

Abstract
Peptide hormones and neurotransmitters are of central importance in most aspects of intercellular communication and are involved in virtually all degenerative diseases. In this review, we discuss physicochemical approaches to the design of novel peptide and peptidomimetic agonists, antagonists, inverse agonists, and related compounds that have unique biological activity profiles, reduced toxic side effects, and, if desired, the ability to cross the blood-brain barrier. Designing ligands for specific biological and medical needs is emphasized, as is the close collaboration of chemists and biologists to maximize the chances for success. Special emphasis is placed on the use of conformational (φ-ψ space) and topographical (χ space) considerations in design.
Keywords: peptide pharmacophores, peptide hormones, peptide neurotransmitters, blood-brain barrier, topographical structures, conformation and biological activity, peptide stability
INTRODUCTION
In multicellular animal life, including human life, the maintenance of good health depends on coordinated communication among the different cell types that make a complex organism function properly. This cooperation of cells that have different functions is made possible by intercellular communication in which hormones and neurotransmitters modulate the expressed genome for homeostasis. Changes in the expressed genome lead to a breakdown in intercellular communication such that the affected cells and organs no longer interact with one another properly, resulting in dysfunctions such as prolonged and neuropathic pain, cancer, heart disease, and diabetes.
In this review, we discuss some of the efforts that have been made to examine the chemical and biological properties of peptide hormones and neurotransmitters. We emphasize (a) how creating appropriate chemical tools on the basis of the structures of these hormones and neurotransmitters can inform us of the biological mechanisms related to various behaviors and biological actions involved in health and disease, and (b) how this approach can lead to drugs that can be used to detect and treat these diseases.
GENERAL CONSIDERATIONS RELATED TO PEPTIDES, ESPECIALLY PEPTIDE HORMONES AND NEUROTRANSMITTERS
Peptide hormones and neurotransmitters act as chemical switches that modulate most biological functions of multicellular organisms. These functions include most behaviors, maintenance of homeostasis, and many aspects of development. Generally, peptide hormones and neurotransmitters are produced and stored for use in granules and are released in response to chemical signals. Because peptide hormones and neurotransmitters are chemical switches, nature must have a chemical method for turning them off; this often is accomplished via proteases in the vicinity of the receptors for the hormone or neurotransmitter. These peptides are also biological switches, so their general properties have led to the widespread myth that peptides are unstable to proteolysis. However, many peptides and proteins remain in the body for long times (hours, days, even months). Furthermore, many chemical approaches can be used to stabilize peptides against proteolysis e.g., (1–3), and, since the design of deamino-oxytocin by du Vigneaud and colleagues (4), peptide hormone and neurotransmitter structure-activity relationship (SAR) studies have made the stabilization of peptides against proteolysis an important consideration. Subsequent approaches (5) include D–amino acid replacements, amide bond replacements, conformational constraints, cyclization, and the use of N-methylation and novel amino acid substitutions. From a practical perspective, the problem of stabilizing peptides against proteolysis has been solved (6).
Most organic molecules do not cross the blood-brain barrier well. For small organic molecules, some general approaches have been suggested, of which Lipinski’s rules (7) are the most widely used. Peptides can cross the blood-brain barrier (e.g., 8–14). However, Lipinski’s rules do not apply, and small-molecule considerations are usually irrelevant.
Finally, the use of peptides and peptidomimetics as drugs has inherent advantages. As mentioned above, peptides and proteins are the major modulators of all aspects of animal behavior including human behavior, and most degenerative diseases result from changes in the expressed genome that lead to dysfunction. Therefore, one can suggest that peptides and proteins have superior abilities in modulating disease states and, in some cases, returning the dysfunctional state to a homeostatic state with little or no toxicity. Furthermore, peptides—even rather small peptides with 2–10 residues—have inherent three-dimensional (3D) properties. Consequently, ancillary sites (15, 16) enable chemical modification (e.g., with fluorophores, drugs, imaging agents) with no or minimal loss in biological activity. Peptides, therefore, have revolutionary potential in serving as drugs for early detection and treatment of disease.
GENERAL CONSIDERATIONS RELATED TO STRUCTURE-ACTIVITY RELATIONSHIPS OF PEPTIDE HORMONES AND NEUROTRANSMITTERS
A central hypothesis of chemical biology and molecular pharmacology is that when there is a change in biological activity, there must be a corresponding change in chemical structure. In the case of peptide hormones and neurotransmitters, proper consideration of this central hypothesis requires careful, comprehensive thinking about both pharmacological and chemical structure. Whereas enzymes modulate biological function by making and breaking covalent bonds, peptide hormones and neurotransmitters modulate biological function by binding to receptors, thereby changing the structure of the receptors by noncovalent chemical interactions. The peptide-receptor complex has a new conformation that depends on (a) the basic interactions with the ligand; (b) whether the ligand is an agonist, a partial agonist/antagonist, a neutral antagonist, or an inverse agonist; and (c) whether the ligand is orthosteric or allosteric. A different downstream biological effect occurs for each of these cases, and a different structure (ligand-receptor complex) can take a variety of pathways (17–21). These and other related issues not discussed here suggest that the current paradigm of examining binding affinities and second-messenger effects often provides insufficient information for choosing ligands for further development.
Comprehensive examination of peptide and protein structure must be addressed in the design of peptides and peptidomimetic ligands. Most peptide hormones and neurotransmitters are linear peptides or cyclic disulfide-containing peptides that may have secondary structure preferences in φ-ψ-ω space (backbone conformation/Ramachandran space) such as α-helical, β-sheet, β-turn, or extended conformations (Supplemental Figure 1; follow the Supplemental Materials link from the Annual Reviews home page at http://www.annualreviews.org). However, they are conformationally flexible enough to assume different conformations depending on their interacting partners (e.g., receptors, cytokines, secretory granules, enzymes, membranes, blood-borne proteins). All these conformational states are important during a peptide’s in vivo life. Thus, SAR design requires taking all these factors into consideration and gathering multiple inputs from pharmacology, computational chemistry, and biophysical analysis (22, 23).
Whereas secondary structure is a critical aspect in peptide- and protein-ligand design, the structure of the peptide in χ space when it interacts with its biological partners is more important. In this regard, the pharmacophore for a peptide ligand depends on the chemical nature of the side-chain groups of specific amino acid residues in the peptide. Therefore, the 3D relationships of these side-chain groups should determine a peptide’s ultimate biological activities. The energy differences among the low-energy conformations are quite small for α amino acids (0–2 kcal mol−1), and the energy barriers are such that, generally, all three low-energy torsional angles are available at physiological temperatures. However, for particular interactions with biological acceptors/receptors, a basic hypothesis of chemical biology states that specific torsional angles are preferred for key pharmacophore elements. Indeed, the design of peptides and peptidomimetics in χ space can be a powerful tool that can provide unique insights into the 3D SAR of peptide hormones and neurotransmitters (for reviews, see References 22–26).
DESIGN OF PEPTIDE AND PEPTIDOMIMETIC HORMONES THAT HAVE SPECIFIC BIOLOGICAL ACTIVITY PROFILES
Many natural peptide hormones and neurotransmitters interact with several different receptors—for example, enkephalin and dynorphin interact with OPRM1 (μ opioid receptor), OPRD1 (δ opioid receptor), and OPRK1 (κ opioid receptor)—so developing selective orthosteric and allosteric agonists and antagonists to understand the significance of particular receptors in the modulation of a specific bioactivity in normal and disease states is critical. (Hereafter, the terms OPRM1, OPRD1, and OPRK1 are used interchangeably with μ opioid receptor, δ opioid receptor, and κ opioid receptor, respectively.)
A major initial goal for peptide hormones and neurotransmitters, once the natural peptide ligand or lead ligand is discovered, is to determine the key residues that are essential for biological activity at the ligand’s receptor (the pharmacophore residues). Determining the key residues can be accomplished via several approaches, one of which is the alanine scan (6). Generally, essential residues are either continuous or discontinuous in the peptide sequence, and other residues (known as the address residues) are involved in enhancing the binding affinity. Efficacy studies are important because partial agonists and weak antagonists can provide important leads in the development of antagonists. Antagonists are essential tools for evaluating the biological functions that are specifically related to a particular hormone or neurotransmitter and to its corresponding receptor. A second goal is to determine whether the hormone or neurotransmitter can interact with the other receptors in a particular receptor subfamily. Because a primary goal of many SAR studies is to obtain receptor-selective ligands, SAR studies must have, from the onset, multiple-receptor binding affinity assays and efficacy (second-messenger) assays that include off-target interactions. In addition, researchers should utilize in vivo assays that examine specific biological outcomes related both to normal function and to appropriate animal models for disease states.
AGONIST DEVELOPMENT: δ OPIOID RECEPTOR
Most natural peptide hormones and neurotransmitters are nonselective agonists, so the primary goal is the development of more potent, selective, amphiphilic, and lipophilic molecules that are stable against proteolytic breakdown, able to cross (or not cross) the blood-brain barrier, and amenable to chemical modification for attachment of fluorescent or other imaging agents while potency and selectivity are maintained. The development of c[D-Pen2, D-Pen5]enkephalin (DPDPE) as a biologically stable, highly selective δ opioid receptor ligand and the development of the related analogs of deltorphin illustrate some of the primary approaches to develop ligands for use in a wide variety of biological and potential medical applications (Figure 1). For more comprehensive discussions, see Reference 6.
Figure 1.
Conversion of enkephalin into potent and δ opioid receptor-selective ligands.
The starting point is the native peptide neurotransmitter [Met5]enkephalin (Figure 1). The Tyr1 α-amino and phenyl hydroxyl groups and the Phe4 aromatic group were established early on as the key pharmacophore residues in enkephalin for agonist activity (27). The enkephalins had moderate binding affinities for all three opioid receptors and had short half-lives in vivo. Thus, obtaining highly potent and selective analogs for μ, δ, and κ opioid receptors became an important goal. Modifying residues in the address and other regions of the linear ligand led to some success (e.g., 28).
Investigators employed a cyclization strategy in which the 2 and 5 positions of enkephalin were substituted with the sulfhydryl amino acids cysteine and penicillamine (Pen = β, β-dimethylcysteine) to make derivatives that were 13-membered medium-sized rings (29). Modeling studies suggested that this would constrain the peptide into a conformation in the β-turn regions of a φ-ψ (Ramachandran) space (30). Comprehensive binding affinity and functional bioassays of several cyclic derivatives with these substitutions were examined (e.g., 31) (Table 1). Those with one or more penicillamine residues were found to be OPRD1 selective, and DPDPE was the most potent and selective (DPDPE does not bind to the OPRK1) (Table 1). Nuclear magnetic resonance (NMR) (32) and computational studies showed that DPDPE has a β-turn conformation in its 13-membered cyclic ring, and its X-ray crystal structure (33) showed that a similar conformation was found in the solid crystal and in the aqueous and dimethylsulfoxide solutions. In vivo studies of DPDPE’s antinociceptive activity demonstrated that it had potent analgesic activities. DPDPE and its radiolabel forms also are stable to enzymatic breakdown and are able to cross the blood-brain barrier (34, 35), and DPDPE has been a useful compound for many in vivo studies.
