

ABSTARCT
Epidemiological and clinical studies have increasingly shown that fine particulate matter (PM2.5) is associated with a number of pathological respiratory diseases, such as bronchitis, asthma, and chronic obstructive pulmonary disease, which share the common feature of airway inflammation induced by particle exposure. Thus, understanding how PM2.5 triggers inflammatory responses in the respiratory system is crucial for the study of PM2.5 toxicity. In the current study, we found that exposing human bronchial epithelial cells (immortalized Beas-2B cells and primary cells) to PM2.5 collected in the winter in Wuhan, a city in southern China, induced a significant upregulation of VEGFA (vascular endothelial growth factor A) production, a signaling event that typically functions to control chronic airway inflammation and vascular remodeling. Further investigations showed that macroautophagy/autophagy was induced upon PM2.5 exposure and then mediated VEGFA upregulation by activating the SRC (SRC proto-oncogene, non-receptor tyrosine kinase)-STAT3 (signal transducer and activator of transcription 3) pathway in bronchial epithelial cells. By exploring the upstream signaling events responsible for autophagy induction, we revealed a requirement for TP53 (tumor protein p53) activation and the expression of its downstream target DRAM1 (DNA damage regulated autophagy modulator 1) for the induction of autophagy. These results thus extend the role of TP53-DRAM1-dependent autophagy beyond cell fate determination under genotoxic stress and to the control of proinflammatory cytokine production. Moreover, PM2.5 exposure strongly induced the activation of the ATR (ATR serine/threonine kinase)-CHEK1/CHK1 (checkpoint kinase 1) axis, which subsequently triggered TP53-dependent autophagy and VEGFA production in Beas-2B cells. Therefore, these findings suggest a novel link between processes regulating genomic integrity and airway inflammation via autophagy induction in bronchial epithelial cells under PM2.5 exposure.
KEYWORDS: autophagy, inflammation, PM2.5, TP53, VEGFA
Introduction
The adverse health effects of exposure to air pollution are of great concern worldwide. Particulate matter (PM), the most harmful component of air pollution, includes particles with a median aerodynamic diameter < 0.1 μm (ultrafine particles, PM0.1), < 2.5 μm (fine particles, PM2.5) and < 10 μm (coarse particles, PM10), which are strongly associated with severe air pollution-induced health threats. Most PM10 particles are deposited in the nasal cavities and upper airways, whereas PM2.5 is more likely to reach the deeper parts of the respiratory tract, penetrate deeply into the lung alveoli and enter the bloodstream. Epidemiological and clinical studies have increasingly shown that PM2.5 exposure is a risk factor mainly associated with respiratory and cardiovascular morbidity and mortality. The pulmonary and cardiovascular effects of air pollution containing PM2.5 include impaired lung function; increased pathological incidence of chronic cough, bronchitis, and asthma; accelerated progression of atherosclerotic plaques; plaque instability; altered coagulation; venous thrombosis; and others.1-3 Therefore, elucidating the toxicological mechanisms that link inhaled particles with damage to the respiratory and cardiovascular systems in vivo and in vitro is an active area of research.
After inhalation and deposition in the epithelium of the respiratory tract and lungs, particles are thought to be capable of moving into interstitial spaces between cells and then inducing the production of a variety of proinflammatory cytokines and adhesion molecules, such as IL1B/IL-1β (interleukin 1 β), IL6/IL-6 (interleukin 6), CXCL8/IL8 (C-X-C motif chemokine ligand 8), TNF/TNFα (tumor necrosis factor), ICAM1 (intercellular adhesion molecule 1), etc. The release of these molecules then facilitates a pulmonary inflammatory response within a short time, which exacerbates the airway symptoms, especially in vulnerable populations with preexisting cardiopulmonary diseases and the elderly.2,4-6 When the response is sustained for a longer period, proinflammatory factors may move to the systemic circulation from the lungs and then cause adverse effects on the cardiovascular system.2,3,6 Therefore, control of the inflammatory response may allow for more effective strategies to abate the cardiopulmonary dysfunctions induced by PM2.5. Unfortunately, the mechanisms underlying the association between air particles and increased airway inflammatory response have not been clearly defined.
Autophagy, an evolutionarily conserved catabolic process, mainly targets long-lived proteins, superfluous or damaged organelles, and other cytoplasmic components for degradation via the lysosomal pathway. Autophagy not only plays a critical role in maintaining intercellular homeostasis, but also is considered a mediator of cellular processes under various stress conditions, including cell survival and death, cellular senescence and immune responses.7-9 A large body of evidence indicates that the autophagic pathway becomes activated and subsequently may become deregulated in response to cellular stressors, which contributes substantially to pathogenic processes underlying pulmonary diseases, cardiovascular disease, inflammatory bowel disease, neuronal degeneration, aging and cancer.10-12 Among these processes, respiratory pathologies such as asthma, chronic obstructive pulmonary disease, respiratory tract infection and pulmonary fibrosis, are closely correlated with autophagy, which has been extensively studied and discussed.13-17 The findings indicate that excessive autophagy in chronic lung diseases not only is essential for airway mucus hyper-secretion and airway remodeling,14,15 but also functions mechanistically to impair the host antiviral defense against respiratory viral infections and to promote airway epithelial fibrosis. Therefore, manipulating autophagic pathways and their regulatory components might lead to the development of therapeutics for lung diseases.16,17 However, the function of autophagy in the context of PM2.5-induced airway responses, especially the production of proinflammatory factors, has not been explored.
In the current study, we discovered a critical role for autophagy in mediating PM2.5-induced VEGFA upregulation in human bronchial epithelial cells. Because VEGFA is a key regulator of blood vessel growth and an inducer of vascular leakage and permeability in the airway, an abnormal elevation of local VEGFA expression contributes substantially to the control of chronic airway inflammation and vascular remodeling in lung pathogenesis.18-21 Therefore, the data from our current study provide, for the first time, an understanding of the complex signaling events that characterize PM2.5-induced inflammatory responses mediated by VEGFA upregulation through autophagic pathways. Furthermore, the activation of the ATR-CHEK1/CHK1-TP53 axis functioned as an upstream signaling event to mediate autophagy and VEGFA induction in PM2.5-treated bronchial epithelial cells. These findings thus represent novel information that the modulation of checkpoint responses is required for mediating autophagy-dependent airway inflammation under PM2.5 exposure. Inhibition of excessive VEGFA expression by targeting checkpoint and autophagic pathways may provide a new approach to attenuate the pulmonary toxicity of PM2.5.
Results
PM2.5 exposure induced upregulation of VEGFA production in human bronchial epithelial cells
Proinflammatory cytokine release in lung epithelial cells contributes substantially to the control of airway inflammatory responses under PM2.5 exposure.2,4-6 To investigate whether PM2.5 samples from Wuhan had the ability to induce pulmonary inflammation, in vitro assays were performed using Beas-2B immortalized human bronchial epithelial cells. Cells were treated with different doses of PM2.5, and then the production of proinflammatory factors (IL1B, IL6, CXCL8, TNF and VEGFA) was measured in the supernatants of cultured Beas-2B cells by ELISA (enzyme-linked immunosorbent assay) 24 h after PM2.5 exposure. As shown in Fig. 1A, VEGFA production was significantly upregulated following PM2.5 treatment at different doses, and the peak induction of VEGFA release was observed upon 100 μg/mL of PM2.5 exposure. Under the same conditions, no detectable TNF production was observed, and the expression levels of IL1B, IL6 and CXCL8 in the cell culture media did not show obvious differences before and after PM2.5 exposure. To further confirm these results, we next examined the cytokine transcriptional responses induced by PM2.5 in Beas-2B cells using a RT-PCR (reverse transcription polymerase chain reaction) assay. Based on the alternative splicing of the 8 coding exons, VEGFA mRNA exists as several different isoforms, of which the VEGFA120, VEGFA164 and VEGFA188 isoforms are predominantly expressed.21 As shown in Fig. 1B, a dose-dependent induction of VEGFA transcription (the VEGFA120 and VEGFA164 isoforms) was observed in Beas-2B cells after PM2.5 treatment, whereas the mRNA levels of IL1B, IL6, CXCL8 and TNF remained unchanged before and after PM2.5 exposure. Together, these data indicate that VEGFA might function as at least one of the most crucial proinflammatory mediators released by Beas-2B cells in response to PM2.5 stimulation.
Figure 1.

