

ABSTRACT
Melanoma is a paradigm of aggressive tumors with a complex and heterogeneous genetic background. Still, melanoma cells frequently retain developmental traits that trace back to lineage specification programs. In particular, lysosome-associated vesicular trafficking is emerging as a melanoma-enriched lineage dependency. However, the contribution of other lysosomal functions such as autophagy to melanoma progression is unclear, particularly in the context of metastasis and resistance to targeted therapy. Here we mined a broad spectrum of cancers for a meta-analysis of mRNA expression, copy number variation and prognostic value of 13 core autophagy genes. This strategy identified heterozygous loss of ATG5 at chromosome band 6q21 as a distinctive feature of advanced melanomas. Importantly, partial ATG5 loss predicted poor overall patient survival in a manner not shared by other autophagy factors and not recapitulated in other tumor types. This prognostic relevance of ATG5 copy number was not evident for other 6q21 neighboring genes. Melanocyte-specific mouse models confirmed that heterozygous (but not homozygous) deletion of Atg5 enhanced melanoma metastasis and compromised the response to targeted therapy (exemplified by dabrafenib, a BRAF inhibitor in clinical use). Collectively, our results support ATG5 as a therapeutically relevant dose-dependent rheostat of melanoma progression. Moreover, these data have important translational implications in drug design, as partial blockade of autophagy genes may worsen (instead of counteracting) the malignant behavior of metastatic melanomas.
KEYWORDS: ATG5, copy number variation, core autophagy genes, lineage-specificity, melanoma, mouse models, patient prognosis, targeted therapy
Introduction
Cutaneous melanoma is the most lethal form of skin cancer and a prime example of genetically complex neoplasms.1,2 Specifically, melanomas are the tumors with the highest mutational rate described to date,3 which adds to an increasing list of chromosomal defects, epigenetic changes and transcriptomic and proteomic alterations.1,4-7 Moreover, melanoma cells are highly dynamic, being able to switch phenotypes and expression profiles depending on the cellular context or tumor stage.8,9 Therefore, a main challenge in this pathology is to separate tumor drivers from inconsequential byproducts of malignant transformation.10
Despite their inherent complexity, a fraction of melanomas retain traits that trace back to melanocytes, their cell of origin.11,12 Best characterized of these lineage-specific features are melanosome biogenesis and cellular pigmentation.11 Melanoma cells tune these programs to maintain stemness and an appropriate rate of proliferation/invasion,13-15 as well as to bypass a variety of therapeutic agents.16-18 Yet, the key oncogene that modulates pigmentation, namely, the microphthalmia-associated transcription factor MITF19 can be inactivated by various transcriptional and post-transcriptional mechanisms during melanoma progression.20-23 Therefore, these findings suggest alternative modulators of the melanocytic lineage in this disease. Performing a computational cross-cancer analysis of transcriptomic profiles followed by gene validation in human tissue specimens and in mouse models, we have recently identified a cluster of lysosomal-associated genes that are particularly enriched in melanoma, in a manner not shared by more than 30 tumor types.24 Mechanistically, melanomas were found to depend on lysosomal-associated degradation to counteract a hyperactive influx of macropinosomes induced at early stages of melanoma development.25,26 This lysosomal-dependent vesicular trafficking was found to represent an inherent vulnerability of melanoma cells,24 a concept supported by independent studies.11,27 The specific contribution of other lysosomal-associated processes such as (macro)autophagy to melanoma initiation, progression and response to therapy is still under evaluation.28,29
Melanomas are long known for their ability to induce autophagosome formation in response to a variety of endogenous stress-inducing factors (e.g., deregulated BRAF>MAPK oncogenic signals)30-32 and external cues (including hypoxia, nutrient deprivation, and a broad spectrum of therapeutic agents, among many others).29,33,34 However, lysosomal-dependent degradation may favor or inhibit cell survival, depending on the identity and/or duration of the stimuli.28,29 Mechanisms underlying these dual functions remain unclear. Multitumor genomic or transcriptomic profiles have not been performed to determine whether melanomas can regulate or target autophagy factors in a lineage-specific manner. In particular, it is unclear whether melanomas spare the core autophagy machinery from mutation, targeting instead other genes (for example, regulators of the autophagy network) as recently reported for glioblastomas and multiple carcinoma types.35 Indeed, the expression and function of factors involved in autophagosome assembly is inconsistent across the melanoma literature.28,29 Thus, autophagy genes such as MAP1LC3 or BECN1 may be up- or downregulated depending on the study.36-38 In turn, ATG7 and ATG5 may have opposing roles in melanoma initiation.39,40 Although ATG7 gene expression has not been formally analyzed in human melanomas, targeted deletions in mice (i.e., in the context of the Tyr:CreERT2;BrafCA;ptenΔ/Δ melanoma model) suggest that the murine Atg7 gene acts as a tumor-promoting factor, with a key role in the resistance to the clinically relevant BRAF inhibitor dabrafenib.39 Inducible mouse models for Atg5 deletion in melanocytes are not available, but expression studies support suppressive functions in early-stage stage human melanoma.30 The extent to which ATG5 contributes to melanoma metastasis is confounded by reports where both depletion and overexpression of this protein compromise the tumorigenic potential of cultured melanoma cell lines.30,39
Here we performed a meta-analysis of large multitumor datasets to identify genomic and transcriptomic changes in the autophagy machinery that can distinguish melanoma from other malignant diseases. We used this strategy to interrogate the mutational status, gene dosage, mRNA expression and prognostic value of main effectors and modulators of autophagosome formation. Chromosomal context was evaluated in the assessment of overall patient survival to separate gene-specific from loci-dependent effects. This approach revealed a distinct impact of heterozygous ATG5 loss as a novel risk factor for melanoma metastasis, a feature not recapitulated by other tumor types. The dose-dependent contribution of ATG5 to melanoma metastasis and response to targeted therapy (BRAF inhibition) was further confirmed in a series of newly generated animal models. These results illustrate how an otherwise heterogeneous autophagy machinery can still bear tumor-selective drivers of metastatic potential and drug resistance.
Results
Selective ATG5 mRNA downregulation in melanoma identified within a heterogeneous expression of the core autophagy machinery
A comparative analysis of expression profiles was performed in melanoma and a broad spectrum of cancer types to identify conserved gene clusters that may inform on novel lineage-specific tumor features. We were particularly interested in the core machinery for autophagosome formation because, not being frequently targeted in various carcinomas,35 it could represent a potential distinctive feature in melanoma, inasmuch as other lysosomal functions related to vesicular trafficking.24 To this end, we selected well-known orthologs of the LC3/Atg8 protein (GABARAPL1 and MAP1LC3A), and a series of additional key modulators of phagophore initiation and maturation (ATG3, ATG4A, ATG5, ATG7, ATG10, ATG12, ATG16L1, BECN1, RB1CC1, ULK1 and ULK2).42 mRNA expression of these genes was first screened through the Cancer Cell Line Encyclopedia (CCLE), which encompasses 61 cell lines from melanoma and 856 lines of over 22 different cancer types.43 Hematopoietic tumors showed a distinct gene expression pattern with a consistent downregulation of GABARAPL1 and MAP1LC3A mRNA (Fig. 1, top panel), which we think justifies future studies. Regarding solid tumors there was a high variability in mRNA expression. In fact, Gene Set Enrichment Analysis (GSEA) failed to identify any significant gene cluster in melanoma (Fig. 1, top panel; false discovery rate (FDR) = 0.28). This was in contrast to the melanoma-enriched endolysosomal factors24 (Fig. 1, bottom panel; FDR < 10−03).
Figure 1.
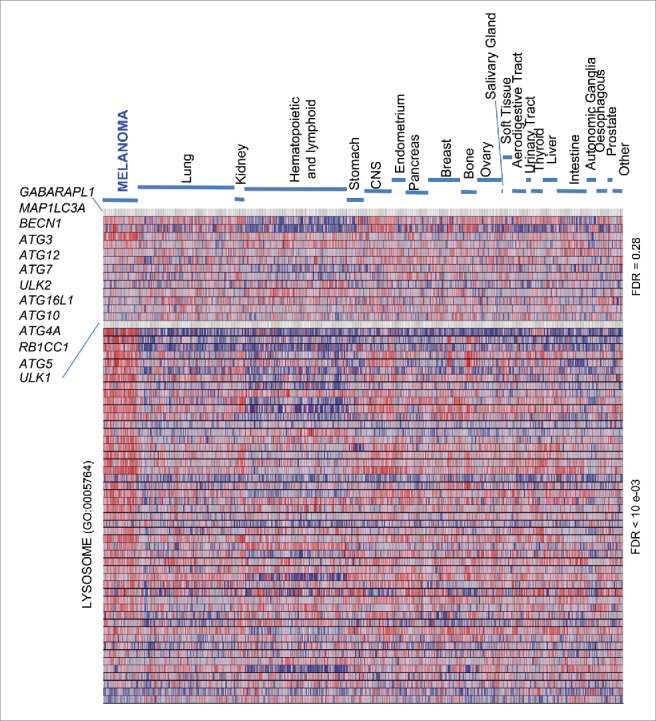
Heterogeneous expression of core autophagy genes in melanoma and a broad spectrum of cancer types. GSEA heat map showing a differential enrichment of the indicated autophagy genes across the Cancer Cell Line Encyclopedia (CCLE), which encompasses a total of 917 cell lines of indicated tumor types. The Lysosome Gene Ontology gene set (GO:0005764), a melanoma-enriched feature, is included to visualize the distinct regulation of lysosomal-associated degradative processes. FDR values for the autophagy vs the lysosome GO set are indicated on the right.
