

ABSTRACT
Autophagy is a highly coordinated process that is controlled at several levels including transcriptional regulation. Here, we identify the transcription factor NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) as a regulator of autophagy gene expression and its relevance in a mouse model of Alzheimer disease (AD) that reproduces impaired APP (amyloid β precursor protein) and human (Hs)MAPT/TAU processing, clearance and aggregation. We screened the chromatin immunoprecipitation database ENCODE for 2 proteins, MAFK and BACH1, that bind the NFE2L2-regulated enhancer antioxidant response element (ARE). Using a script generated from the JASPAR's consensus ARE sequence, we identified 27 putative AREs in 16 autophagy-related genes. Twelve of these sequences were validated as NFE2L2 regulated AREs in 9 autophagy genes by additional ChIP assays and quantitative RT-PCR on human and mouse cells after NFE2L2 activation with sulforaphane. Mouse embryo fibroblasts of nfe2l2-knockout mice exhibited reduced expression of autophagy genes, which was rescued by an NFE2L2 expressing lentivirus, and impaired autophagy flux when exposed to hydrogen peroxide. NFE2L2-deficient mice co-expressing HsAPPV717I and HsMAPTP301L, exhibited more intracellular aggregates of these proteins and reduced neuronal levels of SQSTM1/p62, CALCOCO2/NDP52, ULK1, ATG5 and GABARAPL1. Also, colocalization of HsAPPV717I and HsMAPTP301L with the NFE2L2-regulated autophagy marker SQSTM1/p62 was reduced in the absence of NFE2L2. In AD patients, neurons expressing high levels of APP or MAPT also expressed SQSTM1/p62 and nuclear NFE2L2, suggesting their attempt to degrade intraneuronal aggregates through autophagy. This study shows that NFE2L2 modulates autophagy gene expression and suggests a new strategy to combat proteinopathies.
KEYWORDS: Alzheimer disease, amyloid precursor protein, neurodegenerative diseases, neuroprotection, oxidative stress, proteostasis, Tau
Introduction
The homeostatic control of protein synthesis, folding, trafficking and degradation, i.e. proteostasis, is critical for biological processes, and its alterations lead to pathological outcome, including neurodegenerative diseases.1 The 2 main mechanisms that control protein degradation are the 20S and 26S proteasome and autophagy in its 3 primary modalities, microautophagy, chaperone-mediated autophagy and macroautophagy.2 These processes ensure timely degradation of misfolded, oxidized or altered proteins that otherwise develop proteinopathy. In particular, macroautophagy, usually termed autophagy, participates in the degradation of long-lived proteins that, if improperly cleared, aggregate and cause cellular damage.
The autophagy process requires the coordinated participation of a set of structural proteins that participate in the formation of autophagosomes and autolysosomes, as well as cargo-selective proteins that recognize specific cargos and direct them to degradation.3 Most studies on the regulation of macroautophagy have focused on protein-protein interactions based on MTOR-dependent mechanisms. For instance, upon serum-deprivation or inhibition by rapamycin, low MTOR activity allows ULK1 activation by disruption of the interaction with AMP kinase and results in increased autophagy.4 Because these regulatory mechanisms are cell signaling dependent, they probably represent a quick adaptation to cellular needs of a specific moment. Other reports indicate positive5-8 and negative9,10 transcriptional regulation of autophagy, which might represent a long-term mechanism of adaptation. Two documented transcription factors that fulfill this role are the helix-loop-helix transcription factor TFEB and the forkhead transcription factor FOXO.11,12 Given the complexity of the macroautophagy system and the need for a coordinated interplay between autophagy-regulated and -regulating proteins, it is hypothesized that the expression of autophagy genes is connected through additional common regulatory transcription factor(s).
Considering that autophagy is a proteostatic and defensive mechanism, we sought to determine if this process is regulated by NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2). The transcription factor NFE2L2 is considered a master regulator of cellular homeostasis, as it controls the expression of numerous cytoprotective genes.13,14 These genes share a common cis-acting element in their promoters, named the antioxidant response element (ARE), and the list of cytoprotective genes that contain this enhancer is continuously growing to include those involved in biotransformation, antioxidant defense, and metabolic reprogramming. Regarding autophagy, the cargo recognition protein SQSTM1/p62 contains the motif KIR that interacts with KEAP1 leading to its transport into the phagophore (the autophagosome precursor) and relieving NFE2L2 from inhibition.15,16 In connection with this finding, SQSTM1 contains one ARE and is therefore upregulated by NFE2L2, creating a feed-forward cycle.17 The cargo recognition protein CALCOCO2/NDP52 also appears to be regulated by NFE2L2, leading to the clearance of toxic proteins such as phosphorylated HsMAPT (p-MAPT) in a mouse model of AD, while 3 different ARE sequences were identified in its promoter region.18
Impaired proteostasis is a crucial hallmark of neurodegenerative diseases.19-21 Protein pathology in AD is related to the altered processing of APP, which leads to intracellular accumulation and extracellular deposition of amyloid-β (Aβ) peptides, as well as aggregation of p-MAPT into intracellular neurofibrillary tangles.22 Both APP and HsMAPT are degraded, at least in part, through autophagy and several reports suggest that this process is altered in AD. For instance, dystrophic neurites of AD patients contain more autophagosomes,23 increased levels of MTOR signaling components and lysosomal hydrolases.24
In this study, we sought to determine if genes that participate in autophagy are regulated by NFE2L2. Our results support our hypothesis that NFE2L2 participates in the regulation of autophagy gene expression and suggest a new therapeutic strategy for proteinopathies based on the activation of this transcription factor.
Results
Identification of putative AREs in autophagy genes
To define comprehensively the role of NFE2L2 in the transcriptional regulation of autophagy, we searched the Encyclopedia of DNA Elements at UCSC (ENCODE)25 of the human genome (Feb. 2009) for autophagy genes with putative AREs. This database contains the experimental data from chromatin immunoprecipitation (ChIP) studies of several transcription factors. Although NFE2L2 is not included, we analyzed 2 other ARE binding factors, MAFK and BACH1, for which information is available. We found evidence of MAFK or BACH1 binding in many genes involved directly or indirectly in autophagy. Then, we developed a Python-based bioinformatics analysis script, to compare the consensus human ARE from the JASPAR database26 with putative AREs in the promoter regions of these genes (for details see Material and methods and Tables S1 and S2). We detected 25 putative new AREs (relative score over 80%) in these 16 autophagy genes, besides the internal controls that our algorithm retrieved as the bona fide ARE-genes in NQO1 and HMOX1 (Table 1). Some of these genes, such as SQSTM117 or CALCOCO2 18 were reported to be regulated by NFE2L2, and for SQSTM1 we identified the previously described ARE17 plus 3 new sequences. The other autophagy ARE-genes were identified here for the first time. Moreover, several putative AREs are conserved in mice (Tables S1–S3).
Table 1.
Putative Antioxidant Response Elements (AREs) in the promoter regions of autophagy genes with a relative score higher than 80%. The table also shows the max score and the localization in the human genome.
| GENE (human) | Localization in the human genome | Max score | Relative score | ARE putative binding sequence |
|---|---|---|---|---|
| SQSTM1 | chr5:179247562-179247551 | 19.11 | 1.0 | ATGACTCAGCA (1) |
| chr5:179246594-179246583 | 18.97 | 0.997 | GTGACTCAGCA*(2) | |
| chr5:179247479-179247490 | 11.44 | 0.816 | GCGACCTAGCA (3) | |
| chr5:179246116-179246105 | 13.39 | 0.863 | GTGAGTCAGCG (4) | |
| CALCOCO2 | chr17:46906385-46906374 | 12.65 | 0.845 | ATGAGTAAGCC* |
| chr17:46906430-46906441 | 12.34 | 0.838 | GTGAGGAAGCA | |
| ULK1 | chr12:132381043-132381032 | 12.62 | 0.844 | ATTAGTCAGCA |
| ULK2 | chr17:19783789-19783800 | 15.74 | 0.919 | GTGACAGAGCA |
| ATG2B | chr14:96849918-96849929 | 11.38 | 0.815 | CTGCCTAAGCA |
| ATG4C | chr1:63212369-63212380 | 13.03 | 0.854 | ATGAAATAGCA |
| ATG4D | chr19:10652326-10652315 | 14.96 | 0.9 | ATGACAATGCA |
| chr19:10655902-10655891 | 10.94 | 0.804 | GTGACTTTGAA | |
| ATG5 | chr6:106873861-106873872 | 11.01 | 0.806 | CTGACCTTGCA |
| ATG7 | chr3:11347170-11347159 | 15.94 | 0.924 | ATGAGTCAGCA |
| chr3:11320731-11320720 | 13.65 | 0.869 | CTGACTCAGCT | |
| chr3:11347216-11347227 | 11.08 | 0.807 | AGGACTTGGCA | |
| chr3:11320599-11320610 | 11.07 | 0.807 | ATGATTGAGCT | |
| GABARAPL1 | chr12:10364974-10364963 | 14.58 | 0.891 | GTGACTCAGGA |
| chr12:10366072-10366083 | 18.97 | 0.997 | GTGACTCAGCA | |
| ATG9B | chr7:150723213-150723224 | 14.24 | 0.883 | CTGACTCAGCC |
| ATG10 | chr5:81278192-81278181 | 16.65 | 0.941 | CTGACTCAGCA |
| ATG16L1 | chr2:234134609-234134598 | 16.53 | 0.938 | ATGATTCAGCA |
| chr2:234134578-234134589 | 13.56 | 0.867 | ATGACCTAGCC | |
| WIPI2 | chr7:5239766-5239755 | 16.65 | 0.941 | ATGACTGAGCA |
| LAMP1 | chr13:113951364-113951353 | 12.06 | 0.831 | GTGACCCGGCC |
| AMBRA1 | chr11:46576251-46576240 | 11.51 | 0.818 | TTGACTTAGCT |
| chr11:46572300-46572311 | 10.84 | 0.802 | ATGACTAATCT | |
| NQO1 | chr16:69760919-69760908 | 18.97 | 0.997 | GTGACTCAGCA* |
| HMOX1 | chr22:35768205-35768216 | 12.3 | 0.837 | ATGCCTCAGCC |
| chr22:35768021-35768010 | 18.97 | 0.997 | GTGACTCAGCA* | |
| chr22:35768127-35768116 | 18.97 | 0.997 | GTGACTCAGCA* | |
| chr22:35747111-35747100 | 13.24 | 0.859 | GTTACTCAGCC |
Asterisks denote AREs that have been described previously.
