

Graphical abstract
Keywords: Vaccine, Nanoparticles, SAPN, Rationally designed, Adjuvant
Abstract
Vaccines have been the single most significant advancement in public health, preventing morbidity and mortality in millions of people annually. Vaccine development has traditionally focused on whole organism vaccines, either live attenuated or inactivated vaccines. While successful for many different infectious diseases whole organisms are expensive to produce, require culture of the infectious agent, and have the potential to cause vaccine associated disease in hosts. With advancing technology and a desire to develop safe, cost effective vaccine candidates, the field began to focus on the development of recombinantly expressed antigens known as subunit vaccines. While more tolerable, subunit vaccines tend to be less immunogenic. Attempts have been made to increase immunogenicity with the addition of adjuvants, either immunostimulatory molecules or an antigen delivery system that increases immune responses to vaccines. An area of extreme interest has been the application of nanotechnology to vaccine development, which allows for antigens to be expressed on a particulate delivery system. One of the most exciting examples of nanovaccines are rationally designed protein nanoparticles. These nanoparticles use some of the basic tenants of structural biology, biophysical chemistry, and vaccinology to develop protective, safe, and easily manufactured vaccines. Rationally developed nanoparticle vaccines are one of the most promising candidates for the future of vaccine development.
1. A basic overview of vaccine function
Vaccines are one of the greatest public health innovations in human history. Vaccination provides an extremely effective mechanism to deal with infectious diseases by preventing the development of morbidity and mortality. The World Health Organization estimates that vaccines prevent 2–3 million human deaths annually, and these numbers would rise by at least 6 million if all children received the recommended vaccination schedule [1]. Only two infectious diseases have been eliminated in human history, both the result of a successful vaccination campaign. The first, the human disease small-pox, was officially declared eliminated from the human population in 1979 [2]. The second, the livestock disease rinderpest was declared eliminated in 2011 [3]. While other diseases such as measles and polio are also close to elimination there is still much to be done [2].
Infectious disease vaccines work by serving as a prophylactic controlled exposure to an infectious agent. This initial exposure ideally induces a strong immune response in a vaccinated individual. A vertebrate’s immune system is composed of two different branches, the innate and adaptive immune system. Following exposure to an infectious agent or administration of a vaccine, activation of the innate immune system precedes generation of adaptive immunity. The innate immune system is composed of a diverse array of cell types such as neutrophils, dendritic cells, monocytes, macrophage, and eosinophils all of which function to interact with foreign molecules in a nonspecific manner. Innate immune cells phagocytose infectious agents, secrete inflammatory cytokines, and/or attract and activate other immune cells through the secretion of chemical messengers such as chemokines. These processes lead to initiation of an effective immune response [4].
Vaccines are ultimately dependent on the development of an effective adaptive immune response. Broadly, adaptive immune responses are divided into two different categories, humoral and cellular. Cells of the adaptive immune system respond to specific regions of infectious agents known as epitopes. One or more epitopes are contained on a larger molecule known as an antigen. Humoral immune responses are dependent on the activity of antibodies, secreted glycoproteins from B cells that bind to specific epitopes. A naïve B cell contains B cell receptors on its surface, which vary in their specificities. Upon binding of the B cell receptor to a matching epitope, B cells can mature into plasma cells and begin to secrete epitope specific antibodies that will ideally lead to protection against infection [5].
Cellular immune responses are based on the action of T cells. All nucleated cells have on their surface Major Histocompatibility Complex Class I (MHC-I) molecules. When infected with an intracellular infectious agent, cells are able to present on their surface linear epitopes from those infectious agents complexed with MHC-I to alert the immune system of the infection. Cytotoxic T cells (TC) that contain the matching T cell receptor are able to bind to the MHC-I presenting specific epitopes leading to the death of the infected cell [4].
One of the most important cell types in vaccine development are T helper cells (TH). Antigen Presenting Cells (APCs) such as dendritic cells, macrophage, and B cells are able to phagocytize, process, and present CD4+ epitopes in complex with Major Histocompatibility Complex Class II (MHC-II) on their surface. These epitopes stimulate CD4+ T cells leading to their maturation into TH cells. Active TH cells are able to stimulate cells of both the innate and adaptive immune system through the secretion of cytokines. These cytokines are able to modulate the immune response leading to a stronger and more effective immune response. Based on the profile of the secreted cytokine responses they are either classed as T helper 1 response (TH1) or T helper 2 response (TH2). TH1 responses favor the development of a cellular based immune response, while TH2 responses favor the development of a humoral immune response. Traditionally, vaccine development has focused on the development of strong TH2 responses, but currently a vaccine candidate that has a balanced TH1/TH2 response is considered optimal [5].
After activation, B cells, TH, and TC undergo proliferation to effectively deal with infection. In an ideal situation some of these cells persist after clearance resulting in the development of immunological memory. When a previously exposed host is exposed to an infectious agent again, antigen-specific immune memory cells are activated and proliferate faster and to a greater magnitude, leading to rapid clearance of the infectious agent and mitigation of disease. Strong and effective memory responses protect hosts against subsequent infections leading to lifelong immunity, the hallmark of an effective vaccine [5].
Vaccines not only work on the organismal level, but also on the population level. In the concept known as herd immunity if a certain fraction of the population is immune to an infectious agent the disease will have a very low likelihood of finding another naïve host and spreading. The number of people who need to be vaccinated for herd immunity varies from disease to disease, normally between 60 and 90%. It is extremely important because in any given population some vaccinated individuals will not develop protection based upon genetics, there will be individuals who cannot be vaccinated because of age or disease state, and there will be some unvaccinated individuals [6], [7]. Herd immunity is the altruistic side of vaccination that will ultimately lead to the elimination of pathogens from either the human or animal population.
2. The origin of vaccines
By the 15th century there are documented attempts in Middle Eastern and Asian cultures to prevent small-pox infection by variolation. In these cultures, the pustules from patients with mild cases of small-pox were taken and dried, then used to scratch the surface of another patient’s skin, or inhaled. It was a way to inoculate people against a more severe form of the disease. It was protective, with lower death rates than infection of a naïve person with the small-pox virus. The concept of variolation was brought back to Europe in 1718 by Lady Mary Wortley Montagu, the wife of the British ambassador to the Ottoman Empire. She saw the practice and had her children variolated to prevent them from becoming infected with small-pox [8].
Edward Jenner, a country doctor in late 18th century in England, made two key observations. The first was that milkmaids previously infected with cowpox, a zoonotic disease that is easily transmitted from cows to humans, did not develop smallpox. He also noted that when he variolated patients who recovered from cow-pox they did not develop a response of a typical small-pox lesion. He reasoned that by inoculating people with the material contained in cow-pox pustules he would protect them against subsequent infection with small-pox. He performed the first known vaccine trial in 1796 by taking cow-pox pustules from a milkmaid, and inoculating an 8-year-old boy. He noted that boy felt general malaise for a day, but recovered quickly. He later variolated the child with small-pox, however, the child did not show signs of becoming infected with the disease [9].