Despite the unconserved expression of the autophagy machinery described above, the CCLE revealed a variable, albeit significant, downregulation of ATG5 mRNA, particularly in melanoma cell lines (Fig. 1; P = 8 × 10−4; see Table S1). To confirm these results, a meta-analysis of ATG5 mRNA expression was then performed on 11 independent multicancer data sets (see Supplementary Information for a listing of these datasets and the corresponding identifiers). This included transcriptomic profiles of the Cancer Genome Project repository (Fig. 2A; N = 723; P = 9.5 × 10−6), the NCI60 panel, and a series of transcriptomic profiles available through the Oncomine portal, which in total encompass over 2700 tumor cell lines (see examples in Fig. S1A-C and Table S1 for additional information). This approach confirmed ATG5 among the top 11–35% underexpressed genes in melanoma (see Table S1; data on other tumor types are analyzed below).
Figure 2.
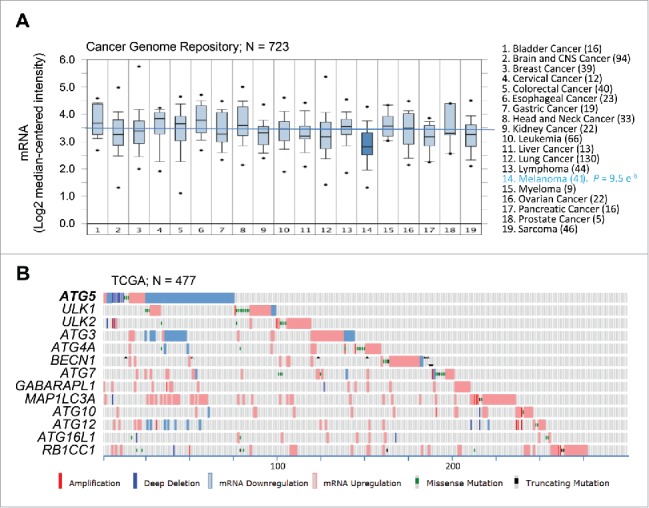
Consistent dowregulation of ATG5 mRNA expression in melanoma cell lines and tumor specimens. (A) Box plots depicting ATG5 mRNA expression (median centered intensity) across the indicated 19 tumor types (numbers of cell lines in parentheses) extracted from Oncomine from the Cancer Genome Project cell line dataset (E-MTAB-783; N = 732). P value for ATG5 downregulation in melanoma is indicated on the right. (B) mRNA expression (defined by RNA-Sequencing normalized to diploid cases by RSEM), as well as allelic number, missense and truncated mutations of core autophagy genes (color coded as indicated) in TCGA melanomas (N = 477 specimens). Shown is information extracted from cBioPortal for melanoma cases (numbered at the bottom) with alterations in the indicated factors.
Next, we interrogated the expression and genomic status of ATG5 and the other core autophagy factors in human melanoma specimens. To this end, we mined the Cancer Genome Atlas (TCGA), the largest clinically annotated repository of information on human tumor biopsies, which for melanoma includes 478 samples.1 Here, ATG5 was also distinct from the other autophagy genes analyzed. Thus, 70% of cases with defects in this gene corresponded to mRNA downregulation (see Fig. 2B). In contrast, the rest of the autophagy genes studied were affected primarily by mRNA upregulation (Fig. 2B). Together, these results support a distinct regulation of ATG5 that separates this gene from other autophagy factors, and melanoma from other tumor types.
Shallow chromosomal loss but not promoter methylation targeting the ATG5 gene in melanoma
Next, we assessed mechanisms underlying ATG5 downregulation in melanomas, and their impact in patient prognosis. Promoter hypermethylation has been previously reported in 9 out of 13 early-stage melanomas.40 Therefore, we interrogated the 478 tumor specimens of the TCGA melanoma data set, 45% of which correspond to patients with lymph node or distal metastases (stage III/IV melanomas). In parallel, HOXD9 was used as a reference for a known melanoma-methylated gene.44 In this larger data set, the ATG5 promoter was found as hypomethylated (Fig. 3A; see different profiles with respect to HOXD9). Moreover, Pearson and Spearman rank bivariate analyses failed to identify any significant correlation between ATG5 promoter methylation and overall patient survival (Fig. 3B, N = 275, P= −0.049, r= −0.024). Therefore, these new data support alternative mechanisms reducing ATG5 mRNA levels in human melanoma tumors.
Figure 3.

ATG5 undergoes heterozygous copy number loss (shallow deletions) instead of hypermethylation in melanoma tumors. (A) mRNA expression (RNA sequence vs RSEM) as a function of promoter methylation status for ATG5 in the TCGA melanomas, with HOXD9 included as reference for a methylation-regulated gene in this tumor type. (B) Lack of correlation between ATG5 promoter methylation and overall patient survival in TCGA melanomas (N = 275). Indicated are Pearson (P) and Spearman rank (r) correlations. (C) ATG5 allelic loss in representative melanoma cell lines defined by FISH. Probes for ATG5 at 6q21 and an unrelated locus at 6p21 were labeled in red (R) and green (G), respectively. (D) ATG5 copy number in the indicated cell line data sets. (E) Graphical representation of the percentage of cases with the indicated alterations in the ATG5 gene in specimens with clinical annotations in the TCGA melanoma database (N = 366). Amp and del stand for amplifications and deletions, respectively.
Fluorescence in situ hybridization (FISH) was then performed to visualize ATG5 copy number. To this end, fluorescently labeled probes were generated from the corresponding bacterial artificial chromosome clones spanning the ATG5 locus at chromosome band 6q21, as well as unrelated areas at 6p21.1 (see Materials and Methods). As experimental systems, we selected melanoma cell lines with mutations in the oncogenes BRAF (SK-Mel-19, SK-Mel-29, G-361) or NRAS (SK-Mel-147), which represent characteristic alterations of this tumor type.45 As shown in Fig. 3C, all these lines showed ATG5 copy number reduction, suggesting that partial allelic loss may account for the reduced transcript expression of this gene described above (Fig. 1 and Fig S1). Partial ATG5 allelic loss (shallow deletion) was further identified in 63% and 70% of 2 independent multitumor cell line panels (CCLE/GSE36133 and GSE7606, respectively; Fig. 3D), and in 57% of the TCGA melanoma tumors (Fig. 3E).
Intriguingly, deep (homozygous) deletions were however, rare, affecting only 2.3% of human melanomas (Fig. 3E). Therefore, complete loss of this gene may be deleterious for melanoma progression. To functionally assess this hypothesis, ATG5 protein expression was depleted in 3 independent human melanoma cell lines by a pool of 3 validated siRNAs (Fig. S2A,B) or by 3 independent shRNA constructs (Fig. S2C,D). These strategies reduced cell proliferation, consistent with previous data in mouse melanoma cells.41 Importantly, ATG5 depletion significantly inhibits the invasive capacity of human melanoma cells (Fig S2E), compromising cell viability particularly under stress conditions that may be encountered during metastasis, such as anoikis due to growth in suspension (Fig. S2F and results not shown).
Heterozygous ATG5 loss predicts poor overall survival in melanoma patients
Given the heterogeneous expression of the autophagy machinery (Fig 1) we then tested to what extent ATG5 copy number could be physiologically relevant for patient prognosis. Ten-year overall survival information was downloaded from NCI datasets for ATCC melanoma patients. Interestingly, patients with partial (shallow) ATG5 allelic loss had the worst prognosis (Fig. 4A; N = 255, P = 0.0152). Overall survival and disease-free survival were more compromised (P = 0.0019 and P = 0.0059, respectively; see Figs. S3A,B) when considering longer observation times (29 y), and both ATG5 copy number variation and mRNA downregulation. This impact on poor prognosis was consistent with shallow ATG5 loss accumulating in advanced stage III/IV melanomas (Fig. 4B, left panel).
Figure 4.