PM2.5 exposure induced upregulation of VEGFA production in human bronchial epithelial cells. (A) Beas-2B cells were left untreated or were treated with different concentrations of PM2.5 (12.5, 25, 50, and 100 μg/mL) for 24 h. The production of VEGFA, IL1B, IL6, CXCL8 and TNF was then detected in the cell culture supernatant using ELISA (*, P < 0.05; **, P < 0.01). (B) Beas-2B cells were left untreated or were treated with PM2.5 as described in (A); then, the transcriptional responses of VEGFA, IL1B, IL6, CXCL8 and TNF were determined with an RT-PCR assay. (C) Beas-2B cells were transfected with a VEGFA promoter-driven luciferase reporter, and then stable transfectants were established. The transfectants were exposed to different doses of PM2.5 (as indicated), and the induction of VEGFA promoter-dependent luciferase activity was determined 12 h after PM2.5 exposure (**, P < 0.01). (D) Beas-2B cells were left untreated or were treated with PM2.5 as described in (A), and the intracellular VEGFA expression levels were examined using a western blot assay. (E) Beas-2B cells were left untreated or were treated with PM2.5 (100 μg/mL) for the indicated time periods; then, the intracellular VEGFA expression levels were measured. (F and G) Beas-2B cells were treated with PM2.5 (100 μg/mL) alone or in combination with PMB (50 μg/mL) for 24 h. Then, VEGFA transcription and protein synthesis and secretion were analyzed using RT-PCR, western blot and ELISA assays, respectively. (H) Primary human bronchial epithelial cells were left untreated or were treated with PM2.5 (20 µg/mL) for 24 h; then, VEGFA transcription and intracellular protein synthesis were detected using RT-PCR and western blot assays, respectively. (I) Primary human bronchial epithelial cells were left untreated or were treated with PM2.5 (20 μg/mL) for the indicated time periods; then, the production of VEGFA was detected in the cell culture supernatant using ELISA (**, P < 0.01).
We then focused our following study on epithelial VEGFA production induced by PM2.5. A VEGFA luciferase reporter plasmid containing ∼3.0 kb of the human VEGFA gene promoter22 was transfected into Beas-2B cells, and stable transfectants were established. Then, the transfectants were treated with PM2.5 as described above. We found that PM2.5 exposure dose-dependently induced an increase in VEGFA promoter-driven luciferase activity in Beas-2B cells (Fig. 1C). This result further confirmed the induction of VEGFA transcription in Beas-2B cells in response to PM2.5 treatment. Next, we determined the intracellular VEGFA expression levels in Beas-2B cells under treatment with different doses of PM2.5 or a single dose of PM2.5 (100 μg/mL) for different time periods with a western blot assay. As shown in Fig. 1D, the dose-dependent induction of intracellular VEGFA expression was readily observed in Beas-2B cells, which was consistent with the results obtained from the luciferase, RT-PCR and ELISA assays presented in Fig. 1A to C. Furthermore, the inducible VEGFA expression was detected 1.5 h after PM2.5 exposure and was sustained for 24 h (Fig. 1E). Together, these results indicate that in response to Wuhan PM2.5 exposure, Beas-2B cells expressed VEGFA at a high level and for a long duration.
To exclude the possible effects of endotoxin contamination on the PM2.5-induced VEGFA production in Beas-2B cells, we next compared the expression levels of VEGFA in Beas-2B cells treated with PM2.5 with or without cotreatment with PMB (polymyxin B), an antibiotic widely used to eliminate the effects of endotoxin contamination. We found that neither VEGFA transcription nor VEGFA protein synthesis or secretion was changed in PM2.5-treated Beas-2B cells in the absence or presence of PMB cotreatment (Fig. 1F, 1G). These data indicate that increased VEGFA production is a specific response induced by PM2.5 and unrelated to endotoxin.
To further confirm the above results related to VEGFA secretion responses in Beas-2B cells, we next examined VEGFA production in primary human bronchial epithelial cells under PM2.5 treatment. As shown in Fig. 1H and 1I, a significant upregulation of VEGFA transcription and VEGFA protein synthesis and secretion was readily observed in the primary cells upon PM2.5 exposure. Taken together, these data indicate that VEGFA functions as an important proinflammatory factor in airway epithelial cells in response to PM2.5 stimulatin.
PM2.5 exposure induced autophagy, which was critical for mediating VEGFA upregulation in human bronchial epithelial cells
To examine the signal transduction pathways leading to VEGFA induction, we first addressed the possible involvement of autophagy, a well-known metabolic process closely related to lung pathogenesis,13-17 in mediating VEGFA upregulation under PM2.5 exposure. Because an increase in the MAP1LC3B/LC3B (microtubule-associated protein light chain 3 β)-II:LC3B-I ratio, the induction of BECN1/BECLIN 1 (Beclin 1) expression and a decrease in SQSTM1/p62 (sequestosome 1) levels are hallmarks of autophagosome accumulation and autophagic degradation,23 we first analyzed the levels of these specific autophagic key proteins using a western blot assay. As shown in Fig. 2A and 2B, both a dose- and time-dependent upregulation of MAP1LC3B and BECN1 expression, as well as degradation of SQSTM1, were observed in Beas-2B cells treated with PM2.5, which reflected enhanced autophagosome synthesis and the activation of autophagic degradation pathways in response to PM2.5 stimulation.
Figure 2.

PM2.5 exposure induced autophagy in human bronchial epithelial cells. (A and B) Beas-2B cells were left untreated or were treated with PM2.5 as described in Fig. 1A and 1E. Then, the expression levels of MAP1LC3B, BECN1 and SQSTM1 were examined. (C) Beas-2B cells were left untreated or were treated with PM2.5 (100 μg/mL) for 24 h; then, autophagy was examined under confocal microscopy after the cells were stained with Cyto-ID Green Autophagy Detection Reagent. (D) Beas-2B cells were treated with PM2.5 and were stained with Cyto-ID Autophagy Detection Reagent as described in (C). Then, the cells were collected and subjected to a flow cytometric analysis to quantitatively measure the autophagic fluorescence intensity inside the cells (**, P < 0.01). (E) TEM of Beas-2B cells untreated (left panel) or treated with 100 μg/mL of PM2.5 for 24 h (middle and right panels). The right panel shows a high-magnification image of the indicated region in the middle panel. Red arrows indicate double-membrane autophagic vesicles. Blue arrows indicate the particles taken up by the cells. (F) Beas-2B cells were treated with PM2.5 (100 μg/mL) alone or in combination with BafA1 (0.1 μM) during the final 4 h before the cells were harvested. Then, the expression levels of MAP1LC3B and SQSTM1 were examined 24 h after PM2.5 exposure. (G) Beas-2B cells were treated as described in Fig. 1F, and the expression levels of MAP1LC3B, BECN1 and SQSTM1 were measured. (H) Beas-2B cells were treated as described in Fig. 1F, and autophagy was examined under confocal microscopy after the cells were stained with Cyto-ID Green Autophagy Detection Reagent. (I) Primary human bronchial epithelial cells were left untreated or were treated with PM2.5 (20 μg/mL) for 24 h. Then, the expression levels of MAP1LC3B, BECN1 and SQSTM1 were measured. (J) Primary human bronchial epithelial cells were treated as described in (I) and autophagy was examined under confocal microscopy after the cells were stained with Cyto-ID Green Autophagy Detection Reagent.
Next, we measured autophagic activity in PM2.5-treated Beas-2B cells using the commercial Cyto-ID Autophagy Detection Kit. As shown in Fig. 2C, when Beas-2B cells were stained with Cyto-ID Green Autophagy Detection Reagent, we observed a significant induction of autophagic activity inside the cells after 24 h of PM2.5 exposure, which was evidenced by a specific green autophagic fluorescence signal that accumulated in spherical vacuoles in the perinuclear region of PM2.5-treated cells. In contrast, no obvious signal was detected in untreated Beas-2B cells under the same conditions.
To further confirm the above results, we then performed a flow cytometry-based quantitative analysis of cell populations loaded with the Cyto-ID Green Autophagy Detection Reagent. The data in Fig. 2D show that Beas-2B cells without PM2.5 treatment were stained only faintly and displayed low fluorescence signal intensity. After exposure to PM2.5 for 24 h, the Cyto-ID-dependent autophagic fluorescence signals increased dramatically in Beas-2B cells. In addition, transmission electron microscopy (TEM) clearly revealed the presence of double-membrane autophagic vesicles containing partially degraded cytoplasmic materials in Beas-2B cells treated with PM2.5 (Fig. 2E). Collectively, these data indicate that PM2.5 exposure induces autophagic activity in Beas-2B cells. TEM images of PM2.5-treated Beas-2B cells also frequently exhibited different components of PM2.5, including soot aggregates, fly ash particles and mineral particles (Fig. 2E), indicating that PM2.5 is taken up into the cells.
Enhanced autophagosome accumulation could result from either an increase in autophagic flux or defects in the downstream process of autophagosome-lysosome fusion, thereby inhibiting autophagic processing and degradation. To determine which was the case for PM2.5, an MAP1LC3B turnover assay was performed in the absence or presence of BafA1 (bafilomycin A1), an autophagy inhibitor that affects the acidification of the lysosome, making it unable to digest MAP1LC3 and other contents. As shown in Fig. 2F, a combination of PM2.5 and BafA1 treatment resulted in a significantly higher accumulation of MAP1LC3B compared with PM2.5 treatment alone. In addition, the decrease in SQSTM1 levels caused by PM2.5 was effectively reversed by BafA1 cotreatment. These data clearly demonstrate that PM2.5 exposure results in an increase in autophagic flux, rather than defects in autophagic degradation, in Beas-2B cells.