Table 1.
Binding affinity assays and biological assays of penicillamine-containing cyclic enkephalins
| Compound | Binding affinity assays | Biological assays | ||
|---|---|---|---|---|
| vs[3H]DADLE δ (nM) |
vs[3H]naloxone μ (nM) |
MVD (nM) |
GPI (nM) | |
| c[D-Pen2,Cys5]enka | 11.7 | 178 | 0.32 | 213 |
| c[D-Pen2,D-Cys]enka | 26 | 157 | 6.3 | 1,350 |
| c[D-Pen2,Cys5]enk-NH2a | 3.4 | 73 | 3.6 | 118 |
| c[D-Pen2,D-Cys5]enk-NH2a | 7.2 | 162 | 16.8 | 117 |
| c[D-Pen2,D-Pen5]enk (DPDPE) | 1.6 | 610 | 4.1 | 7,300 |
| [2S,3S]TMT1-DPDPE | 211 | 720 | 170 | 290 |
| [2S,3R]TMT1-DPDPE | 5.0 | 4,300 | 1.8 | 0% at 60 μM (antagonist) |
| [2R,3S]TMT1-DPDPE | 9% at 10 μM | 0% at 10 μM | 28% at 10 μM | 75% at 10 μM |
| [2R,3R]TMT1-DPDPE | 3,500 | 77,000 | 2,200 | 75% at 82 μM |
Data taken from Reference 29.
Abbreviations: DADLE, [D-Ala2,D-Leu5]enkephalin; DPDPE, c[D-Pen2,D-Pen5]enkephalin; enk, enkephalin; GPI, guinea pig ileum; MVD, mouse vas deferens; TMT, β-methyl-2′,6′-dimethyltyrosine (trimethyltyrosine).
The conformational SAR studies led to further investigations into topographical requirements for DPDPE at the OPRD1, particularly the three preferred side-chain conformations of the Tyr1 and Phe4 aromatic residues that enable DPDPE’s agonist activity. To examine this aspect of 3D bioactivity in 3D space, researchers designed novel amino acid derivatives that retain the key low-energy gauche conformation in χ space [i.e., gauche (−), gauche (+), and trans conformations; see Supplemental Figure 1]. Constraining the key low-energy gauche conformations is necessary because the amino acids Tyr and Phe do not have highly preferred gauche conformations with large energy barriers between them. Computational chemistry was used to design in silico a variety of α amino acids with different kinds of conformational constraints (e.g., 22–24). These novel structures were deemed important for biological activities on the basis of the hypothesis (see above) that changes in the structure of pharmacophore elements can lead to novel compounds with new functional activities. For example, on the basis of conformational and topographical considerations, the Tyr1 residue in DPDPE is particularly interesting because it is not part of the cyclic ring structure and thus has a highly flexible conformation in both φ-ψ and χ space. Therefore, constraining this amino acid in χ space will lead to novel and readily determined conformational preferences.
For the purpose of constraining tyrosine in χ space, β-methyl-2′,6′-dimethyltyrosine (TMT) is particularly useful (Figure 2). This molecule has two chiral carbon atoms and thus four possible isomers (2S,3S; 2S,3R; 2R,3S; and 2R,3R). The low-energy gauche conformations for the 2S,3R compound are given in Figure 2. Similarly, each of the three other isomers also has three low-energy gauche conformations, and each has its own unique topography. All four isomers of TMT were incorporated into the peptide to fully elucidate the topographical preferences for the Tyr pharmacophore moiety at the OPRD1 (36, 37). An asymmetric synthesis of all four isomers of TMT was developed (37), each isomer was incorporated into DPDPE (38), and the binding affinities and in vitro activities of all four isomers of TMT1-DPDPE (38, 39) were examined (Table 1). As predicted, the four TMT1-containing DPDPE analogs have different biological activity profiles (Table 1). Only the [2S,3R]TMT1-DPDPE analog mimics the binding affinity profile of DPDPE. This clearly demonstrated that the trans χ1 conformation for Tyr1 is preferred at the OPRD1. This analog interacts only in the micromolar range at the OPRM1, and in the functional assay it is a weak antagonist (39) at the OPRM1. Conformational analysis using NMR showed that the conformation of the cyclic moiety of DPDPE was not changed by the presence of the modified tyrosine moieties. This was the first example in which a single topographical change in χ space in a peptide hormone, protein hormone, or neurotransmitter ligand could lead to agonist activity at one receptor subtype and to antagonist activity at a different receptor subtype. Because agonist and antagonist interactions with opioid receptors lead to different 3D conformations of the ligand-receptor complex, these results clearly show that changes in topographical space can lead to different biological activities; these findings were confirmed by in vivo assays for pain (39). Interestingly, DPDPE had an effect on downregulation of the receptor that differed from the effect of the nonpeptide δ opioid receptor ligand SNC80 on downregulation of the receptor (40).
Figure 2.
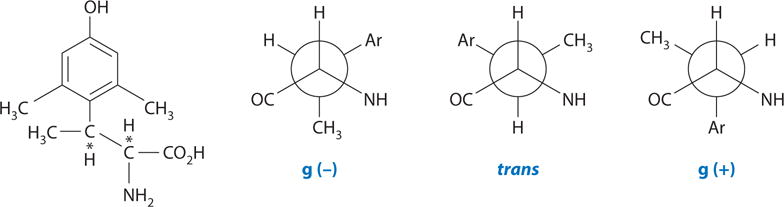
Structure of β-methyl-2′,6′-dimethyltyrosine (trimethyltyrosine, TMT) and its low-energy side-chain gauche conformations for the 2S,3R isomer.
DEVELOPMENT OF δ OPIOID RECEPTOR ANTAGONISTS: TOPOGRAPHICAL CONSIDERATIONS
The availability of all four isomers of β-methyl-2′,6′-dimethyltyrosine (trimethyltyrosine, TMT) discussed above provided an opportunity to examine the topographical requirement of the Tyr residue at the OPRD1. Schiller and coworkers (41) showed that the dipeptide Tyr-Tic was an antagonist at the OPRD1 and that the dipeptide unit was the smallest peptide to retain significant binding to the OPRD1. Salvadori et al. (42) later showed that use of 2′,6′-dimethyltyrosine (DMT) to give DMT-Tic resulted in a more potent and selective δ opioid antagonist. Neither Tyr nor DMT is topographically constrained to any significant extent at χ1, so we decided to investigate the application of TMT (a) to modify the Tyr-Tic antagonist, and (b) to examine all four analogs: [2S,3S]TMT-Tic, [2S,3R]TMT-Tic, [2R,3S]TMT-Tic, and [2R,3R]TMT-Tic (43). The results of binding affinity assays and bioassays at the δ and μ opioid receptors are given in Table 2. In this case, the [2S,3R]-containing dipeptide was the most potent and selective OPRD1 antagonist in the binding affinity assays and in the mouse vas deferens (OPRD1) bioassay. The dipeptide had weak and partial agonist activity in the guinea pig ileum (OPRM1) assay (Table 2). Further extensive studies demonstrated that [2S,3R]TMT-L-Tic-OH was a potent inverse agonist at the hOPRD1 (44) and a potent and selective OPRD1 neutral antagonist in the mouse brain (45). These comprehensive studies demonstrate several important insights that could be obtained using topographical constraints in addition to those of enhanced potency and receptor selectivity already discussed:
The topographical requirements of the tyrosine pharmacophore moiety for agonist and antagonist activity of opioid peptides at the OPRD1 are similar. The difference in efficiency (antagonist versus agonist) must lie elsewhere in the ligand-receptor binding interaction.
At other subtypes of the receptor, a simple topographical difference in χ space can determine whether a ligand is an agonist or antagonist. For example, [2S,3R]TMT-L-Tic-OH is a partial agonist at the OPRM1, albeit a weak one. Thus, conformational constraints in χ space for key residues can modulate agonist versus antagonist activity. This use of χ constrained amino acids provides a novel approach to the design of antagonists for peptide hormone and neurotransmitter receptors. For key pharmacophore residues, agonists and antagonists can have different topographical requirements.
Topographically constrained peptide hormones and neurotransmitters can provide a powerful tool in conjunction with computational chemistry to develop peptide mimetics from peptides in 3D space and to compare peptide and nonpeptide pharmacophores for the same receptor.
Table 2.
Binding affinity assays and biological assays of β-methyl-2′,6′-dimethyltyrosine (trimethyltyrosine, TMT) analogs of Tyr-Tic
| Compound | Binding affinity assays | Biological assays | ||
|---|---|---|---|---|
| vs[3H]pCl-DPDPE(δ) IC50 (nM) | [3H]DAMGO(μ) IC50 (nM) | MVD (δ) | GPI (μ) | |
| H-L-Tyr-L-Tic-OHa | 190 ± 50 | 28,000 ± 2,900 | – | – |
| [2S,3S]TMT-L-Tic-OHb | 120 ± 27 | >80,000 | ND | ND |
| [2S,3R]TMT-L-Tic-OH | 9.3 ± 0.53 | 35,000 ± 18,000 | 1 μM shifts DPDPE 547-fold | 1 μM does not antagonize PL-017 30% at 30 μM |
| [2R,3S]TMT-L-Tic | >10,000 | >80,000 | ND | ND |
| [2R,3R]TMT-L-Tic | >10,000 | >80,000 | ND | ND |
DESIGN OF NONPEPTIDE LIGANDS FOR A RECEPTOR FROM A PEPTIDE NEUROTRANSMITTER LEAD
Once there is detailed knowledge of the topographical requirements for the pharmacophore of a G protein–coupled receptor such as the OPRD1, including the conformation–biological activity relationship in χ space for potent and receptor-selective ligands, it is possible to design de novo a nonpeptide ligand for that receptor directly from the knowledge of the ligand’s 3D requirement, even without the receptor’s X-ray structure. Here, molecular modeling and computational chemistry of the constrained peptide ligands in 3D vector space can be used. In the case of a δ receptor ligand, the key pharmacophore moieties (terminal amino group, phenylhydroxyl group, and phenyl groups) in the 3D space that are derived from studies such as those discussed above allow one to evaluate the use of several different small-molecule scaffolds, primarily heterocyclic scaffolds, as possible replacements for the peptide backbone scaffold. The basic criterion used for rejection or acceptance of a particular scaffold was its ability to form covalent bonds with the pharmacophore elements without greatly disturbing the 3D relationships of the key pharmacophore moieties in topographical vector space. Figure 3 provides a simple picture of the 3D design in silico. The computational studies and 3D model building on various scaffolds (templates) are done in vector space so that the correct 3D relationships are retained in the structures evaluated. These structures are later relaxed to allow examination of the low-energy conformations (46). In the first-order design, investigators chose a scaffold that was as simple as possible: the piperazine scaffold substituted as shown in Figure 3. The placement of the primary amino group was uncertain; thus, the first exploration evaluated the nature of the hydrophobic R groups. The structures examined by total synthesis involved a five-step synthetic procedure that proceeded in high yields (40) with a variety of hydrophobic R groups ranging from H to p-t-butyl (Table 3). The binding affinity assays show that when R is small (H, Me, iBu), the analogs have micromolar to high nanomolar binding affinities at both the μ and δ opioid receptors. However, as the groups become more hydrophobic and bulky, the analogs bind with increased affinity to the OPRD1 and with reduced affinity to the OPRM1; the p-t-butyl-y-hydroxyphenyl analog, I E, shows the highest affinity and greatest δ opioid receptor selectivity (Table 3). I E is a potent agonist in the mouse vas deferens (δ) in vitro assay and a weak agonist in the guinea pig ileum (μ) in vitro assay. I E has a single chiral center, so both the (+) and (−) analogs were prepared, and the (−) analog was found to be more potent and more selective.