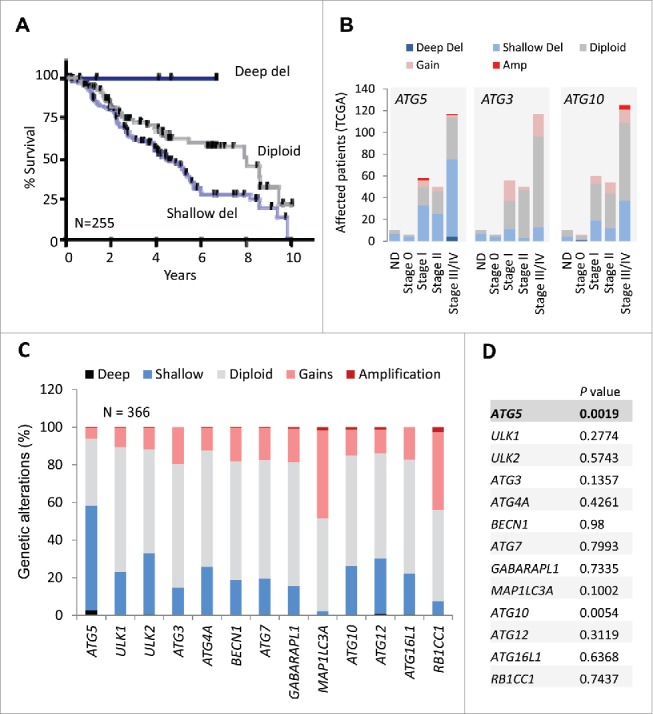
Prognostic value of shallow ATG5 deletions in melanoma not shared by other core autophagy genes. (A) Kaplan Meier curves for overall survival for melanoma patients with diploid, partial losses (shallow deletions) or deep deletions of the ATG5 locus (N = 255). Log-rank p value for shallow deletion vs diploid ATG5 content p = 0.0152. (B) Comparative distribution of TCGA melanoma patients at different stages of tumor progression (stage 0 to stage IV) as function of the genomic status of the ATG5, ATG3 or ATG10 loci (N = 255 melanomas). The corresponding genetic alterations are color coded as indicated. ND: cases with no defined staging classification. (C) Graphical representation of the distribution of genomic changes in the indicated autophagy genes in the TCGA melanomas. (D) Overall survival (log-rank p value) of patients with genetic changes and/or deregulated mRNA expression with respect to cases with diploid status of the indicated autophagy genes.
Next, copy number changes were interrogated for 12 additional autophagy genes in melanomas. Interestingly, none of these factors were found subject to the degree of copy loss identified for ATG5 (Fig. 4C). Instead, these genes maintained a largely diploid status, with MAPL1LC3A and RB1CC1 amplified in about 40% of TCGA melanomas (Fig. 4C). Regarding overall survival, aggregate copy number changes were intriguingly significant for ATG10 (Fig. 4D), which interestingly, is an E2-like ubiquitin ligase that transfers ATG12 to ATG5 during autophagosome formation.46 However, ATG10 did not appear to undergo a significant reduction in mRNA expression (Fig. 1), nor copy loss at advanced stages of the disease (stages III and IV), where ATG5 shallow losses accumulate (Fig. 4B; see also ATG3, another E2-like enzyme as a comparison). Therefore, these results support a distinct downregulation of ATG5 in melanoma as a putative indicator of patient prognosis.
Prognostic value of ATG5 downregulation in melanoma not shared by other 6q21-mapping genes and in other tumor types
The long arm of chromosome 6 (including the ATG5 locus at 6q21), has been reported to undergo copy number losses in melanoma and other tumor types47 (see also www.cancerindex.org). Therefore, we questioned whether the prognostic value found for ATG5 copy number in melanoma was a selective feature of this gene or a broader consequence of larger changes affecting this chromosomal area. To this end, the TCGA melanomas were mined for genomic changes of factors mapping within 1Mb upstream or downstream of ATG5 (Fig. 5A). These include AIM1 (absent in melanoma 1),48 BVES (blood vessel epicardial substance) and POPDC3 (popeye domain containing 3), factors frequently downregulated in gastric cancer.49 In addition, we also included PRDM1 (PR domain 1), a tumor suppressor in NK cell neoplasms;50 as well as QRSL1 (glutaminyl-tRNA synthase [glutamine-hydrolyzing]-like 1) and RTN4IP1 (reticulon 4 interacting protein 1), of unknown contribution to tumorigenicity. Deep and shallow genomic deletions were similar for all the genes analyzed (see examples for ATG5 and PRDM1 in Fig. 5B, and additional information in Fig. 5C). However, none of the genes studied showed mRNA downregulation at the rate shown for ATG5 (Fig. 5C). Moreover, while various post-transcriptional modifications affect the functional status and ultimate expression of these factors, for example for AIM1,48 changes in copy number and mRNA expression were not sufficient per se to define poor overall patient survival (see P values in Fig. 5C).
Figure 5.
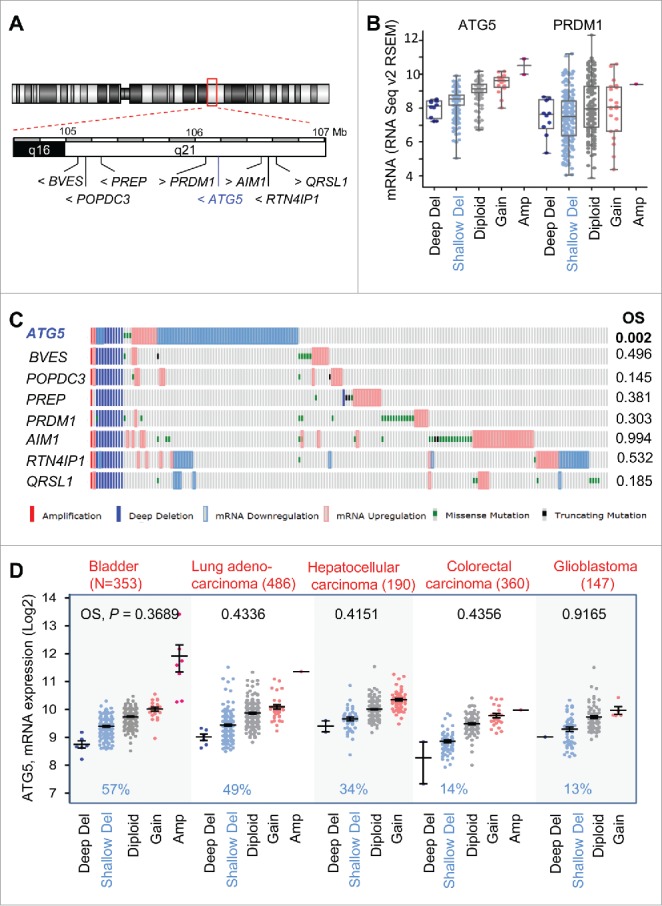
Prognostic features of ATG5 in melanoma not shared by neighboring genes at chromosome 6q21 or in other tumor types. (A) Schematic representation of genes mapping within 1 Mb upstream and downstream from the ATG5 locus at 6q21. (B) Comparative allelic status of ATG5 and PRDM1 in TCGA melanomas with respect to mRNA levels of these genes calculated by RSEM as a function of other diploid factors in this tumor. (C) Allelic status, mRNA expression, missense and truncated mutations (coded as depicted at the bottom) of the indicated genes in TCGA melanomas (N = 477 specimens, showing those with alterations in the indicated factors). Log-rank P values for overall survival (OS) estimated with respect to patients with no alterations in the indicated genes were extracted from cBioPortal for a 30-year observation period and are depicted on the right. (D) mRNA expression and allelic status of ATG5 in the indicated tumor types included in the TCGA. The number of specimens per tumor is indicated in parentheses, and the corresponding rate (%) of cases with shallow deletions of ATG5 are marked in blue. Log-rank p values for overall survival in patients with aggregate mRNA or copy number changes are also listed (in black).
Regarding other cancer types, there was a high variability in ATG5 copy number in the CCLE-contained tumors (not shown). Still, a meta-analysis of TCGA databases (with over 2000 tumor biopsies analyzed) identified ATG5 allelic loss in different tumor types (see examples in Fig. 5D). Tumors with ATG5 copy loss include bladder cancer (57% of N = 353 TCGA cases) as well as carcinomas of lung (49%, N = 486) and liver (34%, N = 190). Nevertheless, ATG5 loss is not a generalized feature of all aggressive cancers, as defined for example for colorectal carcinoma (14%, N = 360), or glioblastoma (13%, N = 147) (Fig. 5D; see also Fig. S4). Moreover, in these tumor types, aggregate changes in ATG5 copy number and mRNA expression failed to reach significant differences in overall survival and disease-free survival (see Fig. 5D for the corresponding p values). Therefore, while allelic losses at chromosomal band 6q21 may occur in multiple neoplasms,51 copy number changes of ATG5 are particularly indicative of poor prognosis in melanoma.
