

ABSTRACT
In humans, loss of TBC1D20 (TBC1 domain family, member 20) protein function causes Warburg Micro syndrome 4 (WARBM4), an autosomal recessive disorder characterized by congenital eye, brain, and genital abnormalities. TBC1D20-deficient mice exhibit ocular abnormalities and male infertility. TBC1D20 is a ubiquitously expressed member of the family of GTPase-activating proteins (GAPs) that increase the intrinsically slow GTP-hydrolysis rate of small RAB-GTPases when bound to GTP. Biochemical studies have established TBC1D20 as a GAP for RAB1B and RAB2A. However, the cellular role of TBC1D20 still remains elusive, and there is little information about how the functional loss of TBC1D20 causes clinical manifestations in WARBM4-affected children. Here we evaluate the role of TBC1D20 in cells carrying a null mutant allele, as well as TBC1D20-deficient mice, which display eye and testicular abnormalities. We demonstrate that TBC1D20, via its RAB1B GAP function, is a key regulator of autophagosome maturation, a process required for maintenance of autophagic flux and degradation of autophagic cargo. Our results provide evidence that TBC1D20-mediated autophagosome maturation maintains lens transparency by mediating the removal of damaged proteins and organelles from lens fiber cells. Additionally, our results show that in the testes TBC1D20-mediated maturation of autophagosomes is required for autophagic flux, but is also required for the formation of acrosomes. Furthermore TBC1D20-deficient mice, while not mimicking severe developmental brain abnormalities identified in WARBM4 affected children, display disrupted neuronal autophagic flux resulting in adult-onset motor dysfunction. In summary, we show that TBC1D20 has an essential role in the maturation of autophagosomes and a defect in TBC1D20 function results in eye, testicular, and neuronal abnormalities in mice implicating disrupted autophagy as a mechanism that contributes to WARBM4 pathogenesis.
KEYWORDS: acrosome, autophagic flux, autophagosome, brain, cataracts, GAPs, GEFs, infertility, lens, lens fiber cells, lysosome, neurodegeneration, neuron, polymicrogyria, RAB1B, RAB GTPase, TBC domain, TBC1D20, testis, Warburg micro syndrome
Introduction
Warburg micro syndrome is a genetically heterogeneous autosomal recessive disorder characterized by congenital eye, brain, and genital abnormalities.1-4 In humans, mutations in the TBC1D20 gene that result in abolished TBC1D20 protein function cause Warburg Micro syndrome 4 (WARBM4; OMIM#615663).4 Children affected with WARBM4 present with profound disabilities including blindness, brain malformations, severe intellectual deficiency, inability to learn how to talk or communicate, and severe motor dysfunction, as well as genital abnormalities characterized by hypogenitalism.4 In blind-sterile (bs) mutant mice, a loss of TBC1D20 protein function causes congenital cataracts and male infertility.4,5 TBC1D20 belongs to a large family of over 40 GAPs characterized by the presence of the evolutionarily conserved catalytically active TBC domain.6 TBC1D20 is an endoplasmic reticulum (ER) type II membrane protein with the TBC domain positioned in the cytosol.7 GAPs, including TBC1D20, increase the intrinsically slow GTP-hydrolysis rate of small RAB-GTPases when bound to GTP, returning the “active” GTP-bound RAB-GTPase to the “inactive” GDP-bound state.6,8 Biochemical screens of active GTP-bound RAB GTPases revealed that TBC1D20 facilitates GTP hydrolysis of only 2 GTPases: RAB1B and RAB2A.4,7,9 Therefore, a role has been established for TBC1D20 to act as a GAP for RAB1B and RAB2A.
While great progress has been made by identifying the genetic cause of WARBM4 and by establishing bs mice as a mouse model which mimics TBC1D20 deficiency in WARBM4-affected children, the cellular function of TBC1D20 still remains elusive. It is not understood how functional loss of TBC1D20 results in clinical presentations in affected WARBM4 children and eye and testicular abnormalities in bs mice. Overexpression of wild-type (WT) TBC1D20 maintains RAB1B in the inactive GDP-bound state and results in disrupted ER-to-Golgi vesicular trafficking, as well as a loss of Golgi structures implying that TBC1D20, as a negative regulator of RAB1B, mediates the secretory pathway and maintenance of Golgi structures.7 However, siRNA-mediated knockdown of TBC1D20 in HeLa cells does not alter the secretory pathway and does not significantly alter Golgi structures.7 These results suggest that other GAPs contribute to the RAB1B inactivation during ER-to-Golgi vesicular trafficking and maintenance of Golgi structures. These data also suggest that TBC1D20 has another not-yet-identified cellular function of essential importance for the eye, brain, and genital development in both humans and mice.
Our recent studies also established that a functional deficiency of TBC1D20 in human and mouse cell lines results in an accumulation of lipid droplets (LDs).4,5 LDs are dynamic organelles that facilitate intracellular storage of lipids and cholesterol.10-13 Excess intracellular lipids and cholesterol are converted into triacylglycerols and cholesterol esters and are stored within the LD core; upon metabolic need LDs are broken down via autophagy for a release of stored lipids and cholesterol.14-16 Cells with abrogated autophagy exhibit accumulation of enlarged LDs phenotypically similar to the enlarged LDs identified in TBC1D20-deficient human and mouse cells.4,5,14,15 In addition to its role in LD turnover, autophagy is a fundamental evolutionarily conserved cellular mechanism that facilitates lysosome-mediated degradation of damaged proteins and organelles.17-20 Autophagy is indispensable for eye, testicular, and brain development and homeostasis21-27 supporting the idea that disrupted autophagy may be contributing to clinical presentations in WARBM4-affected children and TBC1D20-deficient mice. Furthermore, the TBC1D20 substrate RAB1B facilitates the formation of autophagosomes in both yeast and mammalian cells.28-33 Collectively these findings prompted us to hypothesize that TBC1D20 is a mediator of autophagy.
In this study, we present the first evidence that TBC1D20, via its RAB1B GAP function, is a key regulator of autophagosome maturation required for the maintenance of autophagic flux and degradation of autophagic cargo. These findings provide novel insights into the fundamental mechanisms mediating autophagy. Additionally, this study illuminates how TBC1D20-mediated autophagosome maturation is indispensable for lens fiber cell homeostasis and terminal differentiation, the formation of acrosomes and male fertility, and neuronal development and maintenance.
Results
bs MEFs exhibit accumulation of enlarged autophagosomes
As the initial step in determining if TBC1D20 has a role in autophagy, we analyzed MEFs, generated from embryonic day 13.5 (E13.5) WT and TBC1D20-deficient bs littermates, following transfection with a plasmid encoding green fluorescent protein (GFP)-tagged MAP1LC3A/B (microtubule-associated protein 1 light chain 3 α/β; LC3). In bs MEFs it appeared that LC3-positive structures were enlarged and accumulating (Fig. 1B) when compared to WT MEFs (Fig. 1A). Quantification analysis confirmed that a significantly greater percentage (P = 0.0001; n = 30) of the GFP-LC3-positive area per cell in bs MEFs (Fig. 1C). This finding suggested that enlarged LC3-positive autophagosomes were accumulating in bs MEFs. To test this hypothesis, we evaluated WT and bs MEFs by transmission electron microscopy (TEM). Images revealed an accumulation of enlarged electron-light double-membraned structures present in bs MEFs, indicative of autophagosomes (Fig. 1E). Some autophagosomes in bs MEFs were observed as a part of enlarged multi-lamellar structures (Fig. 1E) consistent with our findings following transfection with the GFP-LC3 plasmid (Fig. 1B). TEM analysis did not identify accumulation of any other cellular structures in bs MEFs when compared to WT MEFs (Fig. S1A–B). To confirm this, we immunostained WT and bs MEFs with RAB5 as an early endosome marker, FLAG-RAB11 as a recycling endosome marker, and LAMP2 (lysosomal-associated membrane protein 2) as a lysosome marker (Fig. S1C–H). We measured the percentage area positive for RAB5, FLAG-RAB11, and LAMP2 per cell and did not observe a significant difference between WT and bs MEFs (Fig. S1I).
Figure 1.

Evaluation of autophagosomes in WT and bs MEFs. WT (A) and bs (B) MEFs 24 h following transfection with GFP-LC3 (red). Nuclei were stained with DAPI (blue). Scale bar: 5 μm. (C) Quantification analysis revealed significantly greater (P = 0.0001; n = 30) percentage area positive for GFP-LC3 in bs MEFs when compared to WT MEFs. TEM images of WT (D) and bs (E) MEFs revealed an accumulation of double-membraned autophagosomal structures (red arrowheads) in bs MEFs. Some autophagosomes in bs MEFs (E) were observed as a part of larger multi-lamellar double-membrane structures (blue arrowheads). Scale bar: 100 nm. (F) Immunoblotting of WT and bs MEF cell lysates for LC3; immunoblotting for ACTB served as a loading control. (G) Quantification analysis confirmed a significant accumulation (P = 0.0008; n = 3) of endogenous LC3-II in bs MEF cell lysates when normalized to ACTB whereas levels of LC3-I:ACTB did not significantly differ (P = 0.2381; n = 3). The data in (C) and (G) are presented as the mean ± SEM and significance was established with the Student t test.
The accumulation of autophagosomes in bs MEFs could be the result of an increase in the production of autophagosomes, or alternatively the result of a disruption in autophagic flux and a failure of autophagosomes to fuse with lysosomes, or undergo subsequent degradation. Western blotting of endogenous LC3 in WT and bs MEFs (Fig. 1F) identified significantly greater (P = 0.0008; n = 3) levels of LC3-II in bs MEFs when compared to WT MEFs whereas levels of LC3-I did not significantly differ (P = 0.2381; n = 3) (Fig. 1G). These findings suggested that the accumulation of enlarged autophagosomes in bs MEFs is caused by disrupted autophagic flux.
Accumulation of enlarged autophagosomes in bs MEFs is caused by disrupted autophagic flux
Accumulation of SQSTM1/p62 (sequestosome 1) is an established indicator of disrupted autophagic flux.34-36 In order to confirm that the TBC1D20 functional loss caused disrupted autophagic flux in bs MEFs, we proceeded with the evaluation of SQSTM1. Immunofluorescence identified a significantly greater (P = 0.0008; n = 30) percentage of the SQSTM1-positive area in bs MEFs when compared to WT MEFs (Fig. 2A–C). Immunoblotting of WT and bs lysates for SQSTM1 and ACTB/β-actin as a loading control confirmed significantly greater (P = 0.0002; n = 3) levels of SQSTM1 in bs MEFs when compared to WT MEFs (Fig. 2D–E). To explore this further, we measured levels of SQSTM1 from WT and bs MEFs following treatment for 2 h with DMSO as vehicle control, serum-free and amino acid-free media as an inducer of autophagy by starvation, bafilomycin A1 in complete medium as an inhibitor of autophagy by inhibiting lysosomal function, and bafilomycin A1 in serum-free and amino acid-free media to induce both autophagy by starvation and inhibit autophagy by disrupting lysosomal function (Fig. 2F). Quantification analysis revealed significantly lower levels of SQSMT1 in WT MEFs following starvation (P = 0.0049; n = 3) whereas starvation had no effect on bs MEFs (P = 0.31; n = 3). Additionally, in WT MEFs bafilomycin A1 and bafilomycin A1+starvation treatments resulted in significantly higher levels of SQSTM1 (P = 0.0013 and P = 0.0007 respectively; n = 3) when compared to vehicle-treated WT MEFs (Fig. 2G). By contrast, the levels of SQSTM1 in vehicle-treated bs MEFs did not significantly differ from the levels of SQSTM1 in bs MEFs following treatments with bafilomycin A1 (P = 0.851; n = 3), and bafilomycin A1+starvation (P = 0.3234; n = 3) (Fig. 2F–G). Prolonged treatments (4 h) with DMSO vehicle control, starvation, bafilomycin A1, or bafilomycin A1+starvation did not significantly change SQSTM1 levels in bs MEFs although prolonged starvation in WT MEFs restored SQSTM1 levels (Fig. S2A–B) consistent with previous reports.37 Therefore, in bs MEFs, SQSTM1 levels are insensitive to treatments which increase or decrease autophagic degradation in WT MEFs with properly functioning autophagy.