Validation of putative AREs by ChIP and qRT-PCR
The newly identified ARE sequences were subsequently validated by ChIP assays for NFE2L2. Because of the lack of adequate antibodies to immunoprecipitate endogenous NFE2L2 efficiently, we used HEK293T cells transfected with an expression vector for V5-tagged NFE2L2. Moreover, this construct lacked the KEAP1 regulatory domain (ETGE), facilitating NFE2L2 stabilization, its translocation to the nucleus and binding to target genes.27 ChIPs were performed with anti-V5 antibody, and with anti-IgG as negative control. Immunoprecipitated DNA was analyzed by quantitative real time PCR (qRT-PCR) with specific primers surrounding the putative AREs (Table S4).
Among the 27 putative AREs analyzed in autophagy genes we found enrichment of 11 ARE regions in V5-immunoprecipitated chromatin, indicating that NFE2L2 binds these promoter regions (Fig. 1A-B). Our study also detected enrichment of 2 positive controls from the bona fide NFE2L2 targets NQO1 and HMOX1. No enrichment was detected with specific primers for ACTB, for an upstream region of NQO1 that does not contain AREs,28 or for ATG3, all of which had low scores for putative AREs in the bioinformatics analysis. Negative control assays were further performed without antibodies or on nontransfected cells (data not shown). Next, we sought to determine the existence of other potential AREs in the SQSTM1 promoter aside from the one already described.17 For this purpose we used specific primers surrounding the other potential AREs (Table S4 and Table 1). We observed that NFE2L2 bound to all these sequences (Fig. 1B), demonstrating that they constitute additional AREs in this gene.
Figure 1.
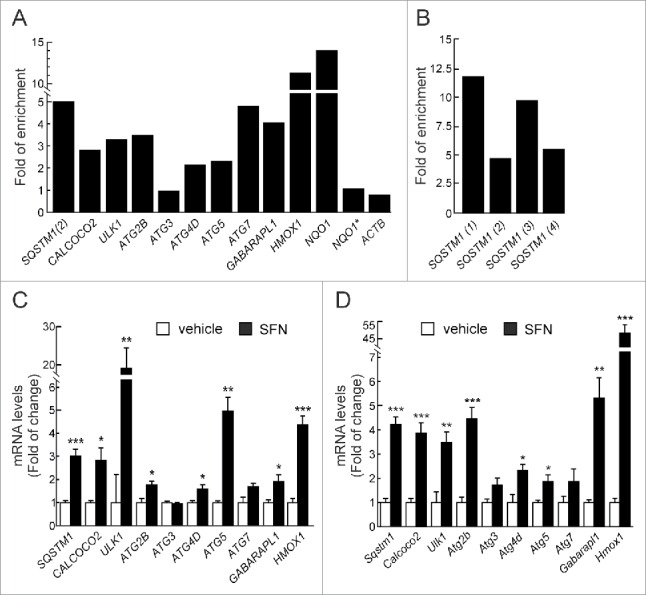
NFE2L2 modulates autophagy gene expression. (A) HEK293T cells were transfected with an expression vector for NFE2L2ΔETGE-V5.27 ChIP analysis was performed with anti-IgG or anti-V5 antibodies and the potential AREs with the highest score were analyzed by qRT-PCR. (B) The same ChIP analysis of putative AREs in the promoter of SQSTM1. The figures show representative data normalized as the fold of enrichment with the anti-V5 antibody vs. the IgG antibody. In (A), ACTB and an upstream region of NQO1 that does not contain any ARE (NQO1*) were analyzed as negative controls. Previously described AREs in HMOX1, NQO1, SQSTM1 and CALCOCO2 were analyzed as positive controls. These experiments were repeated 3 times with similar results. In (B), numbers in brackets indicate the AREs from Table 1 specifically amplified. (C and D) HEK293T and HT22 cells were submitted to sulforaphane (SFN, 15 μM) for 12 h. mRNA levels of the indicated genes were determined by qRT-PCR and normalized by Actb levels. Data are mean ± SEM (n = 3). Statistical analysis was performed with the Student t test. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 vs. untreated conditions.
We went on to further confirm our observations in HEK293T cells, treated with the NFE2L2 activator sulforaphane (SFN) (15 μM, 12 h), which has been used previously to induce autophagy.29-31 Transcript levels of the selected autophagy genes were analyzed by qRT-PCR, demonstrating increased expression of SQSTM1, CALCOCO2, ULK1, ATG2B, ATG4D, ATG5, and GABARAPL1 upon SFN treatment (Fig. 1C). HMOX1 was analyzed and confirmed as a positive control. We next extended our observations to murine cells by analyzing hippocampus-derived HT22 cells treated with SFN (15 μM, 12 h). SFN augmented the expression of all the murine counterparts of the genes identified in the human cells (Fig. 1D). The combined results indicated that the regulation of expression of autophagy genes is conserved in both species.
We next analyzed the expression of these genes in mouse embryonic fibroblasts (MEFs) from wild-type (Nfe2l2-WT) or Nfe2l2-knockout (Nfe2l2-KO) mice by qRT-PCR (Fig. 2A). Impaired expression of Sqstm1, Calcoco2, Ulk1, Atg2b, Atg4d, Atg5, Atg7 and Gabarapl1 was observed in Nfe2l2-KO MEFs. Moreover, reduced expression of Sqstm1, Calcoco2 and Atg7 products was also confirmed at the protein level with available antibodies (Fig. 2B-C).
Figure 2.
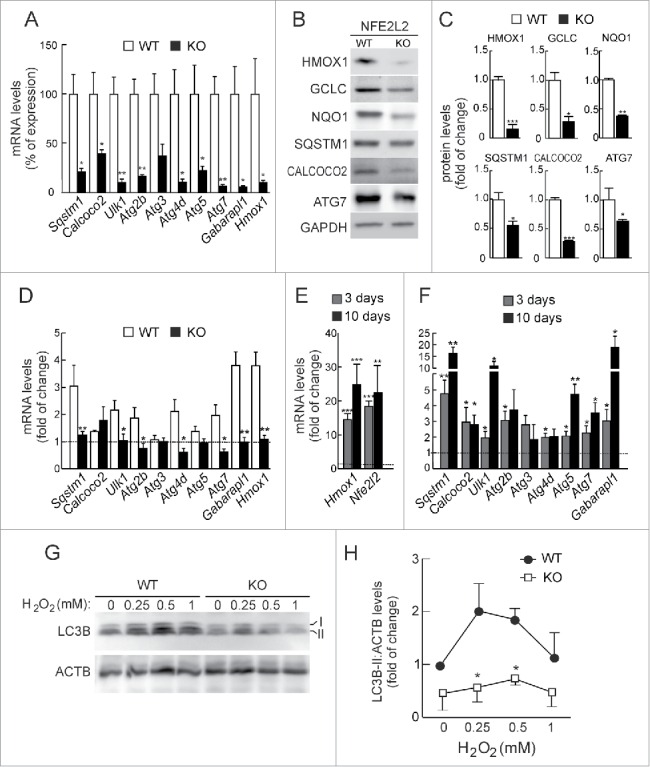
NFE2L2 deficiency results in decreased autophagy gene expression. (A) Expression levels of the indicated genes from Nfe2l2-WT and nfe2l2-KO mouse embryonic fibroblasts (MEFs) were determined by qRT-PCR and normalized by Actb levels. Data are mean ± SEM (n = 3). Statistical analysis was performed with the Student t test. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 vs. Nfe2l2-WT MEFs. (B) Representative immunoblots for the indicated proteins of Nfe2l2-WT and nfe2l2-KO MEFs. (C) Densitometric quantification of representative blots from (B) relative to GAPDH levels. Data are mean ± SEM (n = 3). Statistical analysis was performed using the Student t test. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 vs. Nfe2l2-WT MEFs. (D) Nfe2l2-WT and nfe2l2-KO MEFs were submitted to SFN (15 µM, 6 h). mRNA levels of the indicated genes were determined by qRT-PCR and normalized to Actb levels. (E) nfe2l2-KO MEFs were transduced with NFE2L2ΔETGE-V5 or GFP-expressing lentivirus and mRNA levels of the indicated genes were analyzed by qRT-PCR following 3 and 10 after transduction. For (D) and (E), bars represent the fold of change normalized to the untreated condition (D), or GFP-lentivirus infection (E) depicted with the dashed lines. Data are mean ± SEM (n = 3). Statistical analysis was performed with the Student t test. *, p < 0.05; and **, p < 0.01 vs. control conditions. (F) Nfe2l2-WT and nfe2l2-KO cells were treated with the indicated concentrations of H2O2 during 6 h. Representative immunoblots for the indicated proteins of Nfe2l2-WT and nfe2l2-KO MEFs. (G) Densitometric quantification of representative blots from (F) relative to ACTB/β-actin levels. Data are mean ± SEM (n = 3). Statistical analysis was performed using Student t test. *, p < 0.05 vs. Nfe2l2-WT.
To further define the role of NFE2L2 in the regulation of these autophagy genes we performed chemical and genetic manipulations of this transcription factor. In response to SFN nfe2l2-KO MEFs exhibited a reduced induction of these genes compared to Nfe2l2-WT MEFs (Fig. 2D). In addition, rescue experiments in nfe2l2-KO MEFs employing NFE2L2-ΔEGTE-V5 augmented normal basal levels of expression of autophagy genes when compared with nfe2l2-KO MEFs infected with a control GFP-expressing lentivirus. Indeed, Sqstm1, Calcoco2, Ulk1, Atg5, Atg7 and Gabarapl1 exhibited a robust response (Fig. 2E).