ATG5 is dispensable for cellular pigmentation and nevi formation in vivo
The meta-analyses discussed above in human specimens strongly support an active contribution of ATG5 downregulation to melanoma progression and metastasis, with a specific selection for partial (heterozygous) loss of this gene. Genetically engineered mice were then generated to validate this concept in vivo. First, to define the contribution of ATG5 on the survival and physiological roles (i.e., pigmentation) of normal melanocytes, a floxable Atg5 strain52 (Atg5tm1Myok, herein referred to as Atg5flox/flox for simplicity) was crossed into mice expressing tamoxifen-inducible Cre recombinase under the control of the Tyr (tyrosinase) promoter (Tyr::CreERT2).53 These mice developed normally, and neither mono nor biallelic deletion of Atg5 (herein indicated as Atg5+/Δ or atg5Δ/Δ, respectively) had an impact on fur color (not shown). Therefore, we concluded that in mice, Atg5 is dispensable for the viability or proliferation of normal melanocytes. This is in contrast to deleterious effects of conditional deletion of Atg5 in brain neural cells,54 which as melanocytes, have a neural crest origin.
Next, the Tyr::CreERT2; Atg5flox/flox mice were subsequently interbreed with a strain carrying a constitutively active BrafV600E allele expressed at the endogenous locus (Tyr::CreERT2; BrafCA/CA)55 for the analysis of benign melanocytic lesions (nevi). This is an important pending question as human nevi are constituted of senescent cells56,57 with high ATG5 levels.40 Moreover, ATG5 was previously found to modulate premature senescence driven by oncogenic BRAF in cultured melanocytes.40 Intriguingly, our newly generated Tyr::CreERT2;BrafCA/CA;Atg5flox/flox animals demonstrated that Atg5 copy number had no positive or negative contribution to the development of hyperplastic melanocytic lesions induced by BrafV600E (Fig. 6A, see Fig. 6B for immunohistochemical analysis of ATG5 protein expression in the indicated genetic backgrounds). In these models, tamoxifen was administered systemically to ensure a homogeneous onset of melanocytic lesions (otherwise highly variable). This approach also allowed for a simultaneous analysis of different anatomical areas, therefore discarding possible context-dependent effects of cutaneous melanocytes (see Fig. 6A for hyperpigmented lesions in ears, paws and back skin of representative mice with different Atg5 allelic status). Together, these results emphasize the relevance of addressing gene function in vivo, particularly in the skin, where surrounding keratinocytes and fibroblasts influence melanocyte survival and functional status.58
Figure 6.
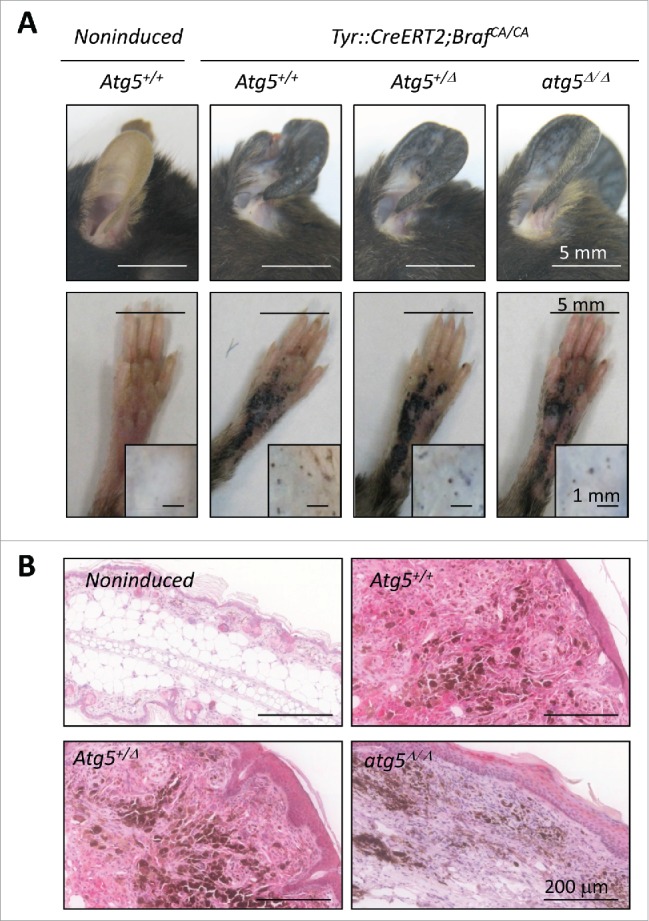
Atg5 is dispensable for nevi formation driven by oncogenic BrafV600E in genetically modified mice. (A) Hyperproliferative pigmented lesions generated in the tamoxifen responsive Tyr::CreERT2;BrafCA/CA mice crossed to Atg5flox/flox animals for assessment of Atg5 gene dosage (Atg5+/−, Atg5+/Δ or atg5Δ/Δ) in the melanocytic compartment. Shown are images of lesions generated in ears, paws or the back skin (the latter in insets) captured 6 mo after tamoxifen induction. Equivalent anatomical areas in tamoxifen-untreated (noninduced) Tyr::CreERT2;BrafCA/CA;Atg5+/− control animals are also included as a reference. (B) Visualization of ATG5 protein levels by immunohistochemical staining (pink) of paraffin-embedded ear sections of animals of the indicated genotypes and treated as in (A). Nuclei were counterstained by hematoxilin. The brown color corresponds to melanin.
Inducible mouse models validate Atg5 heterozygous loss as a key driver of malignancy in melanoma
A third animal model was generated to define the impact of Atg5 copy number in melanoma development. To this end, the Tyr::CreERT2; BrafCA/CA;Atg5flox/flox mice were crossed into a floxable strain that allows for a melanocyte-dependent deletion of the tumor suppressor Pten55 (see Materials and Methods for the MGI-standardized nomenclature of each of the individual strains used). The Tyr::CreERT2;BrafCA/CA;ptenΔ/Δ;Atg5flox/flox animals were then interbred for the analysis of siblings expressing both copies of Atg5 or bearing heterozygous or homozygous deletions of this gene (i.e., Atg5+/−, Atg5+/Δ and atg5Δ/Δ, respectively).
In the context of Tyr::CreERT2;BrafCA/CA;ptenΔ/Δ− driven melanomas, Atg5 copy number (altering as expected the protein expression of classical autophagy factors such as SQSTM1/p62; see Fig. S5A) had a differential impact on tumor growth depending on whether deletions affected one or the 2 alleles of Atg5. Specifically, although melanomas could be developed in all backgrounds (not shown), complete Atg5 knockdown (atg5Δ/Δ) reduced the number and size of cutaneous melanoma lesions in all anatomical areas, while instead, the Atg5+/Δ counterparts showed an accelerating tumor development (Fig. 7A, B; see additional detail in Fig. S5A, where the SOX10 protein was used as a marker for melanocytic cells). This behavior was in contrast to a modest reduction in the growth of localized melanomas reported for heterozygous Atg7 (Atg7+/Δ) in a similar Tyr::CreERT2;BrafCA/CA;ptenΔ/Δstrain.39
Figure 7.

Impact of Atg5 copy number on melanoma initiation and metastasis in vivo (melanocyte-specific and inducible mouse models). (A) Cutaneous melanomas generated in the melanocyte-specific Tyr::CreERT2;BrafCA/CA;Ptenflox/flox; Atg5flox/flox mice, bred to maintain (Atg5+/−) or undergo mono- or bi-allelic deletion of Atg5 (Atg5+/Δ or atg5Δ/Δ, respectively). The upper photographs were captured 3 wk after systemic administration of tamoxifen. Lower panels correspond to paraffin-embedded specimens processed for the detection of ATG5 protein expression (in pink). Nuclei were counterstained with hematoxylin. Brown staining corresponds to melanin. (B) Quantification of the average tumor number and size generated in animals as in (A), represented as mean −/+ SEM. (C) Higher metastatic potential of Tyr::CreERT2;BrafCA/CA;ptenΔ/Δ;Atg5+/Δ melanomas determined by macroscopic examinations of lungs 4 wk after tamoxifen administration and represented −/+ SEM. Here as in (B), P values corresponded to paired t test. (D) Lung micrometastases at low and high magnification (upper and lower panels, respectively) in the indicated mouse genotypes visualized by virtue of melanin-expressing colonies (brown, arrows). (E) Differential response of Tyr::CreERT2;BrafCA/CA;ptenΔ/Δ melanomas to dabrafenib depending on Atg5 copy number. Images correspond to representative lesions in the depilated back skin of the indicated animal groups, 4 wk after tamoxifen induction, and 3 wk of dabrafenib treatment (10 mg/kg orally, once a day). (F) Response rates measured as the reduction in tumor size vs averaged untreated controls of the Tyr::CreERT2;BrafCA/CA;ptenΔ/Δ driven melanomas of Atg5+/−, Atg5+/Δ or Atg5Δ/Δ backgrounds, represented as waterfall plots. (G) Size distribution (mm2) of control and dabrafenib-treated cutaneous lesions of the indicated genotypes.