Next, we compared the levels of PM2.5-induced autophagic responses in the absence or presence of PMB. As expected, the accumulation of MAP1LC3B and BECN1 and the decrease in SQSTM1 levels were similar in Beas-2B cells treated with PM2.5 alone or cotreated with PM2.5 and PMB (Fig. 2G). Moreover, Cyto-ID Autophagy Detection Reagent-stained Beas-2B cells displayed similar autophagic fluorescence signals upon PM2.5 exposure with or without cotreatment with PMB (Fig. 2H). These data demonstrate that autophagy induction is a specific response induced by PM2.5 and is unrelated to endotoxin.
Finally, we examined autophagic activity in primary human bronchial epithelial cells under PM2.5 treatment to further confirm the above results obtained in Beas-2B cells. As shown in Fig. 2I and 2J, the upregulation of MAP1LC3B and BECN1 expression and SQSTM1 degradation, as well as Cyto-ID-dependent autophagic fluorescence signals were readily observed in the primary cells upon PM2.5 exposure. Together, these data indicate that autophagy can be efficiently induced by PM2.5 in human bronchial epithelial cells.
To determine whether autophagy induction is involved in VEGFA upregulation under PM2.5 exposure, Beas-2B cells were left untreated or were pretreated with 3-MA (3-methyladenine), a well-known autophagy inhibitor that functions by blocking autophagosome formation, followed by exposure to PM2.5. As shown in Fig. 3A, the efficiency of 3-MA in inhibiting PM2.5-induced autophagy was verified by the reduction in MAP1LC3B and BECN1 expression in the 3-MA-pretreated Beas-2B cells compared to the cells treated with PM2.5 alone. Then, we found that PM2.5-induced VEGFA production was almost completely blocked by 3-MA pretreatment (Fig. 3A). To further confirm these results, Beas-2B cells stably expressing a VEGFA promoter-driven luciferase reporter were also successively treated with 3-MA and PM2.5. The results showed that 3-MA pretreatment significantly inhibited the induction of VEGFA transcription by PM2.5 exposure (Fig. 3B). At the same time, we also observed the obvious inhibition of PM2.5-induced VEGFA production and secretion in the cell culture supernatants of Beas-2B cells under pretreatment of 3-MA by ELISA (Fig. 3C). These findings together indicate that autophagy plays a critical role in mediating VEGFA upregulation in PM2.5-treated Beas-2B cells. In the following experiment, siRNA specifically targeting ATG5 (autophagy-related 5) or a control siRNA was introduced into Beas-2B cells stably transfected with a VEGFA-promoter driven luciferase reporter, and the induction of VEGFA expression was detected in the presence or absence of ATG5 expression. As shown in Fig. 3D, 3E and 3F, knockdown of ATG5 expression not only blocked the induction of MAP1LC3B and BECN1 expression but also dramatically inhibited the upregulation of VEGFA transcription and protein synthesis induced by PM2.5. In addition, we also observed downregulation of PM2.5-induced VEGFA transcription and protein synthesis by knockdown BECN1 expression in the Beas-2B cells (Fig. 3G, 3H and 3I). Furthermore, when BafA1 was used to block the autophagic processing and degradation induced by PM2.5 in Beas-2B cells, the induction of VEGFA transcription and protein synthesis was also dramatically inhibited under the same conditions (Fig. 3J, 3K and 3L). Together, these results demonstrate that an increase in autophagy flux is required for mediating VEGFA upregulation in airway epithelial cells upon PM2.5 exposure.
Figure 3.

Autophagy induction was critical for mediating VEGFA production in Beas-2B cells upon PM2.5 exposure. (A) Beas-2B cells were pretreated with 3-MA, followed by exposure to PM2.5 (100 μg/mL). Then, the expression of VEGFA, BECN1 and MAP1LC3B was analyzed 24 h after PM2.5 exposure. (B) Beas-2B cells stably transfected with VEGFA promoter-driven luciferase reporter were treated with 3-MA and PM2.5 as described in (A). Then, the induction of VEGFA promoter-dependent luciferase activity was examined 12 h after PM2.5 exposure (**, P < 0.01). (C) Beas-2B cells were treated with 3-MA and PM2.5 as described in (A). Then, the production and secretion of VEGFA in Beas-2B cells were detected in the cell culture supernatant using ELISA 24 h after PM2.5 exposure (**, P < 0.01). (D) Beas-2B cells were transfected with ATG5 siRNA or control siRNA and then exposed to PM2.5 (100 μg/mL) 36 h after transfection. The expression of ATG5, VEGFA, BECN1 and MAP1LC3B was examined 24 h after PM2.5 exposure. (E) Beas-2B cells stably transfected with VEGFA promoter-driven luciferase reporter were transfected with ATG5 siRNA or control siRNA and treated with PM2.5 as described in (D). Then, the induction of VEGFA promoter-dependent luciferase activity was determined 12 h after PM2.5 exposure (**, P < 0.01). (F) Beas-2B cells were transfected and treated with PM2.5 as described in (D). Then, the production and secretion of VEGFA in Beas-2B cells were detected in the cell culture supernatant using ELISA 24 h after PM2.5 exposure (**, P < 0.01). (G) Beas-2B cells were transfected with BECN1 siRNA or control siRNA and then exposed to PM2.5 (100 μg/mL) 36 h after transfection. The expression of BECN1, MAP1LC3B and VEGFA was examined 24 h after PM2.5 exposure. (H) Beas-2B cells stably transfected with VEGFA promoter-driven luciferase reporter were transfected with BECN1 siRNA or control siRNA and treated with PM2.5 as described in (G). Then, the induction of VEGFA promoter-dependent luciferase activity was determined 12 h after PM2.5 exposure (**, P < 0.01). (I) Beas-2B cells were transfected and treated with PM2.5 as described in (G). Then, the production and secretion of VEGFA in Beas-2B cells were detected in the cell culture supernatant using ELISA 24 h after PM2.5 exposure (**, P < 0.01). (J) Beas-2B cells were treated as described in Fig. 2F, and then VEGFA expression levels were measured 24 h after PM2.5 exposure. (K) Beas-2B cells stably transfected with VEGFA promoter-driven luciferase reporter were treated as in Fig. 2F, and the induction of VEGFA promoter-dependent luciferase activity was examined 12 h after PM2.5 exposure (**, P < 0.01). (L) Beas-2B cells were treated as described in Fig. 2F, and then the production and secretion of VEGFA in Beas-2B cells were detected in the cell culture supernatant using ELISA 24 h after PM2.5 exposure (**, P < 0.01).
PM2.5-induced autophagy contributed to increased VEGFA production by activating the SRC-STAT3 pathway
To determine the underlying mechanism by which autophagy mediated VEGFA production in PM2.5-treated epithelial cells, transcription factors that might be involved in regulating VEGFA transcription were analyzed. As shown in Fig. 4A and 4B, PM2.5 exposure induced a strong activation of STAT3 in Beas-2B cells, as evidenced by an increased level of STAT3 phosphorylation and an upregulation of STAT3-dependent luciferase activity after PM2.5 treatment. Furthermore, knockdown of STAT3 expression effectively inhibited the induction of VEGFA expression by PM2.5 (Fig. 4C). These data indicate that STAT3 functions as a critical transcription factor for triggering VEGFA production in PM2.5-treated Beas-2B cells. Under the same PM2.5 exposure conditions, we also found the induction of SRC protein kinase activation in Beas-2B cells (Fig. 4A). Knockdown of SRC expression almost completely blocked STAT3 activation and VEGFA production in Beas-2B cells in response to PM2.5 stimulation (Fig. 4D and 4E). These data indicate that SRC-STAT3 pathway activation might play a critical role in mediating VEGFA production in PM2.5-treated Beas-2B cells. In the following experiment, we further observed similar levels of SRC-STAT3 pathway activation in Beas-2B cells treated with PM2.5 with or without PMB cotreatment (Fig. 4F); these results thus excluded the possibility of endotoxin contamination involvement in SRC-STAT3 pathway activation induced by PM2.5.
Figure 4.