Figure 3.
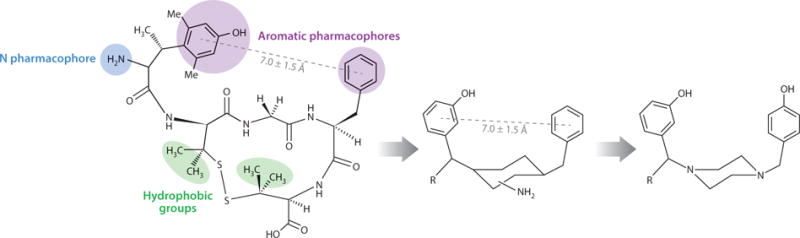
Use of c[D-Pen2, D-Pen5]enkephalin (DPDPE) and [2S,3R]TMT1-DPDPE to design a nonpeptide peptide mimetic for the δ opioid receptors.
Table 3.
Binding affinity assays and biological assays for design of nonpeptide opioid receptor-selective ligands based on TMT1-DPDPE
| Compound | Binding affinity assays | Biological assays | ||
|---|---|---|---|---|
| vs[3H]pCl-DPDPE IC50 (nM) | vs[3H]DAMGO IC50 (nM) | MVD EC50 (nM) | GPI EC50 (nM) | |
| [2S,3R]TMT1-DPDPE | 5.0 | 4,300 | 1.8 | antagonist |
| I A; R = H | 6,400 | 8,100 | – | – |
| I B; R = Me | 610 | 780 | – | – |
| I C; R = i-Bu | 420 | 2,100 | – | – |
| I D; R = C6H3 | 34 | 500 | 174 | 1,250 |
| I E; R = p-tBu-C6H4 | 8.4 | 17,000 | 85 | 39,000 |
| I E (+); R = p-tBu-C6H4 | 42 | 11,000 | 210 | 3,000 |
| I E (−); R = p-tBu-C6H4 | 41 | 7,700 | 360 | 7,600 |
| [MeO-Tyr1]DPDPE | 230 | 11,000 | – | – |
|
II; m-CH3O, R = p-tBu-C6H4 |
1,800 | >8,000 | – | – |
| DPDPE | 1.6 | 610 | 4.1 | 7,300 |
Abbreviations: DAMGO, [D-Ala2,N-MePhe4,Gly-ol]enkephalin; DPDPE, c[D-Pen2,D-Pen5]enkephalin; GPI, guinea pig ileum; MVD, mouse vas deferens; ND, not determined; TMT, β-methyl-2′,6′-dimethyltyrosine (trimethyltyrosine).
Although these results were supportive of the design, several additional SAR experiments were conducted to determine whether the new OPRD1 selective ligand was indeed a mimetic of DPDPE. In addition, the SAR of these ligands were compared with the SAR of other nonpeptide OPRD1 selective ligands, such as SNC80 (Supplemental Figure 2), to see whether the new δ opioid receptor ligand, or SNC80, was a true peptide mimetic (i.e., a mimetic with the same SAR as those of DPDPE). SNC80 has a methoxyl substitute in the aromatic moiety, and this derivative is much more OPRD1 selective than its hydroxyl-substituted analog, BW373U86 [SNC80 is 2,300-fold selective; BW373U86, 31-fold selective (47)]. Therefore, the methoxy derivative of I E (II, Table 3) was prepared. Contrary to what was found in the SNC80 and BW373U86 series, the opposite effect was observed (Table 3). The methoxy analog II was substantially less potent and less OPRD1 selective than the hydroxyl analog. So what is the case for [p-methoxy-L-tyrosine]DPDPE? To find out, investigators prepared [MeO-Tyr]DPDPE (Table 3) and discovered the ligand to be much less potent (230 nM versus 1.6 nM) and less selective (380-fold selective versus 48-fold selective) than DPDPE. Thus, I E has the same SAR as does DPDPE, whereas SNC80 has a different SAR. Furthermore, I E binds with almost the same affinity at both the wild-type receptor and the mutated receptor, whereas SNC80 binds substantially better at the wild-type receptor than at the mutated receptor (W284L). This result indicates that I E is a good peptide mimetic and has the same SAR as do peptides, whereas SNC80 is not a good peptide mimetic. Subsequently, the W284L mutant receptor was shown to retain agonist-specific mechanisms of G protein activation (48), and here again I E was shown to closely parallel DPDPE, but other nonpeptide ligands for the OPRD1 did not (49). A considerable effort was put into improving biological activity, and although some favorable additional leads were found, these efforts eventually were dropped when in vivo studies showed that at higher doses, the improved analogs had undesirable toxicities. Efforts to eliminate these toxicities have not been successful. When similar conformational constraints in χ space for the Tyr2 position of oxytocin (H-c[Cys-Try-Ile-Gln-Asn-Cys]-Pro-Leu-Gly-NH2) were made along with the p-methoxy-Tyr analog, potent analogs with superior properties and unique structural features were obtained (50).
CONVERTING SOMATOSTATIN TO A HIGHLY POTENT OPIOID RECEPTOR LIGAND
Many peptide hormones and neurotransmitters have off-target biological interactions, albeit at reduced binding affinities. This aspect of peptide chemistry and biology has not been well investigated, although this may change in the future because many of these off-target interactions are likely allosteric in nature, and this may provide unique opportunities for modulation of biological activity, especially in disease states. In this regard, Rezik et al. (51) reported that somatostatin had weak biological activity at opioid receptors. This led us to design somatostatin analogs with high potency and selectivity for opioid receptors and with little or no somatostatin-like activities. We started with somatostatin (Figure 4) and reduced the size of the peptide (52) while retaining binding affinity for the somatostatin receptor(s). Further modifications by Maurer et al. (53) showed antagonist activities at the opioid receptor (Figure 4, part I), which led to the design of somatostatin analogs with conformational constraints and other structural modifications that were appropriate for opioid receptors but not somatostatin receptors. Binding affinity and functional activity measurements were used as a guide to enhancing opioid-like binding affinities and biological activities while reducing the binding affinity for somatostatin receptors (54, 55). Both conformational constraints with cyclic structures and topographical constraints in χ space of specific structural features were applied to enhance two aspects of biological activity: (a) selectivity for OPRM1 versus OPRD1 activity, and (b) selectivity for opioid receptors versus somatostatin receptors. Table 4 summarizes some of the key findings from the extensive studies.
Figure 4.
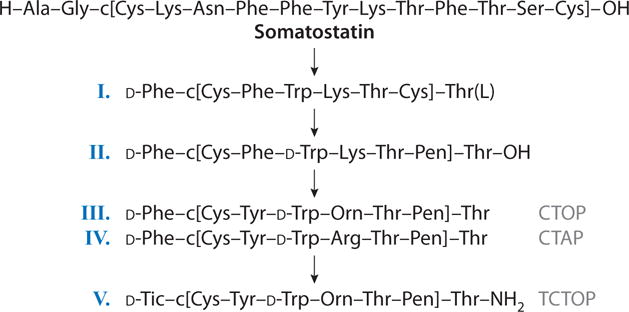
Converting somatostatin-14 to a highly potent μ opioid receptor-selective antagonist.
Table 4.
Binding affinity assays of somatostatin-like analogs with opioid receptors
| Compound | IC50, nM | μ versus δ selectivity | |
|---|---|---|---|
| Binding versus [3H]DPDPE | Binding versus [3H]CTOP | ||
| 1. Somatostatin | 15,000a | 20,000a | 0.7 |
| 2. D-Phe-c[Cys-Phe-D-Trp-Lys-Thr-Pen]Thr-OH | 20,000a | 1,000b | 20 |
| 3. D-Phe-c[Pen-Phe-D-Trp-Lys-Thr-Pen]Thr-OH | 9,800a | 10,800b | none |
| 4. D-Phe-c[Cys-Tyr-D-Trp-Lys-Thr-Pen]Thr-OH | 3,800a | 290b | 13 |
| 5. D-Phe-c[Cys-Tyr-D-Trp-Lys-Thr-Pen]Thr-NH2 | 950a | 3.5b | 270 |
| 6. D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Pen]Thr-NH2 | 2,000 | 5.5 | 360 |
| 7. D-Phe-c[Cys-Tyr-D-Trp-Arg-Thr-Pen]Thr-NH2 (CTAP) | 4,500 | 3.5 | 1,300 |
| 8. D-Phe-c[Cys-Trp-D-Trp-Orn-Thr-Pen]Thr-NH2 (CTOP) | 13,500 | 2.8 | 4,800 |
| 9. D-Tic-c[Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 | 1,300 | 1.2 | 1,100 |
| 10. D-Tic-c[Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 | 16,000 | 1.4 | 11,300 |
vs[3H]DADLE.
vs[3H]naloxone.
Abbreviations: DADLE, [D-Ala2,D-Leu5]enkephalin; DPDPE, c[D-Pen2,D-Pen5]enkephalin.
In particular, placing a Pen residue in position 7 of the octapeptide led to enhanced binding affinity at the δ opioid receptor (Table 4, analog 2), but not when penicillamine was substituted at positions 2 and 7 (Table 4, analog 3). Modifying the C terminus to a carboxamide terminus to stabilize the peptide against proteolysis also enhanced potency at both the μ and δ opioid receptors (Table 4, analog 5) and reduced affinity for the somatostatin activity (54). To further explore the SAR of the new OPRM1-selective analogs, we substituted the exocyclic D-Phe1 residue with the bicyclic χ-constrained amino acid D-tetrahydroisoquinoline carboxylate (D-Tic; see Supplemental Figure 3) to yield analogs 9 and 10 in Table 4. Both analogs are highly potent, with binding affinities of 1.2–1.4 nM at the OPRM1. The Orn5 analog 10 has 11,300-fold selectivity relative to OPRM1 and 15,000-fold selectivity relative to the somatostatin receptor (data not shown; e.g., 55). These peptides are highly stable in vitro and in vivo against proteolytic degradation (56) and have been widely used to study the biological function of the μ opioid receptor.