While somewhat controversial in his time Jenner spent the rest of his life publicizing his technique. At this point the germ theory of disease had not been established, and people did not understand that both small-pox and cow-pox were caused by closely related viruses. Many people had concerns that vaccination with a different disease would not actually lead to protection. It was not until 1837 when England began keeping Cause of Death Records, that William Farr was able to determine that communities that have had high vaccination rates had low rates of death from small-pox. Ultimately, in 1840 variolation was banned in England and vaccination became the standard prophylactic treatment for small-pox [8], [10]. Jenner had succeeded in the development and implementation of the world’s first vaccine (Fig. 1 , Table 1 ).
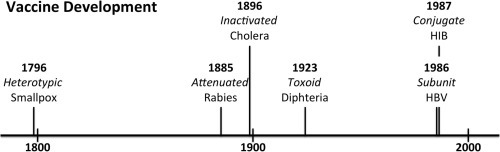
Fig. 1.
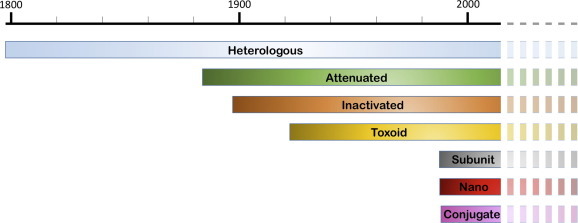
A timeline showing when different classes of vaccines became clinically available and the overlap of availability. Some vaccines like Hepatitis B technically fall into multiple classes such as subunit as well as nanovaccines. Lighter/dashed colors indicate that these classes will most likely remain important for the foreseeable future. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
The major classes of vaccines currently or previously used.
| Class | Description | First disease target | Year first used |
|---|---|---|---|
| Heterologous | A closely related microorganism | Smallpox | 1796 |
| Attenuated | A weakened microorganism | Rabies | 1885 |
| Inactivated | A killed microorganism | Cholera | 1896 |
| Toxoid | Inactivated bacterial toxin | Diphtheria | 1923 |
| Subunit | One or multiple antigens from an infectious agent | Hepatitis B | 1986 |
| Conjugate | A weakly immunogenic polysaccharide antigen attached to an immunogenic protein | Haemophilus influenza type B (HIB) | 1987 |
French Microbiologist and Chemist Louis Pasteur made the next major advance in the development of vaccines. In 1879 while studying chicken cholera, Pasteurella multocida, he had chickens inoculated with a month old culture after a vacation. The inoculated chickens developed minor symptoms of the disease, but recovered. He later inoculated the same chickens with a fresh culture of bacteria and saw that chickens previously inoculated with the old culture were protected from infection, while naïve birds still developed symptoms [10]. Pasteur had stumbled onto the concept of attenuation. If microorganisms are grown in suboptimal conditions, or are treated with certain chemicals they are not as virulent as microorganisms grown under ideal conditions. By exposing the chickens to the attenuated bacteria Pasteur was able to induce protection against subsequent lethal challenge with the virulent P. multocida. In 1881 Pasteur was able to repeat similar findings by attenuating Bacillus anthracis and vaccinating farm animals with the attenuated B. anthracis [11].
In 1879 Pierre Galtier had discovered that something in the saliva of rabid dogs caused rabies in other mammals. By 1884 Pasteur had developed a way to propagate the infectious agent, decreasing the incubation time to days instead of months. He demonstrated that by inoculating dogs and other mammals with his attenuated strain of the rabies virus the animals were protected when challenged with the normal rabies virus. When 9 year old Joseph Meister was bitten by a rabid dog in 1885 Pasteur was able to vaccinate him with the attenuated virus preventing the boy from developing rabies [11] (Table 1). Pasteur was able to demonstrate that an attenuated infectious agent could still result in protection of vaccinated humans against subsequent exposure to the virulent infectious agent. His work led to the development of attenuated vaccines for typhoid fever, cholera, and plague in the late 19th and the early 20th century [10] (Fig. 1).
Scientists in the same era began to determine that some diseases such as diphtheria are caused by the toxins produced by invading bacteria. Scientists recognized that Corynebacterium diphtheria, the causative agent of diphtheria, produced an exotoxin. They also noted that animals that were given sub-lethal doses of purified toxin developed an antidote in their serum. These antidotes quickly became to be understood as antibodies that bind to the toxin, antigen, in a specific conformation leading to neutralization [10], [12]. Using a similar concept as Pasteur, scientists developed a vaccine candidate known as a toxoid vaccine that consisted of diphtheria toxin treated with formalin. While no longer toxic to the animal it is still immunogenic leading to the development of antibodies and ultimately protection against diphtheria [10], [13], [14] (Fig. 1, Table 1).
3. The golden age of vaccinology
Vaccine development was initially slow, partially because of the lack of techniques to culture infectious agents. Viruses are intracellular parasites and need host tissue to reproduce and grow, so viral culture was dependent on the development of an effective tissue culture system. The first attempts at growing animal tissues in vitro occurred in 1885, when Wilhelm Roux was able to sustain part of a chicken embryo in a saline solution for a few days. Over the next 50 years the techniques and ability of scientists to maintain animal cultures began to improve [15], [16], [17]. By the mid-twentieth century tissue culture techniques had matured to the level were scientists were able to propagate viruses in culture, including in human tissues [18], [19].
One of the first diseases targeted for vaccine development was poliomyelitis, polio. Polio is caused by one of three strains of the Enterovirus, Poliovirus. While polio was present in human populations since recorded history, epidemics only began to occur in the late nineteenth/early twentieth centuries. The worst recorded epidemic occurred in the United States in 1952 which resulted in 57,628 cases, 3145 deaths, and 21,269 cases of paralysis [20]. By this point the virus had been already cultured by John Enders, and vaccine development was underway [19]. Two major approaches developed that would ultimately give clues to how vaccinology would progress over the next 60 years.
The first approach was pioneered by Jonas Salk. Salk had previously worked on an inactivated influenza vaccine, and believed the most effective candidate would be an inactivated poliovirus [21]. By this point scientists knew that protection against polio was mediated by Immunoglobulin G (IgG), and that viremia preceded any paralytic effects in patients. Salk hypothesized that by injecting patients with inactivated poliovirus they would develop the IgG response necessary for preventing clinical symptoms of the disease from developing. Salk’s inactivated trivalent polio vaccine (IPV) consisted of one of each of the three different strains of the Poliovirus that had been formalin inactivated. He developed the vaccine in 1952, and by 1953 had performed initial animal and human studies [22]. Over the next two years clinical studies increased in size until the IPV was determined to be safe and between 80 and 90% effective depending on the strain of poliovirus contracted [23]. The vaccine was approved in the United States in 1955. Between 1954 and 1961 the incidence of paralytic polio in the US decreased from 13.9/100,000 to 0.8/100,000 due to the Salk Vaccine [24].