PM2.5-induced autophagy contributed to VEGFA production by activating the SRC-STAT3 pathway. (A) Beas-2B cells were left untreated or were treated with PM2.5 as described in Fig. 1A; then, the activation status of SRC and STAT3 was determined. (B) Beas-2B cells were transfected with STAT3-dependent luciferase reporter, and stable transfectants were established. The transfectants were exposed to different doses of PM2.5 (as indicated), and the induction of STAT3-dependent luciferase activity was examined 12 h after PM2.5 exposure (*, P < 0.05; **, P < 0.01). (C) Beas-2B cells were transfected with STAT3 siRNA or control siRNA; then, cells were treated with PM2.5 (100 μg/mL) 36 h after transfection. The expression of STAT3 and VEGFA was examined 24 h after PM2.5 exposure. (D) Beas-2B cells were transfected with SRC siRNA or control siRNA and then were treated with PM2.5 (100 μg/mL) 36 h after transfection. The expressions of SRC and VEGFA and the activation status of STAT3 was examined 24 h after PM2.5 exposure. (E) Beas-2B cells stably transfected with the STAT3-dependent luciferase reporter were transfected with SRC siRNA or control siRNA and exposed to PM2.5 (100 μg/mL) 36 h after transfection. The induction of the STAT3-dependent luciferase activity was determined 12 h after PM2.5 exposure (**, P < 0.01). (F) Beas-2B cells were treated as described in Fig. 1F, and then the activation status of SRC and STAT3 was determined 24 h after PM2.5 exposure. (H, I and J) Beas-2B cells were treated or transfected as described in Fig. 3A, 3C and 3E. Then, the activation status of SRC and STAT3 was determined 24 h after PM2.5 exposure. p, phosphorylated.
In fact, we also discovered the involvement of other transcription factors, including NFKB/nuclear factor-κB, AP-1/activator protein-1 and EGR1 (early growth response 1), in mediating VEGFA transcription in Beas-2B cells upon PM2.5 exposure (data not shown). However, when autophagy induction in Beas-2B cells was blocked by 3-MA pretreatment, ATG5 siRNA transfection or costimulation with BafA1, only PM2.5-induced STAT3 activation was dramatically suppressed with the impairment of autophagy (Fig. 4G, 4H and 4I), whereas the activation status of other transcription factors did not show obvious changes under the same PM2.5 exposure conditions (data not shown). Moreover, the induction of SRC protein kinase activation was also sharply inhibited in the 3-MA-pretreated, ATG5 siRNA-transfected or BafA1-costimulated Beas-2B cells compared to the control cells (Fig. 4G, 4H and 4I). Taken together, these data lead us to conclude that the induction of autophagy mediates VEGFA production in bronchial epithelial cells by activating the SRC-STAT3 pathway upon PM2.5 exposure.
PM2.5 treatment induced TP53 transactivation, which was critical for mediating autophagy induction in bronchial epithelial cells
After identifying the functional link between autophagy induction and VEGFA production in bronchial epithelial cells, we next explored the upstream signal transduction pathways leading to autophagy induction following PM2.5 exposure. TP53 is a transcription factor that is activated by a wide variety of cellular stresses and subsequently orchestrates biological outputs, including the modulation of autophagy in a transcription-dependent or -independent manner.24 In PM2.5-treated Beas-2B cells, we detected both the dose- and time-dependent upregulation of TP53 phosphorylation at serine 15 (Fig. 5A and B), a representative signaling event indicating the activation of this protein. Under the same conditions, an enhancement of TP53-dependent luciferase activity was readily detected in Beas-2B cells (Fig. 2C), further confirming the elevation of TP53 transcriptional activity in response to PM2.5.
Figure 5.

PM2.5 induced TP53 transactivation, which was critical for mediating autophagy induction in Beas-2B cells. (A and B) Beas-2B cells were left untreated or were treated with PM2.5 as described in Fig. 1A and 1E, and the induction of TP53 activation and DRAM1 expression was examined. (C) Beas-2B cells were transfected with TP53-dependent luciferase reporter, and stable transfectants were established. The transfectants were exposed to different doses of PM2.5 (as indicated), and the induction of TP53-dependent luciferase activity was examined 12 h after PM2.5 exposure (**, P < 0.01). (D) Beas-2B cells were treated as described in Fig. 1F, and the induction of TP53 activation and DRAM1 expression was examined 24 h after PM2.5 exposure. (E) Beas-2B cells were transfected with TP53 siRNA, DRAM1 siRNA or control siRNA; then, they were treated with PM2.5 (100 μg/mL) 36 h after transfection. The activation status of TP53 and the expression levels of DRAM1, BECN1 MAP1LC3B were examined 24 h after PM2.5 exposure. (F and G) Beas-2B cells were transfected and treated as described in (E), and then the autophagy signals were detected as described in Fig. 2C and 2D (**, P < 0.01). p, phosphorylated.
DRAM1 is considered the most important TP53 target gene that functions as a positive regulator of autophagy.25 As shown in Fig. 5A and 5B, we readily observed an obvious upregulation of DRAM1 expression accompanying TP53 activation after PM2.5 exposure. In addition, the responses of TP53 activation and DRAM1 induction after PM2.5 exposure did not show detectable differences in the absence or presence of PMB costimulation (Fig. 5D). Together, these data indicate that PM2.5 treatment effectively induces TP53-DRAM1 pathway activation in Beas-2B cells.
Next, TP53 and DRAM1 siRNAs were separately transfected into Beas-2B cells to determine whether TP53-DRAM1 pathway activation contributes to autophagy induction in the PM2.5-induced response. We observed that the upregulation of both MAP1LC3B and BECN1 upon PM2.5 exposure was completely blocked by knockdown of TP53 or DRAM1 expression (Fig. 5E). Moreover, there was a significant reduction in the autophagic fluorescence signals from the Cyto-ID Autophagy Detection Reagent-stained Beas-2B cells with the impairment of TP53 or DRAM1 expression (Fig. 5F and 5G). Together, these results indicate that TP53-DRAM1 pathway activation is essential for inducing autophagy in PM2.5-treated Beas-2B cells.
ATR was required for the induction of TP53-dependent autophagy in bronchial epithelial cells under PM2.5 exposure
Next, we focused on identifying the signal transduction mechanism responsible for TP53 phosphorylation (at serine 15), transactivation and the subsequent induction of autophagy in response to PM2.5 treatment. Multiple lines of evidence have revealed that TP53 activation is triggered by a variety of signaling pathways under different stress conditions. ATM (ATM serine/threonine kinase) and ATR are class III phosphatidylinositol 3-kinase-related kinase family members that contribute to TP53 activation in response to DNA damage and checkpoint stresses.24,26,27 Because Wuhan PM2.5 induced an obvious G0/G1 arrest in Beas-2B cells (data not shown), we first examined the possible roles of ATM and ATR in mediating TP53 activation under PM2.5 treatment. As shown in Fig. 6A, accompanying TP53 accumulation, both ATM and ATR were strongly activated in Beas-2B cells after PM2.5 exposure at certain doses (Fig. 6A). Furthermore, knockdown of ATR expression resulted in the complete inhibition of the TP53 phosphorylation and DRAM1 upregulation induced by PM2.5. However, none of these responses were observed in ATM siRNA-transfected Beas-2B cells under the same PM2.5 exposure conditions (Fig. 6B). In addition, an attenuation of TP53-dependent luciferase activity was readily detected in Beas-2B cells transfected with ATR siRNA, but not ATM siRNA, in response to PM2.5 stimulation (Fig. 6C). Together, these data indicate that ATR plays a critical role in mediating TP53-DRAM1 pathway activation in the PM2.5-induced response.
Figure 6.
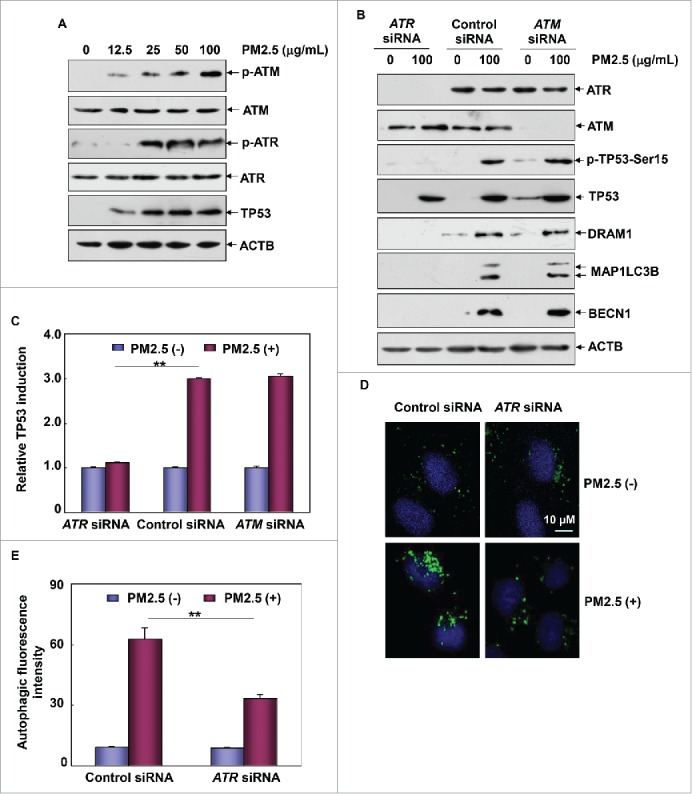
ATR was required for the induction of TP53-dependent autophagy in Beas-2B cells upon PM2.5 exposure. (A) Beas-2B cells were treated as described in Fig. 1A, and then the activation status of ATM and ATR was determined. (B) Beas-2B cells were transfected with ATR siRNA, ATM siRNA or control siRNA; then, cells were treated with PM2.5 (100 μg/mL) 36 h after transfection. The activation status of TP53 and the expression levels of ATR, ATM, DRAM1, BECN1 and MAP1LC3B were examined 24 h after PM2.5 exposure. (C) Beas-2B cells stably transfected with TP53-dependent luciferase reporter were transfected with ATR siRNA, ATM siRNA or control siRNA; then exposed to PM2.5 (100 μg/mL) 36 h after transfection. The induction of TP53-dependent luciferase activity was determined 12 h after PM2.5 exposure (**, P < 0.01). (D and E) Beas-2B cells were transfected with ATR siRNA or control siRNA and then treated with PM2.5 (100 μg/mL) 36 h after transfection. The autophagy signals were detected as described in Fig. 2C and 2D (**, P < 0.01). p, phosphorylated.