DEVELOPMENT OF MELANOTROPIN LIGANDS WITH UNIQUE BIOLOGICAL ACTIVITY PROFILES FOR BIOLOGICAL AND MEDICAL APPLICATIONS
Research on the melanotropin peptide hormones and neurotransmitters that are products of the primordial animal gene proopiomelanocortin provides an excellent example of how an unstable natural peptide hormone/neurotransmitter can be transformed into a bioavailable ligand that crosses the blood-brain barrier while retaining its key peptide/protein scaffold. Examples of such melanotropin peptide hormones and neurotransmitters include α melanocyte-stimulating hormone [also known as α-MSH (Ac-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2)], β-MSH, and γ-MSH. In the process, investigators developed potent and receptor-selective agonists and antagonists that can be used for medical applications. This research has led to human clinical trials at the College of Medicine at the University of Arizona (57–60) and elsewhere.
An initial goal of research on α-MSH was to obtain analogs that were more stable than α-MSH to proteolytic degradation and that maintained enhanced bioavailability in vivo so that its in vivo biological activities could be examined. Investigating the chemical basis for the rapid degradation of α-MSH and prolonging in vivo biological activities by base-catalyzed racemization of α-MSH led to the insight that a D-Phe7 residue was at least in part responsible (61). These and related studies (61, 62) led to the design of [Nle4,D-Phe7]α-MSH (NDP-α-MSH), melanotan-I (MT-I) (Figure 5), which has prolonged biological activities in vitro and in vivo (62, 63) and enhanced stability to proteolysis (64). [Norleucine (Nle) is the isosteric amino acid derivative of Met; see below.] NDP-α-MSH is stable to proteolysis for hours, and this added stability is likely due to the stabilization by β-turn conformation at the D-Phe7 position. To test this hypothesis, Sawyer et al. (65) converted the linear α-MSH to the cyclic peptide by preparing the cyclic disulfide-containing analog of α-MSH, c[Cys4,Cys10]α-MSH (Figure 5), which is a superpotent analog of α-MSH. NDP-α-MSH has been used as the ligand for in vitro and in vivo experiments at the MC1R, MC3R, MC4R, and MC5R receptors because of its potent agonist activity at all four of these melanocortin receptor subtypes. At the time NDP-α-MSH was developed, the MC3R, MC4R, and MC5R receptors had not been discovered. Because this analog was inactive at the adrenal receptor, it was initially considered to be a very selective analog.
Figure 5.
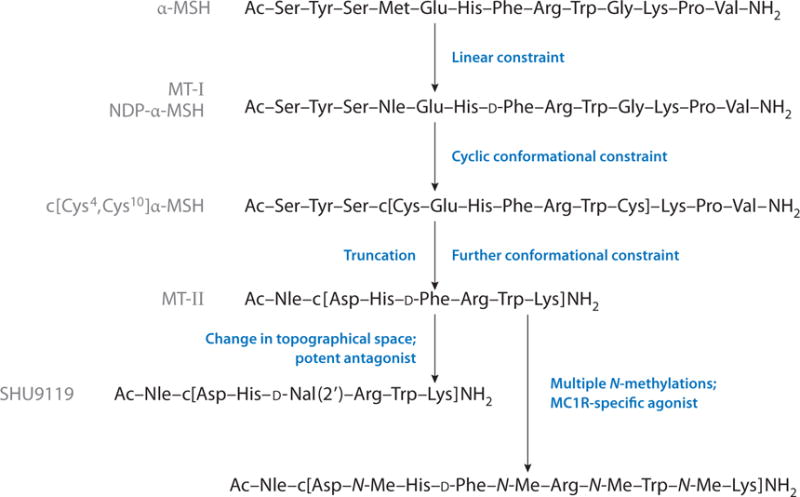
Development of novel analogs of α-MSH with novel biological activity profiles. Abbreviations: α-MSH, α melanocyte-stimulating hormone; MC, melanocortin; MT, melanotan; NDP-α-MSH, [Nle4,D-Phe7]α-MSH.
On the basis of computational studies and extensive SAR studies (e.g., 66), we designed Ac-Nle4-c[Asp5,D-Phe7,Lys10]α-MSH(4–10)-NH2, melanotan-II (MT-II) (67) (Figure 5). This compound is a superpotent agonist at the melanocortin receptors, has the ability to cross the blood-brain barrier, is highly stable against proteolytic degradation, and has prolonged bioactivity in vivo. Most importantly, MT-II exhibits exceptional stability and bioavailability, along with its potent agonist activity at melanocortin receptors MC1R, MC3R, MC4R, and MC5R, each of which utilizes MSH as its native ligand. In our laboratory, other academic institutions, and pharmaceutical and biotechnology companies, MT-II has served as a ligand for numerous biological studies and as a basic template for the further design of peptide and peptide mimetic ligands for melanocortin receptors.
Potent and selective antagonists are essential tools in demonstrating unambiguously that a particular ligand or drug manifests its bioactivity via the particular target in question. Furthermore, potent and specific antagonists often are useful for treatment of diseases involving a particular protein target (e.g., enzymes, receptors, growth factors, immune modulators). Despite years of effort, only minimal success in obtaining a potent melanocortin receptor antagonist had been achieved. In the course of examining topographical approaches, our group (22–24, 68) obtained a receptor-selective peptide and peptidomimetic ligand for the melanocortin receptors by substituting the D-Phe7 residue in MT-II with a 2′-naphthylalanine [D-Nal(2′)] to obtain Ac-Nle4-c[Asp5,D-Nal(2′)7,Lys10]α-MSH(4–10)-NH2, also known as SHU9119 (Figure 5). This analog was a potent antagonist at the MC3R and MC4R receptors but a potent agonist at the MC1R and MC5R receptors. The corresponding D-Nal(1′) analog, in contrast, is a potent agonist at all four melanocortin receptors for MSH ligands. That such a small topographical difference in a peptide hormone and neurotransmitter that has critically important biological functions (e.g., feeding behavior, sexual function, response to pain, learning behavior) demonstrates how minimal changes in the right structure can have profound effects on bioactivity and provides an important lesson in drug design. Since then, a wide variety of potent and variously selective peptide and peptidomimetic agonist and antagonist ligands for the melanocortin receptors MC1R, MC3R, MC4R, and MC5R (see References 69–74 for a few key references) have been developed. A couple of recent studies (75, 76) provide new structural information based on the pharmacophore for future directions in ligand design of melanocortin ligands.
For example, the effects of multiple peptide backbone N-methylations of the agonist Ac-Nle4-c[Asp5,D-Phe7,Lys10]α-MSH(4–10)-NH2, MT-II (75) and the antagonist SHU9119 (see above) have been investigated. In the case of MT-II, the multiple N-methylated analog Ac-Nle-c[Asp-N-MeHis-D-Phe-N-MeArg-N-MeTrp-N-MeLys]NH2 (Figure 5) is a potent and completely specific agonist ligand for the MC1R (75). This compound also has a unique conformation as determined by comprehensive NMR and computational studies that provided unique insights into the specific 3D requirements for melanocortin receptors. In addition, nonpeptide ligands that are related to MSH on the basis of modeling the MT-II conformation have been identified (76). These ligands are allosteric activators/inhibitors of the melanocortin receptors. Such ligands may provide alternate signaling/metabolic pathways that can offer novel approaches to the design and development of drugs with fewer side effects. For example, MT-II given peripherally in human clinical trials proved effective in psychogenic erectile dysfunction and in male and female sexual motivation. In those cases, both central and spinal melanocortin receptors were involved (e.g., 59, 77).
DEVELOPMENT OF GLUCAGON ANTAGONISTS AND INVERSE AGONISTS
Control of glucose homeostasis is complex, but two major hormones are thought to be the major modulators of the process: insulin and glucagon. Enormous efforts have been devoted to the design and study of insulin and insulin derivatives for the treatment of both type 1 and type 2 diabetes (78). Glucagon has received much less attention, but it has significant potential in the treatment of diabetes.
Glucagon (Figure 6) is a linear 29–amino acid peptide that is quite hydrophobic. It has low solubility in aqueous solutions at physiological pH and readily forms fibrils (amyloids) under both acidic and basic conditions. Its synthesis is difficult, so we began to examine its SAR by applying semisynthetic methods to obtain and prepare the des-His1 and the Des-Asn29-Thr29 homoserine lactone glucagon analogs. These studies established that the His1 residue was critical for full agonist activity (e.g., 79) and that glucagon (1–21) was a full agonist (80). Eventually, the approach led to the first glucagon antagonist found: semisynthetic (Des-His1) (Nε-phenylthiocarbamoyl Lys12)-glucagon(81), which could lower blood glucose levels in vivo in diabetic rats (82).
Figure 6.
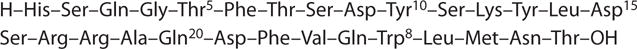
Structure of glucagon.
Further exploration of glucagon SAR by solid-phase synthesis of glucagon analogs revealed the importance of the N-terminal region (e.g., 83), the 10–13 region (84) (Figure 6), and the conformational differences between agonists and antagonists (85). In addition, Wakelam et al. (17) observed that glucagon could activate two signal-transduction systems; this was one of the first observations that a hormone or neurotransmitter could activate more than one downstream signaling system. Its significance was soon demonstrated in work on glucagon desensitization (86).
Further SAR studies led to the development of other glucagon antagonists, and in vitro and in vivo biological experiments demonstrated that these analogs still had partial agonist activity under some conditions (e.g., 87). After considerable effort, we found that by modifying the C-terminal residues and replacing Asp9 with Glu9, a potent pure glucagon antagonist (possibly an inverse agonist) could be obtained (88). Eventually, we discovered other antagonists that could be evaluated in sensitive assays for partial agonist activity (89, 90). These new glucagon antagonist analogs have inverse agonist activity and thus might provide a new mechanism for modulating and regulating in vivo glucose production and diabetic states; their evaluation for treatment of diabetes awaits future studies.
NEW PARADIGMS FOR DRUG DISCOVERY, MULTIVALENT LIGANDS, AND PROLONGED AND NEUROPATHIC PAIN AND TOLERANCE
All pain medicines have serious side effects, especially when used for prolonged periods. Therefore, we need new approaches to designing ligands for pain treatment that do not result in tolerance, dependence, or toxic side effects. Recent observations from comparative genomics and proteomics studies (e.g., 91–93) demonstrate that in many diseases, including prolonged and neuropathic pain, changes in gene expression (specifically, in ascending and descending pain pathways) lead to disease states, and that the treatments themselves can enhance aspects of the disease. For example, prolonged treatment by opioids leads not only to the development of tolerance and dependence but also, in many cases, to the development of the neuropathic pain states of allodynia and hyperalgesia. As a result of these and other biological observations of the nature of neuropathic pain, researchers developed a new approach in designing drugs to treat this disease state: It involves targeting with a single ligand two or more receptors/acceptors that are directly involved in the disease state and that can treat the disease state relative to the normal state (92). We illustrate this approach by discussing the design and development of multivalent ligands that are agonists at the μ and δ opioid receptors and antagonists at the neurokinin-1 (NK-1) receptors in a single molecule.