The second approach was the use of an attenuated oral polio vaccine (OPV). Some scientists believed that an IPV would not induce long term protection; the most famous of this group was Albert Sabin. He proposed using an attenuated virus delivered through an oral route, the natural infection route of the virus. By 1954 Sabin had generated four different OPV candidates, and by 1956 he had determined the three attenuated viral strains that would ultimately wind up in his final vaccine candidate [25], [26]. His OPV candidate underwent clinical studies between 1957 and 1960 in the United States, USSR, and around the world [26]. The OPV was approved in 1961 in the United States, and by 1965 had become the most widely accepted polio vaccine [27], [28]. One of the major advantages of the OPV vaccines is because patients actually become infected with the vaccine they are able to spread the OPV viruses to their surrounding communities as they would if they were infected with a virulent strain of the virus [29]. The major disadvantage is that in a small number of patients spontaneous reversions will occur in the attenuated vaccine strains that lead to a virulent virus that can lead to vaccine derived paralytic poliomyelitis (VDPP) in the patient and their surrounding community [30].
The work of Salk and Sabin both moved the field of vaccinology further. They demonstrated that with two different approaches, it was possible to develop vaccine candidates that were able to prevent polio epidemics, and that each method has its own strengths and weaknesses. After 1965 most of the world moved to using the OPV in place of the IPV [27]. Because of the risk of the development of VDPP from the OPV, countries that have eliminated the disease have tended to move away from the trivalent OPV to IPV again [31]. Currently, the United States uses only the IPV vaccine. This history also highlights the frequently changing needs of vaccine development in light of the biology and epidemiology of the infectious agent.
The “golden age” of vaccinology also saw the generation of three other extremely important attenuated-virus vaccine candidates for childhood diseases. The first was for measles, a highly contagious disease that greater than 90% of the people eventually developed [32]. The second disease was mumps. The final vaccine candidate was for rubella. By the mid-1970s these three vaccines would be combined into a single multivalent attenuated-virus vaccine candidate, the measles, mumps, and rubella vaccine (MMR) [33]. While different corporations produce different formulations of the same product the MMR vaccine has been determined to be safe and effective at reducing global morbidity and mortality from measles, mumps, and rubella [34].
4. The third phase of vaccine development: subunit vaccines
During the 1950s and 1960s scientists’ understanding of molecular microbial genetics increased. New knowledge as to the role of DNA in the cell, the nature of genes, the function of phage, and the discovery of restriction enzymes led to the proposal that DNA molecules could be modified to include foreign DNA in the early 1970s [35]. Within fifteen years techniques were developed to generate recombinant DNA, transform or transduce it into a host cell such as Escherichia coli, Saccharomyces cerevisiae, or a baculovirus-insect cell expression system to produce recombinant proteins. Over the subsequent 40 years more and more expression systems have been developed only increasing the ability to produce recombinant proteins.
This revolution in biology allowed vaccinologists to approach the concept of vaccines in a new light, working under the hypothesis that using specific antigens from various infectious agents, safe, cheap, and effective vaccine candidates could be produced. All of the previous inactivated vaccines, attenuated vaccines, and toxoid vaccines required a culture of a given infectious agent before a vaccine candidate could be developed. By using recombinant DNA technology, protein antigens from organisms that are either unculturable, highly pathogenic, or extremely expensive to culture could be generated. Subunit vaccines reduce the risk of side effects such as spontaneous reversions of attenuated vaccines and denaturing of antigenic peptides with inactivated vaccines [36].
There are subunit vaccine candidates for almost every known human or animal vaccine in existence. However, there is a major problem with these subunit vaccines. Exposure to whole organism vaccines, whether inactivated, attenuated, or from a closely related species, does not just expose a subject to just one copy of an antigen in a vacuum. Whole organism vaccines contain multiple copies of each antigen, as well as other immunostimulatory molecules. The resulting problem is that subunit vaccines in general are not as immunostimulatory [37]. This effect is most likely caused by reduced ability to cross link B cell receptors as well as a reduced ability to stimulate APCs [4]. Many approaches have been taken to develop a subunit vaccine that is not only safe, easy to produce, but also as effective as a whole organism vaccine.
5. Increasing the immune response to subunit vaccines: adjuvants
One way that vaccines can be modified to increase their efficacy is the addition of an adjuvant. The word adjuvant comes from the Latin word adiuvare, which means to help. In the simplest terms an adjuvant is something that helps a vaccine function. That could mean the adjuvant allows the vaccine to be more immunogenic, induce a stronger humoral or a cellular immune response, increase antigen processing by APC, decrease the total amount of vaccine that needs to be injected in a processes known as dose sparing, or aid in the development of a long term memory response [38]. Broadly, there are two main classes of adjuvants, immunopotentiators and delivery systems. Immunopotentiators stimulate the immune system, while delivery systems function to carry and present the vaccine to the hosts’ immune system [39]. While adjuvants have been used with most vaccine formulations almost since the dawn of the vaccine age, they are essential for use with any subunit vaccine. Despite being used for almost a century adjuvants are not well understood, and any new vaccine/immunopotentiator/delivery system formulation needs to be tested for efficacy and toxicity [40].
The first known and most widely used adjuvant are aluminum salts, also known as Alum. Aluminum containing salts are currently the only universally approved adjuvant for human use in the United States. Aluminum potassium sulfate was first identified in 1926 to induce higher titers of antibodies in Guinea pigs when injected in formulation with a diphtheria toxoid, in comparison to the toxoid vaccine injected alone [41]. Subsequently, it has been demonstrated that aluminum-containing salts result in stronger humoral responses to vaccines, but do not affect cellular responses. The exact mechanism of action is not completely understood and still widely debated, and most likely depends on the vaccine, route of injection, and organism the vaccine is being injected into. Generally, it is known that Alum attracts innate immune cells, particularly immature dendritic cells to the site of vaccination. The cells are able to mature and present the vaccine to TH cells in lymph nodes, which are subsequently able to stimulate B cells that have encountered the vaccine to generate high titers of antibodies. Alum also binds to proteins and has the potential, depending on vaccine route, to function as both as a immunopotentiator and as a delivery system [42], [43].
Another major class of adjuvants are the emulsions. The first emulsion adjuvant was generated by Jules Freund in 1944 and contained mineral oil, an emulsifying agent, and dried ground Mycobacterium tuberculosis creating a water-in-oil emulsion [44], [45]. This formulation known as Freund’s Complete Adjuvant (FCA) was shown to stimulate both the humoral and the cellular immune pathways. Ultimately, because of the presence of M. tuberculosis it was determined to be too dangerous to use in humans. This resulted in the generation of Freund’s Incomplete Adjuvant (IFA), which contained everything in FCA except the M. tuberculosis. IFA leads to primarily a humoral response to vaccines. Eventually, IFA was used in formulation with IPV, however, its use was discontinued in humans due to it being too reactive [46].
The initial successes with the immunogenicity of emulsions lead to the desire to create safer alternatives to FCA or IFA that would be as immunogenic, without the side effects. Focus moved to oil-in-water emulsions. In 1997 MF59 (Novartis) the first oil-in-water emulsion was approved in Europe. It is a mixture of squalene, span 85 (a surfactant), and tween 80 in a citrate buffer combined with the antigen [47]. It is believed to function by recruiting innate and adaptive immune cells to the site of injection, but like alum the mechanism is not completely understood. It is known to induce higher titers of antibodies as well as TC responses [47], [48]. Other companies and laboratories have developed oil-in-water emulsions to varying degrees of success. Each one is slightly different, but as of now only MF59 is approved and only for an inactivated influenza vaccine in Europe [45].