Regarding the less understood role of autophagy factors at late stages of melanoma progression, the Tyr::CreERT2;BrafCA/CA;ptenΔ/Δmice revealed that Atg5 heterozygous deletion enhanced lung metastasis with respect to Atg5+/− and particularly to atg5Δ/Δ littermates, as evident by macroscopic examination (Fig. 7C, P = 0.024) or by detection of melanin-expressing cells in serial histological sections (Fig. 7D). Therefore, these data in mice provide a functional explanation for the enrichment in heterozygous deletions found for ATG5 in human melanoma cells and clinical specimens (Fig. 3D,E), and the correlation between ATG5 copy number and melanoma patient prognosis (Fig. 4A).
Heterozygous Atg5 loss reduces the response of BrafV600E;ptenΔ/Δmelanomas to targeted therapy
Next we assessed the potential therapeutic impact of Atg5 copy number on the response to BRAF inhibitors, as these compounds have been well demonstrated to induce autophagosome formation in vitro and in vivo.39,60-62 Atg5+/−, Atg5+/Δ or atg5Δ/Δ littermates of the Tyr::CreERT2;BrafCA/CA;ptenΔ/Δ mice were treated with dabrafenib as an example of a BRAF inhibitor in clinical use.63 Drug administration was initiated once melanocyte hyperproliferation was activated (see Materials and Methods). Dabrafenib significantly reduced tumor growth in the Atg5+/− cohorts, a response enhanced in the atg5Δ/Δ-driven lesions (see examples of representative animals in Fig. 7E and quantification as waterfall plots in Fig. 7F). In contrast, heterozygous Atg5 loss, reflecting the most frequent situation in human melanomas, compromised drug response (Fig. 7E,F), with an accumulation of high-size tumors (Fig. 7G; Fig. S5B; P < 0.001) that required euthanasia (not shown). Together, these findings provide evidence for the relevance of ATG5 copy number, not only in melanoma initiation, but as a key modulator of the metastatic potential of melanoma cells, and their response to clinically relevant compounds.
Discussion
Solid cancers typically share a series of hallmarks (including self-sufficiency in growth signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and deregulation of metabolic pathways, among others)64,65 which can be acquired by deregulation of pathways with essential roles in a broad spectrum of pathologies. For example, hyperactivation of RAS>BRAF>MEK, PIK3CA>AKT, NOTCH or SHH signaling pathways, as well as downregulation of PTEN or functional inhibition of p53 are few examples of “classical” tumorigenic events accumulated, albeit with varied incidence, in multiple neoplasias, including melanoma.2,3 Cancer hallmarks can also be ensued in a tumor-type or lineage-restricted manner.12 Signaling cascades involved in melanosome maturation, and more recently, vesicular trafficking linked to endolysosomal degradation, are characteristically wired in melanomas.11,24,26,27,66 Here we showed that core drivers of (macro)autophagy, another lysosomal-associated mechanism, are, however, expressed in a highly heterogeneous manner, with no gene cluster identifiable as a melanoma-associated signature. Nevertheless, and despite this variability, this study identified selective allelic loss of ATG5 as a putative risk factor for the metastasis of cutaneous melanomas, with no correlation with the mutational status of RAS, BRAF or other melanoma-associated oncogenes. Importantly, the prognostic value of ATG5 copy number in melanoma was not shared by other core autophagy factors and not recapitulated in a broad spectrum of cancer types. These results were obtained by a triple approach: (i) a meta-analysis of 19 large multitumor data sets, which in total encompass more than 2700 cell lines and 2000 clinical specimens in melanoma and over 22 additional cancer types; (ii) functional studies in cultured cells and (iii) melanocyte-specific mouse models that mimic alterations in the BRAF oncogene and the PTEN tumor suppressor characteristic of human melanomas.55 Additionally, these genetically engineered mice further emphasize therapeutic implications of ATG5 dosage in the resistance to dabrafenib, a BRAF inhibitor actively used in melanoma treatment.67
The autophagy core (namely, genes most directly involved in autophagosome/autolysosome formation) was first described in yeast as an ordered step-wise cascade.68 However, this machinery is now known to be extremely intricate, with early phagosome formation modulated by more than 700 protein-protein interactions.46 These networks are in turn, further influenced by multiple context-dependent feedback and feedforward signaling cascades.46,69 Dissecting the impact of autophagy in cancer has the added complication of rather unconserved expression patterns, as recently demonstrated in glioblastomas and 11 different carcinoma types by cross-cancer profiling of molecular alterations.35 Here we extended this heterogeneity to melanomas and a broad spectrum of tumors of various etiologies, emphasizing the need for functional validation in physiologically relevant systems. Indeed, without the inducible mouse models generated in this study, an unsupervised computational analysis may have missed the relevance of ATG5 heterozygosity in melanoma progression and response to targeted therapy. Moreover, the inter-tumoral variability shown here in human cancer databases may also underlie differing pro- or antitumorigenic roles of Atg5 loss for example in models of liver carcinoma vs. lung adenocarcinomas, respectively.59,70
This study also brings caution on the autophagy factors to be selected when aiming to define prognostic relevance within a given tumor type (for example, in screens for tumor biomarkers). In melanoma, we found ATG5 copy number distinctively significant in its correlation to disease-free survival. While additional validation will be required from independent datasets, these results may have important future translational relevance. To date (and despite great progress in the identification of pro-metastatic factors)2,71 melanoma prognosis is largely defined by histopathological criteria such as depth of invasion of the primary lesions, which may be susceptible to subjective interpretation.72,73 In this context, it is interesting to note that the observed impact of ATG5 heterozygosity on overall melanoma patient survival was not reproduced by copy number changes of neighboring genes at the 6q21 locus, such as AIM1, PRDM1, PREP or POPDC3, previously reported as tumor suppressors.48-50 Of interest, 6q21 cytogenetic changes are frequent in other melanoma subtypes, including uveal melanomas, whose mechanisms of metastasis are still poorly understood.74 Therefore, it is tempting to speculate that ATG5 heterozygosity may also contribute to noncutaneous melanomas.
The mouse models described here also illustrate the relevance of assessing tumorigenic roles of autophagy factors in vivo. In particular, finding that Atg5 was dispensable for benign melanocytic lesions in the Tyr::CreERT2;BrafCA/CA animals was rather surprising as this protein is highly expressed in senescent human nevi.40 Intriguingly, this was also the case for Atg7 deletion in a similar inducible mouse strain (i.e. Tyr::CreERT2;BrafCA/CA;atg7Δ/Δ).39 Moreover, in the context of cutaneous melanomas generated in the Tyr::CreERT2;BrafCA/CA;ptenΔ/Δ mice, both complete loss of Atg5 (this study) and Atg7 (ref. 39) reduced tumor onset. This is consistent with the notion derived from multiple studies, including ours, that melanomas cannot sustain a complete blockade of autophagy.28,29 Nevertheless, while Atg7+/Δ had a minor impact on Tyr::CreERT2;BrafCA/CA;ptenΔ/Δ primary melanomas and their response to dabrafenib,39 here we found heterozygous Atg5+/Δ to unexpectedly exacerbate both these processes. Moreover, the potential clinical relevance of Atg5 gene dosage was further emphasized by the enhanced metastatic potential of the Atg5+/Δ melanomas. The contribution of Atg7 to melanoma metastasis has yet to be defined. Nevertheless, comparative analyses of Atg5/ATG5 and Atg7/ATG7, in mouse models and human specimens, are granted as they may prove relevant to identify differential regulators and functional targets of these genes, for example, in the modulation of the tumor secretome.75 In this context, an attractive possibility is that the partial downregulation of ATG5 (but not ATG7) in melanomas reflects a developmental program shared by the lineage-specific drivers MITF11,27 and RAB724,25 which act in part by providing a fine tuning to lysosomal-associated secretory programmes.26
Together, our results identified ATG5 heterozygous loss as a distinctive feature of aggressive melanomas, selectively exploiting cytogenetic alterations at the chromosome 6q21 band, and contributing to disease-free survival in a manner not shared by other tumor types. Moreover, the reduced efficacy of dabrafenib in the case of Atg5+/Δ-driven mouse melanomas raises caution with respect to incomplete inactivation of this gene in clinical trials, as this may result in an unexpected worsening of patient outcome.
Materials and methods
Databases and statistical analyses
The 19 multitumor data sets used in this study for a meta-analysis of the expression and genomic status of the autophagy genes and of ATG5 neighboring factors mapping at chromosome 6q21 are listed in the Supplementary Information (Tables S1 and S2, respectively). These tables also summarize information on number of specimens and overall count of tumor types, as well as platforms employed for estimation of mRNA levels, copy number variation and mutation rates of the indicated genes, with the corresponding GSE identifiers when available. Data were extracted directly from the repositories or via cBioPortal76 or Oncomine platforms. Gene set enrichment analysis (GSEA) was applied to the indicated datasets for the Gene Ontology lysosome GO:0005764 set and core autophagy factors. Genes were ranked based on limma moderated t statistic. After Kolmogorov-Smirnoff testing, gene sets showing FDR <0.25 were considered enriched between classes under comparison. For TCGA, mRNA levels correspond to RNA-sequence data analyzed by expectation maximization (RSEM) and normalized to cases with diploid gene status with the corresponding data set; copy-number was determined using GISTIC 2.0. Heatmaps for copy number variation were prepared with GENE-E. Statistical significance of bivariate analyses (mRNA vs. copy number or methylation vs. survival rates) is presented as a function of Pearson (P) or Spearman rank (r) correlations. For assessment of clinical features (overall survival or disease-free survival), Log-rank test P values ≤ 0.05 were considered significant.