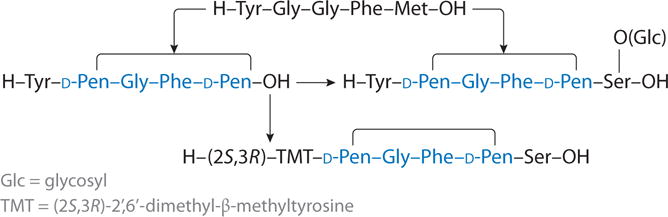
The past decade has seen the discovery that the development of neuropathic pain is accompanied by the upregulation of neurotransmitters and their receptors in pain pathways (both ascending and descending) that are stimulatory for neural transmission and that this upregulation can enhance pain perception. These compounds include peptide hormones; neurotransmitters such as substance P, calcitonin gene-related peptide, cholecystokinin, and MSH; and their receptors. The evidence for substance P and the NK-1 receptors is particularly well established (e.g., 93–95). The new approach involves the design of multivalent ligands that have μ and δ opioid agonist activity and NK-1 receptor antagonist activity. To ensure high potency and good in vivo stability and biodistribution properties, the design utilizes a peptidomimetic that contains at its N terminus the half-biphalin tetrapeptide (e.g., H-Tyr-D-Ala-Gly-Phe-) and at its C terminus a peptidomimetic tripeptide NK-1 receptor antagonist pharmacophore [e.g., -Pro-Leu-Trp-X, where X = 0–3′,5′-Bzl(CF3)2, or N-3′,5′-Bzl(CF3) or N-Bzl] (92, 96) with an intervening amino acid residue that can serve as an address for both the opioid and the NK-1 receptors (see Table 5 for structures of some of the most potent compounds).
Table 5.
Novel multivalent ligands for opioid (μ/δ) and NK-1 receptors and biological activity profiles for novel analgesics for neuropathic pain
| Compound | Binding affinity assays Ki (nM) | Efficacy (second-messenger) assays EC50 (nM)v | Functional assays IC50 (nM), Ki (nM) | |||||
|---|---|---|---|---|---|---|---|---|
| hDOR [3H]DPDPE | rMOR [3H]DAMGO | hNK1 [3H]SP | hDOR | hMOR | MVD(δ) | GPI(μ) | SPGPI Antagonist | |
| 1. H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-3′,5′-Bzl(CF3)2 | 2.8 | 36 | 0.084 | 2.9 | 32 | 22 | 360 | 25 |
| 2. H-Tyr-D-Ala-Gly-Phe-Nle-Pro-Leu-Trp-O-3′,5′-Bzl(CF3)2 | 1.8 | 9.7 | ND | 4.0 | 25 | 17 | 370 | 7.9 |
| 3. H-Tyr-D-Ala-Gly-Phe-Met2-Pro-Leu-Trp-NH-3′,5′-Bzl(CF3)2 | 0.66 | 16.0 | 0.0065 | 8.6 | 7.0 | 15 | 490 | 10 |
| 4. H-Tyr-D-Ala-Gly-Phe-Met2-Pro-Leu-Trp-NH-Bzl | 0.44 | 1.8 | 3.2 | 2.6 | 21 | 4.8 | 61 | 9.9 |
| 5. H-Tyr-D-Ala-Gly-Phe-Nle-Pro-Ser(Glc)-Trp-NH-3′,5′-Bzl(CF3)2 | 3.7 | 8.0 | 0.077 | 7.9 | 18 | 13 | 520 | 1.8 |
Abbreviations: DAMGO, [D-Ala2,N-MePhe4,Gly-ol]enkephalin; DPDPE, c[D-Pen2,D-Pen5]enkephalin; GPI, guinea pig ileum; hDOR, human δ opioid receptor; hMOR, human μ opioid receptor; hNK1, human neurokinin-1 receptor; MVD, mouse vas deferens; ND, not determined; rMOR, rat μ opioid receptor; SP, substance P; SPGPI, substance P guinea pig ileum.
The initial design (Table 5, compound 1) had the structure H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-3′,5′-Bzl(CF3)2, with Met5 serving as an address residue for both the μ/δ opioid pharmacophore and the NK-1 antagonist pharmacophore. To determine that the compound had the biological activities desired at the opioid and NK-1 receptors required a large battery of in vitro binding affinity assays, efficacy (second-messenger) assays, and functional bioassays. Indeed, compound 1 had nanomolar binding affinity for the μ, δ, and NK-1 receptors; potent nanomolar agonist activity at the μ and δ opioid receptors; potent antagonist activity at the human NK-1 receptor; and potent activities in functional assays as well. Compound 2 (Table 5) has a biological activity profile in which the Met5 residue was replaced with the isosteric amino acid derivative of Met, norleucine (Nle). The results of the binding affinity assays, efficacy (second-messenger) assays, and functional assays for compounds 1 and 2 are similar. A major potential problem with the ester compound 1 is its short half-life in rat serum (approximately 1 min) (96, 97) due to cleavage of the ester bond. Therefore, the ester bond was replaced by an amide bond to the 3′,5′-Bzl(CF3)2 and a simple Bzl amide derivative of compound 1 (compounds 3 and 4, respectively). Indeed, this replacement greatly increases the serum stability of compounds 3 and 4 from a half-life of 1 min for compound 1 to a half-life of more than 4 h. Compounds 3 and 4 retain good binding affinity at all three receptors (μ, δ, and NK-1). Furthermore, they retain their potent agonist activity at the μ and δ receptors and potent antagonist activities at the human NK-1 receptor. To enhance their stability and biodistribution properties, including penetration through the blood-brain barrier, we prepared numerous glycosylated analogs of our lead compounds (98), of which the Ser(Glc)7 analog is illustrated in Table 5 (compound 5). Again, the binding affinity assays, efficacy (second-messenger) assays, and tissue functional assays show that these glycosylated analogs in our designed series retain the designed biological activity profiles with excellent potencies.
Detailed examination of the in vivo biological activities of these compounds in various pain assays and evaluation of development of tolerance and various toxicities has been reported (e.g., 99); the compounds show excellent antinociceptive activity in acute and neuropathic pain states. Interestingly, the compounds discussed above cross the blood-brain barrier well. On the basis of our modeling studies associated with the design of these ligands, we proposed that the peptides cross the blood-brain barrier owing to their conformational properties in the presence of biological membranes. Comprehensive NMR studies, in conjunction with computational methods, were conducted to examine the conformations of our new ligands in aqueous solution and in the membrane environments (micelles) (97). The compounds shown in Table 5 have no specific conformations in aqueous solution, as might be expected. However, in the presence of micelles, 2D NMR studies demonstrated that these peptides form highly stable conformations with a large number of nuclear Overhauser effects (NOEs) (as many per residue as is generally found in proteins, or more). Using the constraints that could be placed on the 3D conformation using the NOEs, we were able to establish that these compounds have stable helical-like structures in the presence of micelles (97).
Most of the in vivo studies thus far involve compounds 2 and 3 (Table 5) (see 99 for details). These compounds have potent antinociceptive effects in naive animals against acute pain. Most importantly, these compounds show potent antinociceptive effects in animal models of neuropathic pain, including models in which morphine has little or no antinociceptive activities. In addition, contrary to many μ opioid ligands, they do not result in impairment of motor skills. It also was shown, as mentioned above, that the peptides cross the blood-brain barrier, and good antinociception occurs when the peptides are administered peripherally (intravenously), centrally (intracerebroventricularly), or via spinal (intrathecal) administration. An exciting finding is that these compounds do not lead to the development of tolerance even after long-term use (greater than 10 days), and, on the basis of place preference studies, the animals do not develop dependence, either. The potent antinociceptive potencies of these compounds in animals with acute pain and neuropathic pain without the development of tolerance are exciting, and it is hoped that this new paradigm can be pushed ahead so we can obtain the data necessary for human trials.
OTHER RELATED STUDIES
The multivalent approach to ligand design has been applied, with promising results, to the design of other neuropeptides and neuroreceptors involved in various pain states: cholecystokinin, bradykinin, and MSH, to name a few (e.g., 100, 101). Although space does not allow a discussion of these design approaches, suffice it to say that in some cases, the overlapping-pharmacophore concept works, although it is generally more difficult than adjacent- or tethered-pharmacophore approaches. The field will have to develop much better computational approaches to ligand design and ligand-receptor/acceptor interactions to be more successful in de novo design of this kind. A more difficult problem is to examine the possibility that treating some disease states can reverse phenotype from, for example, a pain and addictive phenotype to a nonpain and nonaddictive phenotype, but it is an exciting prospect.
In this regard, the multivalent approach has been used for cancer detection and treatment with the objective of distinguishing cancer cells from normal cells (e.g., 102, 103). The basic hypothesis of this approach is that one can target simultaneously in a single structure two or more cell surface proteins (e.g., receptors, acceptors, cytokines, enzymes) that would distinguish cancer cells from normal cells. In this case, multivalency is used to obtain the inherent synergies that are expected from multivalent versus monovalent interactions on a single cell surface and to minimize false positives. In previous work, the application of multivalency using a water-soluble polymer to which we had attached multiple copies of an MSH derivative for detection of melanoma cancer was examined through the use of in vitro imaging of human and animal melanoma cancer cells (e.g., 104). However, these conjugates were not useful for in vivo experiments. New scaffolds based on modeling studies placed ligands at sufficient distances for homomeric and heteromeric cell surface protein cross-linking. A variety of scaffolds that are biocompatible, water soluble, and convenient for attaching both peptide and nonpeptide ligands needed for cell surface protein cross-linking were prepared. They also could contain reported groups (fluorescent, phosphorescent, etc.) or anticancer drugs and have been investigated for cancer detection with considerable success (e.g., 105, 106). Both homomultivalent and heteromultivalent ligands for in vitro and in vivo studies have been developed, with binding affinities that are 300-fold more selective than the binding affinities of monomeric ligands (e.g., 105–108). Cells containing two targets can readily be distinguished from cells that contain only one target; this can be accomplished using bivalent ligands attached to a scaffold that has a fluorescent moiety attached to it. The cells containing both receptors are fluorescently labeled within a few minutes, whereas the cells that contain only one receptor are barely visible during the same time period.