In order to determine the impact of NFE2L2-deficiency in the autophagy flux, we analyzed the conversion of LC3B-I to LC3B-II as an indicator of autophagosome formation. The putative AREs in the LC3 coding gene could not be validated by ChIP and we did not detect significant changes in LC3 mRNA levels in NFE2L2-deficient primary cortical neurons (data not shown), suggesting that these changes are not connected with NFE2L2 regulation of this gene. Following serum-deprivation or rapamycin, we detected similar increases in the levels of LC3B-II between Nfe2l2-WT and nfe2l2-KO MEFs, indicating that NFE2L2 has little or no effect in the regulation of autophagy by these stimuli (Fig. S1). Interestingly, H2O2 treatment during 4 h increased LC3B-II levels in both cell types, but to a lesser extent in nfe2l2-KO MEFs (Fig. 2F and 2G). We concluded that NFE2L2 may regulate autophagy under oxidative stress conditions.
NFE2L2 deficiency impairs autophagy in a mouse model of Alzheimer disease
To determine the relevance of NFE2L2 in the modulation of autophagy, we used a mouse model of proteinopathy that reproduces the 2 main hallmarks of AD, namely amyloidopathy and tauopathy. These animals express mutant HsAPPV717I and HsMAPTP301L under the control of the Thy1 promoter32 and develop intracellular HsAPP and HsMAPT aggregates after 12 mo of age. In addition, we generated HsAPP- and HsMAPT-expressing mice in wild-type (AT-Nfe2l2-WT) and nfe2l2-knockout (AT-nfe2l2-KO) genetic backgrounds.
First of all and to specifically determine the impact of NFE2L2-deficiency in neurons, we analyzed autophagy gene expression in hippocampal/cortical primary cultures by qRT-PCR (Fig. 3A). As expected, nfe2l2-KO neurons exhibited impaired expression of Sqstm1, Calcoco2, Ulk1, Atg2b, Atg5, Atg7 and Gabarapl1. Autophagy gene expression was then analyzed in hippocampus and brainstem (Table S5A-C) of Nfe2l2-WT, nfe2l2-KO, AT-Nfe2l2-WT and AT-nfe2l2-KO mice. In agreement with previous studies,33,34 11-13-mo-old nfe2l2-KO mice had minimal changes in expression levels of Nfe2l2-dependent genes compared to Nfe2l2-WT mice, Aox1 (aldehyde oxidase 1) being the most significant. Comparison of AT-Nfe2l2-WT vs. AT-nfe2l2-KO hippocampus also showed a small yet significant change in the expression of Aox1, Sqstm1, Atg2b, and Gabarapl1.
Figure 3.
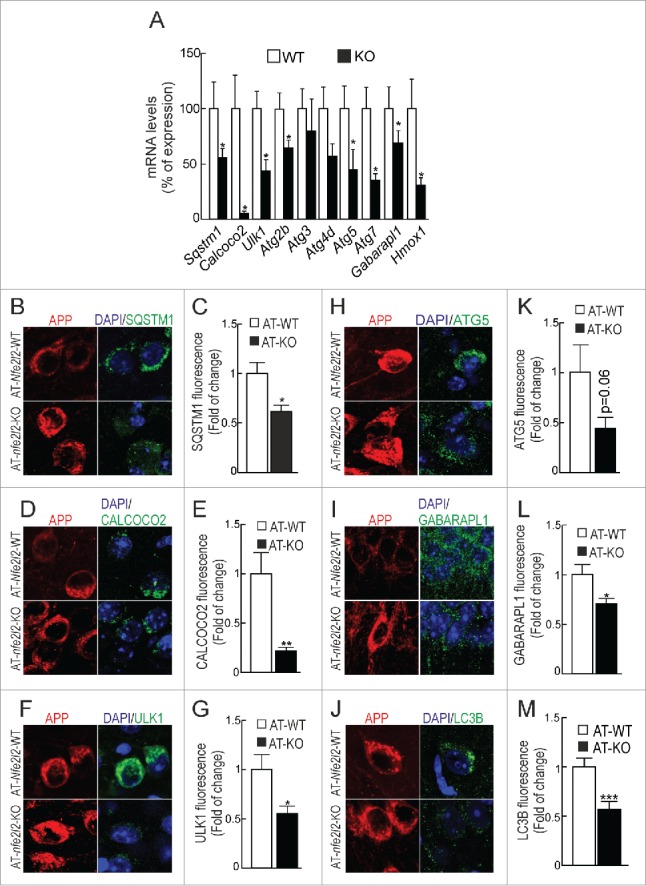
NFE2L2 deficiency impairs autophagy in neurons. (A) Expression levels of the indicated genes from Nfe2l2-WT and nfe2l2-KO primary hippocampal/cortical neurons were determined by qRT-PCR and normalized to Actb levels. Data are mean ± SEM (n = 3). Statistical analysis was performed with the Student t test. *, p <0.05 vs. Nfe2l2-WT neurons. (B-G) Confocal analysis of double immunofluorescence with anti-APP/Aβ (4G8, red) and anti-SQSTM1, CALCOCO2, ULK1, ATG5, GABARAPL1 or LC3B (green) antibodies in brain of AT-Nfe2l2-WT and AT-nfe2l2-KO. Fluorescence intensity was analyzed in the cytosol of 100 randomly chosen APP-positive neurons of each genotype. Quantification was derived from 3 independent experiments with 2 fields per experiment. Fluorescence intensity was quantified in 1.5-µm-thick stacks using ImageJ software. Data are mean ± SEM. Statistical analysis was performed with a Student t test. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 vs. AT-Nfe2l2-WT mice.
Due to the small changes found in whole brain tissue, we reasoned that the effect of NFE2L2 on autophagy might be more evident by looking at the neurons that express mutant HsAPP and HsMAPT proteins. As shown in Fig. 3B-F and Fig. S3, the protein levels of SQSTM1, CALCOCO2, ULK1, ATG5 and GABARAPL1 were reduced in neurons of the AT-nfe2l2-KO mice expressing HsAPP, compared to those of AT-Nfe2l2-WT mice. Similar results were obtained in HsMAPT-expressing neurons (Figs. S3 and S4). LC3B was analyzed as an estimate of overall autophagy. We found that LC3B protein levels were also reduced in neurons of AT-nfe2l2-KO vs. AT-Nfe2l2-WT mice expressing HsAPP (Figs. 3G and S3) or HsMAPT (Figs. S3 and S4). Together, these results point to the relevance of NFE2L2 in regulation of autophagy under proteotoxic stress.
The total levels of APP protein were similar in hippocampal lysates of 12-mo-old AT-Nfe2l2-WT and AT-nfe2l2-KO mice (data not shown). Measurement of soluble and insoluble fractions of Aβ40 and Aβ42 peptides by ELISA revealed similar levels of soluble peptides but reduced insoluble Aβ40 (Fig. 4A) and Aβ42 (Fig. 4B) in the AT-nfe2l2-KO mice. Immunohistochemical staining of hippocampal sections for APP/Aβ (4G8 antibody) revealed more intracellular vesicles in neurons of AT-nfe2l2-KO mice (Fig. 4D and 4E). This was most evident in the subiculum, which is the first brain region to develop amyloidopathy. Taken together, these results indicated that APP proteostasis was impaired in NFE2L2-deficient mice.
Figure 4.

NFE2L2 deficiency leads to intracellular APP/Aβ accumulation. (A-B) Determination of Aβ levels by ELISA in hippocampal brain homogenates of 12–14 mo-old AT-Nfe2l2-WT and AT-nfe2l2-KO mice. Aβ40 and Aβ42 levels were measured in the soluble (FS) and insoluble (FI) fractions (formic acid extracted). Soluble and insoluble Aβ40 (A) and Aβ42 (B) levels were normalized by total protein amount. (C) Ratio IF:SF of Aβ40 and Aβ42 peptides. Data are mean ± SEM (n = 10). Statistical analysis was performed with a Student t test. *, p < 0.05; and **, p < 0.01 comparing AT-Nfe2l2-WT and AT-nfe2l2-KO mice. (D), Immunohistological analysis of APP/Aβ expression (4G8 antibody) in hippocampus of 13-mo-old AT-Nfe2l2-WT and AT-nfe2l2-KO mice. (E) Estimation of the number of APP/Aβ-positive granules in CA1 and subiculum from 12–14-mo-old AT-Nfe2l2-WT and AT-nfe2l2-KO mice. Data are mean ± SEM (n = 8). Statistical analysis was performed with the Student t test. *, p<0.05; and **, p < 0.01 comparing AT-Nfe2l2-WT vs. AT-nfe2l2-KO groups.
Regarding tauopathy, we observed no major differences of total HsMAPT (HT7 antibody) or p-MAPT (AT8 antibody) in hippocampal sections of both Nfe2l2 genotypes (Fig. 5A). However, a modest increase in the levels of both murine endogenous MmMAPT and transgenic HsMAPT (TAU46 antibody) and p-HsMAPT (PHF-1 antibody) proteins was observed in the sarkosyl-insoluble fractions from AT-nfe2l2-KO hippocampal homogenates (Fig. 5B-E). These results reflected the fact that both transgenic and endogenous MAPT tended to accumulate in the insoluble fraction in the absence of NFE2L2.
Figure 5.