Figure 2.

Autophagic flux in WT and bs MEFs. Immunostaining of WT (A) and bs (B) MEFs for SQSTM1. (C) Quantification analysis revealed a significantly greater (P = 0.0008; n = 30) percentage area of SQSTM1 per cell in bs MEFs when compared to WT MEFs. (D) Western blot for SQSTM1 of lysates from WT and bs MEFs; immunoblotting for ACTB served as a loading control. (E) Quantification analyses revealed a significantly greater (P = 0.0002; n = 3) levels of SQSTM1:ACTB in lysates from bs MEFs when compared to WT MEFs. (F) Immunoblotting of WT and bs MEF cell lysates for SQSTM1 following 2-h treatment with DMSO as vehicle control (lanes 1 and 5), serum-free and amino-acid free media (ST) (lanes 2 and 6), 100 nM bafilomycin A1 (BF) in complete medium (lanes 3 and 7), and combined treatment with bafilomycin A1 and serum-free and amino-acid free media (BF+ST) (lanes 4 and 8); immunoblotting for ACTB served as a loading control. (G) Quantification analysis revealed significantly greater levels (P = 0.0027; n = 3) of SQSTM1:ACTB in lysates from bs MEFs when compared to WT following treatment with DMSO (P = 0.0027; n = 3) or ST (P = 0.0001; n = 3). No significant differences were identified between WT and bs MEFs following treatments with BF (P = 0.0969; n = 3), and BF+ST (P = 0.097; n = 3). In WT MEFs ST significantly (P = 0.0049; n = 3) lowered SQSTM1 levels whereas BF and BF+ST significantly increased (P = 0.0013 and P = 0.0007 respectively; n = 3) levels of SQSTM1 when compared to DMSO treatment; by contrast SQSTM1 levels in bs MEFs did not significantly change following DMSO treatment and ST (P = 0.31; n = 3), BF (P = 0.851; n = 3) and ST+BF (P = 0.3234; n = 3).
Autophagosomes in bs MEFs fail to mature
Next, we set out to identify which autophagic step is disrupted as a result of a loss of TBC1D20 function. Initially, we wanted to determine if TBC1D20 facilitates the maturation of autophagosomes or alternatively if TBC1D20 is required for lysosomal function. WT and bs MEFs were transfected with GFP-LC3, a marker for autophagosomes, and immunostained for the lysosomal marker LAMP2. In WT MEFs, GFP-LC3 autophagosomes appeared to associate with LAMP2-positive lysosomes (Fig. S3A-A″). In bs MEFs, we observed enlarged GFP-positive autophagosomes that appeared to only sporadically associate with LAMP2-positive lysosomes (Fig. S3B-B″) suggesting that in these cells autophagosomes failed to fuse with lysosomes. To test this hypothesis further, both WT and bs MEFs were transfected with an mCherry-enhanced green fluorescent protein (EGFP)-LC3 clone which encodes a tandem mCherry-EGFP-LC3 fusion protein. This construct allows for monitoring of the autophagic flux by taking advantage of differential pH properties of EGFP (pH sensitive) and mCherry (unaffected by pH) LC3 tags.38,39 Therefore, red-green colocalization (mCherry+EGFP), visualized as yellow, results from fluorescence of both EGFP and mCherry LC3 tags in pH neutral autophagosomes. Red-only fluorescence (mCherryΔEGFP) in the absence of green indicates that only the mCherry LC3 tag is visible in acidic amphisomes and autolysosomes, while the EGFP tag is no longer detectable due to the tag's pH sensitivity.
In WT MEFs, we identified LC3-positive “yellow” (mCherry+EGFP) structures indicative of autophagosomes (Fig. 3A-A″); in WT MEFs we also identified the presence of “red-only” structures indicative of amphisomes and autolysosomes (mCherryΔEGFP) (Fig. 3A-A″). By contrast, bs MEFs exhibited the presence of enlarged EGFP and mCherry positive “yellow” LC3 puncta without any mCherry-positive “red” puncta present (Fig. 3B-B″). Quantification analysis revealed a significantly greater (P = 0.0001; n = 30) percentage area of mCherry+EGFP per cell as well as a significantly lower (P = 0.0001; n = 30) percentage area of mCherryΔEGFP per cell for bs MEFs when compared to WT MEFs (Fig. 3C). Collectively these findings indicate that in bs MEFs a loss of TBC1D20 function results in a failure of autophagosomes to acidify.
Figure 3.
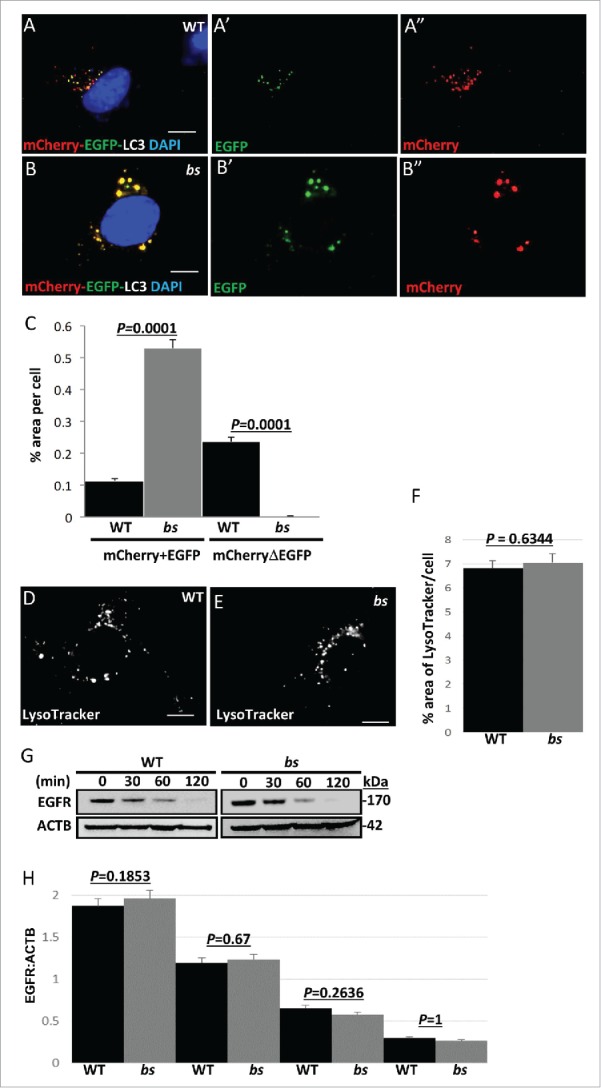
Autophagosome maturation in WT and bs MEFs. (A-A″) WT MEFs expressing tandem mCherry-EGFP-LC3 fusion protein revealed a mix of pH neutral autophagosomes (“yellow” = mCherry+EGFP tags) and acidic amphisomes and autolysosomes (“red-only” = mCherryΔEGFP tag). (B-B”) In bs MEFs mCherry-EGFP-LC3 fusion protein was arrested in enlarged pH neutral autophagosomes (“yellow”) indicating a failure of mCherry-EGFP-LC3 to progress to acidic amphisomes and autolysosomes. Nuclei were stained with DAPI (blue). Scale bars in all images: 5 μm. (C) Quantification analysis of WT and bs MEFs expressing mCherry-EGFP-LC3 tandem protein revealed significantly greater (P = 0.0001; n = 30) percentage area positive for both EGFP and mCherry (“yellow” = mCherry+EGFP) in bs MEFs; quantification analysis also revealed significantly lower (P = 0.0001; n = 30) percentage area of only mCherry that is not positive for EGFP (“red only” = mCherryDEGFP) in bs MEFs. LysoTracker Red-positive structures did not appear to differ between WT (D) and bs (E) MEFs. The area of LysoTracker Red-positive staining per cell (F) did not significantly differ (P = 0.6344; n = 30) between the 2 genotypes. (G) Western blot analysis of cell lysates for EGFR and ACTB from WT and bs MEFs treated with 40 ng/mL EGF and 20 μg/mL cycloheximide for 0, 30, 60 and 120 min. (H) Quantification analysis did not reveal a significant difference in the levels of EGFR relative to ACTB between WT and bs MEFs in untreated cells (P = 0.1854; n = 3), 30 min (P = 0.67; n = 3), 60 min (P = 02636; n = 3) or 120 min (P = 1; n = 3) following the treatment with EGF and cycloheximide. The data in (C), (F) and (H) are presented as the mean ± SEM and significance was established with the Student t test.
To exclude the possibility that in bs MEFs a failure of autophagosomes to acidify is caused by a defect in the overall organelle acidification, we stained WT and bs MEFs with LysoTracker Red and acridine orange which are reagents that stain acidic vesicles. Morphology of acidic vesicles in WT and bs MEFs did not differ following staining with LysoTracker Red (Fig. 3D–E and Fig. S3C–D) or acridine orange (Fig. S3E–F). The percentage of area positive for LysoTracker Red per cell (Fig. 3F) as well as the overall intensity of LysoTracker Red staining (Fig. S3G) measured in individual WT and bs MEFs did not significantly differ (P = 0.6344; n = 30 and P = 0.954; n = 30, respectively). These findings indicate that in bs MEFs acidification of organelles was not disrupted. Next, we wanted to determine if TBC1D20 was required for the function of lysosomes. The activity of CTSB/cathepsin B, which is a lysosomal peptidase, in live cells did not differ between WT and bs MEFs (Fig. S3H–J). Additionally, the activity of CTSD, another lysosomal peptidase, also did not significantly differ (P = 0.5.40; n = 3) between WT and bs MEF cell lysates (Fig. S3K). To explore this further, we wanted to determine if a loss of TBC1D20 function in bs MEFs also disrupts endosomal-lysosomal pathway. EGFR (epidermal growth factor receptor) is a cell surface tyrosine kinase that upon EGF (epidermal growth factor)-mediated ligand binding is rapidly endocytosed and then targeted to lysosomes for degradation.40 In WT and bs MEFs the rate of EGFR degradation following binding to EGF did not significantly differ (Fig. 3G–H), indicating that the endosomal-lysosomal pathway was not disrupted. Collectively these findings provide evidence that a functional loss of TBC1D20 in bs MEFs results in a failure of autophagosomes to mature resulting in disrupted autophagic flux and consequently an accumulation of autophagosomes without disrupting endocytosis or lysosomal function.