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Drug design based on the natural peptide pharmacophore has become an established paradigm in novel drug discovery and development. As highlighted in this review, establishing the pharmacophore by systematic SAR and screening in combination with conformational and topographical side-chain constraints can provide novel structures with novel biological activity profiles and can successfully deliver useful drug leads for various receptors. Obtaining critical 3D structural information for the initial pharmacophore leads, when they are complexed with the receptors, is an additional tool made possible by the increasing power of state-of-the-art computational methods. Although limitations still are prevalent, especially for membrane proteins such as G protein–coupled receptors, progress in crystalizing these proteins in their biologically relevant states is expected. Furthermore, state-of-the-art NMR and other biophysical methods will be able to examine the proteins’ biologically important structural and dynamic properties and thus will provide added information relevant to drug design and development. Recognizing systematic ways of thinking about and developing orthosteric and allosteric ligands for receptors and acceptors will be especially important, as will carefully evaluating the conformations that result from ligand-receptor interactions. In this regard, it also will be critical to recognize and pharmacologically evaluate multiple signaling pathways that result from these ligand-receptor/acceptor interactions. Here again, the advances in a variety of spectroscopic, imaging, and computational methodologies will continue to enable and expand the application of pharmacophore-based design and development of drugs for treatment of disease.
Supplementary Material
SUMMARY POINTS.
Structural and conformational considerations in peptide and peptidomimetic ligand design are critical to pharmacological and medical applications.
The critical importance of χ space in peptide/protein structural evaluation and biological activity, an underdeveloped area of ligand design, is illustrated with several examples of important changes in biological activity.
In the design of bioactive peptide hormones and neurotransmitters, there is enhancement of potency and stability on their interactions with G protein–coupled receptors.
Systematic methods are available to design peptide and peptidomimetic ligands that are receptor-selective agonists, antagonists, and inverse agonists.
The design of multivalent ligands leads to ligands with unique biological activity profiles.
Melanocortin ligands with multiple unique biological activities have led to clinical trials for pigmentation, melanoma, feeding behavior, and sexual behavior and function.
Novel agonist and antagonist ligands for pain, addiction, and tolerance—including applications to prolonged and neuropathic pain—can be designed with minimal or no toxicities.
Applications of conformational and topographical considerations toward the design of novel oxytocin, glucagon, opioids, somatostatin, melanotropins, and other hormones and neurotransmitters can provide ligands with novel biological activity profiles.
FUTURE ISSUES.
The further development of design in χ space for modulation of biological activity and disease is needed in peptide and protein research.
Comprehensive studies of peptides and peptidomimetics that interact with membranes and cross membrane barriers need further development.
Enhanced methods for peptide and peptidomimetic drug delivery will continue to be developed.
Design and use of multivalency for detection and treatment of disease will be an essential aspect of drug design.
Glossary
- Inverse agonist
ligand that binds to a receptor but does not activate it; instead, it lowers the basal signaling system activity
- Orthosteric site
binding site on a receptor or acceptor for the native ligand
- Allosteric site
binding site on a receptor or acceptor for the native ligand that differs from the orthosteric site
- χ space
the space formed by the torsion angles between Cα-Cβ, Cβ-Cγ, and related bonds in the side chains of amino acids
- Topographical space
the 3D structure of the peptide, including disposition of side-chain groups in vector space
- Peptide mimetic
a nonpeptide ligand with the structure-activity relationships of a peptide for the same target
- Allodynia
an enhanced pain state resulting from normally nonpainful stimuli
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review, although they have peer-reviewed support in some of the areas discussed.
Contributor Information
Victor J. Hruby, Email: hruby@email.arizona.edu.
Minying Cai, Email: mcai@email.arizona.edu.
LITERATURE CITED
- 1.Hruby VJ. Organic chemistry and biology: chemical biology through the eyes of collaboration. J Org Chem. 2009;74:9245–64. doi: 10.1021/jo901767e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hruby VJ. Conformational restrictions of biologically active peptides via amino acid side chain groups. Life Sci. 1982;31:189–99. doi: 10.1016/0024-3205(82)90578-1. [DOI] [PubMed] [Google Scholar]
- 3.Kessler H. Conformational and biological activity of cyclic peptides. Angew Chem Int Ed Engl. 1982;21:512–23. [Google Scholar]
- 4.Ferrier BM, Jarvis D, du Vigneaud V. Deamino-oxytocin: its isolation by partition chromatography on Sephadex and crystallization from water, and its biological activities. J Biol Chem. 1965;240:4264–66. [PubMed] [Google Scholar]
- 5.Rudinger J. The design of peptide hormone analogs. In: Ariens EJ, editor. Drug Design. Vol. 2. New York: Academic; 1971. pp. 319–419. [Google Scholar]
- 6.Hruby VJ. Designing peptide receptor agonists and antagonists. Nat Rev Drug Discov. 2002;1:847–58. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 7.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 8.Kastin AJ, Nissen C, Nikolics K, Medzihradszky K, Coy DH, et al. Distribution of 3H-α-MSH in rat brain. Brain Res Bull. 1996;1(1):19–26. doi: 10.1016/0361-9230(76)90045-9. [DOI] [PubMed] [Google Scholar]
- 9.Pan W, Tu H, Kastin AJ. Differential BBB interactions of three ingestive peptides: obestatin, ghrelin, and adiponectin. Peptides. 2006;27:911–16. doi: 10.1016/j.peptides.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 10.Witt KA, Gillespie TJ, Huber JD, Egleton RD, Davis TP. Peptide drug modifications to enhance bioavailability and blood-brain barrier permeability. Peptides. 2001;22:2329–43. doi: 10.1016/s0196-9781(01)00537-x. [DOI] [PubMed] [Google Scholar]
- 11.Egleton RD, Davis TP. Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx. 2005;2:44–53. doi: 10.1602/neurorx.2.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abbruscato TJ, Williams SA, Misicka A, Lipkowski AW, Hruby VJ, Davis TP. Blood-to-central nervous system entry and stability of biphalin, a unique double-enkephalin analog, and its halogenated derivatives. J Pharmacol Exp Ther. 1996;276:1049–57. [PubMed] [Google Scholar]
- 13.Egleton RD, Mitchell SA, Huber JD, Palian MM, Polt R, Davis TP. Improved blood-brain barrier penetration and enhanced analgesia of an opioid peptide by glycosylation. J Pharmacol Exp Ther. 2001;299:967–72. [PubMed] [Google Scholar]
- 14.Davis TP, Abbruscato TJ, Brownson E, Hruby VJ. Conformationally constrained peptide drugs targeted at the blood-brain barrier. In: Rapaka RS, editor. Membranes and Barriers: Targeted Drug Delivery. Vol. 154. Washington, DC: Natl. Inst. Drug Abuse; 1995. pp. 47–60. (NIDA Res Monogr). [PubMed] [Google Scholar]
- 15.Sharma SD, Nikiforovich GV, Jiang J, de Lauro Castrucci AM, Hadley ME, Hruby VJ. Cationized melanotropin analogues: structure function relationships. In: Hodges RS, Smith JA, editors. Peptides: Chemistry, Structure and Biology. Leiden, Neth: ESCOM; 1994. pp. 398–399. [Google Scholar]
- 16.Sharma SD, Nikiforovich GV, Jiang J, de Lauro Castrucci AM, Hadley ME, Hruby VJ. A new class of positively charged melanotropin analogs: a new concept in peptide design. In: Schneider CH, Eberle AN, editors. Peptides 1992. Leiden, Neth: ESCOM; 1993. pp. 95–96. [Google Scholar]
- 17.Wakelam MJO, Murphy GJ, Hruby VJ, Houslay MD. Activation of two signal-transduction systems in hepatocytes by glucagon. Nature. 1986;323:68–71. doi: 10.1038/323068a0. [DOI] [PubMed] [Google Scholar]
- 18.Kenakin T. Allosteric theory: taking therapeutic advantage of the malleable nature of GPCRs. Curr Neuropharmacol. 2007;5:149–56. doi: 10.2174/157015907781695973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8(1):41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis JA, Lebois EP, Lindsley CW. Allosteric modulation of kinases and GPCRs: design principles and structure diversity. Curr Opin Chem Biol. 2008;12(3):269–80. doi: 10.1016/j.cbpa.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 21.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein–coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 22.Hruby VJ, al-Obeidi F, Kazmierski WM. Emerging approaches in the molecular design of receptor-selective peptide ligands: conformational, topographical and dynamic considerations. Biochem J. 1990;268:249–62. doi: 10.1042/bj2680249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hruby VJ. Conformational and topographical considerations in the design of biologically active peptides. Biopolymers. 1993;33:1073–82. doi: 10.1002/bip.360330709. [DOI] [PubMed] [Google Scholar]
- 24.Hruby VJ, Li G, Haskell-Luevano C, Shenderovich MD. Design of peptides, proteins, and peptidomimetics in chi space. Biopolymers. 1997;43:219–66. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 25.Gibson SE, Guillo N, Tozer MJ. Towards control of χ-space: conformationally constrained analogues of Phe, Tyr, Trp and His. Tetrahedron. 1999;55:585–615. [Google Scholar]
- 26.Hruby VJ. Design in topographical space of peptide and peptidomimetic ligands that affect behavior. A chemist’s glimpse at the mind-body problem. Acc Chem Res. 2001;34:389–97. doi: 10.1021/ar990063q. [DOI] [PubMed] [Google Scholar]
- 27.Hruby VJ, Gehrig CA. Recent developments in the design of receptor specific opioid peptides. Med Res Rev. 1989;9:343–401. doi: 10.1002/med.2610090306. [DOI] [PubMed] [Google Scholar]
- 28.Gacel G, Zajac JM, Delay-Goyet P, Daugé V, Roques BP. Investigation of the structural parameters involved in the μ and δ opioid receptor discrimination of linear enkephalin-related peptides. J Med Chem. 1988;31:374–83. doi: 10.1021/jm00397a019. [DOI] [PubMed] [Google Scholar]