One of the most exciting areas in adjuvant development is the use of pathogen associated molecular patterns (PAMPs) as adjuvants either by themselves, or in combination with another type of adjuvant (Table 2 ). PAMPs are repetitive molecules that are present in or on invading microorganisms. Multicellular organisms have evolved pattern recognition receptors (PRR) that are germ-line encoded receptors that recognize PAMPs. Some PRR are present on the cell membrane such as Toll-Like Receptors (TLR), and C-Type Lectin Receptors (CLR). While others are internal receptors such as NOD-Like Receptors (NLR) and RIG-I-Like Receptors (RLR). Each class of PRR is further subdivided into different receptors and each has a specific target and a specific signaling pathway. Many are well understood, which is a major advantage in adjuvant development. By using PAMPs that are well understood, vaccine formulations can be developed that lead to safer and more effective immune stimulation by reducing the side effects of other adjuvants.
Table 2.
The major PAMPs that are currently being used or being investigated as potential adjuvants.
| PAMP | Microorganism of origin | Host receptor | Adaptive immune branch activated |
|---|---|---|---|
| LPS | Gram negative bacteria | TLR4/inflammasome | Humoral/cellular |
| Flagellin | Flagellated bacteria | TLR5/inflammasome | Humoral/cellular |
| ssRNA agonists | ssRNA viruses | TLR7/TRL8 | Humoral/cellular |
| CpG | microorganisms | TLR9 | Humoral/cellular |
One of the best-studied and understood PAMPs is Lipopolysaccharide (LPS) also known as endotoxin (Table 2). LPS is a component of the gram negative bacteria’s outer cell wall, composed of three different domains. The outermost region is known as the O-specific chain, the middle domain is known as the core domain, the final domain known as lipid A [49]. Lipid A is responsible for signaling through TLR4. Upon initial stimulation of TLR4 signaling through TIRAP-MyD88 adapter protein leads to transcription via AP-1 and NF-κB transcription factors of proinflammatory cytokines. As signaling continues TLR4 is endocytosed which leads to signaling through TRAM-TRIF and transcription of interleukin-I genes, leading to antimicrobial activity [50], [51]. These two responses can be activated independently of each other [52]. Wildtype LPS is not a good adjuvant, even though it is highly immunogenic. Too much LPS leads to septic shock, and too little leads to endotoxin tolerance [53], [54]. By chemically modifying Lipid A from Salmonella minnesota 595 to remove a phosphate group and generating slight alterations to the fatty acid chains a potential new adjuvant was generated known as monophosphoryl lipid A (MPLA) [49]. MPLA only signals through the TRAM-TRIF signaling pathway preventing the generation of proinflammatory cytokines [55]. This adjuvant has been shown to be able to increase serum antibody titers as well as inducing strong cellular immune responses [54], [56], [57]. In humans 10,000 times the dose of MPLA is better tolerated than wildtype lipid A [58]. This adjuvant has been approved in combination with Alum for use in the Cervarix human papilloma virus (HPV) vaccine in the United States.
Another PAMP that is a potential adjuvant candidate is flagellin, a component of the bacterial flagellum (Table 2). Each flagellin monomer is subdivided into four different domains D0, D1, D2, and D3 [59]. D0 and D1 are highly conserved regions, while D2 and D3 are considered variable regions. Flagellin stimulates cells through TLR5 via the conserved D1 domain [60], [61], [62]. Upon stimulation TLR5 signals through a MyD88 pathway leading to the activation of the transcription factors AP-1 and NF-κB and the transcription of proinflammatory cytokines [63]. All studies indicate that flagellin functions to increase antibody titers [64], [65]. This increase is attributed to the flagellin activating DC as well as other APCs and the recruitment of B and T cells to the lymph nodes [66], [67], [68], [69], [70], [71]. This leads to interactions between B cells and TH cells leading to increased antibody production [64]. Initial studies seemed to indicate that flagellin is not a strong adjuvant for TC responses, however, it has repeatedly been demonstrated that it does function in this regard as well [72], [73], [74], [75], [76]. Flagellin is also able to activate the inflammasome via amino acids 441–476 which are present on the C terminus in the D0 domain. These amino acids are able to interact with the PRR Naip5 [77], [78]. The role of the inflammasome in regard to adjuvant function is not completely clear.
Unmethylated CpG motifs are also currently being investigated as potential adjuvants (Table 2). Bacteria do not frequently methylate CpG motifs, however, vertebrates do allowing for the differentiation of foreign DNA [79]. During a natural infection process CpG motifs are recognized by TLR9, which is present in endoplasmic reticulum (ER), late endosomal compartment, and the lysosomal compartment. Different organisms localize TLR9 to different cell types, but it is most frequently localized to plasmacytoid DC and B cells in humans [80]. Upon stimulation TLR9 exits the ER and interacts with MyD88, which leads to the activation of signaling pathways leading to the activation of NF-κB and AP-1, the production of TH1 proinflammatory cytokines and increased innate and adaptive immune response [81]. For adjuvant use synthetic CpG oligodeoxynucelotides (CpG ODN) are generated that contain unmethylated CpGs. Generally, CpG ODNs contain a phosphorothioate linkage in place of a phosphodiester linkage, which causes the CpG ODN to have a longer half-life in vivo. CpG ODNs are divided into different classes based upon the number and location of CpG repeats, each has different strengths and weaknesses as well as slightly different biological effects [81]. Clinical trials using CpG as an adjuvant have been performed in a wide range of infectious diseases. All resulted in an increase in the immunogenicity of the vaccine tested, with effects ranging from increased antibody production, faster memory response, or a change in the cytokine profile of the vaccine leading to a stronger immune response [80], [82].
RNA binding TLRs are also being examined as potential targets for adjuvant development (Table 2). TLR7 and TLR8 are both localized to endosomes, with TLR7 primarily present in plasmacytoid DC and B cells and TLR8 primarily present in monocytes/macrophage and myeloid DC. Both receptors bind single stranded RNA sequences from infecting microorganisms and TLR7 the synthetic agonist Imiquimod (R837) with both binding the synthetic agonist Resiquimod (R848). Stimulation of both leads to signaling through MyD88 and activation of NF-κB and AP-1, and ultimately proinflammatory cytokines [83], [84]. The short half-life of RNA necessitates that in the development of successful long lasting vaccine formulation one of the synthetic agonists is used. Initial studies indicated both of these agonists are immunostimulatory when given as a topical ointment, however, when given admixed as an injection extremely high doses were necessary. Subsequent studies have determined that fusion of these agonists to a given antigen, admixture with other PAMPs, and newer agonist designs show promise as successful adjuvant candidates [85], [86].
6. Increasing the immune response to subunit vaccines: repetitive antigen displays
While the addition of a chemical or biological adjuvant to a vaccine is one mechanism to induce stronger immune responses, there are other ways too. One aspect of a whole organism vaccine that makes it so successful is the fact that it expresses multiple antigens in a repetitive array. In most cases there is not just one copy of a B cell antigen on the cell or viral surface. By vaccinating with a recombinant protein this effect does not occur, because the antigens become diluted in the patient’s body fluids. Work beginning in the 1960s would lead to the development of another mechanism to generate more effective vaccine candidate that would more closely mimic a whole organism vaccine.
