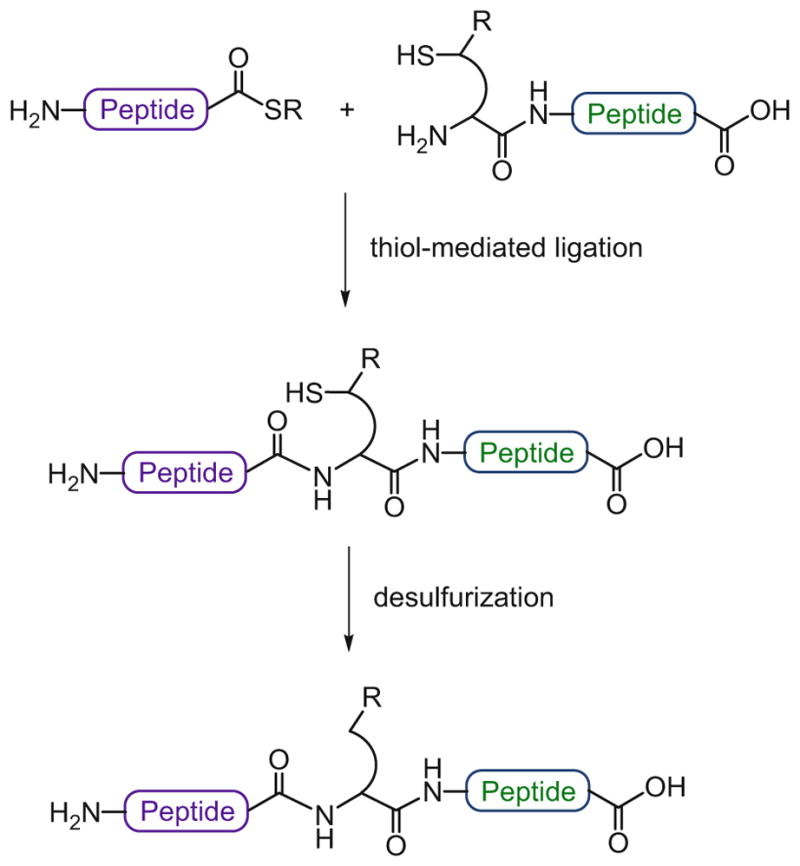
Non-cysteine ligation followed by desulfurization
This idea was first introduced by Yan and Dawson, who took advantage of cysteine-based NCL and then converted the N-terminal cysteine residue into alanine by reduction of the side chain with Raney nickel or Pd/Al2O3 [52]. In essence, this method provided a means to use cysteine as an alanine surrogate, which is a much more abundant amino acid in natural protein sequences. Subsequent work from Pentelute and Kent showed that unprotected cysteine residues could be desulfurized in the presence of methionine and Acm-protected cysteine residues with Raney nickel, although a large excess of the metal was required [53]. In 1998, González and Valencia had introduced a photochemical method for the efficient removal of the thiol group of cysteine without metal reagents, using triethyl phosphite as the nucleophile and triethylborane in acetonitrile as a radical initiator [54]. Driven by our interest in the total synthesis of complex glycoproteins, which often contain extensive and sensitive functional groups, we developed a mild, metal-free protocol for the selective reduction of cysteine to alanine compatible with a wide range of functionalities, including carbohydrate, thiazolidine, biotin, and thioester moieties [55]. This novel desulfurization strategy is aqueous-compatible and employs TCEP as a reducing agent, in conjunction with the water-soluble radical initiator VA-044 and tert-butylthiol as a thiol additive. This mild, radical-based protocol represented a significant improvement over previously reported metal-based NCL desulfurization methods and has been successfully applied in the synthesis of a number of complex proteins and glycoproteins [56–59]. Having identified a highly versatile cysteine reduction method, this ligation/ desulfurization approach opened the door to a new set of thiol-based ligation strategies with a range of different amino acid residues. Thus, through recourse to desulfurization methods, the reach of the NCL logic was extended, in our laboratory and others, to include alanine [52, 55], valine [60, 61], threonine [62], leucine [63, 64], lysine [65, 66], proline [67, 68], phenylalanine [69], methionine [70, 71], serine [72], arginine [73], aspartate [74], tryptophan [75], and glutamate [76] as viable ligation sites. Overall, these non-cysteine-based thiol ligation strategies, which are referred to as extended NCL reactions, followed by post-ligation desulfurization, have greatly facilitated the total chemical synthesis of complex, biologically relevant proteins and glycoproteins.
3.2 Total Synthesis of Homogeneous N-Glycosylated Proteins
Our laboratory has a long-standing interest in the total synthesis of naturally occurring, complex N-linked glycoproteins of important therapeutic value. This pursuit arises from the motivation to understand the structural and functional effects of protein glycosylation at the molecular level and, hopefully, to bring these complex biologics into the realm of structures that can be accessed by chemical means for structure-activity relationship studies. We have primarily focused our synthetic efforts on the human glycoprotein hormones (hGPH), granulocyte–macrophage colony-stimulating factor (GM-CSF) and EPO.
3.2.1 Human Glycoprotein Hormones
We are interested in the synthesis of a family of glycoproteins involved in a variety of important biological processes – the human glycoprotein hormones (hGPH). The members of this family are follicle stimulating hormone (FSH), luteinizing hormone (LH), chorionic gonadotropin (CG), and thyroid stimulating hormone (TSH). These glycoproteins are composed of two non-covalently associated subunits, a common α-subunit (α-hGPH) and a unique hormone-specific β-subunit (β-FSH, β-LH, β-CG, and β-TSH), each of which contains diverse carbohydrates at defined glycosylation sites. As the α-subunit is shared amongst these glycoproteins, we initially focused on the synthesis of the α-subunit (α-GPH) as the first step to access the human glycoprotein hormones.