In the following experiment, we observed the prevention of MAP1LC3B and BECN1 upregulation (Fig. 6B) and a decrease in the Cyto-ID-dependent autophagic fluorescence signals in Beas-2B cells after ATR siRNA transfection (Fig. 6D and 6E), further confirming that ATR, rather than ATM, is required for TP53-dependent autophagy induction in PM2.5-treated Beas-2B cells.
The ATR-CHEK1 axis mediated TP53-dependent autophagy induction in bronchial epithelial cells upon PM2.5 exposure
CHEK1 is a well-known downstream target of ATR that mediates TP53 activation in response to checkpoint stresses.26,27 Therefore, to determine whether CHEK1 is involved in the functional link between ATR and TP53 activation in Beas-2B cells under PM2.5 exposure, we next examined the role of CHEK1 in PM2.5-treated Beas-2B cells. As shown in Fig. 7A, phosphorylation of CHEK1 was significantly upregulated in Beas-2B cells after exposure to certain doses of PM2.5. In addition, knockdown of ATR expression significantly suppressed the CHEK1 activation induced by PM2.5 (Fig. 7B). These results thus suggest a potential role of CHEK1 downstream of ATR in mediating PM2.5-induced responses. In addition, the activation of both ATR and CHEK1 induced by PM2.5 did not show any differences in the absence or presence of PMB costimulation (Fig. 6C), indicating a specific effect of PM2.5 on triggering ATR-CHEK1 axis activation in Beas-2B cells.
Figure 7.

CHEK1 was the downstream target of ATR that mediated TP53-dependent autophagy in Beas-2B cells under PM2.5 exposure. (A) Beas-2B cells were treated as described in Fig. 1A, and the activation status of CHEK1 was determined. (B) Beas-2B cells were transfected with ATR siRNA or control siRNA and treated with PM2.5 (100 μg/mL) 36 h after transfection. The activation status of CHEK1 was determined 24 h after PM2.5 exposure. (C) Beas-2B cells were treated as described in Fig. 1F, and the activation status of the ATR-CHEK1 axis was determined 24 h after PM2.5 exposure. (D) Beas-2B cells were transfected with CHEK1 siRNA or control siRNA and then treated with PM2.5 (100 μg/mL) 36 h after transfection. The activation status of CHEK1 and TP53 and the expression levels of DRAM1, BECN1 and MAP1LC3B were examined 24 h after PM2.5 exposure. (E) Beas-2B cells stably transfected with TP53-dependent luciferase reporter were transfected with CHEK1 siRNA or control siRNA and exposed to PM2.5 (100 μg/mL) 36 h after transfection. The induction of TP53-dependent luciferase activity was determined 12 h after PM2.5 exposure (**, P < 0.01). (F and G) Beas-2B cells were transfected with CHEK1 siRNA or control siRNA and treated with PM2.5 (100 μg/mL) 36 h after transfection. Then, the autophagy signals were detected as described in Fig. 2C and 2D (**, P < 0.01). p, phosphorylated.
Then, we observed that ablation of CHEK1 expression not only effectively impaired the phosphorylation and transactivation of TP53, but also suppressed the induction of DRAM1 expression by PM2.5 (Fig. 7D and 7E), clearly confirming the critical contribution of CHEK1 to TP53 transactivation in the PM2.5-induced response. Most importantly, autophagy induction was also impaired by knockdown of CHEK1 expression, as evidenced by the inhibition of MAP1LC3B and BECN1 induction and the reduction in Cyto-ID-dependent autophagic fluorescence signals in CHEK1 siRNA-transfected Beas-2B cells compared to the control cells (Fig. 7D, 7F and 7G). Taken together, we conclude that CHEK1 is the downstream target of ATR that mediates TP53-dependent autophagy in Beas-2B cells.
The ATR-CHEK1-TP53 signaling pathway activation was critical for mediating autophagy-dependent VEGFA induction in bronchial epithelial cells under PM2.5 exposure
After clarifying the role of the ATR-CHEK1-TP53-DRAM1 pathway activation in mediating autophagy induction in the PM2.5-induced response, we finally determined whether this pathway contributes to regulating autophagy-dependent VEGFA induction under PM2.5 exposure. To this end, TP53, DRAM1, ATR and CHEK1 siRNAs were separately transfected into Beas-2B cells to block the activation of this pathway at different levels. We observed that knockdown of any of the critical signaling molecules in the ATR-CHEK1-TP53-DRAM1 pathway almost completely blocked SRC and STAT3 activation, as well as the VEGFA production induced by PM2.5 (Fig. 8A, 8C and 8E). As expected, the induction of VEGFA transcription was also dramatically suppressed in the various siRNA-transfected Beas-2B cells compared to the control siRNA-transfected cells under the same PM2.5 exposure conditions (Fig. 8B, 8D and 8F). Taken together, these data indicate that the ATR-CHEK1-TP53-DRAM1 pathway activation is critical for mediating autophagy-dependent VEGFA induction in PM2.5-treated Beas-2B cells.
Figure 8.

ATR-CHEK1-TP53 signaling pathway activation was required for mediating autophagy-dependent VEGFA production in Beas-2B cells under PM2.5 exposure. (A, C and E) Beas-2B cells were transfected with TP53 siRNA or DRAM1 siRNA (A), ATR siRNA (C) or CHEK1 siRNA (E) and their respective control siRNAs; then, cells were treated with PM2.5 (100 μg/mL) 36 h after transfection. The activation status of SRC and STAT3 and the expression level of VEGFA were determined 24 h after PM2.5 exposure. (B, D and F) Beas-2B cells stably transfected with a VEGFA promoter-driven luciferase reporter were transfected and treated as described in (A, C and E), respectively. Then, the induction of VEGFA promoter-dependent luciferase activity was determined 12 h after PM2.5 exposure (**, P < 0.01). p, phosphorylated.
Discussion
The association between air particles (especially PM2.5) and increased risk of respiratory and cardiovascular diseases is well established. The induction or exacerbation of airway inflammation is considered a common mechanism underlying the particle exposure-induced development of various adverse health effects in the respiratory and cardiovascular systems.1-6 Thus, understanding how particulate matter triggers inflammatory reactions in the airway is a central issue in particle toxicology.
The pulmonary epithelium represents a primary barrier that prevents the entry of inhaled PM. As a consequence, airway epithelial cells are also a primary target of inhaled noxious PM. For this reason, in vitro experiments involving cultured epithelial cells treated with various types of ambient and laboratory surrogate particles have become a well-accepted technique for investigating the basic mechanisms of particle toxicology. Additionally, many in vitro studies have focused on the upregulation and release of proinflammatory cytokines, such as IL1B, IL6, CXCL8 and TNF, by airway epithelial cells in response to PM2.5 exposure due to the critical role of these cytokines in the initiation and resolution of inflammation.4-6 In fact, elevation of these cytokines has also been observed in the bronchial alveolar lavage fluid of animals,28 further confirming the results obtained from the in vitro studies. In this work, to explore the inflammatory responses induced by the particles, the expression levels of several inflammatory factors were analyzed. We found that the upregulation of VEGFA expression constituted the major signaling event in immortalized or primary human bronchial epithelial cells exposed to PM2.5 collected in Wuhan in the winter (Fig. 1). Because VEGFA is a key regulator of blood vessel growth and an inducer of vascular leakage and permeability in the airway, homeostatic VEGFA expression is a critical means of lung structure maintenance, whereas excessive VEGFA expression is believed to be effective in regulating airway hyperactivity, chronic airway inflammation and vascular remodeling.18-21 Therefore, the data from this study are the first to reveal that the PM2.5-induced acute response in bronchial epithelial cells is mediated by VEGFA-related pathogenesis. Modulating airway VEGFA expression might be helpful to ameliorate the adverse airway responses induced by particle exposure.