TBC1D20, via its RAB1BGAP function, mediates maturation of autophagosomes
We wanted to establish the specific autophagic step mediated by TBC1D20 function. In both yeast and mammalian cells, activation of RAB1B from the GDP-bound to the GTP-bound form is required for the formation of autophagosomes.29,32 We and others have shown that in vitro TBC1D20 mediates the GTP hydrolysis of RAB1B-GTP to the GDP-bound form.4,7,9 Thus, we hypothesized that maturation of autophagosomes is dependent on the TBC1D20-mediated GTP-hydrolysis of RAB1B-GTP to inactivate the RAB1B-GDP form. To test this model, we cotransfected WT and bs MEFs with FLAG-tagged RAB1B and GFP-LC3. In both WT (Fig. 4A–A') and bs (Fig. 4B–B') MEFs FLAG-RAB1B and GFP-LC3 colocalize, suggesting that in both cell lines RAB1B is associated with autophagosomes. Consistent with our results shown in Fig. 1A–C, the percentage area of GFP-LC3 per cell was significantly greater (P = 0.0059; n = 30) in bs+FLAG-RAB1B MEFs when compared to WT+FLAG-RAB1B MEFs (Fig. 4F).
Figure 4.
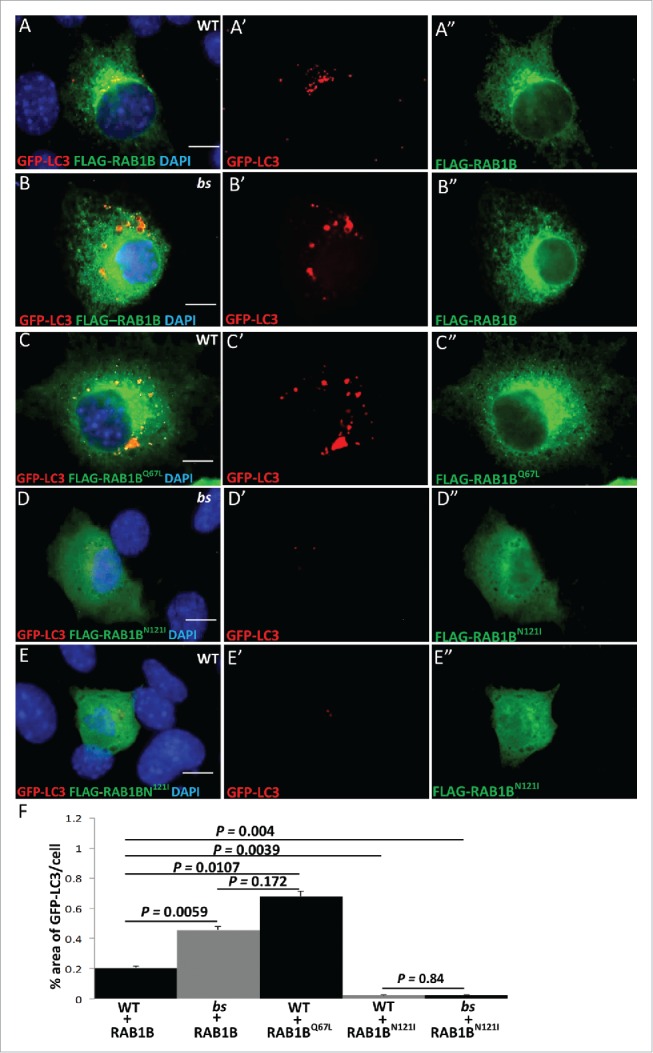
TBC1D20 is a regulator of RAB1B during autophagy. WT (A-A') and bs (B-B') MEFs following transfection with plasmids encoding GFP-LC3 (red) and FLAG-RAB1B (green). WT MEFs (C-C') following transfection with plasmids encoding GFP-LC3 (red) and FLAG-RAB1BQ67L (green). bs (D-D') and WT MEFs (E-E') following transfection with GFP-LC3 (red) and FLAG-RAB1BN121I (green). Nuclei were stained with DAPI (blue). Scale bar in all images: 5 μm. Quantification analysis (F) revealed significantly greater (P = 0.0059; n = 30) percentage area positive for GFP-LC3 in bs MEFs when compared to WT MEFs following transfection with FLAG-RAB1B. Additionally, WT MEFs overexpressing FLAG-RAB1BQ67L also exhibited significantly greater (P = 0.0107; n = 30) percentage area of GFP-LC3 when compared to WT MEFs overexpressing FLAG-RAB1B. The percentage area of GPF-LC3 per cells did not significantly differ (P = 0.172; n = 30) between bs MEFs overexpressing FLAG-RAB1B and WT MEFs overexpressing FLAG-RAB1BQ67L. Both WT and bs MEFs overexpressing FLAG-RAB1BN121I exhibited significantly lower (P = 0.004 and P = 0.0039, respectively) percentage area of GFP-LC3 per cell when compared to WT MEFs transfected with FLAG-RAB1B. The percentage area of GFP-LC3 per cell did not significantly differ (P = 0.84; n = 30) between WT and bs MEFs transfected with FLAG-RAB1BN121I.
Given that a functional loss of TBC1D20 renders RAB1B active in the GTP-bound state, next we wanted to determine if the accumulation of enlarged autophagosomes identified in bs MEFs was caused by the accumulation of RAB1B-GTP. WT MEFs were transfected with FLAG-RAB1BQ67L, a constitutively GTP-bound (active) mutant of RAB1B. Accumulation of enlarged GFP-LC3 positive autophagosomes was observed (Fig. 4C–C') phenotypically reminiscent of the GFP-LC3-positive structures identified in bs MEFs transfected with FLAG-RAB1B (Fig. 4B–B'). The percentage area of GFP-LC3 per cell in WT+FLAG-RAB1BQ67L was significantly greater (P = 0.0107; n = 30) when compared to the percentage area of GFP-LC3 per cell in WT-FLAG-RAB1B (Fig. 4F). However, the percentage area of GFP-LC3 per cell in bs+FLAG-RAB1B and WT+FLAG-RAB1BQ67L did not change significantly (P = 0.172; n = 30) (Fig. 4F). To explore this further, we overexpressed the dominant-negative FLAG-RAB1BN121I mutant of RAB1B in bs and in WT MEFs and in both cases the formation of GFP-LC3-positive structures was abrogated. Collectively these findings established evidence that a failure of TBC1D20 to inactivate RAB1B-GTP causes accumulation of enlarged LC3-positive autophagosomes, whereas a failure of RAB1B-GDP to activate into the GTP-bound state abrogates the formation of LC3-positive autophagosomes.
bs MEFs exhibit accumulation of ubiquitinated autophagic cargo
One of the key roles of autophagy is to facilitate the delivery of ubiquitinated autophagic cargo into the lysosome for degradation, and cells with abrogated autophagy exhibit accumulation of ubiquitinated proteins, lipid droplets, peroxisomes and mitochondria.14,15,17,41-43 Our previous studies have already established that TBC1D20 functional loss in bs MEFs results in accumulation of enlarged LDs.4 To explore this further, we immunostained WT and bs MEFs for ubiquitin. Our results show accumulation of ubiquitin in bs MEFs (Fig. 5B) when compared to WT MEFs (Fig. 5A). Western blotting of WT and bs cell lysates confirmed accumulation of ubiquitinated proteins in bs MEFs (Fig. 5C). Next, we wanted to determine if other autophagic cargo such as peroxisomes and mitochondria also accumulate in bs MEFs. The immunofluorescence analysis for ABCD3 (ATP-binding cassette sub-family D [ALD], member 3), a peroxisomal marker, revealed a significantly increased (P = 0.001; n = 30) percentage of ABCD3-immuno positive area per cell in bs MEFs when compared to WT MEFs (Fig. 5D–F). Immunostaining and western blotting for the mitochondrial markers COX4I1 (cytochrome c oxidase subunit IV isoform 1) and TOMM20 (translocase of outer mitochondrial membrane 20 homolog [yeast]), respectively, also confirmed significant accumulation of mitochondria in bs MEFs, a hallmark of autophagic inhibition (Fig. 5G–I). Staining of WT and bs MEFs with the MitoTracker Red dye revealed that the accumulating mitochondria in bs MEFs remained polarized (Fig. 5J–K).
Figure 5.

Autophagic cargo in WT and bs MEFs. Immunostaining of WT (A) and bs (B) MEFs for ubiquitin. Immunoblotting of WT and bs cell lysates (C) confirmed accumulation of ubiquitinated proteins in bs MEFs; immunoblotting for ACTB served as a loading control. Immunostaining of WT (D) and bs (E) MEFs for ABCD3 as a peroxisomal marker; quantification of ABCD3-positive puncta (F) confirmed a significant (P = 0.0001; n = 30) accumulation of ABCD3 in bs MEFs. Immunostaining of WT (G) and bs (H) MEFs for COX4I1 as a mitochondrial marker. Western blotting of WT and bs lysates for TOMM20 as a mitochondrial marker revealed in bs lysates significantly greater (P = 0.0063; n = 3) levels of TOMM20 relative to ACTB (I). Staining of WT (J) and bs (K) MEFs with the mitochondrial dye MitoTracker Red established that accumulating mitochondria in bs MEFs remained polarized. Nuclei in all images were stained with DAPI. Scale bar: 5 μm. The data in (F) and (I) are presented as the means ± SEM and significance was established with the Student t test.
bs lens fiber cells exhibit disrupted autophagic flux and cataracts
We wanted to determine if disrupted TBC1D20-mediated autophagic flux identified in bs MEFs also causes cataracts in bs lenses. We previously reported that cataracts in bs mice are a progressive disease with first morphological abnormalities noted in lens fiber cells at E17.5.4 To explore this further, we clinically evaluated bs lenses with a slit lamp biomicroscope following postnatal eyelid opening around day 12 (P12). In bs mice, we identified the presence of cataracts within the lens core, whereas the lens cortex remained transparent (Fig. 6B). By P28, bs cataracts progress to total vacuolated cataracts that encompass the entire lens (not shown) consistent with our previous reports.4 Morphologically, at P12 WT lenses exhibited highly organized epithelium at the anterior, elongated differentiated fiber cells present at the cortex, and terminally differentiated anuclear lens fiber cells present within the lens core forming the “organelle-free zone” (OFZ) (Fig. 6C). Morphological analysis of P12 bs lenses identified the presence of organized epithelia at the anterior as well as organized elongated cortical fibers within the cortex (Fig. 6D) consistent with the transparent lens cortex observed following clinical evaluation (Fig. 6B). However, in bs lenses fiber cells present within the lens core were highly degenerated and disorganized (Fig. 6D) consistent with lens core cataracts identified following clinical evaluation (Fig. 6B). We hypothesized that disrupted TBC1D20-mediated flux causes cataracts in bs mice which, if true, indicated we would detect disruption of autophagic flux and accumulation of autophagic cargo in P12 bs cortical fiber cells prior to the onset of cataracts. We proceeded to immunostain P12 WT and bs lenses for SQSTM1 as a marker for autophagic flux and ubiquitin as a marker for autophagic cargo. In WT lenses, SQSTM1-positive (Fig. 6E) and ubiquitin-positive (Fig. 6G) immunostaining was present in lens epithelial cells and cortical fibers in a perinuclear pattern, but was absent in the nuclear fibers within the lens core. By contrast, immunostaining of bs lenses revealed an accumulation of SQSTM1 (Fig. 6F) and ubiquitin (Fig. 6H) in cortical fiber cells, progressively increasing toward the lens core, with the greatest accumulation of both ubiquitin and SQSTM1 in fiber cells within the lens core. Therefore, TBC1D20 functional loss in bs lens fiber cells results in disrupted autophagic flux and accumulation of ubiquitinated autophagic cargo.