Human α-Glycoprotein Hormone (α-hGPH)
In our early studies, a simplified version of this domain incorporating chitobiose as a model glycan was synthesized as a proof of principle. The validated route was then applied to prepare the α-hGPH bearing the dibranched, sialylated and fucosylated N-linked dodecasaccharide. This complex-type N-glycan is a consensus sequence which has also been found to exist on the β-subunits of the human glycoproteins and incorporates the key features associated with these hormones. The challenging synthesis of this sugar, which involves approximately 70 chemical steps [77], utilizes glycal chemistry to build the core trisaccharide and highlights the use of the Sinaÿ radical glycosylation [78] for the simultaneous coupling of both biantennary side-chains of the dodecasaccharide. A seven-step deprotection procedure followed by Kochetkov amination [79] afforded the fully deprotected N-glycan bearing an anomeric amine (not shown).
With the synthetic dodecasaccharide amine in hand, we then built up the corresponding glycopeptide fragments using the Lansbury aspartylation reaction [28] with peptide thioesters α-hGPH[31–58] and α-hGPH[59–81], themselves prepared by Fmoc-based SPPS followed by single amino acid attachment under Sakakibara’s epimerization-free conditions [80] (Scheme 8). After removal of all acid-labile protecting groups with a TFA-based cocktail, the fully deprotected (except Acm-containing cysteines) α-hGPH[82–92] and glycosylated α-hGPH [59–81] and α-hGPH[31–58] were then combined by means of a sequential two-step protocol involving ligation and subsequent cysteine unmasking. Lastly, the peptide thioester α-hGPH[1–30] (prepared by SPPS and direct thioesterification of the C-terminal glycine residue) was coupled with α-hGPH[31–92] via NCL to provide the Acm-protected primary sequence of α-hGPH featuring two defined, complex N-linked glycans [81].
Scheme 8.

Synthesis of the α-subunit of human glycoprotein hormones, α-hGPH[1–92]
Human β-Follicle Stimulating Hormone (β-hFSH)
As our first complete target (with both α and β subunits synthesized), we selected hFSH, a member of the family of the gonadotropic hormones which play a key role in the regulation and maintenance of important reproductive processes. Naturally synthesized and secreted in the anterior pituitary gland, it has been used in clinical settings to treat anovulatory disorders associated with infertility [82]. Currently, hFSH is obtained from recombinant techniques as a complex mixture of glycoforms. This variability may well be the cause of the formidable liabilities and side effects associated with its use [83]. Faced with the unavailability of homogeneous forms of hFSH, chemical synthesis seems to be the most straightforward approach to overcome this limitation.
The β-subunit of hFSH (β-FSH) was synthesized as described in Scheme 9 [84]. The individual peptide and glycopeptide segments were prepared by SPPS (with subsequent Sakakibara elongation or C-terminal glycine thioesterification to obtain the corresponding thioesters) and Lansbury aspartylation. Thus, fragment β-hFSH[66–111], peptide thioester β-hFSH[28–65], and glycosylated β-hFSH[20–27] were combined by NCL followed by cleavage of the N-terminal thiazolidine (Thz) blocking group in a stepwise manner. The last glycopeptide segment β-hFSH [1–19] was then joined under NCL conditions to form the glycosylated β-subunit of hFSH with two N-linked dodecasaccharides at the native glycosylation sites and Acm-protecting groups at the cysteine residues.
Scheme 9.

Synthesis of the β-subunit of human follicle stimulating hormone, β-hFSH[1–111]
Human β-Luteinizing Hormone (β-hLH) and β-Chorionic Gonadotropin Hormone (β-hCG)
Human luteinizing hormone (hLH) and chorionic gonadotropin hormone (hCG) belong to the family of the gonadotropins and are implicated in the stimulation of the gonadal and endocrine functions, as well as in pregnancy. hLH is released by the pituitary gland and hCG is produced in the human placenta. They both have similar physiological functions, and are used clinically as fertility drugs in reproductive medicine [85]. The β-subunit of hCG has been found to be overexpressed in several types of cancers and encompasses an epitope for hCG-based monoclonal antibodies. These features have made hCG a promising target for important biomedical and therapeutic applications [86, 87]. hLH and hCG share a high degree of similarity in their peptide sequence although hCG incorporates an extended C-terminal domain with four O-glycosylation sites. Thus, β-hLH contains 121 residues and only one N-glycosylation site (Asn30), whereas β-hCG comprises 145 amino acids and incorporates both N-linked (Asn13 and Asn30) and O-linked sugars (Ser121, Ser127, Ser132, and Ser138), making it the longest and most complex human glycoprotein hormone. Several studies investigating the importance of the oligosaccharide chains on the biological activity of these hormones suggest a more relevant role of the underlying carbohydrate residues vs the peripheral sugar units [88]. We sought to gain access to homogeneous glycoforms of β-hLH and β-hCG by chemical means for further exploration of the influence of the glycans on hormone action and function. Initially, we accomplished the synthesis of the simpler β-subunit of hLH bearing chitobiose as a model glycan to explore the feasibility of the synthetic route. Upon successful completion of the target molecule by sequential NCL, we then applied this strategy to assemble the more complex hCG β-subunit bearing not only two N-glycans but also four O-linked sugars [89].
The synthesis included two key features to access both terminal domains of the molecule. A successful double Lansbury aspartylation enabled the simultaneous, convergent installation of two chitobiose units onto the N-terminus of the peptide backbone to provide β-hCG[1–37]. The C-terminal fragment bearing closely spaced O-glycosylation sites, β-hCG[110–145], was prepared following a more linear “cassette” approach, whereby the four O-linked glycosylamino acids were sequentially incorporated in a single, solid-supported synthesis. The assembly of the Acm-protected, full-length β-hCG containing two N-linked (chitobiose) and four O-linked glycans (GalNAc) was performed in a modular and convergent fashion by ligation of the individual (glyco)peptide fragments (i.e., O-glycosylated β-hCG[110–145], β-hCG[72–109], β-hCG[38–71], and N-glycosylated β-hCG[1–37]) from the C- to the N-terminus, using a multiple NCL-based strategy (Scheme 10). Importantly, this synthesis of the β-subunit of hCG represents the longest human glycoprotein hormone (hGPH) accessed as a single glycoform by strictly chemical means.