NFE2L2 deficiency leads to accumulation of aggregated HsMAPT. (A) Immunohistochemical analysis of HsMAPT expression in hippocampus of 13-mo-old AT-Nfe2l2-WT and AT-nfe2l2-KO mice stained with anti HT7 or AT8 antibodies that recognize total (HT7) or phosphorylated (AT8) HsMAPT. (B) Hippocampal tissue lysates from AT-Nfe2l2-WT and AT-nfe2l2-KO mice were separated into sarkosyl-soluble (SS) and sarkosyl-insoluble (SI) fractions. Total HsMAPT and MmMAPT levels were determined using anti-TAU46 antibody that recognizes MAPT from both species. (C) Densitometric quantification of representative blots from (B). (D) Hippocampal tissue lysates from AT-Nfe2l2-WT and AT-nfe2l2-KO mice were separated into sarkosyl-soluble (SS) and sarkosyl-insoluble (SI) fractions and analyzed with anti-PHF1 antibody that recognizes human p-HsMAPT. (E) Densitometric quantification of representative blots from (D). For (C) and (E), data are mean ± SEM (n = 6). Statistical analysis was performed with the Student t test. *, p < 0.05; comparing AT-Nfe2l2-WT and AT-nfe2l2-KO groups.
We went on to analyze the localization of HsAPP and HsMAPT proteins in connection to the autophagy marker SQSTM1 in AT-Nfe2l2-WT and AT-nfe2l2-KO mice by double immunofluorescence staining. We observed that in AT-Nfe2l2-WT mice a large number of APP vesicles were also decorated with the NFE2L2-regulated autophagy gene product SQSTM1. In contrast, AT-nfe2l2-KO mice showed less colocalization with SQSTM1, while APP-positive vesicles were larger, and appeared swollen, compared to AT-Nfe2l2-WT mice (Fig. 6A-B). HsMAPT showed a more diffuse pattern but once again, AT-nfe2l2-KO mice exhibited less colocalization between HsMAPT and SQSTM1 (Fig. 6C-D).
Figure 6.

NFE2L2 modulates neuronal autophagy. (A) Confocal analysis of double immunofluorescence with anti-APP/Aβ (4G8, red) and anti-SQSTM1 (green) antibodies in subiculum of AT-Nfe2l2-WT and AT-nfe2l2-KO. (B) Quantification of colocalization between APP/Aβ and SQSTM1 staining expressed as Mander's coefficients. (C) Confocal analysis of double immunofluorescence with anti-HsMAPT (HT7, red) and anti-SQSTM1 (green) in subiculum neurons of AT-Nfe2l2-WT and AT-nfe2l2-KO. (D) Quantification of colocalization between HsMAPT and SQSTM1 expressed as Mander's coefficients. For (B) and (D), Mander's coefficients were derived from 3 independent experiments with 2 fields per experiment. Fluorescence was quantified in 1.5-µm-thick stacks using the JACoP plugin of ImageJ software. Data are mean ± SEM. Statistical analysis was performed with a Student t test. *, p < 0.05; and ***, p < 0.001, comparing M1 and M2 coefficients obtained from AT-Nfe2l2-WT and AT-nfe2l2-KO images.
Evidence of altered NFE2L2 activity and autophagy in AD patients
We compared the levels of NFE2L2 and several autophagy proteins in lysates from brain hippocampus of asymptomatic and AD donors (Fig. 7A-B). The levels of NFE2L2 and one of its downstream targets, HMOX1, were increased in AD patients vs. controls. More importantly, the levels of the autophagy proteins SQSTM1, CALCOCO2 and ATG7 were also increased. We analyzed more specifically the NFE2L2-SQSTM1 pathway in neurons by immunohistochemistry. It was technically not possible to perform double labeling because of the different retrieval requirements for each of the antigens NFE2L2, SQSTM1 and APP, which we circumvented by analyzing 3 adjacent sections (each 4-μm thick) containing the same cells. We observed intracellular APP/Aβ in similar vesicles as observed above in AT-Nfe2l2-WT and AT-nfe2l2-KO mice (Fig. 7C). Interestingly, the neurons in human brain with highest abundance of vesicular APP were localized in the entorhinal cortex, and also expressed high levels of NFE2L2 and SQSTM1 (Fig. 7C). Similarly, some scattered neurons were found in the cortex that presented HsMAPT-positive neurofibrillary tangles, stained with anti-HsMAPT (HT7). Such neurons that intensely stained for HsMAPT were also expressing high levels of nuclear NFE2L2 and SQSTM1 (Fig. 7C). These results confirmed in human brain that NFE2L2 was upregulated in neurons suffering from proteotoxic APP/Aβ or HsMAPT insults.
Figure 7.
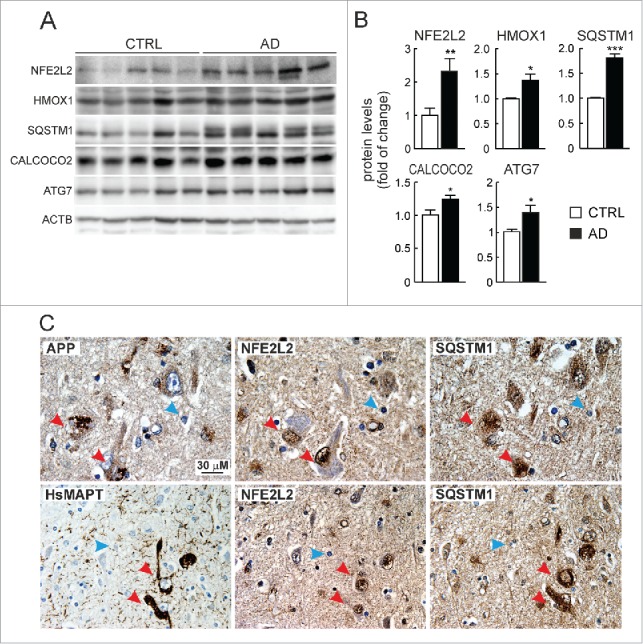
Evidence of NFE2L2 upregulation in neurons of AD subjects expressing high levels of HsAPP and HsMAPT. (A) Immunoblot analysis of NFE2L2, HMOX1, SQSTM1, CALCOCO2, and ATG7 in hippocampus of asymptomatic controls and patients with diagnosed AD who combined amyloidopathy and tauopathy. (B) Densitometric quantification of protein levels from representative immunoblots of (A) relative to ACTB. Data are represented as mean ± SEM (n = 5 controls and n = 5 AD subjects). Statistical analysis was performed with a Student t test. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 comparing control vs. AD groups. (C) Adjacent 4-μm-thick brain sections from AD subjects were immunostained with anti-APP/Aβ (4G8), anti-HsMAPT (HT7), anti-NFE2L2 and anti-SQSTM1 antibodies as indicated. Red arrows point to the same neuron expressing HsAPP or HsMAPT, NFE2L2 and SQSTM1; blue arrows depict internal negative controls for cells that do not express HsAPP or HsMAPT and present basal staining for NFE2L2 and SQSTM1.
Discussion
A functional connection between NFE2L2 transcriptional signature and autophagy can be predicted by the fact that both processes participate in oxidant defense by preventing thiol modification or clearing oxidized proteins, respectively.35 In search for a mechanistic connection between both processes, we report that NFE2L2 activates the expression of some genes involved in autophagy initiation (ULK1), cargo recognition (SQSTM1 and CALCOCO2), autophagosome formation (ATG4D, ATG7 and GABARAPL1), elongation (ATG2B and ATG5), and autolysosome clearance (ATG4D). Therefore, our study adds NFE2L2 as a transcription factor that regulates macroautophagy, next to the TFEB and FOXO family of transcription factors.11,12 The existence of several controls for autophagy gene expression probably assures proteostasis under different circumstances. Thus, TFEB and FOXO3/FOXO3A participate in multiple scenarios including rapamycin-sensitive cell signaling and nutritional stress.36,37 Nonetheless, NFE2L2 may be more relevant in tissues that support highly oxidative metabolism. By this mechanism, oxidative stress-induced NFE2L2 may function under nutrient-rich conditions to upregulate transcription of genes encoding proteins required for autophagy, similar to what has been found for the transcription factors TFEB and FOXO3 under starvation conditions. As the brain is not allowed to starve and functions largely under nutrient-rich conditions when glucose is available, this may be a relevant mechanism to activate autophagy in neurons.
Very few studies have performed direct ChIP sequencing for NFE2L2, because of the low or even misleading specificity of available antibodies.38 Therefore, we undertook a different approach based on the analysis of other proteins that bind ARE, MAFK and BACH1, for which more reliable ChIP sequencing data are available in the ENCODE database. MAFK is a leucine-zipper transcription factor that makes heterodimers with NFE2L2.39 BACH1 is a repressor of several AREs, also containing a leucine-zipper domain and forming heterodimers with MAF proteins.40 Our bioinformatics analysis retrieved 25 new putative AREs located in 16 autophagy genes, many of which were conserved in mice. We validated 11 AREs in 8 autophagy genes as targets of NFE2L2 by additional ChIP analysis in HEK293T cells and by qRT-PCR in human and mouse cells treated with the NFE2L2 activator SFN, which disrupts the normal KEAP1-NFE2L2 interaction by making an adduct with KEAP1 at cysteine residues.41,42 In other studies the effect of SFN was attributed to modulation of signaling events of the MTOR pathway through RPS6KB1/pS6K1,29 activation of AMP kinase,30 or the MAPK/ERK pathways31 rather than to targeting NFE2L2 itself. However, we show that the induction of autophagy genes by SFN is impaired in nfe2l2-KO MEFs, therefore indicating that NFE2L2 is at least partially involved. Moreover, the expression of autophagy genes in these NFE2L2-deficient cells was increased by the re-introduction of NFE2L2 expression with the help of a lentiviral vector.