Mouse models, quantification and treatment
Strains used in this study were as follows: Tyr::CreERT2/1Lru,53 Atg5tm1Myok,54 Braftm1Mmcm (BrafCA/CA)55 and Ptentm2Mak.77 For simplicity these lines are herein referred as to Tyr:CreERT2, Atg5flox/flox, BrafCA/CA, and Ptenflox/flox, respectively. For the analysis of nevi, crosses were set to generate Tyr:CreERT2;BrafCA/CA mice expressing or lacking one or the 2 copies of Atg5 (herein referred to as Atg5+/−, Atg5+/Δ or atg5Δ/Δ, respectively). In turn, melanomas were obtained in the Tyr:CreERT2;BrafCA/CA;pten Δ/Δ background, also with different Atg5 copy number. These benign and malignant lesions were induced in 12- to 14-wk-old animals by 100 μl intraperitoneal injection of tamoxifen (Sigma, T5648; stock 8 mg/ml) for 3 consecutive d. Growth of cutaneous lesions (> 80 tested per background) was monitored by optical inspection and photographic followup at different time points upon induction, with tumor diameters scored by ImageJ. Tumors were also measured with calipers after postmortem depilation. To estimate metastatic potential, animals were euthanized at different time points for necropsy and paraffin embedding of relevant tissues (lymph nodes, lung, liver), for subsequent histopathological evaluation of protein levels of ATG5, the melanocytic lineage (SOX10), or classical autophagy targets (SQSTM1/p62). For assays of drug response in vivo, dabrafenib (Selleckchem, S2807) was diluted in a 5% glucose solution (Sigma, G8270) and administered at 10 mg/kg orally, once a day, during 3 wk, treatment starting one wk after tamoxifen induction. A minimum of 200 lesions were analyzed per experimental condition. Response rates were represented as the reduction in the size (diameter) and tumor number at the indicated time points, and represented with respect to vehicle-treated controls. To account for gender and variability, paired t test was performed by comparing the averages calculated for each gender and same genotype in each experiment. All experiments with mice were performed in accordance with protocols approved by the Institutional Ethics Committee of the CNIO and the Instituto de Salud Carlos III.
Histology and immunohistochemistry
Hematoxylin and eosin stainings were performed in formalin-fixed paraffin-embedded tissues. For immunohistochemistry, we used the Discovery XT automated IHC platform (Roche/Ventana Medical Systems, Tucson, AZ, US). Briefly, paraffin sections were cut at 2 μm. Sections were then deparaffinized in xylene and rehydrated through a graded series of ethanol (100%, 96%, 70%), using standard procedures. Heat-induced antigen retrieval was performed with the Dako PT link and Target retrieval solution (PT100/PT101 and S1699, respectively), essentially as previously described.24 Sections were incubated with the corresponding primary antibody (see below) at room temperature. Depending on the primary antibody, 2 different detection systems were used for visualization: (i) Ventana UltraView Universal DAB visualization system (Roche, 760–500); or (ii) DISCOVERY OmniMap anti-Rb HRP (RUO) detection system (Roche, 760–4311) and an intermediate RbaG linker (Dako, E0466). Sections were finally washed, counterstained with Carazzi´s hematoxylin (Panreac Quimica, 2552981610), dehydrated through a graded alcohol series, cleared in xylene and permanently mounted (Tissue-Tek GLas mounting medium, Sakura Finetek, 1408GLas). For the detection of micrometastases, 100 serial sections were performed per lung specimen. Hematoxilin and eosin-stained sections were then scanned with an automated computerized image analysis system (Ariol SL-50, Genetix Europe Limited, Hampshire, UK) for visualization by independent investigators. Antibodies used were as follows: ATG5 (abcam, ab108327); SOX10 (Santa Cruz Biotechnology, sc-17342); and SQSTM1/p62 (Novus Biologicals, NBP1-49956).
Fluorescence in situ hybridization (FISH)
To visualize ATG5 copy number, FISH was performed on representative human melanoma cell lines (SK-Mel-19, SK-Mel-29, SK-Mel-147, SK-Mel-103 and G-361). Bacterial artificial clones were obtained from the BACPAC Resource Center of the Children's Hospital Oakland Research Institute (CHORI, Oakland, CA, USA) spanning the ATG5 locus at 6q21 (RP11-7G03, RP1-134E15, RP11-352K22 and CTD-2025J24). Clones that hybridize to 6p21.1 (RP11-342H9, RP4-669F6, RP11-169I2) were used as a reference to estimate Chr6 copy number. ATG5 and reference probes were labeled with Spectrum Red and Spectrum Green respectively, by nick translation (Vysis/Abbot Laboratories, 32–801300) according to the manufacturer's specifications. Dual-color FISH was performed in methaphase spreads of the cell lines fixed with carnoy (methanol:acetid acid 3:1). To this end, probes were denatured at 90°C for 5 min, codenatured with the sample at 80°C for 2 min and left overnight to hybridize at 37°C in a humid dark chamber. After post-hybridization washes, slides were counterstained with DAPI in Vectashield mounting medium (Vector Laboratories, H-1000). Fluorescence signals were scored in each sample by counting the number of single-copy genes and control probe signals in 300–500 well-defined nuclei (average of 400 counts). Cell images were captured using a charge-coupled device camera (Photometrics SenSys camera) connected to a computer running the Chromofluor image analysis system (Cytovision, Applied Imaging Ltd, UK).
Supplementary Methods include details on siRNA- and shRNA-based ATG5 depletion in human melanoma cell lines, and subsequent analyses of invasive potential and proliferative capacity in adherent and nonadherent conditions (anoikis) included in Supplementary Figures.
Supplementary Material
Abbreviations
- AIM1
absent in melanoma 1
- AKT
AKT serine/threonine kinase
- ATG3
autophagy-related 3
- ATG4A
autophagy-related 4A cysteine peptidase
- Atg8
yeast autophagy-related 8
- ATG16L1
autophagy-related 16 like 1
- BECN1
Beclin 1
- BRAF
B-Raf proto-oncogene, serine/threonine kinase
- BVES
blood vessel epicardial substance
- CCLE
Cancer Cell Line Encyclopedia
- FDR
false discovery rate
- FISH
fluorescence in situ hybridization
- GABARAPL1
GABA type A receptor associated protein like 1
- GSEA
gene set enrichment analysis
- MAP1LC3A
microtubule-associated protein 1 light chain 3 α
- MAP1LC3B
microtubule- associated protein 1 light chain 3 β
- MAP2K/MEK
mitogen-activated protein kinase kinase
- MAPK
mitogen-activated protein kinase
- MITF
melanogenesis associated transcription factor
- PIK3CA
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α
- POPDC3
popeye domain containing 3
- PRDM1
PR domain 1
- PREP
prolyl endopeptidase
- PTEN
phosphatase and tensin homolog
- QRSL1
glutaminyl-tRNA synthase (glutamine-hydrolyzing)-like 1
- RB1CC1
RB1 inducible coiled-coil 1
- RTN4IP1
reticulon 4 interacting protein 1
- RSEM
RNA-sequencing by expectation maximization
- SEM
standard error of the mean
- SHH
sonic hedgehog
- SOX10
SRY-box 10
- SQSTM1/p62
sequestosome 1
- TCGA
the Cancer Genome Atlas
- Tyr
tyrosinase
- ULK1/2
unc-51 like autophagy activating kinase 1/2
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank all the colleagues in the CNIO Melanoma Group, in particular former member Alicia González-Serrano for their help and support; Sandra Barral (Columbia Univ, USA) for aid in statistical analyses; Juan Cruz Cigudosa (CNIO, Molecular Cytogenetics Unit) and Lydia Sanchez (CNIO, Histology and Immunohistochemistry Unit) for technical assistance with FISH and expression studies in tumor specimens, respectively. We also thank Jose A Esteban (CBMSO, Madrid) and Simón Méndez-Ferrer (CNIC) for critical reading of this manuscript, and Noburu Mizushima (Tokyo Medical and Dental University), Lionel Larue (Inst. Curie, France), and Martin McMahon (UCSF, USA) for Atg5flox/fox; Tyr:CreERT2, and BRAFCA mouse strains, respectively.