Patients infected with hepatitis B virus (HBV) produce a noninfectious particle in their blood [87], [88]. This particle is actually composed of a protein antigen (S or HBsAg) that is present in the envelope of the HBV. HBsAg is produced in large quantities during infection and is able to self-assemble into a 22 nm virus-like particle (VLP) [89]. VLPs are particles that resemble the size and shape of viruses but do not contain any viral genetic material, meaning they are not infectious [90] (see Fig. 2 B). Quickly it was identified that the VLP was about 1000 times more immunogenic than non-assembled HBsAg [91] and these particles were identified as potential vaccine candidates [87].
Fig. 2.
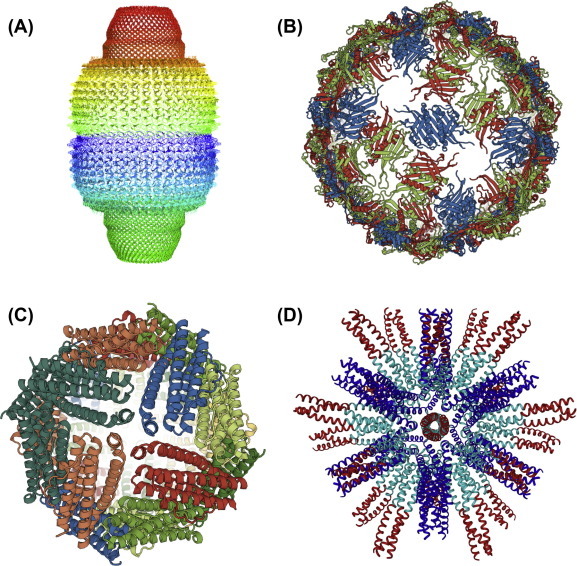
Types of protein assemblies that are used as vaccines carriers. (A) Vaults: The example shows the rat liver vault comprising 78 identical major vault protein chains (PDB-RCSB code 4V60). The view is perpendicular to the 39-fold symmetry axis. (B) VLPs: The example shows the RNA bacteriophage Q beta forming a T = 3 icosahedral particle (PDB-RCSB code 1QBE). The view is down the twofold axis of the icosahedron. (C) Ferritin: Shown is the octahedral structure of ferritin from the marine pennate diatom Pseudo-nitzschia multiseries (PmFTN) (PDB-RCSB code 4ZKW). The view is down the fourfold axis of the octahedron. (D) SAPNs: The example shows a SAPN composed of a pentameric (blue) and trimeric (cyan) coiled-coil domain displaying the trimeric coiled-coil epitope HRC (red) from the SARS coronavirus. The view is down the threefold axis of the icosahedron. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
At the time there was no culturable method for HBV, and only humans and certain non-human primates (NHP) could be infected. It was proposed and eventually carried out that the best mechanism for developing a HBV vaccine was to harvest plasma from asymptomatic volunteers, chemically treat the plasma to inactivate it like it was an infectious particle, and use it as a vaccine candidate [92], [93], [94], [95], [96]. While these initial studies were successful in high risk populations, they were occurring in the late 1970s and early 1980s which coincided with the beginning of the Acquired Human Immunodeficiency (AIDS) epidemic. The vaccine was approved for human use in 1981, but fears about the use of a human blood product, lack of volunteers, and cost led to abandonment of this vaccine and its eventual withdrawal from the United States by 1986 [97].
A recombinant HBsAg based HBV vaccine became a top priority at the dawn of recombinant vaccines. HBsAg produced in E. coli was not immunogenic which led to a search for a different expression host [98], [99], [100]. In 1982, HBsAg was produced in its naturally glycosylated immunogenic form from S. cerevisiae [101] . The vaccine was shown to be immunogenic and safe [102], [103], [104]. The vaccine was approved in 1986 and still remains the standard HBV vaccine (Fig. 1, Table 1). The recombinant HBsAg has the distinction of not only becoming the first approved subunit vaccine, but also the first approved VLP. The knowledge used during the development of the recombinant HBsAg vaccine was eventually applied to other diseases such as HPV, leading to the development of clinically approved HPV vaccines [105]. It also became the foundation for other vaccines such as the most advanced malaria vaccine candidate RTS,S [106].
VLPs also offer the advantage of being repetitive antigen displays. It has been consistently demonstrated that when antigens are displayed repetitively there is an increase of antibody titers [90], [107]. This occurs because of increased crosslinking of the B cell receptors leading to B cell activation [5], [108], [109]. VLPs, because they are made of viral proteins, are processed by APC cells as a virus. Increased and efficient processing leads to activation of T cells and also increased immune responses [90]. One problem with VLPs is that not every virus that a vaccine is needed for has a capsid protein that can self-assemble into an immunogenic VLP. The second problem is not every infectious disease is a virus. For some diseases, successful fusion constructs have been generated such as RTS,S for malaria that uses the HBsAg as a carrier [106]. Generation of fusion constructs is not always successful, however [90]. These problems have led for the development of new technologies that would incorporate the strengths of VLPs while minimizing their weakness.
7. Nanotechnology and nanovaccines
An exciting area in the development of vaccines is the use of nanotechnology. On December 29, 1959 physicist Richard Feynman gave a talk entitled “There’s Plenty of Room at the Bottom.” He predicted in the future scientists would be able to manipulate matter on the atomic level leading to the development of tiny manufacturing machines [110]. In 1981 K. Eric Dexler using the concepts of Feynman, and the advances in genetic engineering proposed that proteins could be designed to carry out atomic level fabrications [111]. Dexler eventually published a book, The Engines of Creation: The Coming Era of Nanotechnology and founded the Foresight institute to direct the development of nanotechnology over the coming years [112]. Dexler’s work is considered the true beginning of the field of nanotechnology, a field that has undergone exponential growth over the subsequent three decades.
Today nanotechnology is defined by the United States National Nanotechnology Initiative as the field that deals with nanoscale objects that are 1–100 nm in size. A nanoparticle is defined as an individual nanoscale object that is able to function as an independent unit [113]. These nanoscale objects are composed of a wide range of materials including organic polymers, inorganic polymers, and biological macromolecules. Nanoscale assemblies have a diverse array of biomedical implications including use as biological sensors, cell targeting systems, drug delivery systems, and as subunit vaccine carriers [114].
One of the major benefits in the use of nanotechnology for the development of vaccines is the resulting nanovaccines can be designed to be effective antigen delivery systems. Attempts have been made with many different types of nanomaterials to develop effective vaccine candidates. Nanotechnology is not a uniform field, but rather a mixture of many different nanoscale materials that vary in their chemical composition and behaviors. The diversity in the field has led to the development of different approaches for nanovaccines. Each group has its own advantages and its own disadvantages making it unlikely that one approach will work for the development of each necessary vaccine.