In the attempt to figure out the signal transduction mechanism leading to VEGFA upregulation in bronchial epithelial cells, we discovered the critical contribution of autophagy to this process (Figs. 2 and 3). In fact, the implications of the autophagy pathway in immune dysfunction and the pathogenesis of inflammatory disorders (such as intestinal bowel disease) have been reported in previous studies.19,29,30 Moreover, due to the common feature of inflammatory responses in lung diseases, several investigations have focused on the functional link between autophagy and inflammation-associated pulmonary pathogenesis. For example, autophagy is essential for airway mucus secretion and therefore acts as an important cofactor in asthma, chronic obstructive pulmonary disease, pulmonary fibrosis and airway remodeling.14 Moreover, epithelial autophagy is responsible for the generation of fibrosis via the EMT (epithelial-to-mesenchymal transition) process in airways.16 In addition, the induction of autophagy directly impairs the expression of antiviral type III IFNL1/IFNλ1 (interferon, lambda 1) and enhances rhinovirus infection in airway epithelial cells, thus functioning as a novel mechanism to hinder the host antiviral defense against respiratory viral infection in the lung.17 Furthermore, autophagy is believed to serve as a cellular signaling event to promote the transforming growth factor β 1-mediated airway remodeling and loss of lung function in asthma.15 In the current study, we additionally elucidated the involvement of autophagy in the PM2.5-induced VEGFA production response in airway epithelial cells, further confirming the association between autophagy and the inflammatory state in the pulmonary system.
The regulation of the production or secretion of proinflammatory cytokines constitutes one of the mechanisms by which autophagy impacts the onset or progression of airway inflammatory responses.31 However, our understanding of how autophagy regulates cytokine production is still rudimentary. As described above, autophagy interferes with host-antirhinovirus infection in the airway by suppressing IFNL1 expression. In this process, DDX58 (DEXD/H-box helicase 58) recognizes viral RNA in the cytoplasm of infected cells and then binds to MAVS (mitochondrial antiviral signaling protein) to induce the production of interferon and the host antiviral defense. Interestingly, the ATG5 protein interacts with DDX58 and MAVS when the autophagic pathway is activated by rhinovirus infection. This interaction is responsible for the suppression of IFNL1 expression and the subsequent increase in rhinovirus replication in human airway epithelial cells.17 In the current study, after revealing the involvement of autophagy in VEGFA induction in PM2.5-treated epithelial cells, we performed many experiments to determine which signal transduction pathway(s) were activated by autophagy, leading to VEGFA induction. Although most of the proinflammatory transcription factors (STAT3, NFKB, AP-1 and EGR1) that we analyzed were found to mediate VEGFA production by PM2.5 (data not shown), only STAT3 activation was controlled by autophagy induction (Fig. 4). Thus, this study is the first to elucidate the underlying mechanism that mediates autophagy-dependent VEGFA induction through STAT3 signaling. In fact, a previous study demonstrates the pivotal role of autophagy in facilitating interferon gamma production by activating the JAK2 (Janus kinase 2)-STAT1 pathway.32 However, we did not observe the activation of JAK2 in response to PM2.5 exposure in epithelial cells (data not shown), whereas SRC protein kinase was activated after autophagy induction and then mediated STAT3 phosphorylation. Therefore, we have elucidated the mechanism by which autophagy regulates proinflammatory VEGFA production in response to PM2.5 by activating the SRC-STAT3 pathway (Fig. 4).
Autophagy plays a critical role in the maintenance of intracellular homeostasis under both physiological and pathological conditions; therefore, the number of factors and signaling pathways implicated in autophagy modulation is continually increasing.7 TP53 is a stress-responsive transcription factor involved in regulating autophagy in both transcription-dependent and -independent manners.33 The lysosomal protein DRAM1 is a transcriptional target of TP53 that can induce autophagosome accumulation and therefore orchestrates the induction of autophagy.25 However, it is noteworthy that previous reports demonstrate the role of the TP53-DRAM1 pathway activation in modulating the autophagic process under genotoxic stress only, which is critical for cell-fate determination.25,34 Therefore, data in this study obtained in PM2.5-treated epithelial cells, expand on the former mechanism by functionally characterizing the TP53-DRAM1-autophagy pathway under conditions of proinflammatory stress (Figs. 5 and 8).
Another novel finding of this work is that the ATR-CHEK1 axis is implicated in regulating autophagy and the subsequent proinflammatory cytokine production in the PM2.5-induced cellular response. It is well accepted that the ATR-CHEK1 pathway is usually activated in response to DNA damage signals and guards the integrity of the genomic DNA by regulating and coordinating multiple cellular processes, including cell cycle arrest, inhibition of replication origin firing, protection of stressed replication forks, and DNA repair.26,27 In the light of recent studies, functional links between the immune response and the ATR-CHEK1-mediated processes that regulate genomic integrity have also been revealed.35-38 For example, the ATR-CHEK1 pathway was found to be activated in the Simian virus 40 large T antigen-induced DNA damage response in human fibroblasts, resulting in the induction of IRF1 (interferon regulatory factor 1) and the transactivation of IFNB1 (interferon β 1) to mediate the antiviral response.35 Intriguingly, in germinal center B cells, downregulation of the ATR-CHEK1 checkpoint axis is required for efficient somatic hypermutation and IgG diversification.36 Data from several other reports have revealed that the DNA damage responses mediated by ATR, ATM and CHEK1 may alert the immune system to the presence of potentially dangerous cells by upregulating the expression of ligands that can induce the activation of innate and adaptive immune cells.37,38 Our current study provides novel evidence for a critical role of the ATR-CHEK1 axis activation in mediating the proinflammatory response in bronchial epithelial cells under PM2.5 exposure (Figs. 6 to 8), further confirming the functional link between checkpoint pathways and immune responses under particle-induced stress. In addition, because neither the exact mechanism nor the cellular consequences of autophagy activation in response to DNA damage signals is fully understood, our finding that autophagy represents a bridge between the ATR-CHEK1 axis activation and VEGFA production provides new insights into these issues.
In summary, the data in this study elucidate the molecular mechanism involved in mediating VEGFA production in airway epithelial cells under PM2.5 exposure. These in vitro results contribute to the long-term goal of toxicology studies on PM, which focus on identifying specific proinflammatory signaling pathways. Determining whether these in vitro results are applicable to PM-induced lung inflammation and pathogenesis in vivo is worthy of additional investigation.
Materials and methods
Plasmids, antibodies and reagents
The VEGFA promoter-driven luciferase reporter plasmid and TP53-dependent luciferase reporter plasmid were provided by Dr. Chuanshu Huang (New York University, USA). STAT3-dependent luciferase reporter plasmid was provided by Dr. Ming Shi in our institute. The siRNAs and regents used were as follows: ATR siRNA (Cell Signaling Technology, 6288), ATM siRNA (Cell Signaling Technology, 6328), ATG5 siRNA (Cell Signaling Technology, 6348), DRAM1 siRNA (Riobo Technology, 1314.14), CHEK1 (CHK1) siRNA (Riobo Technology, 13285.14), BECN1 siRNA (Cell Signaling Technology, 6222), 3-MA (Sigma-Aldrich, M9281), PMB (Sigma-Aldrich, P1004) and BafA1 (LC Laboratories, B1080); Primary antibodies used were as follows: BECN1 (Cell Signaling Technology, 3495), MAP1LC3B (Cell Signaling Technology, 3868), phospho-TP53 (Ser15; Cell Signaling Technology, 9284), TP53 (Cell Signaling Technology, 2524), phospho-SRC (Tyr416; Cell Signaling Technology, 6943), SRC (Cell Signaling Technology, 2109), phospho-STAT3 (Tyr705; Cell Signaling Technology, 9145), STAT3 (Cell Signaling Technology, 9139), phospho-ATM (Ser1981; Cell Signaling Technology, 5883), ATM (Cell Signaling Technology, 2873), phospho-ATR (Ser428; Cell Signaling Technology, 2853), ATR (Cell Signaling Technology, 2790), phospho-CHEK1 (Ser345; Cell Signaling Technology, 2348), CHEK1 (Cell Signaling Technology, 2360), SQSTM1 (Cell Signaling Technology, 8025), ACTB (Cell Signaling Technology, 4970), ATG5 (Cell Signaling Technology, 9980) and DRAM1 (Santa Cruz Biotechnology, 98654).
PM2.5 sample collection and analysis
The PM2.5 sample was collected in the Hanyang District of Wuhan, a city in China with serious air pollution. Particulate air samples (ambient and nominally 10X concentrated) were collected using an aerosol concentration enrichment system with Teflon filters (GelmanTeflo, 37 mm, 0.2-μm pore). The samples were collected on the roof of the buildings on the busy streets every weekday for 6 h (between 9 a.m. and 3 p.m.) for 3 mo between November 2011 and February 2012. The filter samples were stored at a constant temperature and relative humidity (−20 ± 0.5°C, 40 ± 5% RH) until analysis. The filter was cut into small pieces 1 cm2 in area, immersed in sterilized water and then sonicated 3 times to extract the water-soluble components. After the water-extracted samples were collected and frozen, the particle sample was stored at −20°C until use. The analysis of PM2.5 components was performed using high-performance liquid chromatography (Waters, 2695).