- 29.Mosberg HI, Hurst R, Hruby VJ, Gee K, Yamamura HI, et al. Bis-penicillamine enkephalins possess highly improved specificity toward δ opioid receptors. Proc Natl Acad Sci USA. 1983;80:5871–74. doi: 10.1073/pnas.80.19.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramachandran GN, Sasisekharan V. Conformation of polypeptides and proteins. Adv Protein Chem. 1968;23:283–437. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 31.Akiyama K, Gee KW, Mosberg HI, Hruby VJ, Yamamura HI. Characterization of [3H][2-D-penicillamine,5-D-penicillamine]-enkephalin binding to δ opiate receptors in the rat brain and neuroblastoma–glioma hybrid cell line (NG 108-15) Proc Natl Acad Sci USA. 1985;82:2543–47. doi: 10.1073/pnas.82.8.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hruby VJ, Kao LF, Pettitt BM, Karplus M. The conformational properties of the delta opioid peptide [D-Pen2, D-Pen5]enkephalin in aqueous solution determined by NMR and energy minimization calculations. J Am Chem Soc. 1988;110:3351–59. [Google Scholar]
- 33.Flippen-Anderson JL, Hruby VJ, Collins N, George C, Cudney B. X-ray structure of [D-Pen2, D-Pen5]enkephalin, a highly potent, δ opioid receptor-selective compound: comparisons with proposed solution conformations. J Am Chem Soc. 1994;116:7523–31. [Google Scholar]
- 34.Weber SJ, Greene DL, Sharma SD, Yamamura HI, Kramer TH, et al. Distribution and analgesia of [3H][D-Pen2, D-Pen5]enkephalin and two halogenated analogues after intravenous administration. J Pharmacol Exp Ther. 1991;259:1109–17. [PubMed] [Google Scholar]
- 35.Williams SA, Abbruscato TJ, Hruby VJ, Davis TP. Passage of a δ-opioid receptor selective enkephalin, [D-penicillamine2,5]enkephalin, across the blood-brain and the blood-cerebrospinal fluid barriers. J Neurochem. 1996;66:1289–99. doi: 10.1046/j.1471-4159.1996.66031289.x. [DOI] [PubMed] [Google Scholar]
- 36.Hruby VJ, Tóth G, Gehrig CA, Kao LF, Knapp R, et al. Topographically designed analogues of [D-Pen2, D-Pen5]enkephalin. J Med Chem. 1991;34:1823–30. doi: 10.1021/jm00110a010. [DOI] [PubMed] [Google Scholar]
- 37.Qian X, Russell KC, Boteju LW, Hruby VJ. Stereoselective total synthesis of topographically constrained designer amino acids: 2′, 6′-dimethyl-β-methyltyrosines. Tetrahedron. 1995;51:1033–54. [Google Scholar]
- 38.Qian X, Shenderovich MD, Kövér KE, Davis P, Horváth R, et al. Probing the stereochemical requirements for receptor recognition of δ opioid agonists through topographic modifications in position 1. J Am Chem Soc. 1996;118:280–90. [Google Scholar]
- 39.Bilsky EJ, Qian X, Hruby VJ, Porreca F. Antinociceptive activity of [β-methyl-2′,6′-dimethyltyrosine1]-substituted cyclic[ D-Pen2, D-Pen5]enkephalin and [D-Ala2, Asp4]deltorphin analogs. J Pharmacol Exp Ther. 2000;293:151–58. [PubMed] [Google Scholar]
- 40.Okura T, Cowell SM, Varga E, Burkey TH, Roeske WR, et al. Differential down-regulation of the human δ-opioid receptor by SNC80 and [D-Pen2, D-Pen5]enkephalin. Eur J Pharmacol. 2000;387:R11–13. doi: 10.1016/s0014-2999(99)00761-x. [DOI] [PubMed] [Google Scholar]
- 41.Schiller PW, Nguyen TM-D, Weltrowska G, Wilkes BC, Marsden BJ, et al. Differential stereochemical requirements of μ versus δ opioid receptors for ligand binding and signal transduction: development of a class of potent and highly δ-selective peptide antagonists. Proc Natl Acad Sci USA. 1992;89:11871–75. doi: 10.1073/pnas.89.24.11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salvadori S, Attila M, Balboni G, Bianchi C, Bryant SD, et al. δ opioidmimetic antagonists: prototypes for designing a new generation of ultraselective opioid peptides. Mol Med. 1995;1(6):678–89. [PMC free article] [PubMed] [Google Scholar]
- 43.Liao S, Lin J, Shenderovich MD, Han Y, Hosohata K, et al. The stereochemical requirements of the novel δ-opioid selective dipeptide antagonist TMT-Tic. Bioorg Med Chem Lett. 1997;7:3049–52. [Google Scholar]
- 44.Hosohata K, Burkey TH, Alfaro-Lopez J, Hruby VJ, Roeske WR, Yamamura HI. (2S,3R)TMT-L-Tic-OH is a potent inverse agonist at the human δ-opioid receptor. Eur J Pharmacol. 1999;380:R9–10. doi: 10.1016/s0014-2999(99)00516-6. [DOI] [PubMed] [Google Scholar]
- 45.Hosohata K, Varga EV, Alfaro-Lopez J, Tang X, Vanderah TW, et al. (2S,3R)β-Methyl-2′,6′-dimethyltyrosine-L-tetrahydroisoquinoline-3-carboxylic acid [(2S,3R)TMT-L-Tic-OH] is a potent, selective δ-opioid receptor antagonist in mouse brain. J Pharmacol Exp Ther. 2003;304:683–88. doi: 10.1124/jpet.102.042929. [DOI] [PubMed] [Google Scholar]
- 46.Liao S, Alfaro-Lopez J, Shenderovich MD, Hosohata K, Lin J, et al. De novo design, synthesis, and biological activities of high-affinity and selective non-peptide agonists of the δ-opioid receptor. J Med Chem. 1998;41:4767–76. doi: 10.1021/jm980374r. [DOI] [PubMed] [Google Scholar]
- 47.Knapp RJ, Santoro G, De Leon IA, Lee KB, Edsall SA, et al. Structure-activity relationships for SNC80 and related compounds at cloned δ and μ opioid receptors. J Pharmacol Exp Ther. 1996;277:1284–91. [PubMed] [Google Scholar]
- 48.Hosohata Y, Varga EV, Stropova D, Li X, Knapp RJ, et al. Mutation W284L of the human δ opioid receptor reveals agonist specific conformational mechanisms of G protein activation. Life Sci. 2001;68:2233–42. doi: 10.1016/s0024-3205(01)01011-6. [DOI] [PubMed] [Google Scholar]
- 49.Li X, Knapp RJ, Stropova D, Varga E, Wang Y, et al. Trp284 of the human δ opioid receptor is required for SNC121 binding but not binding of pCl-DPDPE, deltorphin II or naltrindole. Analgesia. 1995;1:539–42. [Google Scholar]
- 50.Liao S, Shenderovich MD, Zhang Z, Maletinska L, Slaninova J, Hruby VJ. Substitution of the sidechain-constrained amino acids β-methyl-2′,6′-dimethyl-4′-methoxytyrosine in position 2 of a bicyclic oxytocin analogue provides unique insights into the bioactive topography of oxytocin antagonists. J Am Chem Soc. 1998;120:7393–94. [Google Scholar]
- 51.Rezik M, Havlicek V, Leybin L, LaBella FS, Friesen H. Opiate-like naloxone-reversible actions of somatostatin given intracerebrally. Can J Physiol Pharmacol. 1978;56:227–31. doi: 10.1139/y78-033. [DOI] [PubMed] [Google Scholar]
- 52.Veber DF, Holly FW, Nutt RF, Bergstrand SJ, Brady SF, et al. Highly active cyclic and bicyclic somatostatin analogues of reduced ring size. Nature. 1979;280:512–14. doi: 10.1038/280512a0. [DOI] [PubMed] [Google Scholar]
- 53.Maurer R, Gaehwiler BH, Buescher HH, Hill RC, Roemer D. Opiate antagonistic properties of an octapeptide somatostatin analog. Proc Natl Acad Sci USA. 1982;79:4815–17. doi: 10.1073/pnas.79.15.4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pelton JT, Gulya K, Hruby VJ, Duckles SP, Yamamura HI. Conformationally restricted analogs of somatostatin with high μ-opiate receptor specificity. Proc Natl Acad Sci USA. 1985;82:236–39. doi: 10.1073/pnas.82.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kazmierski WM, Wire WS, Lui GK, Knapp RJ, Shook JE, et al. Design and synthesis of somatostatin analogues with topographical properties that lead to highly potent and specific μ opioid receptor antagonists with greatly reduced binding at somatostatin receptors. J Med Chem. 1988;31:2170–77. doi: 10.1021/jm00119a019. [DOI] [PubMed] [Google Scholar]
- 56.Hawkins KN, Knapp RJ, Lui GK, Gulya K, Kazmierski WM, et al. [3H]-[H-D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2] ([3H]CTOP), a potent and highly selective peptide for μ opioid receptors in rat brain. J Pharmacol Exp Ther. 1989;248:73–80. [PubMed] [Google Scholar]
- 57.Dorr RT, Lines R, Levine N, Brooks C, Xiang L, et al. Evaluation of Melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sci. 1996;58:1777–84. doi: 10.1016/0024-3205(96)00160-9. [DOI] [PubMed] [Google Scholar]
- 58.Hadley ME, Hruby VJ, Blanchard J, Dorr RT, Levine N, et al. Discovery and development of novel melanogenic drugs, Melanotan-I and -II. In: Borchardt R, Freidinger RM, Sawyer TK, editors. Integration of Pharmaceutical Discovery and Development: Case Histories. New York: Plenum; 1998. pp. 575–95. [DOI] [PubMed] [Google Scholar]
- 59.Wessells H, Gralnek D, Dorr R, Hruby VJ, Hadley ME, Levine N. Effect of an alpha-melanocyte stimulating hormone analog on penile erection and sexual desire in men with organic erectile dysfunction. Urology. 2000;56:641–46. doi: 10.1016/s0090-4295(00)00680-4. [DOI] [PubMed] [Google Scholar]
- 60.Dorr RT, Dvorakova K, Brooks C, Lines R, Levine N, et al. Increased eumelanin expression and tanning is induced by a superpotent melanotropin [Nle4-D-Phe7]α-MSH in humans. Photochem Photobiol. 2000;72:526–32. doi: 10.1562/0031-8655(2000)072<0526:ieeati>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 61.Engel MH, Sawyer TK, Hadley ME, Hruby VJ. Quantitative determination of amino acid racemization in heat-alkali-treated melanotropins: implications for peptide hormone structure-function studies. Anal Biochem. 1981;116:303–11. doi: 10.1016/0003-2697(81)90361-4. [DOI] [PubMed] [Google Scholar]
- 62.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, et al. 4-Norleucine, 7-D-phenylalanine-α-melanocyte-stimulating hormone: a highly potent α-melanotropin with ultralong biological activity. Proc Natl Acad Sci USA. 1980;77:5754–58. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hadley ME, Anderson B, Heward CB, Sawyer TK, Hruby VJ. Calcium-dependent prolonged effects on melanophores of [4-norleucine, 7-D-phenylalanine]-α-melanotropin. Science. 1981;213:1025–27. doi: 10.1126/science.6973820. [DOI] [PubMed] [Google Scholar]
- 64.de Lauro Castrucci AM, Hadley ME, Sawyer TK, Hruby VJ. Enzymological studies of melanotropins. Comp Biochem Physiol B. 1984;78:519–24. doi: 10.1016/0305-0491(84)90090-7. [DOI] [PubMed] [Google Scholar]
- 65.Sawyer TK, Hruby VJ, Darman PS, Hadley ME. [half-Cys4,half-Cys10]-α-melanocyte-stimulating hormone: a cyclic α-melanotropin exhibiting superagonist biological activity. Proc Natl Acad Sci USA. 1982;79:1751–55. doi: 10.1073/pnas.79.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hruby VJ, Wilkes BC, Cody WL, Sawyer TK, Hadley ME. Melanotropins: structural, conformational, and biological considerations in the development of superpotent and superprolonged analogs. Pept Protein Rev. 1984;3:1–64. [Google Scholar]
- 67.al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. Design of a new class of superpotent cyclic α-melanotropins based on quenched dynamic simulations. J Am Chem Soc. 1989;111:3413–16. [Google Scholar]