One approach is the use of different polymers and copolymers. A polymer is a molecule made up of one repeating subunit, while a copolymer is a molecule made up of multiple repeating subunits. Polymeric vaccines are made up of a broad range of compounds including (d,l-lactide-co-glycolide) (PLGA), poly(lactic acid) (PLA), poly(g-glutamic acid) (g-PGA), poly(ethylene glycol) (PEG), and polystyrene [115]. Polymers can either function to encapsulate antigens, have antigens conjugated to their surface via a linker, have antigens adsorbed to their surface, or be admixed with the antigen. The chemistry of the polymer, and its interactions with the antigen determines the most effective mechanism for vaccine delivery.
The PLGA nanoparticle vaccines are the prototypical encapsulated vaccine. PLGA has been widely studied for about 60 years for a variety of biomedical applications including drug delivery and diagnostics [116]. A nanoparticle consisting of PLGA has a core made up of its desired cargo, which is encapsulated by the polymer. Since 1969 PGLA has been approved by the Food and Drug Administration (FDA) for use as a drug delivery system. The benefits of PLGA nanoparticles for any biomedical applications is they slowly degrade when exposed to water, meaning they can protect hydrophobic, hydrophilic, small molecule, or biological macromolecule cargo and release it over an extended period of time [117]. In preclinical studies this has been demonstrated to be an effective carrier in mouse models for vaccines composed of HBsAg, tetanus toxoid, Helicobacter pylori lysate, Listeria monocytogenes antigens, malaria antigens, and B. anthracis spores [116], [118], [119], [120], [121], [122], [123], [124], [125]. These vaccines are able to generate long lasting humoral and cellular immune response. They can also be given in an oral, aerosolized, subcutaneous, intraperitoneal, or intermuscular manner because of the nature of PLGA.
Further strategies can be taken with polymer based nanoparticles to make them more effective vaccine candidates such as the synthetic vaccine particles (SVP). These particles can be used to carry not only antigens in their cores but also PAMPs as adjuvants. They can be encoated with phospholipids and decorated with B cell antigens leading to potentially more efficient vaccine candidates that function as repetitive antigen displays, delivery systems, and as immunopotentiators [126], [127].
Polymers can also serve as inert cores for antigen conjugation or adsorption. One such example are polystyrene beads. Nanoparticles with a solid inert core are also known as nanobeads. Proof of principle ovalbumin vaccines have been developed [128], [129], [130]. These vaccines function as repetitive antigen displays, but still need an external adjuvant added to generate strong immune responses. In comparison with other vaccination techniques they are not better at stimulating immune response [129]. Polymers such as polystyrene are thought to be biocompatible, however, they are not biodegradable. Toxicology studies in cell culture models and small animals indicate that they do not generate free radicals and do not appear to cause adverse effects [128], [129], [131]. Long-term health effects are not known, however. While nanobeads with inert polymer cores function as repetitive antigen displays they still need to be adjuvanted and their long-term health effects are unknown making them not ideal candidates for vaccine development.
Inorganic molecules can also be used as delivery systems for nanovaccines. One of the best studied are gold nanoparticles (AuNP). Many materials take on unique chemical and physical properties on the nanoscale level. Gold is easily modified into nanoparticles of different sizes and shapes. Gold also takes on unique optic properties based upon nanoparticle size and shape that has attracted attention for use in cellular imaging, drug delivery, and photothermal therapy [132], [133]. Studies have determined that AuNP appear to be inert and biocompatible [134]. AuNP can be used in a variety of ways in the development of vaccines with antigens adsorbed or conjugated to their surface [115]. Size and shape of AuNP has been linked to their function as vaccine candidates. Different geometric shapes can lead to differential APC phagocytosis and antigen presentation. These findings also indicate that AuNP can function as effective delivery systems for vaccines [135]. Successful, preclinical vaccine candidates have been developed for diseases such as influenza, West Nile Virus, and plague [135], [136], [137]. There has also been interest and initial studies on the development of both prophylactic and therapeutic cancer vaccines based off of AuNP [138], [139]. Despite the fact that AuNP can be easily modified into the most immunogenic shape and size in all published studies an external adjuvant needed to be added to generate a protective or effective vaccine candidate.
Carbon nanotubes (CNTs) are another class of inorganic nanoparticles of interest in vaccine development. These are sheets of graphene that are folded into a tube shape, that can occur in a single layer, single-walled carbon nanotubes (SWNT), or in multiple layers, multi-walled carbon nanotubes (MWNT). CNTs have many advantages that make them an ideal candidate for vaccine development. They have a large surface area that makes them excellent potential repetitive antigen displays. Adsorption and or conjugation of antigens to their surface is fairly easy [140]. Studies have indicated that they are highly immunogenic in and off themselves, which could potentially indicate that besides functioning as antigen delivery systems they also could function as immunopotentiators [140], [141], [142], [143]. One of the major problems with the application of CNT to vaccinology is that their toxicology is not established and studies are highly conflicting.
Other inorganic nanoscale assemblies based on silica, aluminum, and calcium phosphate, have been used in an attempt to develop effective vaccine candidates [144]. However, many of the same problems occur with all classes. The primary issue is that the long-term health effects are not known. Initial studies have focused on in vitro models and small animal models. For every class of inorganic nanoparticle, studies have indicated that there is some form of toxicology [145]. While there are benefits to each class, the lack of understanding of their long-term health effects makes developing a clinical product difficult at this current time.
8. Biologically derived nanovaccines
As first proposed by Dexler, the use of biological molecules has many advantages for development of nanotechnology, particularly nanovaccines. The first approved subunit vaccine, HBV vaccine, is technically a nanoparticle that is composed completely of protein. The VLPs made by the HBsAg self-assemble into 22 nm particles, which falls well into the 1–100 nm designation of nanoscale assemblies. Other biologically based technologies have also been developed to repetitively display antigens, with potentially lower levels of toxicity in comparison to inorganic and synthetic polymer nanoparticles. By capitalizing on biological macromolecules scientists are able to develop vaccine candidates that more closely resemble microorganisms, meaning they should function more efficiently inside the host.
One example of biologically derived nanoparticles are liposomes, which are phospholipid bilayers surrounding an aqueous chamber. In 1965 it was noted that phospholipids could assemble into these structures [146]. By 1974 the first study came out that indicated that liposomes could function as an adjuvant to increase the immune response to diphtheria toxoid vaccine contained in their aqueous chamber [147]. In the subsequent years, studies have established liposomes as safe and effective antigen delivery system as well as adjuvant. Liposomes are extremely plastic, meaning size, shape, charge, phospholipid composition, and supporting molecular composition can be altered to elicit the desired effect. They can function as repetitive antigen displays when antigens are either incorporated into the phospholipid bilayer, or adsorbed onto the membrane. Liposomes can also carry T cell epitopes as cargo in their aqueous chamber [148].
Studies indicate that liposomes are functional for two main reasons. The fact that they are composed of phospholipids increases the ability of APC cells to phagocytose the particles leading to increased antigen processing and presentation [149]. Studies have indicated that cationic liposomes are the most effective adjuvants because they remain longer associated with cell membranes because of electrostatic interactions [150], [151]. The longer a liposome is associated with the membrane the higher the probability that APC will phagocytose and present the antigens. Liposomes can also carry external adjuvants in their chambers, or have adjuvants like PAMPS incorporated into their membranes leading to stronger immune responses [148], [152], [153].