Reduced clearance of aggregation-prone proteins is a hallmark of many diseases, including AD, and in many a deficit in autophagy was reported.43 In agreement, impaired autophagy of the AT-nfe2l2-KO mice correlated with altered APP processing, stalled in multiple swollen vacuoles with reduced levels of SQSTM1. Moreover, it was surprising to find lower total levels of Aβ in AT-nfe2l2-KO compared to AT-Nfe2l2-WT. Similar results have been reported for ATG7-deficient mice, which show impaired secretion, reduced total Aβ levels and fewer amyloid plaques.44 Several studies have found that autophagosomes are a place for generation of Aβ45-47 and BACE1/β secretase and gamma secretase appear to be localized at least in part in autophagosomes.48-50 Thus, a reduction of autophagosomes might lead to lower Aβ load. In support of this hypothesis, we found reduced levels of autophagy markers (SQSTM1, CALCOCO2, ULK1, AGT5, GABARAPL1 and LC3B) in the AT-nfe2l2-KO mice (Fig. 3).
Our results are also consistent with a recent study in which the amyloidogenic mouse model APP-PSEN1 lacking NFE2L2 exhibits enhanced accumulation of APP in multivesicular bodies, endosomes, and lysosomes and shows an altered production of amyloid proteins due to impaired autophagic flux.33 In these mice, NFE2L2-deficiency leads to increased amyloid plaque accumulation, whereas our AT-nfe2l2-KO mice exhibited a tendency to develop fewer amyloid plaques compared to AT-Nfe2l2-WT counterparts despite presenting with a worse cognitive performance in the Morris water maze and reduced long-term potentiation (A.I.R and A.C., to be reported elsewhere). These findings may be reconciled considering that the APP-PSEN1 mice exhibit a very strong amyloidogenic processing of APP (PSEN1-driven APP processing), while our model exhibits mild amyloidogenic processing (APP-only), suggesting a reduced availability of extracellular Aβ peptides for plaque formation. In this regard, our results are consistent with those reported in the “APP-only” transgenic mice with ATG7-deficiency.51 In this case, impaired autophagy influences unconventional secretion of Aβ to the extracellular space and thereby affects Aβ plaque formation. All in all, available data comply with the hypothesis that autophagic elimination of APP is crucial to prevent amyloidopathy and suggest a role of NFE2L2 in APP clearance.
Although HsMAPT can be degraded through several proteolytic pathways, it is increasingly recognized that impaired autophagy also plays a central role in tauopathy.52 Thus, MTOR signaling regulates HsMAPT phosphorylation and autophagic degradation,53 whereas inhibition of MTOR with rapamycin has the opposite effect.54,55 Our results are fully consistent with at least partial NFE2L2-dependent autophagic degradation of HsMAPT, given its colocalization with SQSTM1 in autophagic vacuoles. Our observations also fit with a previous report indicating that HsMAPT is a cargo of CALCOCO2 to be delivered to the lysosome in autophagosomes and that deficiency in this protein aggravates tauopathy in mice.18
It is not fully clear yet if the NFE2L2 signature is altered in the degenerative or even in the aging brain. In agreement with a previous report,33 we found minimal changes of ARE-gene expression between 11-13-mo-old Nfe2l2-WT and nfe2l2-KO mice. Also, as reported for the APP-PSEN1 mice,33 our results show modest or no changes in autophagy genes in the hippocampus and brainstem of AT-nfe2l2-KO vs. AT-Nfe2l2-WT mice when the whole brain parenchyma is analyzed. Therefore, the strong evidence for a role of NFE2L2 in autophagy genes gathered in cell culture was not reflected in vivo when we analyzed the whole brain parenchyma. However, in double-immunofluorescence assays, we did find differences in the levels of autophagy proteins when we compared HsAPP-HsMAPT-expressing neurons from AT-Nfe2l2-WT vs the AT-nfe2l2-KO brains. Therefore, the effect of NFE2L2 was detected specifically in the neurons that are under proteotoxic attack, and probably for this reason could find a role that was not be detected in the whole brain since most cells are not expressing HsAPP or HsMAPT.
Regarding AD patients, while there is overwhelming evidence of impaired autophagy,56-59 the role of NFE2L2 is controversial. A highly cited study showed that NFE2L2 is predominantly localized in the cytoplasm of AD hippocampal neurons, suggesting reduced NFE2L2 transcriptional activity in the brain.60 However, other studies have reported increased ARE-regulated proteins in AD brains, such as HMOX1, NQO1, or SQSTM1.61-64 Our results, both with protein lysates of hippocampus and with immunohistochemistry are in line with these latter reports, because we found increased nuclear NFE2L2 expression in proteinopathic neurons. One possible explanation for this discrepancy is that NFE2L2 levels might change during disease progression and in different brain regions. It is interesting to add that the rodent Octodon degus naturally develops with aging proteinopathic hallmarks of AD that correlate with increased Nfe2l2 mRNA levels,65 further suggesting upregulation of NFE2L2 to combat proteinopathy.
Our work supports the observation that phosphorylated SQSTM1 is increased in AD samples.64 This modified form of SQSTM1 has high binding affinity to KEAP1 and drives this NFE2L2 repressor to autophagosomes.16 It is then plausible to think that SQSTM1-mediated removal of KEAP1 would result in NFE2L2 accumulation, which in turn would also induce expression of SQSTM1 as well as other autophagy genes providing a feed-forward loop.
A corollary of our study is that NFE2L2 might be a therapeutic target for proteinopathies such as AD. On the one hand, we found that NFE2L2-deficiency impairs autophagy in AT-nfe2l2-KO mice (Figs. 3, S3 and S4). We interpret these data as a need of NFE2L2 for optimal autophagy response to proteinopathy. On the other hand, in AD patients, NFE2L2 and at least HMOX1, SQSTM1, CALCOCO2 and ATG7 are upregulated in the hippocampus as determined by immunoblot (Fig. 7A-B), and NFE2L2 and SQSTM1 proteins are increased in HsAPP- and HsMAPT-injured neurons as determined by immunohistochemistry (Fig. 7C). Thus, it follows that the diseased brain is upregulating NFE2L2 in an attempt to maintain proteostasis. In fact, the still living proteinopathic neurons that we can see by immunohistochemsitry in the postmortem samples exhibit increased levels of NFE2L2 and SQSTM1, suggesting that they may better suited to sustain proteotoxicity. This is just a correlation since we cannot compare with the neurons that have already disappeared, but altogether, our rationale suggests that NFE2L2 activates an autophagy defensive program to maintain proteostasis.
However, in order to consider NFE2L2 as a therapeutic target to combat proteinopathies, the key question is whether it will be useful to increase NFE2L2 levels beyond the physiological response. In other words, is the physiological NFE2L2 response to proteinopathy at saturating levels and would supraphysiological upregulation of NFE2L2 have beneficial or deleterious effects? A partial answer to these questions may be provided by the analysis of epidemiological data, which indicate that the NFE2L2 gene is highly polymorphic and some single nucleotide polymorphisms found in its promoter regulatory region may provide a range of “physiological” variability in gene expression at the population level. For instance, Von Otter et allink 66 found that one haplotype allele of the NFE2L2 promoter was associated with 2 y earlier age at AD onset. These results are also consistent in other neurodegenerative proteinopathies. Thus, Von Otter et al performed a meta-analysis of NFE2L2 haplotypes and found association of the haplotype of the fully functional promoter with decreased risk and delayed onset of Parkinson disease.67 Observations along the same lines have been made in amyotrophic lateral sclerosis.68 Given the population variability in NFE2L2 expression it is conceivable that a small pharmacological upregulation of NFE2L2, comparable to the maximal physiological response found at the population level, will indeed reinforce proteostasis. In this regard, several preclinical studies have so far attempted to upregulate NFE2L2 in an effort to mitigate neurodegenerative disease-associated phenotypes. For instance, the life expectancy of transgenic mice expressing human mutant SNCAA53T is largely increased when crossed with mice overexpressing NFE2L2 in astrocytes69 and the NFE2L2 activator dimethyl fumarate, already in clinical use for multiple sclerosis, reduces SNCA toxicity.70 Other NFE2L2 activators are being studied for alleviation of proteinopathies such as spinal and bulbar muscular atrophy.71 Future work will be needed to determine if pharmacological induction of NFE2L2 may be a valid strategy to facilitate degradation of toxic proteins in the brain.
Material and methods
Bioinformatics analysis
Putative AREs in 26 autophagy-related gene promoters were identified in The Encyclopedia of DNA Elements at UCSC (ENCODE)25 for the human genome (Feb. 2009) taking as reference the available information from chromatin immunoprecipitation (ChIP) of ARE binding factors MAFK and BACH1. The putative MAFK and BACH1 binding regions were localized in 200- to 400-base pair-long DNase-sensitive and H3K27Ac-rich regions, i.e., most likely regulatory promoter regions. In addition, a frequency matrix of the consensus ARE sequence based on the JASPAR database26 was converted to a position-specific scoring matrix (PSSM) (Table S1) by turning the frequencies into scores through the log2(odd-ratio) (odd ratio: observed frequency/expected frequency). One unit was added to each frequency to avoid log(0). Then a script was generated with the Python 3.4 program (Table S2) to scan the promoter sequences with candidate AREs retrieved from ENCODE with the PSSM. The max score was calculated by adding the independent scores for each of the 11 base pairs of the consensus ARE sequence with the PSSM. The relative score (scorerelative) was calculated from this max score (score of the sequencemax) as: scorerelative = (score of the sequencemax – scoremin possible)/(scoremax possible – scoremin possible). The min possible score (scoremin possible) is calculated as the lowest possible number obtained for a sequence from the PSSM and the max possible score (scoremax possible) is the highest possible score that can be obtained. We considered putative ARE sequences those with a scorerelative over 80%, which is a commonly used threshold for the computational framework for transcription factor binding site/TFBS analyses using PSSM. The conservation of the NFE2L2 putative binding sequences in mice was analyzed by comparing the mouse genome through 2 different methods from the UCSC Genome Browser: BLAT search genome useful for high conservation and Comparative Genomics searching conservation in the mouse genome using Multiz Align (multiz 100way) (Tables S1, S2 and S3).