Figure 6.

Autophagy in P12 WT and bs lenses. Clinical evaluation with a slit lamp biomicroscope of WT (A) (n = 6) eyes revealed a transparent lens core (black arrowhead) and a transparent lens cortex (blue arrow); in bs (B) (n = 6) eyes the lens core exhibited cataracts (black arrowhead) whereas the lens cortex remained transparent. H&E evaluation of WT (C) lenses revealed organized epithelium at the lens anterior (black arrow), elongated lens fiber cells (blue arrow) within the cortex and anucleated lens fiber cells within the core (black arrowhead). H&E evaluation of bs (D) lenses (n = 6) identified organized lens epithelial cells at the anterior (black arrow), elongated fiber cells within the lens cortex (blue arrow) and highly degenerated fibers within the lens core (arrowhead). Immunostaining of WT lenses (n = 6) for SQSTM1 (E) and ubiquitin (G) revealed the presence of both SQSTM1 and ubiquitin in lens epithelial cells (white arrows) and cortical fibers (blue arrows) without any staining present in the nuclear fibers (white arrowheads). By contrast immunostaining of bs lenses (n = 6) revealed accumulation of SQSTM1 (F) and ubiquitin (H) in bs cortical lens fibers (blue arrows) and nuclear lens fiber cells (white arrowheads). DAPI staining of WT (n = 6) (I) and bs (n = 6) (J) lenses revealed nuclei in cortical fibers (blue arrows). In WT lenses fibers within the lens core did not stain for DAPI indicating the removal of nuclei (I, white arrowheads) whereas bs fiber cells within the lens core stained for DAPI indicating a failure in the removal of nuclei (J, white arrowhead). Images (I') and (J') are a higher magnification of images (I) and (J) and arrows in (J) point to smaller nuclei retained in bs lens fiber cells within the OFZ. In (E) and (F) nuclei were stained with DAPI (blue). Scale bars: 50 μm. OFZ, organelle-free zone.
Our analysis of P12 bs lenses also revealed that bs lens fiber cells within the lens core retained nuclei (Fig. 6J). Lens fiber cells undergo terminal differentiation where a programmed loss of nuclei and organelles facilitates the formation of organelle-free fiber cells within the lens core, a process required for the vision process.44-46 Retained DAPI-positive nuclei in fiber cells within the bs lens core appeared smaller when compared to the nuclei in cortical fiber cells (Fig. 6J') suggesting that in bs lens fiber cells the denucleation process may have been initiated but not completed. To explore this further, we evaluated lens fiber cell nuclei in bs lens fiber cells at E15.5 prior to the onset of morphological abnormalities. We did not identify any morphological difference between nuclei in lens fiber cells of E15.5 WT and bs mice (Fig. S4A–B). We have shown previously that E15.5 bs lens fiber cells do not exhibit any morphological abnormalities and undergo normal differentiation.4 However, we have not excluded the possibility that aberrant expression of crystallins, the predominant structural proteins in lens fiber cells, may affect the denucleation process observed at P12 bs lens fiber cells (Fig. 6J–J'). Immunostaining for CRYBB1/βB1-crystallin as a marker for lens fiber cell-specific crystallins did not identify any differences in CRYBB1-crystallin expression between WT and bs E15.5 lens fiber cells (Fig. S4C–D).
bs testes exhibit disrupted autophagic flux and a failure in the formation of acrosomes
Previous studies established that infertility in bs male mice is caused by a failure in the formation of acrosomes.4,47 Recently, a hypothesis was proposed that acrosomes form following the fusion of autolysosomes and proacrosomal vesicles from the trans-Golgi.27 Given that TBC1D20 facilitates the formation of autolysosomes in bs MEFs, we proceeded to evaluate if autophagosomes and lysosomes form in bs germ cells. Immunostaining of WT seminiferous tubules for LC3, as an autophagosome marker, and LAMP1 (lysosomal-associated membrane protein 1), as a lysosome marker, revealed a crescent-shaped pattern indicative of acrosome structures (Fig. 7A and 7C; Fig. S5A and 5C). Additionally, immunostaining of WT seminiferous tubules for SQSTM1 as a marker for autophagic flux and ubiquitin as a marker for autophagic cargo also revealed crescent-shaped staining (Fig. 7E and 7G; Fig. S5E and S5G). By contrast, immunostaining of bs seminiferous tubules revealed diffuse and disorganized LC3-positive and LAMP1-positive staining (Fig. 7B and 7D; Fig. S5B and 5D) as well as accumulation of SQSTM1 and ubiquitin (Fig. 7F and 7H; Fig S5F and S5H). These findings suggest that TBC1D20-mediated maturation of autophagosomes is required for maintenance of autophagic flux, but it is also required for the formation of acrosomes.
Figure 7.
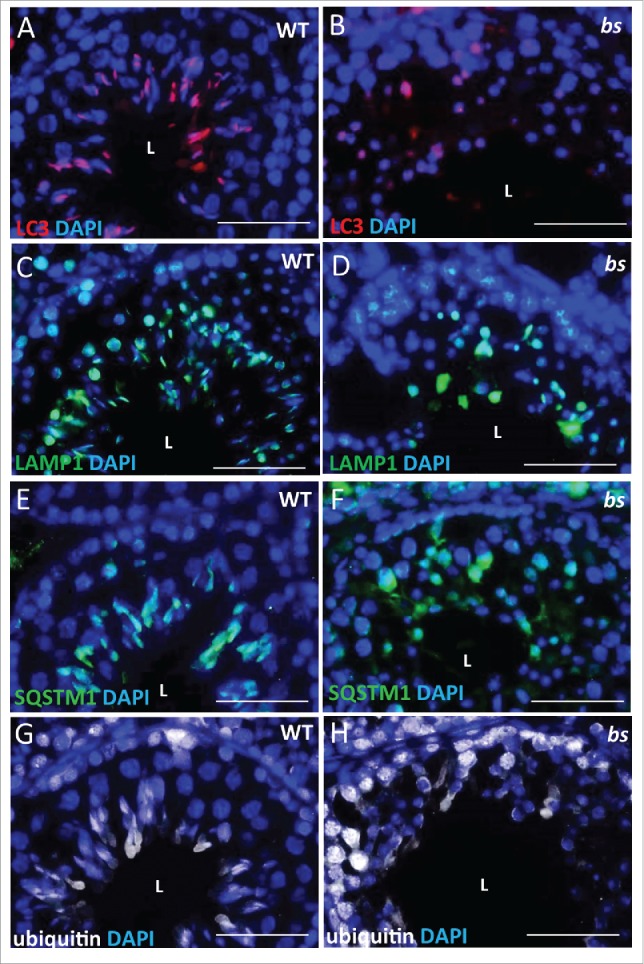
Evaluation of WT and bs seminiferous tubules. Immunostaining of WT seminiferous tubules (n = 10) for LC3 (A), LAMP1 (C), SQSTM1 (E) and ubiquitin (G) revealed crescent-shaped staining indicative of acrosomes. By contrast immunostaining of bs seminiferous tubules (n = 10) for LC3 (B), LAMP1 (D), SQSTM1 (F) and ubiquitin (H) revealed diffuse and disorganized staining. Nuclei were stained with DAPI. L, lumen. Scale bar: 50 μm.
bs mice exhibit disrupted neuronal autophagic flux and adult-onset motor dysfunction
While the functional loss of TBC1D20 in WARBM4-affected children causes severe developmental brain malformations, we did not identify gross morphological brain abnormalities in bs mice.4 However, disrupted neuronal autophagy in mice following ablation of either Atg5 or Atg7 causes adult-onset motor dysfunction.48,49 This prompted us to hypothesize that in bs mice, abortive autophagy caused by functional loss of TBC1D20 may lead to disrupted neuronal autophagic flux and adult-onset motor dysfunction. Hindlimb clasping and altered gait phenotypes have been identified previously as markers of motor dysfunction caused by neurodegeneration.50-52 At 4 wk of age, hind clasping between WT and bs mice did not differ (n = 5) and no differences in gait were noted between WT and bs mice (n = 5) (not shown). However, by 5 mo of age, bs mice (n = 3) started to exhibit the hindlimb clasping phenotype when suspended by the tail (Fig. 8B; Video S1) in contrast to splaying of hindlimbs in age-matched WT littermates (n = 3) (Fig. 8A; Video S2). In addition, by 5 mo of age, bs mice (n = 3) started to exhibit abnormal gait characterized by lowered abdomen and feet positioned outward generating a large angle between the feet position and the direction of motion (Fig. 8D; Video S3) unlike WT animals that displayed alternate limb movements with feet positioned forward (n = 3) (Fig. 8C; Video S4). Western blot analysis identified significant (P = 0.0055; n = 3) accumulation of SQSTM1 (Fig. 8E) and significant (P = 0.01; n = 3) accumulation of ubiquitin (Fig. 8F) in lysates from bs brains indicating disrupted autophagic flux and accumulation of autophagic cargo in bs brains.
Figure 8.