Virosomes could be considered a cross between VLPs and liposomes. Traditionally, each virosome is composed of the phospholipids contained in the envelope of influenza virus along with the viral envelope proteins hemagglutinin and neuraminidase. With this design the particles function as immunogenic influenza viruses, but because they do not contain a genome they are unable to replicate. They demonstrate increased fusion with APC and subsequently, elevated antigen presentation [154]. The phospholipids and envelope proteins were originally isolated from influenza viral cultures grown in embryonated eggs, but now they are synthetically produced and assembled. As with liposomes antigens can be added to the membrane, adsorbed onto the membrane, or carried as cargo in the virosome [155].
Two different virosomes have been used successfully since in the 1990s in humans. The first is Inflexal, which is an influenza vaccine [156]. The second is Epaxal, which is formalin inactivated hepatitis A that is adsorbed to an influenza virosome [157]. Both vaccines have been shown to be effective with good safety profiles. They have also demonstrated that virosomes can function both as antigen delivery system as well as adjuvants. Currently, vaccines for malaria, hepatitis C, various cancers, HIV, and Candida albicans are in various stages of clinical development using influenza virosomes as a core [158].
Nanoparticles composed of protein that can self-assemble into repetitive antigen displays are an area of intense interest in the development of nanovaccines. One class of these nanoscale assemblies are derived from the protein ferritin (Fig. 2C). Ferritin is an iron metabolism protein that is present in bacteria, animals, and plants. Under normal conditions 24 monomers of ferritin will self-assemble into a spherical particle with octahedral symmetry containing an open central cavity. This structure is known as a ferritin cage [159]. Ferritin cages have an overall diameter of 12 nm, while the cores have a diameter of 8 nm [160]. Under starvation, bacterial ferritin can assemble into a particle with tetrameric symmetry composed of 12 ferritin monomers [161]. Ferritin cages have been used for a wide variety of biomedical applications including medical imaging and drug delivery [159].
Studies have determined that ferritin cages can function as vaccine delivery systems. Hemagglutinin from H1N1 subtype of influenza has been fused to ferritin from H. pylori leading to the generation of an influenza vaccine candidate. The resulting particle expresses eight trimeric hemagglutinin molecules in their native conformation on the vertices of the particle. When vaccinated into ferrets the animals developed antibodies to both the head domain and the stalk domain of hemagglutinin. Challenged animals were protected [162]. The head domain of hemagglutinin from influenza is immunodominant, but it is subject to antigenic drift necessitating seasonal changes in vaccine formulations. In a desire to generate a vaccine candidate that focuses on the conserved stem domain, a recombinant ferritin molecule was generated that lacked the head domain. This construct was able to raise stalk specific antibodies and completely protect mice against challenge, and partially protect ferrets [163].
While ferritin cages are exciting potential repetitive display antigen candidates, they do have weaknesses. Ferritin cages have a very rigid assembly. If an antigen needs to be presented in a certain conformation to be immunogenic, ferritin cages may not be able to present it in the most immunogenic form. Ferritin cages seem like excellent candidates for the development of some vaccines, but will not be able to be used for every antigen.
Another naturally occurring nanoscale assembly that has attracted attention are vault proteins (Fig. 2A). These proteins are naturally occurring in most eukaryotes, and may play a role in nuclear transport, signal transduction, and innate immune responses, but no exact function has been established [164]. Each vault is a 70 nm organelle-like structure that is composed of three proteins: the major vault protein (MVP) makes up 70% of the complex, telomerase associated protein-1, poly ADP-ribose polymerase (PARP), and non-coding RNAs [165]. Vaults can be designed to express heterologous proteins inside their central cavity. This is easily done by adding a specific sequence to the protein that has been determined to associate the PARP protein with MVP [165]. Vaults are not immunogenic themselves and do not lead to the development of autoimmune responses making them a good vaccine delivery system [165], [166]. Using the model protein ovalbumin, vaults were successful at inducing TC and TH responses in vaccinated animals. They were less successful at inducing humoral immune responses [167]. Vaults have been developed as vaccine candidates for Chlamydia muridarum infections. Inoculation induced both TH and TC responses leading to protection against challenge [168]. Despite the success with vaults their biggest weakness is that the antigen is contained within the vault cavity, which prevents it from stimulating B cells leading to strong humoral responses. Currently, vaults could function as strong vaccine candidates for intracellular pathogens, or for diseases such as cancer, but further development is needed for their use against a wide range of pathogens.
9. Rationally designed protein nanovaccines
While naturally occurring nanoscale assemblies can be good vaccine candidates for certain infectious diseases, they may not be applicable to a wide range of infectious organisms. Another approach in development of nanovaccines is the use of rationally designed proteins. By combining knowledge of structural biology, biophysics, protein engineering, infectious disease biology, and molecular biology new proteins can be developed to function as ideal vaccine candidates. This approach is considered a bottom-up approach, taking small parts and generating a more complex assembly [169]. VLPs and other approaches are a top-down approach and take a complex protein assembly and strip it down to its basic components, which in the terms of a vaccine would be the smallest part that induces protective immunity [170].
There are many benefits and challenges associated with vaccine development from bottom-up rationally designed proteins. The major benefit is that these designed proteins can function as repetitive antigen displays for a wide range of antigens. They are not limited like ferritin cages or vaults in what conformations of antigens they can present. They also can be optimized to induce both strong cellular and humoral immune responses by the addition of new antigenic domains. To be functional these proteins need to be able to self-assemble using non-covalent interactions into nanoscale assemblies. This self-assembly needs to occur without an added enzyme in a specific and reproducible manner [171]. The major drawback of this approach is protein sequence, no matter how well designed, it does not always perfectly correlate with structure and function [172]. This problem leads to the need for each design to be empirically tested and optimized to achieve the desired structure and ultimately the desired protective effect in vaccinated animals.
A technique that has been applied to the development of rationally designed protein nanoparticles is the layer-by-layer (LbL) fabrication of polypeptide films on solid CaCO3 cores. In this method polypeptide layers of alternating opposite charges are deposited on a CaCO3 nanoparticle core. The electrostatic interactions between the opposite charged polypeptide layers keep particles together. Contained within the different layers different B and T cell antigens can be added allowing for the development of vaccine candidates for different disease states [173]. A malaria and a respiratory syncytial virus (RSV) vaccine candidate designed using this approach were immunogenic and capable of raising neutralizing antibodies for both either malaria or RSV respectively and the malaria candidate was able to generate cellular responses as well [174], [175].
One approach for the development of a rationally designed protein nanoparticle are nanofibers, nanoscale assemblies that are derived from the β-sheet secondary structure. The initial design of the nanofiber core consisted of 11 amino acids (Q11) that could self-assemble into unbranched antiparallel β-sheets [176], [177]. Initially designed for the purpose of regenerative medicine, nanofibers were determined to be non immunogenic and well tolerated in animals [177]. When a 17 amino acid fragment of ovalbumin with established B and T epitopes was added to the N terminus of the construct, nanofibers that self-assembled functioned as repetitive antigen displays. Antibody titers from vaccinated animals were greater than animals vaccinated with the ovalbumin fragment with FCA [178]. Another study also indicated that when animals are vaccinated with nanofibers that contain only TC epitopes from ovalbumin they develop strong TC responses [179]. These initial prototypical studies determined that nanofibers could function as successful vaccine candidates.