Scheme 10.

Synthesis of β-subunit of human chorionic gonadotropin hormone, β-hCG[1–145]
Having accomplished the total synthesis of both α- and β-subunits of these glycosylated proteins, we are now investigating the late-stage challenges towards preparing homogeneous, folded heterodimeric hFSH, hLH, and hCG in our laboratory.
3.2.2 Granulocyte–Macrophage Colony-Stimulating Factor (GM-CSF)
Our continuing interest in complex, therapeutic-level glycoproteins led us to pursue the synthesis of homogeneous glycoforms of GM-CSF with the aim of studying the effect of glycosylation on the biological activity of this target. GM-CSF is a glycoprotein cytokine which promotes cell growth and proliferation and functions as an immune modulator. It is used to stimulate the immune system after bone-marrow transplant and chemotherapy and is being investigated as a vaccine immunoadjuvant [90]. The GM-CSF structure comprises 127 amino acids with both N- and O-glycosylation sites. Our aim was to establish a synthetic strategy to enable access to a number of homogeneous glycosylated variants of GM-CSF. We designed a route with maximal convergence whereby the full-length molecule would be assembled by merging three (glyco)peptide fragments using exclusively alanine ligations.
In the synthesis of a bis-N-glycosylated version containing the chitobiose disaccharide, glycopeptide GM-CSF[34–80] and peptide GM-CSF[81–127] were first connected via NCL to give, following Thz cleavage, GM-CSF[34–127] (Scheme 11) [91]. This fragment was then combined with GM-CSF[1–33], containing a chitobiose moiety, to provide complex glycopeptide intermediate GM-CSF[1–127]. The late-stage transformations towards the doubly-glycosylated GM-CSF congener involved reduction of the two free thiol groups via metal-free desulfurization (MFD) to reveal the native alanine residues, followed by deprotection of the four Acm-bearing cysteine residues. A monoglycosylated variant bearing a single chitobiose (at Asn37) was prepared via a similar synthetic route. Final folding of both fully synthetic analogues under glutathione redox buffering conditions provided the structurally defined GM-CSF glycoforms bearing either one (Asn37) or two (Asn27 and Asn37) chitobiose units at asparagine wild-type sites. Evaluation of the in vitro and in vivo biological activity of these synthetic GM-CSF samples, including a non-glycosylated congener, showed that they all function in a similar manner to commercially available GM-CSF, which is obtained recombinantly as a mixture of glycoforms.
Scheme 11.

Synthesis of the bis-glycosylated GM-CSF glycoform
3.2.3 Erythropoietin (EPO)
Erythropoietin (EPO) is a glycoprotein hormone primarily implicated in the regulation of red blood cell production in the bone marrow, a process known as erythropoiesis [92–94]. Recombinantly derived EPO has been used for the treatment of anemia associated with renal failure and cancer chemotherapy [95]. However, production of recombinant EPO results in a complex mixture of glycoforms, where the primary protein sequence and the glycosylation sites are highly conserved but the carbohydrate domains present significant heterogeneity. Despite studies suggesting that specific sugar modifications affect the stability and erythropoietic activity of EPO [96, 97], the unavailability of single glycoforms has complicated elucidation of the relationship between EPO glycosylation and its biological activity. Chemical synthesis uniquely holds the potential to provide access to homogeneous EPO for systematic evaluation of the effect of the glycan structure on biological activity.
EPO is composed of a 166-amino acid backbone incorporating three N-linked glycans (Asn24, Asn38, and Asn83) and an O-linked glycan (Ser126). In our first total synthesis of homogeneously glycosylated EPO, we chose to include three simpler chitobiose units and the naturally-abundant glycophorin tetrasaccharide at the four native glycosylation sites [98]. From a retrosynthetic perspective, we envisioned gaining access to the full-length glycoprotein, EPO[1–166], by assembling four glycopeptide domains through iterative NCL-based ligations (Scheme 12).
Scheme 12.
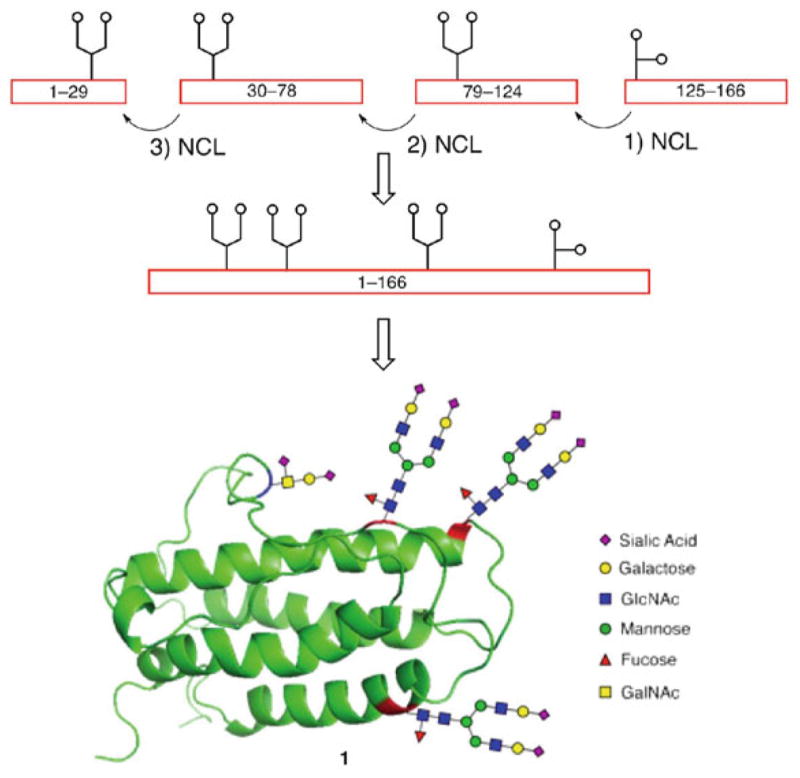
Proposed synthetic strategy for convergent synthesis of erythropoietin, EPO[1–166] and ribbon diagram of the fully glycosylated protein
The O-linked glycophorin was introduced into the peptide chain as a glycosylated serine cassette [99] via addition of a single amino acid at each end of the glycophorin-linked Ser residue followed by NCL of the resulting glycosylated tripeptide with the adjacent peptide segment to provide EPO[125–166] (Scheme 13).