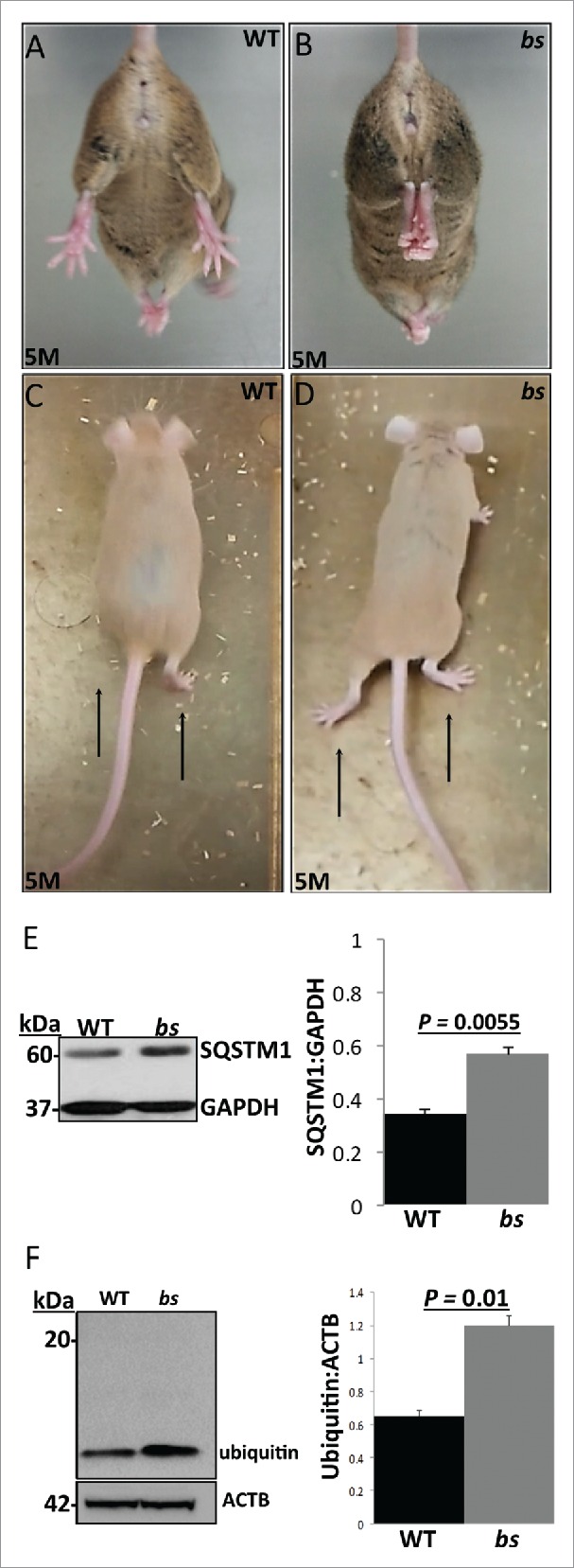
Neuronal phenotypes in WT and bs mice. At 5 mo of age bs mice (n = 3) (B) exhibit hindlimb clasping phenotypes in contrast to splaying of hindlimbs in age-matched WT littermates (n = 3) (A). At 5 mo bs mice exhibited abnormal gait (D) characterized by lower abdomen and feet positioned outward in contrast to age-matched WT animals (C) that display alternate limb movements with feet positioned forward. Western blot analysis of lysates from adult WT and bs brains revealed a significant (P = 0.0055; n = 3) accumulation of SQSTM1 (E) as well as a significant (P = 0.01; n = 3) accumulation of ubiquitin (F) in bs brains; immunoblotting for GAPDH in (E) and ACTB in (F) served as loading controls.
Discussion
In this study, we present evidence that TBC1D20, via its RAB1B GAP function, is a key regulator of autophagosome maturation. Our model (Fig. 9) proposes that TBC1D20–mediated inactivation of RAB1B is required for subsequent fusion of autophagosomes with late endosomes to form amphisomes, but also for a direct fusion of autophagosomes with lysosomes to form autolysosomes. As such, TBC1D20 facilitates autophagic flux, delivery, and subsequent degradation of autophagic cargo in autolysosomes. Our results show that in bs MEFs the TBC1D20 functional loss arrests autophagosome maturation, resulting in accumulation and enlargement of autophagosomes, a failure in the formation of amphisomes or autolysosomes causing disrupted autophagic flux, and consequently accumulation of ubiquitinated autophagic cargo marked for degradation. It was proposed previously that the maturation of intracellular vesicles is mediated by membrane recruitment and activation of a downstream RAB-GTPase with attendant inactivation and membrane disassociation of a preceding RAB-GTPase.53 During autophagy, several RAB-GTPases and associated regulatory proteins play key roles.54-56 Specifically, RAB1, RAB5, RAB7, RAB11, RAB23, RAB32, and RAB33B contribute to autophagosome formation, RAB7, RAB8B, and RAB24 contribute to autophagosome maturation, and while RAB8A and RAB25 also contribute to autophagy their roles are currently unknown.54-56 Therefore, the sequential order of different RAB-GTPases during autophagy still remains poorly understood. Our results suggest that the inactivation of RAB1B-GTP by TBC1D20 may be a required step for the concomitant or subsequent recruitment of another RAB for the formation of amphisomes and/or autolysosomes; however, the identities of upstream or downstream RAB-GTPases at this point remain unclear.
Figure 9.

A proposed model for TBC1D20 function during autophagy. TBC1D20 mediates maturation of autophagosomes via its RAB1B GAP function, a process required for the fusion of autophagosomes with late endosomes to form amphisomes as well as a direct fusion of autophagosomes with lysosomes to form autolysosomes, thereby facilitating autophagic flux and delivery and subsequent degradation of autophagic cargo in autolysosomes.
TBC1D20 is a ubiquitously expressed protein, suggesting that TBC1D20 universally mediates autophagy in all cells, yet the functional loss of TBC1D20 in WARBM4-affected children and in TBC1D20-deficient bs mice leads to abnormalities in lenses, testes, and the brain. This observation suggests that TBC1D20-mediated autophagy is indispensable for the development and homeostasis of these tissues. The lens is a transparent ocular tissue composed of a single anterior layer of proliferating epithelial cells that upon exit from the cell cycle undergo differentiation into elongated lens fiber cells that migrate toward the lens cortex.57 Cortical lens fiber cells continue to migrate toward the lens core and as they start to reach the lens core, lens fiber cells undergo terminal differentiation and begin to lose their organelles resulting in the formation of organelle-free lens core fibers—a process indispensable for lens transparency and the vision process.44-46 Cataracts are defined as a loss of lens transparency, and the formation of insoluble opaque protein aggregates is the disease hallmark.58,59 While disruption of various cellular processes can lead to cataracts, it was shown previously that disrupted autophagy, as a result of mutations in key autophagic regulators FYCO1 and EPG5, causes Mendelian forms of congenital cataracts, and congenital cataracts in individuals with Vici syndrome, respectively.24,25 Our results presented here show that in mice TBC1D20-mediated autophagic flux maintains lens transparency by facilitating the removal of ubiquitinated autophagic cargo from lens fiber cells. We propose that lens fiber cells, as post-mitotic cells, cannot dilute damaged proteins and organelles marked for degradation through cell division and thus require the TBC1D20-mediated autophagic flux for the continuous removal of autophagic cargo. Therefore, we propose that a failure in the removal of ubiquitinated proteins from lens fiber cells will result in an accumulation of insoluble protein aggregates resulting in a loss of lens transparency.
Our results also provide evidence that TBC1D20 facilitates the removal of nuclei during lens fiber cell terminal differentiation. In mice, the removal of nuclei and organelles from the fibers within the lens core is initiated around E17.5 following the completion of lens development.44 We have shown here and previously that in bs mice prior to E17.5 lenses undergo normal development and differentiation4 suggesting that TBC1D20 may have a role during terminal lens fiber cell differentiation. Whereas the mechanism mediating lens fiber cell organelle removal remains poorly understood, it is well established that nuclear degradation in differentiating lens fiber cells is mediated by DNASE2B/DLAD, a lysosomal endonuclease active at low pH. Lenses ablated for Dnase2b exhibit retention of fragmented nuclei in lens fiber cells60,61 that are phenotypically similar to the fragmented nuclei in fiber cells of bs mice. The mechanism by which nuclei are delivered into the lysosome for DNASE2B-mediated degradation remains unclear. It was shown recently that another lysosomal acidic endonuclease, DNASE2A, shares a high degree of functional homology with DNASE2B, and facilitates degradation of intracellular damaged DNA following autophagosome-mediated recruitment and delivery of damaged DNA into the lysosomes.62 Therefore, we propose that during terminal differentiation of lens fiber cells, following nuclear envelope disassembly, nuclei undergo fragmentation and subsequent recruitment into the autophagosomes where TBC1D20-mediated autophagosome maturation facilitates the delivery of fragmented nuclei into the lysosomes for degradation by DNASE2B.
It should be pointed out that denucleation of lens fiber cells is not disrupted in lenses conditionally ablated for Atg5 or Pik3c3, a finding which led to the conclusion that autophagy does not contribute to the formation of organelle-free lens fiber cells within the lens core.63 However, in chick lenses, autophagy and mitophagy contribute to lens fiber cell organelle degradation.64 Additionally, the inactivation of the MAPK/JNK pathway induces upregulation of autophagy and facilitates the premature removal of organelles during terminal differentiation of lens fiber cells.65 Collectively these studies support our findings that autophagy contributes to organelle removal during terminal lens fiber cell differentiation. It is possible that differences in lens phenotypes between mice with Atg5- or Pik3c3-ablated lenses and bs mice may be caused by gene modifiers66 given that these strains are on different genetic backgrounds.4,48 Further studies are needed to clarify the role of autophagy as well as the role of TBC1D20 during terminal lens fiber cell differentiation.56
The acrosome is a testis-specific organelle required for male fertility. Initially, it was proposed that the acrosome is a modified lysosome.67 Subsequent studies revealed that proacrosomic vesicles facilitate the formation of acrosomes and are derived from trans-Golgi stacks.68 Recent reports have classified acrosomes as a lysosome-related organelle (LRO) which encompasses a group of tissue-specific organelles that contain many enzymes also identified in lysosomes of somatic cells.69 However, recent studies have also led to a hypothesis that acrosomes form following fusion of autolysosomes and proacrosomal vesicles from the trans-Golgi stacks.27 In support of this hypothesis are findings that a failure in autophagosome formation caused by Atg7 ablation in germ cells abrogates acrosome formation,27 but also that a functional loss of trans-Golgi proteins GOPC and PICK1 that facilitate the formation and trafficking of proacrosomal vesicles also disrupts the formation of acrosomes.70,71 In bs germ cells, proacrosomal vesicles form and are transported to the perinuclear region, but fail to mature into acrosomes47 suggesting that TBC1D20 functional loss disrupts the formation of acrosomes downstream from proacrosomal vesicle formation and transport. Our results show that in bs germ cells autophagosomes and lysosomes form but fail to organize into crescent-shaped acrosome structures. Given that in MEFs TBC1D20 function is required for the formation of autolysosomes, these findings suggest that in bs testes a failure of autolysosomes to form most likely is the cause of disrupted acrosome biogenesis. Therefore, in testes, TBC1D20-mediated maturation of autophagosomes is required for the autophagic flux but is also required for the formation of acrosomes.
While bs mice recapitulate eye and testicular abnormalities identified in WARBM4-affected children, bs mice do not recapitulate severe developmental brain abnormalities.4 However, our results show that in bs mice, TBC1D20 functional loss results in motor dysfunction most likely caused by disrupted autophagic flux and accumulation of autophagic cargo. Our results are consistent with recent reports showing that mice deficient for ZFYVE26 and EPG5, which are critical regulators of autophagy, exhibit abortive autophagy and progressive adult-onset motor dysfunction72,73 but do not recapitulate the congenital/juvenile brain abnormalities identified in children with ZFYVE26 and EPG5 mutations.24,74 Furthermore, disrupted neuronal autophagy in mice caused by ablation of Atg5 or Atg7 also leads to progressive adult-onset motor dysfunction and neurodegeneration.48,49 It should be pointed out that in humans autophagic dysfunction contributes to adult-onset neurodegenerative disorders characterized by motor dysfunction including Parkinson disease and amyotrophic lateral sclerosis.75-77 It is possible that in humans, but not in mice, autophagy is required for brain development whereas in both humans and mice autophagy is required for neuronal homeostasis. However, it was recently reported that Atg5-mediated autophagy also contributes to neurogenesis.78 These findings suggest that autophagy contributes to both neurogenesis and neuronal homeostasis. Even though bs mice do not exhibit gross morphological brain abnormalities, it is possible that a functional loss of TBC1D20 in mice alters neurogenesis resulting in subtle cortical abnormalities. At this point, it still remains unknown how TBC1D20 facilitates human brain development and if TBC1D20-mediated autophagosome maturation contributes to this process.