Funding
M.S.S. is funded by grants from the Spanish Ministry of Economy and Innovation (projects SAF2011-28317, SAF2014-56868-R and RTC-2014-2442-1), as well as a Team Science Award by the Melanoma Research Alliance, and grants from the Worldwide Cancer Research and the Asociacion Española Contra el Cancer (AECC). M.G-F was funded by a Juan de la Cierva postdoctoral fellowship from the Spanish Ministry of Education and P.K and M.C. by predoctoral fellowships from Fundación La Caixa.
References
- [1].TCGA . Genomic Classification of Cutaneous Melanoma. Cell 2015; 161:1681-96; PMID:26091043; http://dx.doi.org/ 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev 2012; 26:1131-55; PMID:22661227; http://dx.doi.org/ 10.1101/gad.191999.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al.. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499:214-8; PMID:23770567; http://dx.doi.org/ 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hodis E, Watson IR, Kryukov GV, Arold S, Imielinski M, Thereurillat J-P, Nickerson E, Auclair D, Li L, Place C, et al.. A Landscape of Driver Mutations in Melanoma. Cell 2012; 150:251-63; PMID:22817889; http://dx.doi.org/ 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science 2013; 339:957-9; PMID:23348506; http://dx.doi.org/ 10.1126/science.1229259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al.. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012; 44:1006-14; PMID:22842228; http://dx.doi.org/ 10.1038/ng.2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Whiteman DC, Pavan WJ, Bastian BC. The melanomas: a synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell Melanoma Res 2011; 24:879-97; PMID:21707960; http://dx.doi.org/ 10.1111/j.1755-148X.2011.00880.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Villanueva J, Herlyn M. Melanoma and the tumor microenvironment. Curr Oncol Rep 2008; 10:439-46; PMID:18706274; http://dx.doi.org/ 10.1007/s11912-008-0067-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010; 141:583-94; PMID:20478252; http://dx.doi.org/ 10.1016/j.cell.2010.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aplin A, Bosenberg M, Soengas M, Kos L, Arnheiter H, Kelsh R. Unmet needs in melanoma research. Pigment Cell Melanoma Res 2014; 27:1003; PMID:25346049; http://dx.doi.org/ 10.1111/pcmr.12321 [DOI] [PubMed] [Google Scholar]
- [11].Akavia UD, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, Causton HC, Pochanard P, Mozes E, Garraway LA, Pe'er D. An integrated approach to uncover drivers of cancer. Cell 2010; 143:1005-17; PMID:21129771; http://dx.doi.org/ 10.1016/j.cell.2010.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer 2006; 6:593-602; PMID:16862190; http://dx.doi.org/ 10.1038/nrc1947 [DOI] [PubMed] [Google Scholar]
- [13].Pinner S, Jordan P, Sharrock K, Bazley L, Collinson L, Marais R, Bonvin E, Goding C, Sahai E. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res 2009; 69:7969-77; PMID:19826052; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vance KW, Goding CR. The transcription network regulating melanocyte development and melanoma. Pigment Cell Res 2004; 17:318-25; PMID:15250933; http://dx.doi.org/ 10.1111/j.1600-0749.2004.00164.x [DOI] [PubMed] [Google Scholar]
- [15].Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J, Hutchinson P, Tse G, et al.. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013; 24:466-80; PMID:24075834; http://dx.doi.org/ 10.1016/j.ccr.2013.08.018 [DOI] [PubMed] [Google Scholar]
- [16].Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE, et al.. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013; 504:138-42; PMID:24185007; http://dx.doi.org/ 10.1038/nature12688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al.. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014; 4:94-109; PMID:24265153; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Foppen MH, et al.. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014; 5:5712; PMID:25502142; http://dx.doi.org/ 10.1038/ncomms6712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al.. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005; 436:117-22; PMID:16001072; http://dx.doi.org/ 10.1038/nature03664 [DOI] [PubMed] [Google Scholar]
- [20].Thurber AE, Douglas G, Sturm EC, Zabierowski SE, Smit DJ, Ramakrishnan SN, Hacker E, Leonard JH, Herlyn M, Sturm RA. Inverse expression states of the BRN2 and MITF transcription factors in melanoma spheres and tumour xenografts regulate the NOTCH pathway. Oncogene 2011; 30:3036-48; PMID:21358674; http://dx.doi.org/ 10.1038/onc.2011.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bell RE, Levy C. The three M's: melanoma, microphthalmia-associated transcription factor and microRNA. Pigment Cell Melanoma Res 2011; 24:1088-106; PMID:22004179; http://dx.doi.org/ 10.1111/j.1755-148X.2011.00931.x [DOI] [PubMed] [Google Scholar]
- [22].Javelaud D, Alexaki VI, Pierrat MJ, Hoek KS, Dennler S, Van Kempen L, Bertolotto C, Ballotti R, Saule S, Delmas V, et al.. GLI2 and M-MITF transcription factors control exclusive gene expression programs and inversely regulate invasion in human melanoma cells. Pigment Cell Melanoma Res 2011; 24:932-43; PMID:21801332; http://dx.doi.org/ 10.1111/j.1755-148X.2011.00893.x [DOI] [PubMed] [Google Scholar]
- [23].Koludrovic D, Davidson I. MITF, the Janus transcription factor of melanoma. Future Oncol 2013; 9:235-44; PMID:23414473; http://dx.doi.org/ 10.2217/fon.12.177 [DOI] [PubMed] [Google Scholar]
- [24].Alonso-Curbelo D, Riveiro-Falkenbach E, Perez-Guijarro E, Cifdaloz M, Karras P, Osterloh L, Megias D, Canon E, Calvo TG, Olmeda D, et al.. RAB7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell 2014; 26:61-76; PMID:24981740; http://dx.doi.org/ 10.1016/j.ccr.2014.04.030 [DOI] [PubMed] [Google Scholar]
- [25].Alonso-Curbelo D, Osterloh L, Canon E, Calvo TG, Martinez-Herranz R, Karras P, Martinez S, Riveiro-Falkenbach E, Romero PO, Rodriguez-Peralto JL, et al.. RAB7 counteracts PI3K-driven macropinocytosis activated at early stages of melanoma development. Oncotarget 2015; 6:11848-62; PMID:26008978; http://dx.doi.org/ 10.18632/oncotarget.4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Alonso-Curbelo D, Soengas MS. Hyperactivated endolysosomal trafficking in melanoma. Oncotarget 2015; 6:2583-4; PMID:25682879; http://dx.doi.org/ 10.18632/oncotarget.3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ploper D, Taelman VF, Robert L, Perez BS, Titz B, Chen HW, Graeber TG, von Euw E, Ribas A, De Robertis EM. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc Natl Acad Sci U S A 2015; 112:E420-9; PMID:25605940; http://dx.doi.org/ 10.1073/pnas.1424576112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Checinska A, Soengas MS. The gluttonous side of malignant melanoma: basic and clinical implications of macroautophagy. Pigment Cell Melanoma Res 2012; 24:1116-32; http://dx.doi.org/ 10.1111/j.1755-148X.2011.00927.x [DOI] [PubMed] [Google Scholar]
- [29].Maes H, Agostinis P. Autophagy and mitophagy interplay in melanoma progression. Mitochondrion 2014; 19Pt A:58-68; PMID:25042464; http://dx.doi.org/ 10.1016/j.mito.2014.07.003 [DOI] [PubMed] [Google Scholar]
- [30].Maddodi N, Huang W, Havighurst T, Kim K, Longley BJ, Setaluri V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J Invest Dermatol 2010; 130:1657-67; PMID:20182446; http://dx.doi.org/ 10.1038/jid.2010.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326-36; PMID:17235397; http://dx.doi.org/ 10.1172/JCI28833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Goodall ML, Wang T, Martin KR, Kortus MG, Kauffman AL, Trent JM, Gately S, MacKeigan JP. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy 2014; 10:1120-36; PMID:24879157; http://dx.doi.org/ 10.4161/auto.28594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ma Y, Hendershot LM. The stressful road to antibody secretion. Nat Immunol 2003; 4:310-1; PMID:12660729; http://dx.doi.org/ 10.1038/ni0403-310 [DOI] [PubMed] [Google Scholar]
- [34].Tormo D, Checinska A, Alonso-Curbelo D, Perez-Guijarro E, Canon E, Riveiro-Falkenbach E, Calvo TG, Larribere L, Megias D, Mulero F, et al.. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell 2009; 16:103-14; PMID:19647221; http://dx.doi.org/ 10.1016/j.ccr.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, Marra MA, Gorski SM. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015; 11:1668-87; PMID:26208877; http://dx.doi.org/ 10.1080/15548627.2015.1067362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lazova R, Klump V, Pawelek J. Autophagy in cutaneous malignant melanoma. J Cutan Pathol 2010; 37:256-68. PMID: 19615007; http://dx.doi.org/20004946 10.1111/j.1600-0560.2009.01359.x [DOI] [PubMed] [Google Scholar]