Scheme 13.

Synthesis of EPO[125–166]
The chitobiose-containing N-glycopeptide constructs were prepared by our one-flask Lansbury aspartylation/deprotection protocol [30] from the corresponding peptide thioester fragments, themselves obtained by Fmoc-based SPPS followed by single amino acid attachment. On the basis of our previous experience toward a non-glycosylated version of EPO [59], we envisioned a synthetic strategy for merging the four component glycopeptides via a series of iterative alanine ligations, followed by late-stage desulfurization to convert the non-native cysteines into the requisite alanine residues. This first-generation synthesis featured a kinetically controlled NCL (KCL)-based approach [100] to enable the one-flask coupling of three fully elaborated EPO fragments in a highly convergent fashion (Scheme 14). In this KCL route, the aryl thioester of EPO[1–29] undergoes transthioesterification with the N-terminal cysteine of EPO[30–78] at a faster rate than the alkyl thioester, resulting in selective ligation between Cys-29 and Cys-30. Thus, EPO[1–29] and EPO[30–78] were first merged by NCL; upon completion of this ligation, EPO[79–166], itself prepared by NCL between EPO[79–124] and EPO[125–166], was added to assemble the protein sequence. The synthesis was completed by MFD, followed by removal of the Acm groups to afford the primary structure of homogeneously glycosylated EPO.
Scheme 14.

Initial convergent synthesis of EPO
An alternative, more linear strategy was also developed, whereby the full protein chain was assembled via NCL as the final step of the synthetic sequence. The main advantage of this route is that it avoids the need for post-ligation transformations on the full-length sequence prior to protein folding. The approach employs peptides which slightly differ from the fragments used in the convergent route in that the ligation site at the N-terminus is shifted by one residue. Thus, glycopeptide fragments EPO[125–166], EPO[79–124], and EPO[29–78] were combined by successive NCLs to afford the EPO[29–166] domain (Scheme 15). At this point, desulfurization of the non-native cysteines that participated in the ligations, and subsequent Acm deprotection on the remaining cysteine residues set the stage for the merger of the final peptide fragment, EPO[1–28], by NCL. The fully synthetic, homogeneously glycosylated EPO[1–166] thus obtained was then subjected to disulfide oxidation and protein folding to provide a simplified variant of folded EPO which, despite having truncated N-linked glycans, demonstrated erythropoietic activity.
Scheme 15.

Linear synthesis of erythropoietin, EPO[1–166]
Notwithstanding the major breakthrough of generating a fully synthetic version of EPO as a single glycoform, our ultimate goal was to achieve the total synthesis of “wild-type” EPO with consensus carbohydrate structures of realistic, biological-level complexity at all four native glycosylation sites. Therefore, we set out on this effort with the objective of incorporating the elaborate biantennary dodecasaccharide at the three N-glycosylation sites and glycophorin at Ser126 [101]. We initially envisioned adapting our previously described linear strategy, developed in the context of the chitobiose glycoform, to this more complex target. However, the greater steric bulk of the full N-glycans rendered ligation of EPO[29–78] and EPO[79–166] fruitless. Accordingly, the synthetic route was modified and a different ligation site, more distant from the problematic, large dodecasaccharide, was selected. In the newly designed approach, the domain related to pre-EPO[29–124] was reconfigured into three novel fragments, N-glycosylated EPO[29–59] and EPO[60–97], and peptide segment EPO[98–124], which were sequentially assembled, starting from the O-glycosylated, C-terminal domain EPO[125–166] via a series of smooth alanine ligations followed by thiazolidine unmasking (Scheme 16). The resulting fragment EPO[29–166] was then subjected to fourfold MFD to reveal the required alanines. After cleavage of the Acm protecting groups, cysteine-based NCL between EPO[29–166] and EPO[1–28] afforded the complete primary sequence of EPO containing wild-type complex glycan structures at each of the four glycosylation sites. This unfolded material gave rise to a mass spectrum of marginal quality (see Supporting Information in Wang et al. [101]). Not surprisingly, the material assigned as the EPO primary sequence did not even exhibit in vitro activity.
Scheme 16.

Synthesis of fully glycosylated EPO[1–166] bearing wild-type glycans at each of the four glycosylation sites
Upon exposure of the EPO primary sequence to folding conditions, a product was obtained which was reported [101] to exhibit strong erythropoietic activity and, thus, presumed to correspond to a biologically active version of compound 1 (Scheme 12) (although our synthetic EPO showed suitable activity in an in vivo assay, literature precedent suggests that heterogeneous mixtures of tetra-antennary structures have approximately 7× the in vivo activity of analogous bi-antennary EPO glycoforms [102–104]). More recently, an approximately 1-year-old sample of this material, bearing identical mobility properties to those reported from the original sample (see Wang et al. [101]), was found to retain approximately 15% of the in vitro activity of Procrit. It has been found that thawing and re-freezing of the sample tend to erode its erythropoietic activity. In retrospect, however, the inability to obtain a supportive mass spectrum of this material raises concerns with respect to the nature of the folded product. Related sialic acid-containing recombinantly derived EPO mixtures of unspecified structure gave rise to useful mass spectra in the context of glycoform profiling studies ([105, 106] and references therein). The corresponding author of reference [101] assumes full responsibility for not taking cognizance of these precedents. Further clarification of the matter of folding of the presumed precursor would require access to freshly prepared material, which can only be accomplished by repetition of the total synthesis. This has recently been undertaken in order to settle these questions.