In summary, we show that TBC1D20 has an essential role for maturation of autophagosomes, and a defect in TBC1D20 function disrupts maturation of autophagosomes resulting in abortive autophagy, which in turn results in the eye, testicular, and brain abnormalities seen in mice. Based on our findings we propose that disrupted autophagy contributes to the pathogenesis of Warburg Micro syndrome. In support of this hypothesis is a recent report showing that the RAB3GAP1/2 complex is involved in the formation of autophagosomes.79 RAB3GAP1/2 complex has a role as an atypical GAP composed of a catalytically active RAB3GAP1 subunit and a catalytically inactive RAB3GAP2 subunit.80,81 Mutations in RAB3GAP1 and RAB3GAP2 resulting in abolished function of the RAB3GAP complex, cause Warburg Micro syndrome 1 (OMIM #600118) and Warburg Micro syndrome 2 (OMIM#614225), respectively.1,2 Recently it was also shown that the dysfunction of the RAB3GAP complex as well as dysfunction of TBC1D20 causes RAB18 dysregulation indistinguishable from RAB18 deficiency, which causes Warburg Micro syndrome 3 (OMIM#614222).82 Children with mutations in RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 present with indistinguishable eye, brain, and genital clinical manifestation further supporting the idea that abortive autophagy contributes to the pathogenesis of all 4 forms of Warburg Micro syndrome.
Materials and methods
Mice and clinical evaluations
Genotyping and the maintenance of the bs allele has been maintained on the F2 CAST/EiJ XAKR/J background by brother to sister breeding as previously described.4 Clinical evaluation of mouse eyes was done with a Topcon SL-D8Z slit lamp biomicroscope (Topcon, Tokyo, Japan) with a Nikon SLR-based Photo Slit Lamp (Nikon, Tokyo, Japan) imaging system following mydriasis with 1% atropine sulfate (Bausch & Lomb, AB05007).
Mouse embryonic fibroblasts (MEFs), plasmids, and transfections
MEFs were isolated from E13.5 mouse embryos and maintained as previously described.83 GFP-LC3 and mCherry-EGFP-LC3 (Addgene, 22418; depositing lab Jayanta Debnath) vectors were described previously.84 Rab1b cDNA clone RefSeq mRNA (NM_030981) cloned in pCMV-SPORT6 was obtained from ThermoFisher Scientific (MGC:87903; IMAGE:6026121 clone). The ORF was PCR amplified using (F) 5′-AAAGCGGCCGCATGAACCCCGAATATGACTACCT-3′ AND (R) 5′-AAATCTAGACTAGCAACAGCCACCGCCAGC-3′ primers following subsequent digestion with NotI and XbaI (ThermoFisher Scientific, ER0595 and ER0681) and it was ligated into pFLAG.CMV.2 vector (Sigma-Aldrich, E7033). FLAG-RAB1BQ67L and FLAG-RAB1BN121I clones were generated by site-directed mutagenesis of the WT FLAG-RAB1B clone with the Phusion SDM Kit (ThermoFisher Scientific, F541) following the manufacturer's instructions with primers (F) 5′-P-GACACAGCGGGCCTGGAACGGTTCCGGACC-3′ and (R) 5′-P-CCAGATCTGAAGTTTGATAGTTTTGCCAGTC-3′ and F 5′-P-CTCCTGGTGGGCATCAAGAGCGACCTCACC-3′ and (R) 5′-P-CTTATTGACGTTCTCGCTGGCATAGCGGTC-3′ respectively. All clone sequences were confirmed via Sanger sequencing. Trasfections of MEFs were done using Lipofectamine 3000 (ThermoFisher Scientific, L3000008) following the manufacturer's recommendations for MEFs.
Immunocytochemistry
For immunocytochemistry, WT and bs MEFs were grown on coverslips until reaching 50–70% confluency, fixed for 10 min in 4% paraformaldehyde (Electron Microscopy Sciences, 15710) washed 3× for 5 min with 1× phosphate-buffered saline (PBS; ThermoFisher Scientific, 20012043), then blocked in 10% normal serum (ThermoFisher Scientific, PCN5000) for 30 min, and incubated with a primary antibody in SignalStain Ab Diluent (Cell Signaling Technology, 8112) overnight at 4°C in a humidified chamber overnight. Slides were washed 2x for 5 min in 1X PBS (ThermoFisher Scientific, 20012043) plus 0.025% Triton X-100 (Sigma-Aldrich, T8787) then incubated with a secondary antibody in SignalStain Ab Diluent for 1 h at room temperature. DAPI (ThermoFisher Scientific, 62248) was used to stain DNA. Primary antibodies used for immunocytochemistry are the following: SQSTM1 (Novus, NBP1-48320), ubiquitin and COX4I1 (Cell Signaling Technology, 3936 and 11967), LAMP2 (Developmental Studies Hybridoma Bank, ABL-93), FLAG (Sigma-Aldrich, F3165), and ABCD3 (Abcam, ab85550); secondary antibodies used for immunocytochemistry are the following: F(ab')2-goat anti-mouse IgG (H+L) secondary antibody, Alexa Fluor® 488 conjugate (ThermoFisher Scientific, A-11017), goat anti-mouse IgG (H+L), Alexa Fluor® 546 conjugate ThermoFisher Scientific, A-11030) and goat anti-rabbit IgG (H+L), Alexa Fluor® 546 conjugate (ThermoFisher Scientific, A-11035). All slides were mounted using Fluoromount-G (Southernbiotech, 0100-01), and imaged using a Nikon DS-Fi1 camera on a Nikon Eclipse 80i microscope (Nikon, Tokyo, Japan). For labeling acidic organelles, WT and bs MEFs were incubated with 100 nM LysoTracker® Red DND-99 (ThermoFisher Scientific, L7528) or with 2.5 μM Acridine Orange Staining Solution (ImmunoChemistry Technologies, 6130) in full medium for 1 h prior to fixation. Comparison between WT and bs cells were performed on at least 30 different cells (n = 30) from each genotype following a minimum of 3 separate experiments using ImageJ.85 The levels of fluorescence in WT and bs MEFs following staining with LysoTracker Red were measured from individual cells of both genotypes as a corrected total cell fluorescence using ImageJ.85 Significance was calculated via a Student t test (GraphPad, La Jolla, CA), where P < 0.05 was considered significant.
Western blotting, lysosome activity assays, and EGFR degradation assay
Stimulation and inhibition of autophagy were done in WT and bs MEFs following treatments for 2 or 4 h with the following: 1) DMSO (Sigma-Aldrich, C6295) as a control; 2) 100 nM bafilomycin A1 (Sigma-Aldrich, B1793), 3) serum-free and amino-acid free DMEM (Wako, 048-33575), and 4) combined 100 nM bafilomycin A1 and serum-free and amino-acid free DMEM. Cell lysates were generated from MEFs and tissues and western blots performed as previously described.4,86 Primary antibodies used for western blots are the following: LC3, ubiquitin and GAPDH (Cell Signaling Technology, 2775, 3969, and 5274), SQSTM1 and TOMM20 (Novus, NBP1-48320, H00009804-M01), ACTB (Abcam, ab20272), and EGFR (Santa Cruz Biotechnology, 1005); secondary antibodies are the following: peroxidase-conjugated AffiniPure Donkey anti-Rabbit IgG (H+L) and peroxidase-conjugated AffiniPure donkey anti-mouse IgG (H+L) (Jackson ImmunoResearch Laboratories, 111-035-144 and 715-035-150). The detection was performed using the ECL Western Blot Analysis System (GE Healthcare Life Sciences, RPN2108) following the manufacturer's instructions. Each lane on the blots corresponds to an individual lysate sample, and each blot is representative of at least 3 independent experiments. Mitochondria in live cells was assayed using the MitoTracker Red CMXRos (ThermoFisher Scientific, M7512) dye. Lysosome activity in cell lysates was assayed using the Cathepsin D Activity Assay Kit (Abcam, ab65302) following the manufacturer's instructions. Lysosomal activity in live cells was assayed using the Magic Red® Cathepsin B Detection Kit (ImmunoChemistry Technologies, 938) following the manufacturer's instruction for analysis via fluorescent microscopy of adherent cells as well as by measuring fluorescence intensity via plate reader. For the EGFR degradation assay we followed a previously published protocol.87 Briefly, WT and bs MEFs were cultured in serum-free DMEM for 3 h, followed by incubation with serum-free DMEM containing 40 ng/mL of EGF (R&D systems, 236-EG-200) and 20 μg/mL cycloheximide (Sigma-Aldrich, C1988) for 0 min, 30 min, 60 min and 120 min. After each time point, cells were lysed and protein lysates were subjected to western blotting as described above. Each lane on the blots corresponds to an individual lysate sample, and each blot is representative of at least 3 independent experiments. For all experiments comparison between WT and bs MEFs was performed following a minimum of 3 (n = 3) separate experiments and quantified using ImageJ.85 Significance was calculated via a Student t test (GraphPad, La Jolla, CA), where P < 0.05 was considered significant.
TEM
WT and bs MEFs were washed in 0.1 M Sörensens phosphate buffer (0.133 M Na2HPo4, 0.133 M KH2PO4, pH 7.4, 0.05 mM CaCl2). Fixation was performed in the dish with 1% glutaraldehyde (Sigma-Aldrich, 340855)+4% paraformaldehyde (Electron Microscopy Sciences, 15710) in 0.1 M Sörensens phosphate buffer for 10 min. Cells were then washed in 0.1 M Sörensens buffer 3X for 5 min each. Post fixation, cells were scraped from the plate, gently pelleted at 3000 g for 5 min, and the pellet fractions placed in freshly made reduced osmium tetroxide (1% OsO4 + 1.25% potassium hexacyanoferrate in distilled water 2 h on ice), dehydrated through graded methanol and processed into Embed 812 resin for TEM (Electron Microscopy Sciences, 14120) observation of ultrathin (70-nm) sections.
Histology and immunohistochemistry
Tissues were collected from WT and bs littermates, fixed, embedded in paraffin and sectioned at 4-µm thickness and stained for H&E as previously described.4 For immunohistochemistry sections were subjected to antigen retrieval in 10 mM sodium citrate, 0.05% Tween™ 20 (Fisher, BP337-500), pH 6.0 with a 20-min incubation at 95–99°C followed by washing 2x 5 min in 1X PBS (ThermoFisher Scientific, 20012043) and in 1x PBS (ThermoFisher Scientific, 20012043) plus 0.025% Triton X-100 for 10 min, then blocked in 10% normal goat serum (ThermoFisher Scientific, 50197Z) for 30 min, and incubated with a primary antibody in SignalStain Ab Diluent (Cell Signaling Technology, 8112) overnight at 4°C in a humidified chamber overnight. Slides were washes 2x 5 min in PBS plus 0.025% Triton X-100 then incubated with a secondary antibody in SignalStain Ab Diluent for 1 h at room temperature. DAPI was used to stain DNA. For immunohistochemistry antibodies for were the following: LC3 and ubiquitin (Cell Signaling Technology, 3668, 3963); SQSTM1 and LAMP1 (Novus, NBP1-48320, NB120-19294), CRYBB1/β-crystallin (Santa Cruz Biotechnology, sc-22745); secondary antibodies are the following: F(ab')2-goat anti-mouse IgG (H+L) secondary antibody, Alexa Fluor® 488 conjugate (ThermoFisher Scientific, A-11017), goat anti-mouse IgG (H+L), Alexa Fluor® 546 conjugate ThermoFisher Scientific, A-11030) and goat anti-rabbit IgG (H+L), Alexa Fluor® 546 conjugate (ThermoFisher Scientific, A-11035). All evaluations were performed on sections from tissue from at least 6 (n = 6) different animals of each genotype. All slides were mounted using Fluoromount-G (Southernbiotech, 0100-01), and imaged using a Nikon DS-Fi1 camera on a Nikon Eclipse 80i microscope (Nikon, Tokyo, Japan).