Cell culture, transfection and PM2.5 treatment
Beas-2B human bronchial epithelial cells were kindly provided by Dr. Chuanshu Huang. The cells were maintained in DMEM (Gibco, 12800-017) with 10% fetal bovine serum (Life Technologies, 16010-109) supplemented with antibiotic/antimycotic (Life Technologies, 15240-062). Transfections were performed with the LipofectAMINE 2000 (Invitrogen, 11668-019) or LipofectAMINE™ RNAi MAX (Invitrogen, 13778-150) according to the manufacturer's instructions. Prior to cell exposure, particle samples were weighed, resuspended in cell culture medium, and diluted to the final concentrations of 12.5, 25, 50 and 100 μg/mL for cell treatment after sonication.
Primary human bronchial epithelial cells (ScienCell, 3210) were maintained in BEpiCM (a specific medium for culturing primary bronchial epithelial cells; ScienCell, 3211) with 10% BEpiCGS (bronchial epithelial cell growth supplement; ScienCell, 3262) supplemented with antibiotic/antimycotic (ScienCell, 0503). The cells were exposed to 20 μg/mL of PM2.5 to analyze intercellular responses.
Western blot assay
After treatment, Beas-2B or primary bronchial epithelial cells were harvested and lysed with ice-cold cell lysis buffer (10 mM Tris-HCl, pH 7.4, 1% SDS [Sigma, 71725], and 1 mM Na3VO4), and the homogenate was centrifuged to collect the supernatant. The harvested samples were quantified using a protein assay kit, separated by SDS-PAGE, and transferred to a PVDF membrane. After being blocked with skim milk, the blots were probed with the appropriate primary antibodies overnight at 4°C before being washed and incubated with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology, 7074 and 7076). Bands were detected as described in our previous report.39
Luciferase reporter assay
Cells were cotransfected with an experimental reporter (either a TP53-dependent luciferase reporter, STAT3-dependent luciferase reporter or the VEGFA promoter-driven luciferase reporter), a control reporter (Renilla luciferase reporter; Promega, E2241) and the pcDNA3.1 plasmid (Invitrogen, V79020), and then the stable transfectants were established. Luciferase activity was tested 12 h after PM2.5 exposure using Firefly-Renilla Dual-Luciferase Reporter Assay System (Promega, E1910). The data were obtained by normalizing the activity of the experimental reporter to that of the internal control. The results were presented as the relative induction by normalizing the luciferase activity in the PM2.5-treated cells to the luciferase activity in untreated control cells, as previously described.22
RNA isolation and RT-PCR assay
Total RNA was extracted with TRIzol reagent (Sigma-Aldrich, T9424), and cDNA was synthesized with the ThermoScriptTM RT-PCR system (Thermo Fisher Scientific, K1622). To analyze the induction of VEGFA, IL1B, IL6, CXCL8 and TNF transcription, the following oligonucleotides were synthesized and used as specific primers to amplify the target cDNAs: VEGFA: 5′-tgcacccatggcagaaggagg-3′ (forward) and 5′-tcaccgcctcggcttgtcaxca-3′ (reverse); IL1B: 5′-gctccgggactcacagcaaaaa-3′ (forward) and 5′-ttggggaactgggcagactcaa-3′ (reverse); IL6: 5′-cgccttcggtccagttgcctt-3′ (forward) and 5′-tgccagtgcctctttgctgctt-3′ (reverse); CXCL8: 5′-acaagagccaggaagaaaccac-3′ (forward) and 5′-aaacttctccacaaccctctgc-3′ (reverse); and TNF: 5′-cagagggaagagttccccagg-3′ (forward) and 5′-ccttggtctggtaggagacgg-3′ (reverse). The primers used to amplify ACTB cDNA were 5′-tgacgtggacatccgcaaag-3′ (forward) and 5′-ctggaaggtggacagcgagg-3′ (reverse).
ELISA
Cytokine production in the cell culture supernatants was quantified with human VEGFA (eBioscience, BMS277/2), IL1B (eBioscience, BMS224/2), IL6 (eBioscience, BMS213HS), CXCL8 (eBioscience, BMS204/3) and TNF (eBioscience, BMS223INST) immunoassay kits. Briefly, cells were seeded into 24-well plates and cultured to 70% to 80% confluence. The cells were subjected to PM2.5 exposure at different doses, and the supernatants were collected 24 h later to analyze the release of cytokines in response to PM2.5 treatment.
Autophagy assay
Cellular autophagy was monitored using the following techniques: western blot analysis of specific key proteins (MAP1LC3B, BECN1 and SQSTM1), transmission electron microscopy (TEM, Hitachi, H7650), confocal microscopy (ZEISS, LSM510 META) and flow cytometry (BD Biosciences, FACSCalibur)-based quantitative analysis. The presence of autophagosomes in bronchial epithelial cells induced by PM2.5 was directly assessed by TEM-based analysis. Then, the Cyto-ID Autophagy Detection Kit (Enzo Life Sciences, 175-0050) was used to monitor specific autophagic fluorescence signals under confocal microscopy or to quantitatively measure the autophagic fluorescence intensity by flow cytometric analysis. The 488 nm excitable Cyto-ID Green Autophagy Detection Reagent (dye) supplied with the kit becomes brightly fluorescent in vesicles produced during autophagy and thus serves as a convenient tool to detect autophagic signals at the cellular level. For the quantitative analysis, cells were incubated with Cyto-ID Reagent, trypsinized following PM2.5 treatment, washed in Assay Buffer and then resuspended in the same buffer. The samples were analyzed in the green (FL1) channel of a flow cytometer to obtain the mean fluorescence intensity, as previously described.40 For the immunoblotting assay, the induction of cellular autophagy in response to PM2.5 exposure was determined as an increase in the endogenous MAP1LC3B-II:LC3B-I ratio, the upregulation of BECN1 expression and the dynamic degradation of SQSTM1. Autophagic flux was assessed by comparing the extent of MAP1LC3B-II accumulation following PM2.5 treatment with or without the concomitant use of BafA1, an inhibitor that prevents autophagosome-lysosome fusion and thereby inhibits autophagic processing and clearance.
Statistics
The data were tested for significance employing the Student t test and presented as mean ±SD. The level of significance was set at P < 0.05.
Abbreviations
- 3-MA
3-methyladenine
- ACTB
actin, beta
- ATG5
autophagy-related 5
- ATM
ATM serine/threonine kinase
- ATR
ATR serine/threonine kinase
- BafA1
bafilomycin A1
- BECN1
Beclin 1; CHEK1, checkpoint kinase 1
- CXCL8
C-X-C motif chemokine ligand 8
- DRAM1
DNA damage regulated autophagy modulator 1
- ELISA
enzyme-linked immunosorbent assay
- IFNL1
interferon, lambda 1
- IL1B
interleukin 1 beta
- IL6
interleukin 6
- MAP1LC3B
microtubule-associated protein 1 light chain 3 beta
- PM
particulate matter
- PMB
polymyxin B
- RT-PCR
reverse transcription polymerase chain reaction
- SQSTM1
sequestosome 1
- SRC
SRC proto-oncogene, non-receptor tyrosine kinase
- STAT3
signal transducer and activator of transcription 3
- TNF
tumor necrosis factor
- TP53
tumor protein p53
- VEGFA
vascular endothelial growth factor A
Disclosure of potential conflicts of interest
The authors have no conflict of interest.