4 Conclusion
The total synthesis of glycosylated proteins has long posed a daunting challenge to practitioners of organic chemistry. Recent key developments in this field, including NCL, non-cysteine-based ligations, MFD, and novel glycosylation methodologies, have led to the realization of the long-standing goal of gaining synthetic access to homogeneous peptides, proteins, and glycoproteins. Several truncated-carbohydrate versions of important biologically active glycoproteins have been synthesized in homogeneous form by a combination of these methods. These include GM-CSF, TSH, and EPO. An application to EPO bearing sialic acid-containing biantennary 12-mer N-glycosides has also been reported. The nature of the previously described biologically active version of this compound warrants further investigation because we did not obtain a useful mass spectrum to support its structure.
In parallel to efforts to achieve the total synthesis of glycoproteins by strictly chemical means, a number of powerful chemical and biological technologies have emerged for making tailored, full-size glycoproteins bearing glycan-defined domains for the study of specific biological questions. These highly enabling strategies, which have been extensively reviewed elsewhere, include the chemoselective, site-specific glycosylation of recombinant proteins via the so-called “tag and modify” approach [107], in vitro chemoenzymatic glycosylation remodeling of heterogeneous glycoproteins [108], direct enzymatic glycosylation of proteins [109], specific glyco engineering of host biosynthetic pathways [108], and in vivo suppressor tRNA technology [110, 111].
All these approaches complement each other and it is envisaged that, either alone or in combination, they will continue to enable the generation of glycosylated proteins with predesigned modifications and increasing complexity for the elucidation of the molecular basis of protein glycosylation. This realization will certainly contribute to the development of novel and improved glycoprotein-derived therapeutics in the next few years.
Acknowledgments
We thank the National Institutes of Health, William and Alice Goodwin and the Commonwealth Foundation for Cancer Research for generous financial support. AF-T gratefully acknowledges the European Commission (Marie Curie International Outgoing Fellowship) for funding. We acknowledge our colleagues of the Danishefsky laboratory whose work is presented in this manuscript. We thank Professors Yasuhiro Kajihara and Carlo Unverzagt for provocative exchanges and, particularly, for citing precedence on the feasibility of obtaining informative mass spectra of related EPO structures. We thank Rebecca Wilson for assistance in the preparation of the manuscript.
Contributor Information
Alberto Fernández-Tejada, Email: aftejada@cib.csic.es, Laboratory for Bioorganic Chemistry, Molecular Pharmacology and Chemistry Program, 1275 York Avenue, New York, NY 10065, USA. Chemical and Physical Biology, CIB-CSIC, Ramiro de Maeztu 9, Madrid 28040, Spain.
John Brailsford, Laboratory for Bioorganic Chemistry, Molecular Pharmacology and Chemistry Program, 1275 York Avenue, New York, NY 10065, USA.
Qiang Zhang, Laboratory for Bioorganic Chemistry, Molecular Pharmacology and Chemistry Program, 1275 York Avenue, New York, NY 10065, USA.
Jae-Hung Shieh, Cell Biology Program, Sloan Kettering Institute for Cancer Research, 1275 York Avenue, New York, NY 10065, USA.
Malcolm A.S. Moore, Cell Biology Program, Sloan Kettering Institute for Cancer Research, 1275 York Avenue, New York, NY 10065, USA
Samuel J. Danishefsky, Email: s-danishefsky@ski.mskcc.org, Laboratory for Bioorganic Chemistry, Molecular Pharmacology and Chemistry Program, 1275 York Avenue, New York, NY 10065, USA
References
- 1.Walsh G, Jefferis R. Nat Biotechnol. 2006;24:1241. doi: 10.1038/nbt1252. [DOI] [PubMed] [Google Scholar]
- 2.Apweiler R, Hermjakob J, Sharon N. BBA-Gen Subj. 1999;1473:4. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 3.Dwek RA. Chem Rev. 1996;96:683. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 4.Varki A. Glycobiology. 1993;3:97. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohtsubo K, Marth JD. Cell. 2006;126:855. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 6.Miller LH, Good MF, Milon G. Science. 1994;264:1878. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 7.Dube DH, Bertozzi CR. Nat Rev Drug Discov. 2005;4:477. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 8.Hudak JE, Bertozzi CR. Chem Biol. 2014;21:16. doi: 10.1016/j.chembiol.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalziel M, Crispin M, Scanlan CN, Zitzmann N, Dwek RA. Science. 2014;343:1235681. doi: 10.1126/science.1235681. [DOI] [PubMed] [Google Scholar]