- 68.Hruby VJ, Lu D, Sharma SD, de Lauro Castrucci AM, Kesterson RA, et al. Cyclic lactam α-melanotropin analogues of Ac-Nle4-cyclo[Asp5, D-Phe7,Lys10]α-melanocyte-stimulating hormone-(4-10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–61. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 69.Grieco P, Balse PM, Weinberg D, MacNeil T, Hruby VJ. D-amino acid scan of γ-melanocyte-stimulating hormone: importance of Trp8 on human MC3 receptor selectivity. J Med Chem. 2000;43:4998–5002. doi: 10.1021/jm000211e. [DOI] [PubMed] [Google Scholar]
- 70.Cai M, Cai C, Mayorov AV, Xiong C, Cabello CM, et al. Biological and conformational study of β-substituted prolines in MT-II template: steric effects leading to human MC5 receptor selectivity. J Pept Res. 2004;63:116–31. doi: 10.1111/j.1399-3011.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- 71.Ballet S, Mayorov AV, Cai M, Tymecka D, Chandler KB, et al. Novel selective human melanocortin-3 receptor ligands: use of the 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one (Aba) scaffold. Bioorg Med Chem Lett. 2007;17:2492–98. doi: 10.1016/j.bmcl.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bednarek MA, MacNeil T, Tang R, Fong TM, Cabello MA, et al. Cyclic analogs of α-melanocyte-stimulating hormone (αMSH) with high agonist potency and selectivity at human melanocortin receptor 1b. Peptides. 2008;29:1010–17. doi: 10.1016/j.peptides.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 73.Hruby VJ, Cai M, Cain JP, Mayorov AV, Dedek MM, Trivedi D. Design, synthesis and biological evaluation of ligands selective for the melanocortin-3 receptor. Curr Top Med Chem. 2007;7:1085–97. doi: 10.2174/156802607780906645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cai M, Nyberg J, Hruby VJ. Melanotropins as drugs for the treatment of obesity and other feeding disorders: potential and problems. Curr Top Med Chem. 2009;9:554–63. doi: 10.2174/156802609788897817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doedens L, Opperer F, Cai M, Beck JG, Dedek M, et al. Multiple N-methylation of MT-II backbone amide bonds leads to melanocortin receptor subtype hMC1R selectivity: pharmacological and conformational studies. J Am Chem Soc. 2010;132(23):8115–28. doi: 10.1021/ja101428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cain JP, Mayorov AV, Cai M, Wang H, Tan B, et al. Design, synthesis, and biological evaluation of a new class of small molecule peptide mimetics targeting the melanocortin receptors. Bioorg Med Chem Lett. 2006;16:5462–67. doi: 10.1016/j.bmcl.2006.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wessells H, Fuciarelli K, Hansen J, Hadley ME, Hruby VJ, et al. Synthetic melanotropic peptide initiates erections in men with psychogenic erectile dysfunction: double-blind, placebo controlled crossover study. J Urol. 1998;160:389–93. [PubMed] [Google Scholar]
- 78.Kahn CR, Weir GC, editors. Joslin’s Diabetes Mellitus. 13th Philadelphia: Lippincott Williams & Wilkins; 1994. p. 1068. [Google Scholar]
- 79.Hruby VJ, Wright DE, Lin MC, Rodbell M. Semisynthetic glucagon derivatives for structure-function studies. Metabolism. 1976;25:1323–25. doi: 10.1016/s0026-0495(76)80133-3. [DOI] [PubMed] [Google Scholar]
- 80.Wright DE, Hruby VJ, Rodbell M. A reassessment of structure-function relationships in glucagon: glucagon1−21 is a full agonist. J Biol Chem. 1978;253:6338–40. [PubMed] [Google Scholar]
- 81.Bregman MD, Hruby VJ. Synthesis and isolation of a glucagon antagonist. FEBS Lett. 1979;101:191–94. doi: 10.1016/0014-5793(79)81324-1. [DOI] [PubMed] [Google Scholar]
- 82.Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi DB. Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist. Science. 1982;215:1115–16. doi: 10.1126/science.6278587. [DOI] [PubMed] [Google Scholar]
- 83.McKee RL, Pelton JT, Trivedi D, Hruby VJ, Sueiras-Diaz J, Coy DH. Receptor binding and adenylate cyclase activities of glucagon analogs modified in the N-terminal region. Biochemistry. 1986;25:1650–56. doi: 10.1021/bi00355a031. [DOI] [PubMed] [Google Scholar]
- 84.Krstenansky JL, Trivedi D, Hruby VJ. Importance of the 10-13 region of glucagon for its receptor interactions and activation of adenylate cyclase. Biochemistry. 1986;25:3833–39. doi: 10.1021/bi00361a014. [DOI] [PubMed] [Google Scholar]
- 85.Hruby VJ, Krstenansky J, Gysin B, Pelton JT, Trivedi D, McKee RL. Conformational considerations in the design of glucagon agonists and antagonists: examination using synthetic analogs. Biopolymers. 1986;25:S135–55. [PubMed] [Google Scholar]
- 86.Murphy GJ, Hruby VJ, Trivedi D, Wakelam MJO, Houslay MD. The rapid desensitization of glucagon-stimulated adenylate cyclase is a cyclic AMP–independent process that can be mimicked by hormones which stimulate inositol phospholipid metabolism. Biochem J. 1987;243:39–46. doi: 10.1042/bj2430039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grady T, Fickova M, Tager HS, Trivedi D, Hruby VJ. Stimulation and inhibition of cAMP accumulation by glucagon in canine hepatocytes. J Biol Chem. 1987;262:15514–20. [PubMed] [Google Scholar]
- 88.Azizeh BY, Van Tine BA, Sturm NS, Hutzler AM, David C, et al. [des His1,des Phe6,Glu9]glucagon amide: a newly designed “pure” glucagon antagonist. Bioorg Med Chem Lett. 1995;5:1849–52. [Google Scholar]
- 89.Azizeh BY, Van Tine BA, Trivedi D, Hruby VJ. Pure glucagon antagonists: biological activities and cAMP accumulation using phosphodiesterase inhibitors. Peptides. 1997;18:633–41. doi: 10.1016/s0196-9781(97)00131-9. [DOI] [PubMed] [Google Scholar]
- 90.Hruby VJ, Ahn JM, Trivedi D. The design and biological activities of glucagon agonists and antagonists, and their use in examining the mechanisms of glucose action. Curr Med Chem Immunol Endocr Metab Agents. 2001;1:199–215. [Google Scholar]
- 91.King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure in underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- 92.Hruby VJ, Porreca F, Yamamura HI, Tollin G, Agnes RS, et al. New paradigms and tools in drug design for pain and addiction. AAPS J. 2006;8(3):E450–60. doi: 10.1208/aapsj080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.King T, Gardella LR, Wang R, Vandanyan A, Ossipov MH, et al. Role of NK-1 neurotransmission on opioid-induced hyperalgesia. Pain. 2005;116(3):276–88. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gu G, Kondo I, Hua XY, Yaksh TL. Resting and evoked spinal substance P release during chronic intrathecal morphine infusion: parallels with tolerance and dependence. J Pharmacol Exp Ther. 2005;314(3):1362–69. doi: 10.1124/jpet.105.087718. [DOI] [PubMed] [Google Scholar]
- 95.Powell KJ, Quirion R, Jhamandas K. Inhibition of neurokinin-1–substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Euro J Neurosci. 2003;18(6):1572–83. doi: 10.1046/j.1460-9568.2003.02887.x. [DOI] [PubMed] [Google Scholar]
- 96.Yamamoto T, Nair P, Vagner J, Largent-Milnes T, Davis P, et al. A structure-activity relationship study and combinatorial synthetic approach of C-terminal modified bifunctional peptides that are δ/μ opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2008;51:1369–76. doi: 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yamamoto T, Nair P, Jacobsen NE, Davis P, Ma SW, et al. The importance of micelle-bound states for the bioactivities of bifunctional peptide derivatives for δ/μopioid receptor agonists and neurokinin-1 receptor antagonists. J Med Chem. 2008;51:6334–47. doi: 10.1021/jm800389v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamamoto T, Nair P, Jacobsen NE, Vagner J, Kulkarni V, et al. Improving metabolic stability by glycosylation: bifunctional peptide derivatives that are opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2009;52:5164–75. doi: 10.1021/jm900473p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Largent-Milnes TM, Yamamoto T, Nair P, Hruby VJ, Lai J, et al. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br J Pharmacol. 2010;161:986–1001. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Agnes RS, Lee YS, Davis P, Ma SW, Badghisi H, et al. Structure-activity relationships of bifunctional peptides based on overlapping pharmacophores at opioid and cholecystokinin receptors. J Med Chem. 2006;49:2868–75. doi: 10.1021/jm050921q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Agnes RS, Ying J, Kövér KE, Lee YS, Davis P, et al. Structure-activity relationships of bifunctional cyclic disulfide peptides based on overlapping pharmacophores at opioid and cholecystokinin receptors. Peptides. 2008;29:1413–23. doi: 10.1016/j.peptides.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gillies RJ, Hruby VJ. Expression-driven reverse engineering of targeted imaging and therapeutic agents. Expert Opin Ther Targets. 2003;7:137–39. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]
- 103.Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Hitting multiple targets with multimeric ligands. Expert Opin Ther Targets. 2004;8:565–86. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 104.Sharma SD, Jiang J, Hadley ME, Bentley DL, Hruby VJ. Melanotropic peptide-conjugated beads for microscopic visualization and characterization of melanoma melanotropin receptors. Proc Natl Acad Sci USA. 1996;93:13715–20. doi: 10.1073/pnas.93.24.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vagner J, Xu L, Handl HL, Josan JS, Morse DL, et al. Heterobivalent ligands crosslink multiple cell-surface receptors: the human melanocortin-4 and δ-opioid receptors. Angew Chem Int Ed. 2008;47:1685–88. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Josan JS, Morse DL, Xu L, Trissal M, Baggett B, et al. Solid-phase synthetic strategy and bioevaluation of a labeled δ-opioid receptor ligand Dmt-Tic-Lys for in vivo imaging. Org Lett. 2009;11(12):2479–82. doi: 10.1021/ol900200k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu L, Vagner J, Alleti R, Rao V, Jagadish B, et al. Synthesis and characterization of a Eu-DTPA-PEGO-MSH(4) derivative for evaluation of binding of multivalent molecules to melanocortin receptors. Bioorg Med Chem Lett. 2010;20:2489–92. doi: 10.1016/j.bmcl.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brabez N, Lynch RM, Xu L, Gillies RJ, Chassaing G, et al. Design, synthesis, and biological studies of efficient multivalent melanotropin ligands: tools toward melanoma diagnosis and treatment. J Med Chem. 2011;54(20):7375–84. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.