Hindlimb clasping and gait phenotypes
WT and bs mice were initially evaluated at 4 wk of age (n = 5) and subsequently at 5 mo (n = 3). Hindlimb clasping and gait phenotypes were evaluated as previously described.50-52 Briefly, for the hindlimb clasping phenotype a mouse was suspended by the tail, observed for 30 sec and scored. If both hindlimbs were entirely retracted and touching the abdomen for 50% of the time suspended it was considered to exhibit the hindlimb clasping phenotype. For the evaluation of the gait phenotype a mouse was placed in a cage-free of bedding and scored for the walking pattern. If mice exhibited lowering of the abdomen and positioning feet outwards they were considered to exhibit abnormal gait.
Statistical analyses
All analyses were performed following a minimum of 3 (n = 3) independent experiments. Significance was calculated via a Student t test (GraphPad, La Jolla, CA), where P < 0.05 was considered significant.
Supplementary Material
Abbreviations
- ABCD3
ATP-binding cassette sub-family D (ALD), member 3
- ACTB/β-actin
actin, β
- bs
blind-sterile
- COX4I1
cytochrome c oxidase subunit IV isoform 1
- EGFP
enhanced green fluorescent protein
- EGFR
epidermal growth factor receptor
- ER
endoplasmic reticulum
- GAPs
GTPase activating proteins
- GFP
green fluorescent protein
- LDs
lipid droplets
- LAMP1
lysosomal-associated membrane protein 1
- LAMP2
lysosomal-associated membrane protein 2
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MEFs
mouse embryonic fibroblasts
- OMIM
Online Mendelian Inheritance in Man
- SQSTM1
sequestosome 1
- TBC1D20
TBC1 domain family, member 20
- TEM
transmission electron microscopy
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
- WARBM4
Warburg Micro syndrome 4
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health grants EY018872, P30EY001931 (D.J.S.), AI104928 (W.T.J.), and a Dr. Michael J. Dunn Summer Medical Student Research Fellowship Award, Medical College of Wisconsin (A.K.P).
References
- [1].Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K, Maina EN, Morgan NV, Tee L, Morton J, et al.. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet 2005; 37:221-3; PMID:15696165; http://dx.doi.org/ 10.1038/ng1517 [DOI] [PubMed] [Google Scholar]
- [2].Borck G, Wunram H, Steiert A, Volk AE, Korber F, Roters S, Herkenrath P, Wollnik B, Morris-Rosendahl DJ, Kubisch C. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum Genet 2011; 129:45-50; PMID:20967465; http://dx.doi.org/ 10.1007/s00439-010-0896-2 [DOI] [PubMed] [Google Scholar]
- [3].Bem D, Yoshimura S, Nunes-Bastos R, Bond FC, Kurian MA, Rahman F, Handley MT, Hadzhiev Y, Masood I, Straatman-Iwanowska AA, et al.. Loss-of-function mutations in RAB18 cause Warburg micro syndrome. Am J Hum Genet 2011; 88:499-507; PMID:21473985; http://dx.doi.org/ 10.1016/j.ajhg.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liegel R, Handley M, Ronchetti A, Brown S, Langemeyer L, Linford A, Chang B, Morris-Rosendahl D, Carpanini S, Posmyk R, et al.. Loss-of-Function Mutations in TBC1D20 Cause Cataracts and Male Infertility in blind sterile Mice and Warburg Micro Syndrome in Humans. Am J Hum Genet 2013; 93:1-14; http://dx.doi.org/ 10.1016/j.ajhg.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Park A, Liegel RP, Ronchetti A, Ebert AD, Geurts A, Sidjanin DJ. Targeted disruption of Tbc1d20 with zinc-finger nucleases causes cataracts and testicular abnormalities in mice. BMC Genet 2014; 15:135; PMID:25476608; http://dx.doi.org/ 10.1186/s12863-014-0135-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Frasa MA, Koessmeier KT, Ahmadian MR, Braga VM. Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat Rev Mol Cell Biol 2012; 13:67-73; PMID:22251903; http://dx.doi.org/ 10.1038/nrm3364 [DOI] [PubMed] [Google Scholar]
- [7].Haas AK, Yoshimura S, Stephens DJ, Preisinger C, Fuchs E, Barr FA. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key Rabs required to maintain a functional Golgi complex in human cells. J Cell Sci 2007; 120:2997-3010; PMID:17684057; http://dx.doi.org/ 10.1242/jcs.014225 [DOI] [PubMed] [Google Scholar]
- [8].Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 2013; 93:269-309; PMID:23303910; http://dx.doi.org/ 10.1152/physrev.00003.2012 [DOI] [PubMed] [Google Scholar]
- [9].Sklan EH, Serrano RL, Einav S, Pfeffer SR, Lambright DG, Glenn JS. TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J Biol Chemstry 2007; 282:36354-61; PMID:17901050; http://dx.doi.org/ 10.1074/jbc.M705221200 [DOI] [PubMed] [Google Scholar]
- [10].Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 2008; 9:125-38; PMID:18216769; http://dx.doi.org/ 10.1038/nrm2336 [DOI] [PubMed] [Google Scholar]
- [11].Guo Y, Cordes KR, Farese RV Jr., Walther TC. Lipid droplets at a glance. J Cell Sci 2009; 122:749-52; PMID:19261844; http://dx.doi.org/ 10.1242/jcs.037630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Walther TC, Farese RV. Lipid Droplets and Cellular Lipid Metabolism. Annu Rev Biochem 2012; 81:687-714; PMID:22524315; http://dx.doi.org/ 10.1146/annurev-biochem-061009-102430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol 2006; 7:373-8; PMID:16550215; http://dx.doi.org/ 10.1038/nrm1912 [DOI] [PubMed] [Google Scholar]
- [14].Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 2009; 458:1131-5; PMID:19339967; http://dx.doi.org/ 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 2013; 20:3-11; PMID:22595754; http://dx.doi.org/ 10.1038/cdd.2012.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dong H, Czaja MJ. Regulation of lipid droplets by autophagy. Trends Endocrinol Metab 2011; 22:234-40; PMID:21419642; http://dx.doi.org/ 10.1016/j.tem.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 2014; 53:167-78; PMID:24462201; http://dx.doi.org/ 10.1016/j.molcel.2013.12.014 [DOI] [PubMed] [Google Scholar]
- [18].Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 2014; 16:495-501; PMID:24875736; http://dx.doi.org/ 10.1038/ncb2979 [DOI] [PubMed] [Google Scholar]
- [19].Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol 2011; 76:397-402; PMID:21813637; http://dx.doi.org/ 10.1101/sqb.2011.76.011023 [DOI] [PubMed] [Google Scholar]
- [20].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713-20; PMID:23817233; http://dx.doi.org/ 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Oz-Levi D, Ben-Zeev B, Ruzzo EK, Hitomi Y, Gelman A, Pelak K, Anikster Y, Reznik-Wolf H, Bar-Joseph I, Olender T, et al.. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet 2012; 91:1065-72; PMID:23176824; http://dx.doi.org/ 10.1016/j.ajhg.2012.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vantaggiato C, Crimella C, Airoldi G, Polishchuk R, Bonato S, Brighina E, Scarlato M, Musumeci O, Toscano A, Martinuzzi A, et al.. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain 2013; 136:3119-39; PMID:24030950; http://dx.doi.org/ 10.1093/brain/awt227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, Kasai-Yoshida E, Sawaura N, Nishida H, Hoshino A, et al.. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013; 45:445-9, 9e1; PMID:23435086; http://dx.doi.org/ 10.1038/ng.2562 [DOI] [PubMed] [Google Scholar]
- [24].Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, Simpson MA, Yau S, Bertini E, McClelland V, et al.. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 2013; 45:83-7; PMID:23222957; http://dx.doi.org/ 10.1038/ng.2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen J, Ma Z, Jiao X, Fariss R, Kantorow WL, Kantorow M, Pras E, Frydman M, Pras E, Riazuddin S, et al.. Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am J Hum Genet 2011; 88:827-38; PMID:21636066; http://dx.doi.org/ 10.1016/j.ajhg.2011.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shiels A, Bennett TM, Knopf HL, Yamada K, Yoshiura K, Niikawa N, Shim S, Hanson PI. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet 2007; 81:596-606; PMID:17701905; http://dx.doi.org/ 10.1086/519980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang H, Wan H, Li X, Liu W, Chen Q, Wang Y, Yang L, Tang H, Zhang X, Duan E, et al.. Atg7 is required for acrosome biogenesis during spermatogenesis in mice. Cell Res 2014; 24:852-69; PMID:24853953; http://dx.doi.org/ 10.1038/cr.2014.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rivera-Molina FE, Novick PJ. A Rab GAP cascade defines the boundary between two Rab GTPases on the secretory pathway. Proc Natl Acad Sci U S A 2009; 106:14408-13; PMID:19666511; http://dx.doi.org/ 10.1073/pnas.0906536106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lynch-Day MA, Bhandari D, Menon S, Huang J, Cai H, Bartholomew CR, Brumell JH, Ferro-Novick S, Klionsky DJ. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci U S A 2010; 107:7811-6; PMID:20375281; http://dx.doi.org/ 10.1073/pnas.1000063107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kakuta S, Yamamoto H, Negishi L, Kondo-Kakuta C, Hayashi N, Ohsumi Y. Atg9 vesicles recruit vesicle-tethering proteins Trs85 and Ypt1 to the autophagosome formation site. J Biol Chem 2012; 287:44261-9; PMID:23129774; http://dx.doi.org/ 10.1074/jbc.M112.411454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Barrowman J, Bhandari D, Reinisch K, Ferro-Novick S. TRAPP complexes in membrane traffic: convergence through a common Rab. Nat Rev Mol Cell Biol 2010; 11:759-63; PMID:20966969; http://dx.doi.org/ 10.1038/nrm2999 [DOI] [PubMed] [Google Scholar]
- [32].Zoppino FC, Militello RD, Slavin I, Alvarez C, Colombo MI. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 2010; 11:1246-61; PMID:20545908; http://dx.doi.org/ 10.1111/j.1600-0854.2010.01086.x [DOI] [PubMed] [Google Scholar]
- [33].Tan D, Cai Y, Wang J, Zhang J, Menon S, Chou HT, Ferro-Novick S, Reinisch KM, Walz T. The EM structure of the TRAPPIII complex leads to the identification of a requirement for COPII vesicles on the macroautophagy pathway. Proc Natl Acad Sci U S A 2013; 110:19432-7; PMID:24218626; http://dx.doi.org/ 10.1073/pnas.1316356110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181-97; PMID:19200883; http://dx.doi.org/ 10.1016/S0076-6879(08)03612-4 [DOI] [PubMed] [Google Scholar]