- 10.Bosques CJ, Tschampel SM, Woods RJ, Imperiali B. J Am Chem Soc. 2004;126:8421. doi: 10.1021/ja049266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imperiali B, O’Connor SE. Curr Opin Chem Biol. 1999;3:643. doi: 10.1016/s1367-5931(99)00021-6. [DOI] [PubMed] [Google Scholar]
- 12.Wormald MR, Petrescu AJ, Pao YL, Glithero A, Elliott T, Dwek RA. Chem Rev. 2002;102:371. doi: 10.1021/cr990368i. [DOI] [PubMed] [Google Scholar]
- 13.Li H, d’Anjou M. Curr Opin Biotechnol. 2009;20:678. doi: 10.1016/j.copbio.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Spiro RG. Glycobiology. 2002;12:43R. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 15.Rudd PM, Dwek RA. Crit Rev Biochem Mol Biol. 1997;32:1. doi: 10.3109/10409239709085144. [DOI] [PubMed] [Google Scholar]
- 16.Rich JR, Wither SG. Nat Chem Biol. 2009;5:206. doi: 10.1038/nchembio.148. [DOI] [PubMed] [Google Scholar]
- 17.Bertozzi CR, Kiessling LL. Science. 2001;291:2357. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 18.Hojo H, Nakahara Y. Biopolymers. 2007;88:308. doi: 10.1002/bip.20699. [DOI] [PubMed] [Google Scholar]
- 19.Buskas T, Ingale S, Boons GJ. Glycobiology. 2006;16:113R. doi: 10.1093/glycob/cwj125. [DOI] [PubMed] [Google Scholar]
- 20.Merrifield RB. J Am Chem Soc. 1963;85:2149. [Google Scholar]
- 21.Yamamoto N, Ohmori Y, Sakakibara T, Sasaki K, Juneja LR, Kajihara Y. Angew Chem Int Ed. 2003;42:2537. doi: 10.1002/anie.200250572. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto N, Takayanagi A, Yoshino A, Sakakibara T, Kajihara Y. Chem Eur J. 2007;13:613. doi: 10.1002/chem.200600179. [DOI] [PubMed] [Google Scholar]
- 23.Mathieux N, Paulsen H, Meldal M, Bock K. J Chem Soc Perkin Trans. 1997;1:2359. [Google Scholar]
- 24.Herzner H, Reipen T, Schultz M, Kunz H. Chem Rev. 2000;100:4495. doi: 10.1021/cr990308c. [DOI] [PubMed] [Google Scholar]
- 25.Gaidzik N, Westerlind U, Kunz H. Chem Soc Rev. 2013;42:4431. doi: 10.1039/c3cs35470a. [DOI] [PubMed] [Google Scholar]
- 26.Wilson RM, Danishefsky SJ. J Am Chem Soc. 2013;135:14462. doi: 10.1021/ja405932r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakshminarayanan V, Thompson P, Wolfert MA, Buskas T, Bradley JM, Pathangey LB, Madsen CS, Cohen PA, Gendler SJ, Boons GJ. Proc Natl Acad Sci U S A. 2012;109:261. doi: 10.1073/pnas.1115166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen-Anisfeld ST, Lansbury PT. J Am Chem Soc. 1993;115:10531. [Google Scholar]
- 29.Bodanszky M, Natarajan S. J Org Chem. 1975;40:2495. doi: 10.1021/jo00905a016. [DOI] [PubMed] [Google Scholar]
- 30.Wang P, Aussedat B, Vohra Y, Danishefsky SJ. Angew Chem Int Ed. 2012;51:11571. doi: 10.1002/anie.201205038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ullmann V, Rädisch M, Boos I, Freund J, Pöhner C, Schwarzinger S, Unverzagt C. Angew Chem Int Ed. 2012;51:11566. doi: 10.1002/anie.201204272. [DOI] [PubMed] [Google Scholar]
- 32.Aussedat B, Vohra Y, Park PK, Fernández-Tejada A, Alam SM, Dennison SM, Jaeger FH, Anasti K, Stewart S, Blinn JH, Liao HX, Sodroski JG, Haynes BF, Danishefsky SJ. J Am Chem Soc. 2013;135:13113. doi: 10.1021/ja405990z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Payne RJ, Wong CH. Chem Comm. 2010;46:21. doi: 10.1039/b913845e. [DOI] [PubMed] [Google Scholar]
- 34.Dawson P, Muir T, Clark-Lewis I, Kent SBH. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 35.Muir TW. Annu Rev Biochem. 2003;72:249. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 36.Macmillan D, Bertozzi CR. Angew Chem Int Ed. 2004;43:1355. doi: 10.1002/anie.200352673. [DOI] [PubMed] [Google Scholar]
- 37.Kent SBH. Chem Soc Rev. 2009;38:338. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 38.Unverzagt C, Kajihara Y. Chem Soc Rev. 2013;42:4408. doi: 10.1039/c3cs35485g. [DOI] [PubMed] [Google Scholar]
- 39.Shin Y, Winans KA, Backes BJ, Kent SBH, Ellman JA, Bertozzi CR. J Am Chem Soc. 1999;121:11684. [Google Scholar]
- 40.Marcaurelle LA, Mizoue LS, Wilken J, Oldham L, Kent SB, Handel TM, Bertozzi CR. Chem Eur J. 2001;7:1129. doi: 10.1002/1521-3765(20010302)7:5<1129::aid-chem1129>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 41.Mezzato S, Schaffrath M, Unverzagt C. Angew Chem Int Ed. 2005;44:1650. doi: 10.1002/anie.200461125. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto N, Tanabe Y, Okamoto R, Dawson PE, Kajihara Y. J Am Chem Soc. 2008;130:501. doi: 10.1021/ja072543f. [DOI] [PubMed] [Google Scholar]
- 43.Mende F, Seitz O. Angew Chem Int Ed. 2011;50:1232. doi: 10.1002/anie.201005180. [DOI] [PubMed] [Google Scholar]
- 44.Kan C, Danishefsky SJ. Tetrahedron. 2009;65:9047. doi: 10.1016/j.tet.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan Y, Chen J, Wan Q, Wilson RM, Danishefsky SJ. Biopolymers. 2010;94:373. doi: 10.1002/bip.21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warren JD, Miller JS, Keding SJ, Danishefsky SJ. J Am Chem Soc. 2004;126:6576. doi: 10.1021/ja0491836. [DOI] [PubMed] [Google Scholar]