Cell culture and reagents
Mouse embryonic fibroblasts (MEFs), mouse hippocampus-derived HT22 cells and human embryonic kidney HEK293T cells were grown in Dulbecco's modified Eagle's medium (Sigma-Aldrich, D5648) supplemented with 10% fetal bovine serum (HyClone, CH30160.03) and 80 μg/ml gentamicin (Laboratorios Normon, 763011.1). Hippocampal/cortical neurons were obtained from E17-E18 mouse embryos. Briefly, embryonic hippocampi were incubated in 0.25% trypsin (Gibco Life Technologies, 12604–013) plus 1 mg/ml DNAse-I (Roche, 10104159001) at 37°C for 20 min. After mechanical dissociation and centrifugation (900 × g, 5 min) cells were plated in minimum essential medium (Gibco Life Technologies, 17504–044) supplemented with 20% glucose (Sigma-Aldrich, 16301), 5% horse serum (Gibco Life Technologies, 26050–088), 5% fetal bovine serum and 80 μg/ml gentamicin, and incubated for 24 h. After plating, the medium was changed to phenol red-free neurobasal medium (Gibco Life Technologies, 10888–022) supplemented with B27 (Gibco Life Technologies, 17504–044) and the neurons were maintained under these conditions for 10 days in vitro. R,S-sulforaphane (SFN; LKT Laboratories, Inc., S8044).
Production of lentiviral stocks and infection of nfe2l2-KO MEFs
Recombinant lentiviral stocks were produced in HEK 293T/17 cells by cotransfecting 10 μg of transfer vector (GFP or NFE2L2ΔEGTE-V5), 6 μg of envelope plasmid pMD2.G (Addgene, 12259; deposited by Dr. Didier Trono) and 6 μg of packaging plasmid pSPAX2 (Addgene, 12260; deposited by Dr. Didier Trono), using Lipofectamine 2000 Reagent (Invitrogen Life Technologies, 116668–019). After 12 h at 37°C the medium was replaced with fresh Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and virus particles were harvested at 24 h and 48 h post-transfection. The nfe2l2-KO MEFs were incubated with the lentivirus for 24 h, and mRNA was extracted 3 and 10 d after lentiviral transduction.
Chromatin immunoprecipitation (ChIP) assay
HEK293T cells were grown on 10-cm plates until they reached 85% confluence and transfected with an NFE2L2 expression plasmid that lacks the high-affinity binding site for KEAP1 and contains a V5 tag (NFE2L2ΔETGE-V5).27 Briefly, cells were crosslinked with 1% formaldehyde (Fluka, 47630) and the reaction was stopped with 125 mM glycine (Bio-Rad, 161–0718). Cells were then washed twice with cold phosphate-buffer (10 mM PO43-, pH 7,4, 137 mM NaCl, 2.7 mM KCl), lysed and sonicated in order to obtain adequate fragment sizes of DNA. Supernatant was diluted 10-fold with ChIP dilution buffer (0.01% sodium dodecyl sulfate [Sigma-Aldrich, 71725], 1.1% Triton X-100 [Sigma-Aldrich, T8787], 1.2 mM ethylenediaminetetraacetic acid, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [Sigma-Aldrich, P7626], 1 µg/ml leupeptin [Sigma-Aldrich, L8511]) and pre-cleared with protein G Sepharose (GE Healthcare, 17-0618-01). ChIP was carried out with anti-V5 antibody (Life Technologies, 37–7500) or anti-IgG (Abcam, ab18413). DNA was eluted and purified, analyzing the presence of previously identified putative AREs by quantitative real-time polymerase chain reaction (qRT-PCR) with specific primers (Table S4). Samples from at least 3 independent immunoprecipitations were analyzed.
Transgenic mice
Animals were housed at room temperature under a 12-h light-dark cycle. Food and water were provided ad libitum and mice were cared for according to a protocol approved by the Ethical Committee for Research of the Universidad Autónoma de Madrid following institutional, Spanish and European guidelines (Boletín Oficial del Estado of 18 March 1988; and 86/609/EEC, 2003/65/EC European Council Directives). HsAPPV717I mice expressing in heterozygosis the HsAPP695 isoform with the V717I mutation under the control of the Thy1 promoter, were backcrossed with C57/BL6j-Nfe2l2+/+ or C57/BL6j-nfe2l2−/− mice for over 5 generations. Similarly, HsMAPTP301L mice, expressing in homozygosis the longest isoform of HsMAPT with the P301L mutation (Tau.4R/2N-P301L) under control of the mouse Thy1 gene promoter, were crossed with C57/BL6j-Nfe2l2+/+ or C57/BL6j-nfe2l2−/− for over 5 generations. HsAPP-HsMAPT-Nfe2l2+/+ (AT-Nfe2l2-WT) and HsAPP-HsMAPT-nfe2l2−/− (AT-nfe2l2-KO) mice were obtained by crossing the proper founder mice from above. Genotypic characterization of the AT-Nfe2l2-WT transgenic mice was described previously.32,72,73 Animals were anesthetized with 8 mg/kg ketamine and 1.2 mg/kg xylazine and perfused with phosphate-buffered saline. The brains were sectioned along the sagittal axis and the right hemispheres were post-fixed in 4% paraformaldehyde during 16 h and cryoprotected by soaking in 30% sucrose solution in phosphate buffer until they sank. The left hemispheres were rapidly dissected and frozen for biochemical analysis.
Human patients
All procedures were approved by the ethics committee of the Tissue Bank of Fundación CIEN (Madrid, Spain). Frozen post-mortem brain tissues were obtained from 5 control subjects (age range 59–78 y) and 5 patients with Alzheimer disease (age range 73–94 y) within a 5-h post-mortem interval, according to the standardized procedures of the Tissue Bank of Fundación CIEN. Information on patient samples, including APOE genotype, is provided in Table S6. The brain samples used in this study belong to patients with a clinical history that indicates lack of familial cases of AD. The control subjects had no background of neuropsychiatric disease and a full neuropathological examination excluded relevant brain pathology. Alzheimer disease diagnosis was confirmed by HT100 staining of frozen tissue sections from the same cases used in the immunofluorescence studies.
Immunohistochemistry of human tissue
Four-µm-thick paraffin-fixed consecutive brain sections were immunostained as described previously.74 The primary antibodies and optimal dilutions are summarized in Table S7. For NFE2L2 immunostaining, brain sections were stained with Histostain-bulk-SP IHC kit broad spectrum (Thermo Fisher Scientific, 959943B) using primary anti-NFE2L2 antibody (Thermo Fisher Scientific, PA13831; 1:200). A negative control omitting primary anti-NFE2L2 antibody yielded no signal (data not shown).
HsMAPT tissue extraction
Mouse brain tissues were homogenized in 0.2 ml of ice-cold buffer (0.1 M 3-[N-morpholino]propanesulfonic acid, pH 7.0, 1 mM ethylenediaminetetraacetic acid, 0.5 mM MgSO4, 1 M sucrose [Panreac AppliChem, A2211]) containing 1 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride and 10 μg/ml each of aprotinin (Sigma-Aldrich, A1153) and 1 μg/ml leupeptin (Sigma-Aldrich, L8511). The homogenates were cleared by centrifugation at 50,000 × g for 20 min at 4°C, and the supernatants were collected as soluble fractions. To prepare the sarkosyl-insoluble fraction, the pellets were resuspended in lysis buffer (0.1 M 3-[N-morpholino]propanesulfonic acid, pH 7.0, 10% sucrose, 2 mM ethylene glycol tetraacetic acid [Sigma-Aldrich, E3889], 0.5 mM MgSO4, 500 mM NaCl, 1 mM MgCl2, 10 mM NaH2PO4, 20 mM NaF) containing 1% N-lauroylsarcosine (sarkosyl; Sigma-Aldrich, L9150) with protease inhibitors (Sigma-Aldrich, L8511, A1153 and P7626), vortexed for 1 min at room temperature, incubated at 4°C for 16 h, and then centrifuged at 200,000 × g for 30 min at room temperature. The supernatant fractions were collected as sarkosyl-soluble fractions, and the pellets, sarkosyl-insoluble fractions, were resuspended in sodium dodecyl sulfate protein loading buffer and incubated at 95°C for 5 min.
Immunoblotting
Immunoblots were performed as described previously.75 The primary antibodies used are presented in Table S7. Membranes were analyzed using the appropriate peroxidase-conjugated secondary antibodies (anti-mouse and anti-rabbit from GE Healthcare UK Limited, NA931V and NA934V, and anti-goat from Santa Cruz Biotech, sc-2020). Proteins were detected by enhanced chemiluminescence (GE Healthcare, RPN2232).
Immunohistochemistry and immunofluorescence of mouse tissues
Sagittal series of 30-µm-thick sections were obtained in a freezing microtome and stained as indicated previously.76 The primary antibodies used are described in Table S7. Antigen retrieval for APP and Aβ detection was performed by incubating the sections for 5 min in formic acid.
Analysis of mRNA levels
Total RNA extraction, reverse transcription and quantitative PCR were done as detailed elsewhere.76 Primer sequences are shown in Tables S8 and S9. To ensure that equal amounts of cDNA were added to the PCR, the housekeeping gene ACTB was amplified. Data analysis was based on the ΔΔCT method with normalization of the raw data to housekeeping genes. All PCRs were performed in triplicate.
Aβ determination by ELISA
Aβ measurements were performed using 2 ELISA kits, one for each fragment (Aβ1–40 and Aβ1–42) (Novex, Thermo Fisher Scientific, KHB3481 and KHB3441, respectively) following the manufacturer's instructions. The samples were sonicated (5 sec) in 10 vol of protein lysis buffer (20 mM [4-{2-hydroxyethyl}-1-piperazineethanesulfonic acid] pH 7.9, 100 mM NaCl, 1 mM ethylene glycol tetraacetic acid, 1 mM ethylenediaminetetraacetic acid, 1% Triton X-100, 5 mM dithiothreitol, 2.5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, and complete protease inhibitor cocktail). The lysate was centrifuged (18,000 × g, for 10 min at 4°C). The supernatant fraction was considered soluble and the pellet fractions were further extracted in formic acid by sonication, and centrifuged. Results were expressed as pg/mg of protein measured by the BCA method (Thermo Fisher Scientific, 23225 )] using BSA as standard.