- [35].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/ 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014; 10:431-41; PMID:24394643; http://dx.doi.org/ 10.4161/auto.27344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- [39].Hansen TE, Johansen T. Following autophagy step by step. BMC Biol 2011; 9:39; PMID:21635796; http://dx.doi.org/ 10.1186/1741-7007-9-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sorkin A, Duex JE. Quantitative analysis of endocytosis and turnover of epidermal growth factor (EGF) and EGF receptor. Curr Protoc Cell Biol 2010; Chapter 15:Unit 15 4; PMID:20235100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al.. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/ 10.1083/jcb.200412022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J, et al.. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci 2013; 126:939-52; PMID:23239026; http://dx.doi.org/ 10.1242/jcs.114819 [DOI] [PubMed] [Google Scholar]
- [43].Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A 2008; 105:20567-74; PMID:19074260; http://dx.doi.org/ 10.1073/pnas.0810611105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bassnett S. On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res 2009; 88:133-9; PMID:18840431; http://dx.doi.org/ 10.1016/j.exer.2008.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wride MA. Lens fibre cell differentiation and organelle loss: many paths lead to clarity. Philos Trans R Soc Lond B Biol Sci 2011; 366:1219-33; PMID:21402582; http://dx.doi.org/ 10.1098/rstb.2010.0324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kuwabara T, Imaizumi M. Denucleation process of the lens. Invest Ophthalmol 1974; 13:973-81; PMID:4430579 [PubMed] [Google Scholar]
- [47].Sotomayor RE, Handel MA. Failure of acrosome assembly in a male sterile mouse mutant. Biol Reprod 1986; 34:171-82; PMID:3955134; http://dx.doi.org/ 10.1095/biolreprod34.1.171 [DOI] [PubMed] [Google Scholar]
- [48].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/ 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- [49].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/ 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- [50].Chu J, Hong NA, Masuda CA, Jenkins BV, Nelms KA, Goodnow CC, Glynne RJ, Wu H, Masliah E, Joazeiro CA, et al.. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc Natl Acad Sci U S A 2009; 106:2097-103; PMID:19196968; http://dx.doi.org/ 10.1073/pnas.0812819106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu X, Dobbie M, Tunningley R, Whittle B, Zhang Y, Ittner LM, Gotz J. ENU mutagenesis screen to establish motor phenotypes in wild-type mice and modifiers of a pre-existing motor phenotype in tau mutant mice. J Biomed Biotechnol 2011; 2011:130947; PMID:22219655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp 2010; PMID:20495529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev 2011; 91:119-49; PMID:21248164; http://dx.doi.org/ 10.1152/physrev.00059.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ 2014; 21:348-58; PMID:24440914; http://dx.doi.org/ 10.1038/cdd.2013.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 2013; 14:759-74; PMID:24201109; http://dx.doi.org/ 10.1038/nrm3696 [DOI] [PubMed] [Google Scholar]
- [56].Bento CF, Puri C, Moreau K, Rubinsztein DC. The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci 2013; 126:1059-69; PMID:23620509; http://dx.doi.org/ 10.1242/jcs.123075 [DOI] [PubMed] [Google Scholar]
- [57].Menko AS. Lens Epithelial Cell Differentiation. Exp Eye Res 2002; 75:485-90; PMID:12457861; http://dx.doi.org/ 10.1006/exer.2002.2057 [DOI] [PubMed] [Google Scholar]
- [58].Moreau KL, King JA. Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med 2012; 18:273-82; PMID:22520268; http://dx.doi.org/ 10.1016/j.molmed.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Boscia F, Grattagliano I, Vendemiale G, Micelli-Ferrari T, Altomare E. Protein oxidation and lens opacity in humans. Invest Ophthalmol Vis Sci 2000; 41:2461-5; PMID:10937554 [PubMed] [Google Scholar]
- [60].Shiokawa D, Tanuma S. DLAD, a novel mammalian divalent cation-independent endonuclease with homology to DNase II. Nucleic Acids Res 1999; 27:4083-9; PMID:10497274; http://dx.doi.org/ 10.1093/nar/27.20.4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Evans CJ, Aguilera RJ. DNase II: genes, enzymes and function. Gene 2003; 322:1-15; PMID:14644493; http://dx.doi.org/ 10.1016/j.gene.2003.08.022 [DOI] [PubMed] [Google Scholar]
- [62].Lan YY, Londono D, Bouley R, Rooney MS, Hacohen N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep 2014; 9:180-92; PMID:25284779; http://dx.doi.org/ 10.1016/j.celrep.2014.08.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Morishita H, Eguchi S, Kimura H, Sasaki J, Sakamaki Y, Robinson ML, Sasaki T, Mizushima N. Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem 2013; 288:11436-47; PMID:23479732; http://dx.doi.org/ 10.1074/jbc.M112.437103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Costello MJ, Brennan LA, Basu S, Chauss D, Mohamed A, Gilliland KO, Johnsen S, Menko AS, Kantorow M. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp Eye Res 2013; 116:141-50; PMID:24012988; http://dx.doi.org/ 10.1016/j.exer.2013.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Basu S, Rajakaruna S, Reyes B, Van Bockstaele E, Menko AS. Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy 2014; 10:1193-211; PMID:24813396; http://dx.doi.org/ 10.4161/auto.28768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Montagutelli X. Effect of the genetic background on the phenotype of mouse mutations. J Am Soc Nephrol 2000; 11 Suppl 16:S101-5; PMID:11065339 [PubMed] [Google Scholar]
- [67].Hartree EF. The acrosome-lysosome relationship. J Reprod Fertil 1975; 44:125-6; PMID:1171229; http://dx.doi.org/ 10.1530/jrf.0.0440125 [DOI] [PubMed] [Google Scholar]
- [68].Martinez-Menarguez JA, Geuze HJ, Ballesta J. Evidence for a nonlysosomal origin of the acrosome. J Histochem Cytochem 1996; 44:313-20; PMID:8601690; http://dx.doi.org/ 10.1177/44.4.8601690 [DOI] [PubMed] [Google Scholar]
- [69].Berruti G, Paiardi C. Acrosome biogenesis: Revisiting old questions to yield new insights. Spermatogenesis 2011; 1:95-8; PMID:22319656; http://dx.doi.org/ 10.4161/spmg.1.2.16820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yao R, Ito C, Natsume Y, Sugitani Y, Yamanaka H, Kuretake S, Yanagida K, Sato A, Toshimori K, Noda T. Lack of acrosome formation in mice lacking a Golgi protein, GOPC. Proc Natl Acad Sci U S A 2002; 99:11211-6; PMID:12149515; http://dx.doi.org/ 10.1073/pnas.162027899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Xiao N, Kam C, Shen C, Jin W, Wang J, Lee KM, Jiang L, Xia J. PICK1 deficiency causes male infertility in mice by disrupting acrosome formation. J Clin Invest 2009; 119:802-12; PMID:19258705; http://dx.doi.org/ 10.1172/JCI36230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Khundadze M, Kollmann K, Koch N, Biskup C, Nietzsche S, Zimmer G, Hennings JC, Huebner AK, Symmank J, Jahic A, et al.. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet 2013; 9:e1003988; PMID:24367272; http://dx.doi.org/ 10.1371/journal.pgen.1003988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhao H, Zhao YG, Wang X, Xu L, Miao L, Feng D, Chen Q, Kovacs AL, Fan D, Zhang H. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol 2013; 200:731-41; PMID:23479740; http://dx.doi.org/ 10.1083/jcb.201211014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hanein S, Martin E, Boukhris A, Byrne P, Goizet C, Hamri A, Benomar A, Lossos A, Denora P, Fernandez J, et al.. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet 2008; 82:992-1002; PMID:18394578; http://dx.doi.org/ 10.1016/j.ajhg.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, et al.. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006; 38:1184-91; PMID:16964263; http://dx.doi.org/ 10.1038/ng1884 [DOI] [PubMed] [Google Scholar]
- [76].Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al.. Hereditary early-onset Parkinson disease caused by mutations in PINK1. Science 2004; 304:1158-60; PMID:15087508; http://dx.doi.org/ 10.1126/science.1096284 [DOI] [PubMed] [Google Scholar]
- [77].Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, et al.. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011; 68:1440-6; PMID:22084127; http://dx.doi.org/ 10.1001/archneurol.2011.250 [DOI] [PubMed] [Google Scholar]
- [78].Lv X, Jiang H, Li B, Liang Q, Wang S, Zhao Q, Jiao J. The crucial role of Atg5 in cortical neurogenesis during early brain development. Sci Rep 2014; 4:6010; PMID:25109817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Spang N, Feldmann A, Huesmann H, Bekbulat F, Schmitt V, Hiebel C, Koziollek-Drechsler I, Clement AM, Moosmann B, Jung J, et al.. RAB3GAP1 and RAB3GAP2 modulate basal and rapamycin-induced autophagy. Autophagy 2014; 10:2297-309; PMID:25495476; http://dx.doi.org/ 10.4161/15548627.2014.994359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Fukui K, Sasaki T, Imazumi K, Matsuura Y, Nakanishi H, Takai Y. Isolation and characterization of a GTPase activating protein specific for the Rab3 subfamily of small G proteins. J Biol Chem 1997; 272:4655-8; PMID:9030515; http://dx.doi.org/ 10.1074/jbc.272.8.4655 [DOI] [PubMed] [Google Scholar]
- [81].Nagano F, Sasaki T, Fukui K, Asakura T, Imazumi K, Takai Y. Molecular cloning and characterization of the noncatalytic subunit of the Rab3 subfamily-specific GTPase-activating protein. J Biol Chem 1998; 273:24781-5; PMID:9733780; http://dx.doi.org/ 10.1074/jbc.273.38.24781 [DOI] [PubMed] [Google Scholar]
- [82].Handley MT, Carpanini SM, Mali GR, Sidjanin DJ, Aligianis IA, Jackson IJ, FitzPatrick DR. Warburg Micro syndrome is caused by RAB18 deficiency or dysregulation. Open Biol 2015; 5:150047; PMID:26063829; http://dx.doi.org/ 10.1098/rsob.150047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hassemer EL, Le Gall SM, Liegel R, McNally M, Chang B, Zeiss CJ, Dubielzig RD, Horiuchi K, Kimura T, Okada Y, et al.. The waved with open eyelids (woe) locus is a hypomorphic mouse mutation in Adam17. Genetics 2010; 185:245-55; PMID:20194968; http://dx.doi.org/ 10.1534/genetics.109.113167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 2009; 10:173-9; PMID:19148225; http://dx.doi.org/ 10.1038/embor.2008.238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671-5; PMID:22930834; http://dx.doi.org/ 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Waguri S, Komatsu M. Biochemical and morphological detection of inclusion bodies in autophagy-deficient mice. Methods Enzymol 2009; 453:181-96; PMID:19216907; http://dx.doi.org/ 10.1016/S0076-6879(08)04009-3 [DOI] [PubMed] [Google Scholar]
- [87].Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, Cohen MA, Chakraborty S, Wang H, Spooner E, et al.. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep 2013; 5:1302-15; PMID:24290752; http://dx.doi.org/ 10.1016/j.celrep.2013.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.