- [37].Miracco C, Cevenini G, Franchi A, Luzi P, Cosci E, Mourmouras V, Monciatti I, Mannucci S, Biagioli M, Toscano M, et al.. Beclin 1 and LC3 autophagic gene expression in cutaneous melanocytic lesions. Hum Pathol 2010; 41:503-12; PMID:20004946; http://dx.doi.org/ 10.1016/j.humpath.2009.09.004 [DOI] [PubMed] [Google Scholar]
- [38].Sivridis E, Koukourakis MI, Mendrinos SE, Karpouzis A, Fiska A, Kouskoukis C, Giatromanolaki A. Beclin-1 and LC3A expression in cutaneous malignant melanomas: a biphasic survival pattern for beclin-1. Melanoma Res 2011; 21:188-95; PMID:21537144; http://dx.doi.org/ 10.1097/CMR.0b013e328346612c [DOI] [PubMed] [Google Scholar]
- [39].Xie X, Koh JY, Price S, White E, Mehnert JM. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov 2015; 5:410-23; PMID:25673642; http://dx.doi.org/ 10.1158/2159-8290.CD-14-1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu H, He Z, von Rutte T, Yousefi S, Hunger RE, Simon HU. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med 2013; 5:202ra123; PMID:24027027; http://dx.doi.org/ 10.1126/scitranslmed.3005864 [DOI] [PubMed] [Google Scholar]
- [41].Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014; 26:190-206. PMID:25117709; http://dx.doi.org/20811353 10.1016/j.ccr.2014.06.025. [DOI] [PubMed] [Google Scholar]
- [42].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/ 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al.. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483:603-7; PMID:22460905; http://dx.doi.org/ 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Marzese DM, Scolyer RA, Huynh JL, Huang SK, Hirose H, Chong KK, Kiyohara E, Wang J, Kawas NP, Donovan NC, et al.. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum Mol Genet 2014; 23:226-38; PMID:24014427; http://dx.doi.org/ 10.1093/hmg/ddt420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, et al.. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001; 409:207-11; PMID:11196646; http://dx.doi.org/ 10.1038/35051606 [DOI] [PubMed] [Google Scholar]
- [46].Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68-76; PMID:20562859; http://dx.doi.org/ 10.1038/nature09204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Trent JM, Rosenfeld SB, Meyskens FL. Chromosome 6q involvement in human malignant melanoma. Cancer Genet Cytogenet 1983; 9:177-80; PMID:6850557; http://dx.doi.org/ 10.1016/0165-4608(83)90039-0 [DOI] [PubMed] [Google Scholar]
- [48].Hoshimoto S, Kuo CT, Chong KK, Takeshima TL, Takei Y, Li MW, Huang SK, Sim MS, Morton DL, Hoon DS. AIM1 and LINE-1 epigenetic aberrations in tumor and serum relate to melanoma progression and disease outcome. J Invest Dermatol 2012; 132:1689-97; PMID:22402438; http://dx.doi.org/ 10.1038/jid.2012.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kim M, Jang HR, Haam K, Kang TW, Kim JH, Kim SY, Noh SM, Song KS, Cho JS, Jeong HY, et al.. Frequent silencing of popeye domain-containing genes, BVES and POPDC3, is associated with promoter hypermethylation in gastric cancer. Carcinogenesis 2010; 31:1685-93; PMID:20627872; http://dx.doi.org/ 10.1093/carcin/bgq144 [DOI] [PubMed] [Google Scholar]
- [50].Karube K, Nakagawa M, Tsuzuki S, Takeuchi I, Honma K, Nakashima Y, Shimizu N, Ko YH, Morishima Y, Ohshima K, et al.. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood 2011; 118:3195-204; PMID:21690554; http://dx.doi.org/ 10.1182/blood-2011-04-346890 [DOI] [PubMed] [Google Scholar]
- [51].Santos GC, Zielenska M, Prasad M, Squire JA. Chromosome 6p amplification and cancer progression. J Clin Pathol 2007; 60:1-7; PMID:16790693; http://dx.doi.org/ 10.1136/jcp.2005.034389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/ 10.1038/nature03029 [DOI] [PubMed] [Google Scholar]
- [53].Yajima I, Belloir E, Bourgeois Y, Kumasaka M, Delmas V, Larue L. Spatiotemporal gene control by the Cre-ERT2 system in melanocytes. Genesis 2006; 44:34-43; PMID:16419042; http://dx.doi.org/ 10.1002/gene.20182 [DOI] [PubMed] [Google Scholar]
- [54].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/ 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- [55].Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009; 41:544-52; PMID:19282848; http://dx.doi.org/ 10.1038/ng.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436:720-4; PMID:16079850; http://dx.doi.org/ 10.1038/nature03890 [DOI] [PubMed] [Google Scholar]
- [57].Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, et al.. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol 2006; 8:1053-63; PMID:16964246; http://dx.doi.org/ 10.1038/ncb1471 [DOI] [PubMed] [Google Scholar]
- [58].Haass NK, Smalley KS, Li L, Herlyn M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res 2005; 18:150-9; PMID:15892711; http://dx.doi.org/ 10.1111/j.1600-0749.2005.00235.x [DOI] [PubMed] [Google Scholar]
- [59].Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V, et al.. A dual role for autophagy in a murine model of lung cancer. Nat Commun 2014; 5:3056; PMID:24445999; http://dx.doi.org/ 10.1038/ncomms4056 [DOI] [PubMed] [Google Scholar]
- [60].Thorburn A, Morgan MJ. Targeting Autophagy in BRAF-Mutant Tumors. Cancer Discov 2015; 5:353-4; PMID:25847956; http://dx.doi.org/ 10.1158/2159-8290.CD-15-0222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Martin S, Dudek-Peric AM, Maes H, Garg AD, Gabrysiak M, Demirsoy S, Swinnen JV, Agostinis P. Concurrent MEK and autophagy inhibition is required to restore cell death associated danger-signalling in Vemurafenib-resistant melanoma cells. Biochem Pharmacol 2015; 93:290-304; PMID:25529535; http://dx.doi.org/ 10.1016/j.bcp.2014.12.003 [DOI] [PubMed] [Google Scholar]
- [62].Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, Piacentini M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ 2015; 22:946-58; PMID:25361077; http://dx.doi.org/ 10.1038/cdd.2014.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Flaherty KT. Targeting metastatic melanoma. Annu Rev Med 2012; 63:171-83; PMID:22034865; http://dx.doi.org/ 10.1146/annurev-med-050410-105655 [DOI] [PubMed] [Google Scholar]
- [64].Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57-70; PMID:10647931; http://dx.doi.org/ 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- [65].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- [66].Zhang T, Zhou Q, Ogmundsdottir MH, Moller K, Siddaway R, Larue L, Hsing M, Kong SW, Goding CR, Palsson A, et al.. Mitf is a master regulator of the v-ATPase, forming a control module for cellular homeostasis with v-ATPase and TORC1. J Cell Sci 2015; 128:2938-50; PMID:26092939; http://dx.doi.org/ 10.1242/jcs.173807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Luke JJ, Hodi FS. Ipilimumab, vemurafenib, dabrafenib, and trametinib: synergistic competitors in the clinical management of BRAF mutant malignant melanoma. Oncologist 2013; 18:717-25. PMID: 23709751; http://dx.doi.org/17909521 10.1634/theoncologist.2012-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-9; PMID:17909521; http://dx.doi.org/ 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- [69].Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol 2015; 25:354-63. PMID: 25759175; http://dx.doi.org/25695667 10.1016/j.tcb.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang L, Wang Y, Lu Y, Zhang Q, Qu X. Heterozygous deletion of ATG5 in Apc(Min/+) mice promotes intestinal adenoma growth and enhances the antitumor efficacy of interferon-gamma. Cancer Biol Ther 2015; 16:383-91; PMID:25695667; http://dx.doi.org/ 10.1080/15384047.2014.1002331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bis S, Tsao H. Melanoma genetics: the other side. Clin Dermatol 2013; 31:148-55; PMID:23438378; http://dx.doi.org/ 10.1016/j.clindermatol.2012.08.003 [DOI] [PubMed] [Google Scholar]
- [72].Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol 2014; 9:239-71; PMID:24460190; http://dx.doi.org/ 10.1146/annurev-pathol-012513-104658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9-29; PMID:24399786; http://dx.doi.org/ 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- [74].Harbour JW. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res 2012; 25:171-81; PMID:22268848; http://dx.doi.org/ 10.1111/j.1755-148X.2012.00979.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kraya AA, Piao S, Xu X, Zhang G, Herlyn M, Gimotty P, Levine B, Amaravadi RK, Speicher DW. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy 2015; 11:60-74; PMID:25484078; http://dx.doi.org/ 10.4161/15548627.2014.984273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Marino S, Krimpenfort P, Leung C, van der Korput HA, Trapman J, Camenisch I, Berns A, Brandner S. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development 2002; 129:3513-22; PMID:12091320 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.