Funding
This project is supported by the National Key Research and Development Programs on Fundamental Sciences (973 Project, 2011CB503803), National Natural Science Foundation of China (No. 31570758, 31270797, 31171342) to Dr. Lun Song
References
- [1].Suhaimi NF, Jalaludin J. Biomarker as a research tool in linking exposure to air particles and respiratory health. Biomed Res Int 2015; 2015:962853; PMID:25984536; http://dx.doi.org/ 10.1155/2015/962853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang C, Tu Y, Yu Z, Lu R. PM2.5 and cardiovascular diseases in the elderly: an overview. Int J Environ Res Pub Health 2015; 12:8187-97; PMID:26193289; http://dx.doi.org/ 10.3390/ijerph120708187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nogueira JB. Poluição atmosférica e doenças cardiovasculares. Rev Port Cardiol 2009; 28:715-33; PMID:19697799 [PubMed] [Google Scholar]
- [4].Veranth JM, Moss TA, Chow JC, Labban R, Nichols WK, Walton JC, Watson JG, Yost GS. Correlation of in vitro cytokine responses with the chemical composition of soil-derived particulate matter. Environ Health Perspect 2005; 114:341-49; PMID:16507455; http://dx.doi.org/ 10.1289/ehp.8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhou Z, Liu Y, Duan F, Qin M, Wu F, Sheng W, Yang L, Liu J, He K. Transcriptomic analyses of the biological effects of airborne PM2.5 exposure on human bronchial epithelial cells. PloS one 2015; 10:e0138267; PMID:26382838; http://dx.doi.org/ 10.1371/journal.pone.0138267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Siponen T, Yli-Tuomi T, Aurela M, Dufva H, Hillamo R, Hirvonen MR, Huttunen K, Pekkanen J, Pennanen A, Salonen I, et al.. Source-specific fine particulate air pollution and systemic inflammation in ischaemic heart disease patients. Occup Environ Med 2015; 72:277-83; PMID:25479755; http://dx.doi.org/ 10.1136/oemed-2014-102240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res 2012; 22:43-61; PMID:21912435; http://dx.doi.org/ 10.1038/cr.2011.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu Y, Eissa NT. Autophagy in innate and adaptive immunity. Proc Am Thorac Soc 2010; 7:22-8; PMID:20160145; http://dx.doi.org/ 10.1513/pats.200909-103JS [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Das G, Shravage BV, Baehrecke EH. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol 2012; 4:a008813; PMID:22661635; http://dx.doi.org/ 10.1101/cshperspect.a008813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mah LY, Ryan KM. Autophagy and cancer. Cold Spring Harb Perspect Biol 2012; 4:a008821; PMID:22166310; http://dx.doi.org/ 10.1101/cshperspect.a008821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lapaquette P, Guzzo J, Bretillon L, Bringer MA. Cellular and molecular connections between autophagy and inflammation. Mediat Inflamm 2015; 2015:398483; PMID:26221063; http://dx.doi.org/ 10.1155/2015/398483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jo EK, Shin DM, Choi AM. Autophagy: cellular defense to excessive inflammation. Microbes Infect 2012; 14:119-25; PMID:21924374; http://dx.doi.org/ 10.1016/j.micinf.2011.08.014 [DOI] [PubMed] [Google Scholar]
- [13].Mizumura K, Choi AMK, Ryter SW. Emerging role of selective autophagy in human diseases. Front Pharmacol 2014; 5:00244; PMID:25414669; http://dx.doi.org/ 10.3389/fphar.2014.00244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dickinson JD, Alevy Y, Malvin NP, Patel KK, Gunsten SP, Holtzman MJ, Stappenbeck TS, Brody SL. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy 2015; 2015:1056967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Poon A, Eidelman D, Laprise C, Hamid Q. ATG5, autophagy and lung function in asthma. Autophagy 2012; 8:694-95; PMID:22498476; http://dx.doi.org/ 10.4161/auto.19315 [DOI] [PubMed] [Google Scholar]
- [16].Cho IH, Choi YJ, Gong JH, Shin D, Kang MK, Kang YH. Astragalin inhibits autophagy-associated airway epithelial fibrosis. Resp Res 2015; 16:51; PMID: 25895672; http://dx.doi.org/ 10.1186/s12931-015-0211-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wu Q, Jiang D, Huang C, van Dyk LF, Li L, Chu HW. Trehalose-mediated autophagy impairs the anti-viral function of human primary airway epithelial cells. PloS one 2015; 10:e0124524; PMID:25879848; http://dx.doi.org/ 10.1371/journal.pone.0124524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee CG, Ma B, Takyar S, Ahangari F, DelaCruz C, He CH, Elias JA. Studies of vascular endothelial growth factor in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 2011; 8:512-15; PMID:22052929; http://dx.doi.org/ 10.1513/pats.201102-018MW [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tuder RM, Yun JH. Vascular endothelial growth factor in the lung: friend or foe. Curr Opin Pharmacol 2008; 8:255-60; PMID:18468486; http://dx.doi.org/ 10.1016/j.coph.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Matsune S. Allergic rhinitis and vascular endothelial growth factor. J Nippon Med Sch 2012; 79:170-75; PMID:22791116; http://dx.doi.org/ 10.1272/jnms.79.170 [DOI] [PubMed] [Google Scholar]
- [21].Haigh JJ. Role of VEGF in organogenesis. Organogenesis 2008; 4:247-56; PMID:19337405; http://dx.doi.org/ 10.4161/org.4.4.7415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dong W, Li Y, Gao M, Hu M, Li X, Mai S, Guo N, Yuan S, Song L. IKKalpha contributes to UVB-induced VEGF expression by regulating AP-1 transactivation. Nucleic Acids Res 2012; 40:2940-55; PMID:22169952; http://dx.doi.org/ 10.1093/nar/gkr1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Loos B, Toit Ad, Hofmeyr J-HS. Defining and measuring autophagosome flux-concept and reality. Autophagy 2014; 10:2087-96; PMID:25484088; http://dx.doi.org/ 10.4161/15548627.2014.973338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hao Q, Cho W. Battle against cancer: an everlasting saga of p53. Int J Mol Sci 2014; 15:22109-27; PMID:25470027; http://dx.doi.org/ 10.3390/ijms151222109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121-34; PMID:16839881; http://dx.doi.org/ 10.1016/j.cell.2006.05.034 [DOI] [PubMed] [Google Scholar]
- [26].Benada J, Macurek L. Targeting the checkpoint to kill cancer cells. Biomolecules 2015; 5:1912-37; PMID:26295265; http://dx.doi.org/ 10.3390/biom5031912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Can Res 2015; 21:4780-85; PMID:26362996; http://dx.doi.org/ 10.1158/1078-0432.CCR-15-0479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Driscoll KE. TNF alpha and MIP-2: role in particle-induced inflammation and regulation by oxidative stress. Toxicol Lett 2000; 112–113:177-84; PMID:10720729 [DOI] [PubMed] [Google Scholar]
- [29].Kaser A, Blumberg RS. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semi Immunol 2009; 21:156-63; PMID:19237300; http://dx.doi.org/ 10.1016/j.smim.2009.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brest P, Corcelle EA, Cesaro A, Chargui A, Belaïd A, Klionsky DJ, Vouret-Craviari V, Hebuterne X, Hofman P, Mograbi B. Autophagy and crohn's disease: at the crossroads of infection, inflammation, immunity, and cancer. Curr Mol Med 2010; 10:486-502; PMID:20540703; http://dx.doi.org/ 10.2174/156652410791608252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Harris J. Autophagy and cytokines. Cytokine 2011; 56:140-44; PMID:21889357; http://dx.doi.org/ 10.1016/j.cyto.2011.08.022 [DOI] [PubMed] [Google Scholar]
- [32].Chang YP, Chen CL, Chen SO, Lin YS, Tsai CC, Huang WC, Wang CY, Hsieh CY, Choi PC, Lin CF. Autophagy facilitates an IFN-gamma response and signal transduction. Microbes Infect 2011; 13:888-94; PMID:21664983; http://dx.doi.org/ 10.1016/j.micinf.2011.05.008 [DOI] [PubMed] [Google Scholar]
- [33].Sui X, Jin L, Huang X, Geng S, He C, Hu X. p53 signaling and autophagy in cancer: a revolutionary strategy could be developed for cancer treatment. Autophagy 2014; 7:565-71; PMID:21099252; http://dx.doi.org/ 10.4161/auto.7.6.14073 [DOI] [PubMed] [Google Scholar]
- [34].Crighton D, Wilkinson S, Ryan KM. DRAM links autophagy to p53 and programmed cell death. Autophagy 2014; 3:72-74; PMID:17102582; http://dx.doi.org/ 10.4161/auto.3438 [DOI] [PubMed] [Google Scholar]
- [35].Forero A, Giacobbi NS, McCormick KD, Gjoerup OV, Bakkenist CJ, Pipas JM, Sarkar SN. Simian virus 40 large T antigen induces IFN-stimulated genes through ATR kinase. J Immunol 2014; 192:5933-42; PMID:24799566; http://dx.doi.org/ 10.4049/jimmunol.1303470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Frankenberger S, Davari K, Fischer-Burkart S, Bottcher K, Tomi NS, Zimber-Strobl U, Jungnickel B. Checkpoint kinase 1 negatively regulates somatic hypermutation. Nucleic Acids Res 2014; 42:3666-74; PMID:24423870; http://dx.doi.org/ 10.1093/nar/gkt1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005; 436:1186-90; PMID:15995699; http://dx.doi.org/ 10.1038/nature03884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tang MLF, Khan MKN, Croxford JL, Tan KW, Angeli V, Gasser S. The DNA damage response induces antigen presenting cell-like functions in fibroblast. Eur J Immunol 2014; 44:1108-18; PMID:24375454; http://dx.doi.org/ 10.1002/eji.201343781 [DOI] [PubMed] [Google Scholar]
- [39].Gao M, Li X, Dong W, Jin R, Ma H, Yang P, Hu M, Li Y, Hao Y, Yuan S, et al.. Ribosomal protein S7 regulates arsenite-induced GADD45alpha expression by attenuating MDM2-mediated GADD45alpha ubiquitination and degradation. Nucleic Acids Res 2013; 41:5210-22; PMID:23563151; http://dx.doi.org/ 10.1093/nar/gkt223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tan Q, Wang H, Hu Y, Hu M, Li X, Aodengqimuge, Ma Y, Wei C, Song L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci 2015; 106:1023-32; PMID:26041409; http://dx.doi.org/ 10.1111/cas.12712 [DOI] [PMC free article] [PubMed] [Google Scholar]