Image analysis and statistics
Different band intensities corresponding to immunoblot detection of protein samples were quantified using MCID software (MCID, Cambridge, UK). Fluorescence intensity and M1 and M2 colocalization coefficients were measured by Mander's analysis using the ImageJ JACoP plug-in. Student t test was used to assess differences between groups. P<0.05 was considered significant. Unless otherwise indicated, all experiments were performed at least 3 times and all data presented in the graphs are the mean of at least 3 independent samples. Results are expressed as mean ± SEM.
Supplementary Material
Abbreviations
- ACTB
actin β
- AD
Alzheimer disease
- Aox1
aldehyde oxidase 1
- APP
amyloid β precursor protein
- ARE
antioxidant response element
- ATG5
autophagy-related 5
- AT-nfe2l2-KO
transgenic mice expressing HsAPPV717I and HsMAPTP301L in an nfe2l2-knokcout genetic background
- AT-Nfe2l2-WT
transgenic mice expressing HsAPPV717I and HsMAPTP301L in a wild-type genetic background
- Aβ
amyloid-β peptide
- BACH1
BTB domain and CNC homolog 1
- CALCOCO2/NDP52
calcium binding and coiled-coil domain 2
- ChIP
chromatin immunoprecipitation
- ENCODE
encyclopedia of DNA elements at the University of California, Santa Cruz
- FI
formic acid extracted insoluble fraction
- FOXO
forkhead box O
- FS
formic acid-extracted soluble fraction
- GABARAPL1
GABA type A receptor associated protein like 1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- HsMAPT/TAU
human microtubule associated protein tau
- HMOX1
heme oxygenase 1
- JASPAR
transcription factor binding profile database
- KEAP1
kelch like ECH associated protein 1
- MAFK
MAF bZIP transcription factor K
- MAP1LC3/LC3B
microtubule associated protein 1 light chain 3
- MEFs
mouse embryo fibroblasts
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NQO1
NAD(P)H quinone dehydrogenase 1
- NFE2L2
nuclear factor, erythroid 2 like 2
- nfe2l2-KO
nfe2l2-knockout mice
- Nfe2l2-WT
wild-type mice
- PSSM
position-specific scoring matrix
- qRT-PCR
quantitative real-time polymerase chain reaction
- SFN
sulforaphane
- SQSTM1/p62
sequestosome 1
- TFEB
transcription factor EB
- ULK1
unc-51 like autophagy activating kinase 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Patricia Rada for her help with animals, Dr. Mar Perez for providing the anti-PHF-1 antibody and Prof. Luis Del Peso for his advice with Python and ChIP validation.
Funding
This work was funded by SAF2013-43271-R of the Spanish Ministry of Economy and Competitiveness.
References
- [1].Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell 2011; 146:682-95; PMID:21884931; http://dx.doi.org/ 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- [2].Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 2015; 47:e147; PMID:25766616; http://dx.doi.org/ 10.1038/emm.2014.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-21; PMID:11099404; http://dx.doi.org/ 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/ 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, et al.. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014; 516:108-11; PMID:25383523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013; 494:323-7; PMID:23389440; http://dx.doi.org/ 10.1038/nature11895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al.. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015; 524:361-5; PMID:26168401; http://dx.doi.org/ 10.1038/nature14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chauhan S, Goodwin JG, Chauhan S, Manyam G, Wang J, Kamat AM, Boyd DD. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol Cell 2013; 50:16-28; PMID:23434374; http://dx.doi.org/ 10.1016/j.molcel.2013.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bowman CJ, Ayer DE, Dynlacht BD. Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat Cell Biol 2014; 16:1202-14; PMID:25402684; http://dx.doi.org/ 10.1038/ncb3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015; 11:867-80; PMID:25836756; http://dx.doi.org/ 10.1080/15548627.2015.1034410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, et al.. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015; 6:6670; PMID:25858807; http://dx.doi.org/ 10.1038/ncomms7670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 2014; 39:199-218; PMID:24647116; http://dx.doi.org/ 10.1016/j.tibs.2014.02.002 [DOI] [PubMed] [Google Scholar]
- [14].Suzuki T, Yamamoto M. FRBM Special issue “Nrf2 regulated Redox Signaling and Metabolism in Physiology and Medicine” Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med 2015 [DOI] [PubMed] [Google Scholar]
- [15].Jiang T, Harder B, Vega MR, Wong PK, Chapman E, Zhang DD. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med 2015; 88(Pt B):199-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al.. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013; 51:618-31; PMID:24011591; http://dx.doi.org/ 10.1016/j.molcel.2013.08.003 [DOI] [PubMed] [Google Scholar]
- [17].Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 2010; 285:22576-91; PMID:20452972; http://dx.doi.org/ 10.1074/jbc.M110.118976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun 2014; 5:3496; PMID:24667209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/ 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- [20].Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, Komatsu M, Abeliovich A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener 2012; 7:48; PMID:22998728; http://dx.doi.org/ 10.1186/1750-1326-7-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/ 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- [22].Jaworski T, Dewachter I, Seymour CM, Borghgraef P, Devijver H, Kugler S, Van Leuven F. Alzheimer's disease: old problem, new views from transgenic and viral models. Biochimica et biophysica acta 2010; 1802:808-18; PMID:20332023; http://dx.doi.org/ 10.1016/j.bbadis.2010.03.005 [DOI] [PubMed] [Google Scholar]
- [23].Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 2007; 120:4081-91; PMID:18032783; http://dx.doi.org/ 10.1242/jcs.019265 [DOI] [PubMed] [Google Scholar]
- [24].Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 2008; 28:6926-37; PMID:18596167; http://dx.doi.org/ 10.1523/JNEUROSCI.0800-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. http://www.webcitation.org/query?url=https%3A%2F%2Fgenome.ucsc.edu%2F&date=2015-07-29 [Google Scholar]
- [26]. http://www.webcitation.org/query?url=http%3A%2F%2Fjaspar.genereg.net%2F&date=2015-07-29 [Google Scholar]
- [27].McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem 2003; 278:21592-600; PMID:12682069; http://dx.doi.org/ 10.1074/jbc.M300931200 [DOI] [PubMed] [Google Scholar]
- [28].Potteti HR, Reddy NM, Hei TK, Kalvakolanu DV, Reddy SP. The NRF2 activation and antioxidative response are not impaired overall during hyperoxia-induced lung epithelial cell death. Oxid Med Cell Longev 2013; 2013:798401; PMID:23738042; http://dx.doi.org/ 10.1155/2013/798401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hac A, Domachowska A, Narajczyk M, Cyske K, Pawlik A, Herman-Antosiewicz A. S6K1 controls autophagosome maturation in autophagy induced by sulforaphane or serum deprivation. Euro J Cell Biol 2015; 94(10):470-81; PMID:26054233 [DOI] [PubMed] [Google Scholar]
- [30].Lee JH, Jeong JK, Park SY. Sulforaphane-induced autophagy flux prevents prion protein-mediated neurotoxicity through AMPK pathway. Neuroscience 2014; 278:31-9; PMID:25130556; http://dx.doi.org/ 10.1016/j.neuroscience.2014.07.072 [DOI] [PubMed] [Google Scholar]
- [31].Jo C, Kim S, Cho SJ, Choi KJ, Yun SM, Koh YH, Johnson GV, Park SI. Sulforaphane induces autophagy through ERK activation in neuronal cells. FEBS Lett 2014; 588:3081-8; PMID:24952354; http://dx.doi.org/ 10.1016/j.febslet.2014.06.036 [DOI] [PubMed] [Google Scholar]
- [32].Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, Van Leuven F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol 2008; 172:786-98; PMID:18258852; http://dx.doi.org/ 10.2353/ajpath.2008.070904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer's disease-like pathology in the APP/ PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging 2015; 36:664-79; PMID:25316599; http://dx.doi.org/ 10.1016/j.neurobiolaging.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kraft AD, Lee JM, Johnson DA, Kan YW, Johnson JA. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J Neurochem 2006; 98:1852-65; PMID:16945104; http://dx.doi.org/ 10.1111/j.1471-4159.2006.04019.x [DOI] [PubMed] [Google Scholar]
- [35].Dodson M, Redmann M, Rajasekaran NS, Darley-Usmar V, Zhang J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem J 2015; 469:347-55; PMID:26205490; http://dx.doi.org/ 10.1042/BJ20150568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy 2011; 7:1379-81; PMID:21785263; http://dx.doi.org/ 10.4161/auto.7.11.17166 [DOI] [PubMed] [Google Scholar]
- [37].Fullgrabe J, Klionsky DJ, Joseph B. Histone post-translational modifications regulate autophagy flux and outcome. Autophagy 2013; 9:1621-3; PMID:23934085; http://dx.doi.org/ 10.4161/auto.25803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lau A, Tian W, Whitman SA, Zhang DD. The predicted molecular weight of Nrf2: it is what it is not. Antioxid Redox Signal 2013; 18:91-3; PMID:22703241; http://dx.doi.org/ 10.1089/ars.2012.4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol 1995; 15:4184-93; PMID:7623813; http://dx.doi.org/ 10.1128/MCB.15.8.4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Igarashi K, Hoshino H, Muto A, Suwabe N, Nishikawa S, Nakauchi H, Yamamoto M. Multivalent DNA binding complex generated by small Maf and Bach1 as a possible biochemical basis for beta-globin locus control region complex. J Biol Chem 1998; 273:11783-90; PMID:9565602; http://dx.doi.org/ 10.1074/jbc.273.19.11783 [DOI] [PubMed] [Google Scholar]