- 47.Wan Q, Chen J, Yuan Y, Danishefsky SJ. J Am Chem Soc. 2008;130:15814. doi: 10.1021/ja804993y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan Y, Chen J, Wan Q, Tan Z, Chen G, Kan C, Danishefsky SJ. J Am Chem Soc. 2009;131:5432. doi: 10.1021/ja808705v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haase C, Seitz O. Angew Chem Int Ed. 2008;47:1553. doi: 10.1002/anie.200704886. [DOI] [PubMed] [Google Scholar]
- 50.Macmillan D, Anderson DW. Org Lett. 2004;6:4659. doi: 10.1021/ol048145o. [DOI] [PubMed] [Google Scholar]
- 51.Wu B, Chen J, Warren JD, Chen G, Hua Z, Danishefsky SJ. Angew Chem Int Ed. 2006;45:4116. doi: 10.1002/anie.200600538. [DOI] [PubMed] [Google Scholar]
- 52.Yan LZ, Dawson PE. J Am Chem Soc. 2001;123:526. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- 53.Pentelute BL, Kent SBH. Org Lett. 2007;9:687. doi: 10.1021/ol0630144. [DOI] [PubMed] [Google Scholar]
- 54.González A, Valencia G. Tetrahedron Asymmetry. 1998;9:2761. [Google Scholar]
- 55.Wan Q, Danishefsky SJ. Angew Chem Int Ed. 2007;46:9248. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 56.Shang S, Tan Z, Danishefsky SJ. Proc Natl Acad Sci U S A. 2011;108:5986. doi: 10.1073/pnas.1103118108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong S, Shang S, Li J, Tan Z, Dean T, Maeda A, Gardella TJ, Danishefsky SJ. J Am Chem Soc. 2012;134:15122. doi: 10.1021/ja306637u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Dong S, Townsend SD, Dean T, Gardella TJ, Danishefsky SJ. Angew Chem Int Ed. 2012;51:12263. doi: 10.1002/anie.201207603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brailsford JA, Danishefsky SJ. Proc Natl Acad Sci U S A. 2012;109:7196. doi: 10.1073/pnas.1202762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen J, Wan Q, Yuan Y, Zhu J, Danishefsky SJ. Angew Chem Int Ed. 2008;47:8521. doi: 10.1002/anie.200803523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haase C, Rohde H, Seitz O. Angew Chem Int Ed. 2008;47:6807. doi: 10.1002/anie.200801590. [DOI] [PubMed] [Google Scholar]
- 62.Chen J, Wang P, Zhu J, Wan Q, Danishefsky SJ. Tetrahedron. 2010;66:2277. doi: 10.1016/j.tet.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harpaz Z, Siman P, Kumar KSA, Brik A. ChemBioChem. 2010;11:1232. doi: 10.1002/cbic.201000168. [DOI] [PubMed] [Google Scholar]
- 64.Tan Z, Shang S, Danishefsky SJ. Angew Chem Int Ed. 2010;49:9500. doi: 10.1002/anie.201005513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang R, Pasunooti KK, Li F, Liu X-W, Liu C-F. J Am Chem Soc. 2009;131:13592. doi: 10.1021/ja905491p. [DOI] [PubMed] [Google Scholar]
- 66.Ajish Kumar KS, Haj-Yahya M, Olschewski D, Lashuel HA, Brik A. Angew Chem Int Ed. 2009;48:8090. doi: 10.1002/anie.200902936. [DOI] [PubMed] [Google Scholar]
- 67.Shang S, Tan Z, Dong S, Danishefsky SJ. J Am Chem Soc. 2011;133:10784. doi: 10.1021/ja204277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Townsend SD, Tan Z, Dong S, Shang S, Brailsford JA, Danishefsky SJ. J Am Chem Soc. 2012;134:3912. doi: 10.1021/ja212182q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crich D, Banerjee A. J Am Chem Soc. 2007;129:10064. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]
- 70.Tam JP, Yu Q. Biopolymers. 1998;46:319. doi: 10.1002/(SICI)1097-0282(19981015)46:5<319::AID-BIP3>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 71.Saporito A, Marasco D, Chambery A, Botti P, Monti SM, Pedone C, Ruvo M. Biopolymers. 2006;83:508. doi: 10.1002/bip.20582. [DOI] [PubMed] [Google Scholar]
- 72.Okamoto R, Kajihara Y. Angew Chem Int Ed. 2008;47:5402. doi: 10.1002/anie.200801097. [DOI] [PubMed] [Google Scholar]
- 73.Malins LR, Cergol KM, Payne RJ. ChemBioChem. 2013;14:559. doi: 10.1002/cbic.201300049. [DOI] [PubMed] [Google Scholar]
- 74.Thompson RE, Chan B, Radom L, Jolliffe KA, Payne RJ. Angew Chem Int Ed. 2013;52:9723. doi: 10.1002/anie.201304793. [DOI] [PubMed] [Google Scholar]
- 75.Malins LR, Cergol KM, Payne RJ. Chem Sci. 2014;5:260. [Google Scholar]
- 76.Cergol KM, Thompson RE, Malins LR, Turner P, Payne RJ. Org Lett. 2014;16:290. doi: 10.1021/ol403288n. [DOI] [PubMed] [Google Scholar]
- 77.Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ. J Am Chem Soc. 2009;131:5792. doi: 10.1021/ja809554x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang YM, Mallet JM, Sinaÿ P. Carbohydr Res. 1992;236:73. doi: 10.1016/0008-6215(92)85007-m. [DOI] [PubMed] [Google Scholar]
- 79.Likhosherstov LM, Novikova OS, Derevitskaja VA, Kochetkov NK. Carbohydr Res. 1986;146:C1. doi: 10.1016/0008-6215(90)84093-a. [DOI] [PubMed] [Google Scholar]
- 80.Sakakibara S. Biopolymers. 1995;37:17. doi: 10.1002/bip.360370105. [DOI] [PubMed] [Google Scholar]
- 81.Aussedat B, Fasching B, Johnston E, Sane N, Nagorny P, Danishefsky SJ. J Am Chem Soc. 2012;134:3532. doi: 10.1021/ja2111459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Howles CM. Hum Reprod Update. 1996;2:172. doi: 10.1093/humupd/2.2.172. [DOI] [PubMed] [Google Scholar]
- 83.Pang SC. Womens Health. 2005;1:87. doi: 10.2217/17455057.1.1.87. [DOI] [PubMed] [Google Scholar]
- 84.Nagorny P, Sane N, Fasching B, Aussedat B, Danishefsky SJ. Angew Chem Int Ed. 2012;51:975. doi: 10.1002/anie.201107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stenman UH, Tiitinen A, Alfthan H, Valmu L. Hum Reprod Update. 2006;12:769. doi: 10.1093/humupd/dml029. [DOI] [PubMed] [Google Scholar]
- 86.Talwar GP, Vyas HK, Purswani S, Gupta JC. J Reprod Immunol. 2009;83:158. doi: 10.1016/j.jri.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 87.Iversen PL, Mourich DV, Moulton HM. Curr Opin Mol Ther. 2003;5:156. [PubMed] [Google Scholar]
- 88.Valmu L, Alfthan H, Hotakainen K, Birken S, Stenman UH. Glycobiology. 2006;16:1207. doi: 10.1093/glycob/cwl034. [DOI] [PubMed] [Google Scholar]
- 89.Fernández-Tejada A, Vadola PA, Danishefsky SJ. J Am Chem Soc. 2014;136:8450. doi: 10.1021/ja503545r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, Zhang Y, Zhang L, Yuan ZR, Tan HS, Das G, Devadas S. Cell Res. 2006;16:126. doi: 10.1038/sj.cr.7310017. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Q, Johnston EV, Shieh J-H, Moore MAS, Danishefsky SJ. Proc Natl Acad Sci U S A. 2014;111:2885. doi: 10.1073/pnas.1400140111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krantz SB. Blood. 1991;77:419. [PubMed] [Google Scholar]
- 93.Lacombe C, Mayeux P. Haematologica. 1998;83:724. [PubMed] [Google Scholar]
- 94.Koury MJ. Exp Hematol. 2005;33:1263. doi: 10.1016/j.exphem.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 95.Tonelli M, Hemmelgarn B, Reiman T, Manns B, Reaume MN, Lloyd A, Wiebe N, Klarenbach S. CMAJ. 2009;180:E62. doi: 10.1503/cmaj.090470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mattio M, Ceaglio N, Oggero M, Perotti N, Amadeo I, Orozco G, Forno G, Kratje R, Etcheverrigaray M. Biotechnol Prog. 2011;27:1018. doi: 10.1002/btpr.633. [DOI] [PubMed] [Google Scholar]
- 97.Higuchi M, Oh-eda M, Kuboniwa H, Tomonoh K, Shimonaka Y, Ochi N. J Biol Chem. 1992;267:7703. [PubMed] [Google Scholar]
- 98.Wang P, Dong S, Brailsford JA, Iyer K, Townsend S, Zhang Q, Hendrickson RC, Shieh JH, Moore MAS, Danishefsky SJ. Angew Chem Int Ed. 2012;51:11576. doi: 10.1002/anie.201206090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schwarz JB, Kuduk SD, Chen X-T, Sames D, Glunz PW, Danishefsky SJ. J Am Chem Soc. 1999;121:2662. [Google Scholar]
- 100.Bang D, Pentelute BL, Kent SBH. Angew Chem Int Ed. 2006;45:3985. doi: 10.1002/anie.200600702. [DOI] [PubMed] [Google Scholar]
- 101.Wang P, Dong S, Shieh J-H, Peguero E, Hendrickson R, Moore MAS, Danishefsky SJ. Science. 2013;342:1357. doi: 10.1126/science.1245095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Takeuchi M, Inoue N, Strickland TW, Kubota M, Wada M, Shimizu R, Hoshi S, Kozutsumi H, Takasaki S, Kobata A. Proc Natl Acad Sci U S A. 1989;86:7819. doi: 10.1073/pnas.86.20.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kochendoerfer GG, Chen S-Y, Mao F, Cressman S, Traviglia S, Shao H, Hunter CL, Low DW, Cagle EN, Carnevali M, Gueriguian V, Keogh PJ, Porter H, Stratton SM, Wiedeke MC, Wilken J, Tang J, Levy JJ, Miranda LP, Crnogoran MM, Kalbag S, Potti P, Schindler-Horvat J, Savatski L, Adamson JW, Kung A, Kent SBH, Bradburne JA. Science. 2003;299:884. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- 104.Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, Bobrowicz P, Stadheim TA, Li H, Choi B-K, Hopkins D, Wischnewski H, Roser J, Mitchell T, Strawbridge RR, Hoopes J, Wildt S, Gerngross TU. Science. 2006;313:1441. doi: 10.1126/science.1130256. [DOI] [PubMed] [Google Scholar]
- 105.Sanz-Nebot V, Benavente F, Vallverdú A, Guzman NA, Barbosa J. Anal Chem. 2003;75:5220. doi: 10.1021/ac030171x. [DOI] [PubMed] [Google Scholar]
- 106.Harazono A, Hashii N, Kuribayashi R, Nakazawa S, Kawasaki N. J Pharm Biomed Anal. 2013;87:65. doi: 10.1016/j.jpba.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 107.Chalker JM, Bernardes GJ, Davis BG. Acc Chem Res. 2011;44:730. doi: 10.1021/ar200056q. [DOI] [PubMed] [Google Scholar]
- 108.Wang LX, Lomino JV. ACS Chem Biol. 2012;7:110. doi: 10.1021/cb200429n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang LX, Amin MN. Chem Biol. 2014;21:51. doi: 10.1016/j.chembiol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang L, Schultz PG. Angew Chem Int Ed. 2005;44:34. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 111.Liu L, Bennett CS, Wong CH. Chem Comm. 2006;42:21. doi: 10.1039/b513165k. [DOI] [PubMed] [Google Scholar]