- [41].Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev 2006; 38:769-89; PMID:17145701; http://dx.doi.org/ 10.1080/03602530600971974 [DOI] [PubMed] [Google Scholar]
- [42].Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 2004; 24:10941-53; PMID:15572695; http://dx.doi.org/ 10.1128/MCB.24.24.10941-10953.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Dis 2012; 11:709-30; PMID:22935804; http://dx.doi.org/ 10.1038/nrd3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC. Abeta secretion and plaque formation depend on autophagy. Cell Rep 2013; 5:61-9; PMID:24095740; http://dx.doi.org/ 10.1016/j.celrep.2013.08.042 [DOI] [PubMed] [Google Scholar]
- [45].Agholme L, Hallbeck M, Benedikz E, Marcusson J, Kagedal K. Amyloid-beta secretion, generation, and lysosomal sequestration in response to proteasome inhibition: involvement of autophagy. J Alzheimer's Dis 2012; 31:343-58; PMID:22555375 [DOI] [PubMed] [Google Scholar]
- [46].Son SM, Song H, Byun J, Park KS, Jang HC, Park YJ, Mook-Jung I. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy 2012; 8:1842-4; PMID:22931791; http://dx.doi.org/ 10.4161/auto.21861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zheng L, Terman A, Hallbeck M, Dehvari N, Cowburn RF, Benedikz E, Kågedal K, Cedazo-Minguez A, Marcusson J. Macroautophagy-generated increase of lysosomal amyloid beta-protein mediates oxidant-induced apoptosis of cultured neuroblastoma cells. Autophagy 2011; 7:1528-45; PMID:22108004; http://dx.doi.org/ 10.4161/auto.7.12.18051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, et al.. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol 2005; 171:87-98; PMID:16203860; http://dx.doi.org/ 10.1083/jcb.200505082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, Cuervo AM, Nixon RA. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol 2004; 36:2531-40; PMID:15325590; http://dx.doi.org/ 10.1016/j.biocel.2004.05.010 [DOI] [PubMed] [Google Scholar]
- [50].Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW, Mahuran DJ. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem 2003; 278:26687-94; PMID:12736250; http://dx.doi.org/ 10.1074/jbc.M304009200 [DOI] [PubMed] [Google Scholar]
- [51].Nilsson P, Sekiguchi M, Akagi T, Izumi S, Komori T, Hui K, Sörgjerd K, Tanaka M, Saito T, Iwata N, et al.. Autophagy-related protein 7 deficiency in amyloid beta (Abeta) precursor protein transgenic mice decreases Abeta in the multivesicular bodies and induces Abeta accumulation in the Golgi. Am J Pathol 2015; 185:305-13; PMID:25433221; http://dx.doi.org/ 10.1016/j.ajpath.2014.10.011 [DOI] [PubMed] [Google Scholar]
- [52].Wang Y, Mandelkow E. Degradation of tau protein by autophagy and proteasomal pathways. Biochemical Society transactions 2012; 40:644-52; PMID:22817709; http://dx.doi.org/ 10.1042/BST20120071 [DOI] [PubMed] [Google Scholar]
- [53].Caccamo A, Magri A, Medina DX, Wisely EV, Lopez-Aranda MF, Silva AJ, Oddo S. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer's disease and other tauopathies. Aging cell 2013; 12:370-80; PMID:23425014; http://dx.doi.org/ 10.1111/acel.12057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liu Y, Su Y, Wang J, Sun S, Wang T, Qiao X, Run X, Li H, Liang Z. Rapamycin decreases tau phosphorylation at Ser214 through regulation of cAMP-dependent kinase. Neurochem Int 2013; 62:458-67; PMID:23357480; http://dx.doi.org/ 10.1016/j.neuint.2013.01.014 [DOI] [PubMed] [Google Scholar]
- [55].Ozcelik S, Fraser G, Castets P, Schaeffer V, Skachokova Z, Breu K, Clavaguera F, Sinnreich M, Kappos L, Goedert M, et al.. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PloS one 2013; 8:e62459; PMID:23667480; http://dx.doi.org/ 10.1371/journal.pone.0062459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005; 1:11-22; PMID:16874045; http://dx.doi.org/ 10.4161/auto.1.1.1513 [DOI] [PubMed] [Google Scholar]
- [57].Martinez-Vicente M. Autophagy in neurodegenerative diseases: From pathogenic dysfunction to therapeutic modulation. Semin Cell Dev Biol 2015; 40:115-26; PMID:25843774; http://dx.doi.org/ 10.1016/j.semcdb.2015.03.005 [DOI] [PubMed] [Google Scholar]
- [58].Wong YC, Holzbaur EL. Autophagosome dynamics in neurodegeneration at a glance. J Cell Sci 2015; 128:1259-67; PMID:25829512; http://dx.doi.org/ 10.1242/jcs.161216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Goetzl EJ, Boxer A, Schwartz JB, Abner EL, Petersen RC, Miller BL, Kapogiannis D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015; 85:40-7; PMID:26062630; http://dx.doi.org/ 10.1212/WNL.0000000000001702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 2007; 66:75-85; PMID:17204939; http://dx.doi.org/ 10.1097/nen.0b013e31802d6da9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Raina AK, Templeton DJ, Deak JC, Perry G, Smith MA. Quinone reductase (NQO1), a sensitive redox indicator, is increased in Alzheimer's disease. Redox Rep 1999; 4:23-7; PMID:10714272; http://dx.doi.org/ 10.1179/135100099101534701 [DOI] [PubMed] [Google Scholar]
- [62].Wang Y, Santa-Cruz K, DeCarli C, Johnson JA. NAD(P)H:quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with Aalzheimer's disease. Neurobiol Aging 2000; 21:525-31; PMID:10924765; http://dx.doi.org/ 10.1016/S0197-4580(00)00114-7 [DOI] [PubMed] [Google Scholar]
- [63].Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, Kelly J, Arvanitakis Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging 2006; 27:252-61; PMID:16399210; http://dx.doi.org/ 10.1016/j.neurobiolaging.2005.01.016 [DOI] [PubMed] [Google Scholar]
- [64].Tanji K, Miki Y, Ozaki T, Maruyama A, Yoshida H, Mimura J, Matsumiya T, Mori F, Imaizumi T, Itoh K, et al.. Phosphorylation of serine 349 of p62 in Alzheimer's disease brain. Acta neuropathologica communications 2014; 2:50; PMID:24886973; http://dx.doi.org/ 10.1186/2051-5960-2-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Inestrosa NC, Reyes AE, Chacon MA, Cerpa W, Villalon A, Montiel J, Merabachvili G, Aldunate R, Bozinovic F, Aboitiz F. Human-like rodent amyloid-beta-peptide determines Alzheimer pathology in aged wild-type Octodon degu. Neurobiol Aging 2005; 26:1023-8; PMID:15748782; http://dx.doi.org/ 10.1016/j.neurobiolaging.2004.09.016 [DOI] [PubMed] [Google Scholar]
- [66].von Otter M, Landgren S, Nilsson S, Zetterberg M, Celojevic D, Bergstrom P, Minthon L, Bogdanovic N, Andreasen N, Gustafson DR, et al.. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer's disease and age-related cataract. Mechanisms Ageing Dev 2010; 131:105-10; PMID:20064547; http://dx.doi.org/ 10.1016/j.mad.2009.12.007 [DOI] [PubMed] [Google Scholar]
- [67].von Otter M, Bergstrom P, Quattrone A, De Marco EV, Annesi G, Soderkvist P, Wettinger SB, Drozdzik M, Bialecka M, Nissbrandt H, et al.. Genetic associations of Nrf2-encoding NFE2L2 variants with Parkinson's disease - a multicenter study. BMC Med Genet 2014; 15:131; PMID:25496089; http://dx.doi.org/ 10.1186/s12881-014-0131-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bergstrom P, von Otter M, Nilsson S, Nilsson AC, Nilsson M, Andersen PM, Hammarsten O, Zetterberg H. Association of NFE2L2 and KEAP1 haplotypes with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2014; 15:130-7; PMID:24102512; http://dx.doi.org/ 10.3109/21678421.2013.839708 [DOI] [PubMed] [Google Scholar]
- [69].Gan L, Vargas MR, Johnson DA, Johnson JA. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci 2012; 32:17775-87; PMID:23223297; http://dx.doi.org/ 10.1523/JNEUROSCI.3049-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lastres Becker I, Garcia-Yague AJ, Scannevin RH, Casarejos MJ, Kugler S, Rabano A, et al.. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson's disease. Antioxid Redox Signal 2016; 25(2):61-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bott LC, Badders NM, Chen KL, Harmison GG, Bautista E, Shih CC, Katsuno M, Sobue G, Taylor JP, Dantuma NP, et al.. A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum Mol Genet 2016; PMID:26962150; http://dx.doi.org/ 10.1093/hmg/ddw073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem 2005; 280:3963-73; PMID:15509565; http://dx.doi.org/ 10.1074/jbc.M409876200 [DOI] [PubMed] [Google Scholar]
- [73].Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, et al.. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 1999; 274:6483-92; PMID:10037741; http://dx.doi.org/ 10.1074/jbc.274.10.6483 [DOI] [PubMed] [Google Scholar]
- [74].Lastres-Becker I, Innamorato NG, Jaworski T, Rabano A, Kugler S, Van Leuven F, Cuadrado A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014; 137:78-91; PMID:24277722; http://dx.doi.org/ 10.1093/brain/awt323 [DOI] [PubMed] [Google Scholar]
- [75].Rojo AI, Medina-Campos ON, Rada P, Zuniga-Toala A, Lopez-Gazcon A, Espada S, Pedraza-Chaverri J, Cuadrado A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. Free Radic Biol Med 2012; 52:473-87; PMID:22142471; http://dx.doi.org/ 10.1016/j.freeradbiomed.2011.11.003 [DOI] [PubMed] [Google Scholar]
- [76].Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia 2010; 58:588-98; PMID:19908287; http://dx.doi.org/ 10.1002/glia.20947 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.