Nanofibers have been successfully used in infectious disease models as well. A vaccine candidate against malaria was able to induce high titers of antibodies that functioned to prevent infection in an in vitro assay [180]. Attempts have been made to increase TH responses to nanofibers by adding the pan-allelic DR epitope (PADRE) a universal TH epitope. A heterogeneous mixture of Q11, Q11-B-cell epitope, and Q11-PADRE was used to develop a Staphylococcus aureus vaccine candidate. Addition of PADRE resulted in an antibody titer increase in a PADRE concentration related manner [181]. While there have been some successes with the nanofiber technology there have been instances were nanofibers did not elicit strong immune responses [182].
While nanofibers are a successful application of rationally designed protein nanoparticles many problems exist with their use. The core sequence is only 11 amino acids long. This is extremely short preventing the addition of large antigens. Allowing for only the addition of short regions of antigens severely limits their immunogenicity. These short peptides need to be chemically synthesized, cannot be epitope tagged, and expressed in a protein expression system [178], [179], [180], [181], [182], [183]. This fact limits their ability to be scaled up in a cost effective manner.
One of the most appealing protein structural motifs for rational design of nanovaccines are coiled-coil oligomerization domains. These motifs consist of two or more α-helices wrapped around each other. Coiled-coils are one of the most common, best understood, and stable oligomerization domains [184]. One application of the coiled-coil oligomerization domain to vaccine design are Synthetic Virus Like Particles (SVLP). These particles are based on chemically synthesized monomers that consist of a lipid attached to a coiled-coil trimeric domain that is chemically attached to an antigen. Based on basic biophysical properties 60–90 monomers can self-assemble into a 20–25 nm particle that contains a lipid core and a surface decorated by antigens. SVLP function as repetitive antigen displays and are capable of inducing humoral responses in vaccinated animals [185], [186].
Perhaps, one of the most promising nanoscale assemblies for vaccine development are Self-Assembling Protein Nanoparticles (SAPNs; Fig. 2D). SAPNs are rationally designed repetitive antigen displays that can function for a diverse array of antigens. Disease antigens can be added to either the C or the N terminus of each SAPN monomer, and in the prototypical SAPN 60 monomers will self-assemble into a particle with regular symmetry [187], [188], [189], [190]. Each assembled SAPN is decorated on its surface with 60 copies of an antigen, making them excellent repetitive antigen displays (Fig. 2D). Not only do SAPNs function as repetitive antigen displays, but the individual particles resemble small viruses in both shape as well as in size at about 25 nm. Together these factors make them excellent immunostimulatory particles and prime vaccine candidates.
One of the major benefits of the SAPN design is the wide variety of coiled-coil oligomerization domains. In nature coiled-coils exist that consist of 2, 3, 4, 5, and 6 α-helices while 7 membered coiled-coil domains have been synthesized in the laboratory [191]. B-cell epitopes can be conformational, meaning the epitope is based on the structure of the antigen, not the linear sequence [192]. The toolbox of well-established well-understood coiled-coil oligomerization domains allows for the presentation of antigens, in particular for oligomeric antigens, in their native conformations. The best immunogenic presentation can be chosen to develop the most effective vaccine candidate. Choosing different oligomerization domains will ultimately change the geometry of each SAPN particle, however, they will still self-assemble and function as effective repetitive antigen display. Another enticing feature of the SAPN is the ability to add universal TH epitopes to the core [193].
SAPNs have been used as effective vaccine candidates for a variety of diseases. Successful candidates have been developed for the viral diseases Avian Influenza, SARS, and HIV [194], [195], [196]. In each viral disease that SAPN technology has been applied to high antibody titers have been observed, and in most cases these antibodies were demonstrated to be functional in in vitro assays. One of the most challenging classes of infectious agents to develop vaccines for are parasitic diseases. Parasites are complex organisms with multiple life stages, immune avoiding mechanisms, and generally infect multiple hosts [197]. SAPNs are effective in the development of parasitic vaccine candidates. SAPN candidates induce strong humoral and cellular immune responses against malaria in murine models leading to protection against challenge [193], [198], [199]. SAPNs have also been shown to be immunogenic and able to induce protection against challenge in a murine model of Toxoplasma gondii [200]. This success further demonstrates the strength of the SAPN as a vaccine technology.
10. Future directions
Since Jenner developed the first vaccine candidate in 1796 great advances have been made in the field of infectious disease vaccinology. Despite the advances that have been made there is still much work to be done. The field has moved from the traditional method of vaccine development, where an organism had to be cultured, attenuated, or killed before it could become an effective vaccine candidate. Today, through modern biological techniques extremely targeted safer vaccines can be developed much more quickly than ever before. The major problem with these candidates is that they tend to be less immunogenic and ultimately less protective than whole organism vaccines. Resolving these issues is the current main focus of vaccine development.
There is a current need for the rapid and effective development of infectious disease vaccines. Besides the big three diseases in need of vaccines, HIV, tuberculosis, and malaria, there are outbreaks of new and emerging diseases such as Ebola and Zika Virus that threaten global public health. New technologies may provide a mechanism for quick development and rapid implementation of vaccines. By further developing vaccine technologies to include more immunostimulatory molecules as well as more epitopes better vaccine candidates will be generated.
Vaccinology as a discipline has traditionally been focused on the development of vaccines for the prevention of infectious diseases. This unique relationship has allowed vaccinology to develop hand in hand with immunology and infectious disease biology. As knowledge of these disciplines has increased it has allowed for the development of new vaccine technologies to be applied to other disease states such as cancer. As development of vaccine technologies continues to be refined and improved more applications will become available to disease states such as cancer, addiction, and obesity. Potentially, in the near future scientists will be able to apply these new technologies to the development of prophylactic treatments to a broad range of diseases leading to further decreases in global morbidity and mortality from everything from infectious diseases to cancer. Vaccinology in its first 200 years of development has only scratched the surface of what is possible.
Acknowledgements
The authors would like to thank Charles Giardina for his assistance in developing this manuscript. Support by the NIH/NIDA (award 1DP1DA033524) to PB for this work is gratefully acknowledged. PB has an interest in the company Alpha-O Peptides that has patents or patents pending on the SAPN technology.
References
- 1.Andre F.E., Booy R., Bock H.L., Clemens J., Datta S.K., John T.J. Vaccination greatly reduces disease, disability, death and inequity worldwide. Bull. World Health Organ. 2008;86:140–146. doi: 10.2471/BLT.07.040089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenwood B. The contribution of vaccination to global health: past, present and future. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 2014;369 doi: 10.1098/rstb.2013.0433. 20130433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morens D.M., Holmes E.C., Davis A.S., Taubenberger J.K. Global Rinderpest eradication: lessons learned and why humans should celebrate too. J. Infect. Dis. 2011;204:502–505. doi: 10.1093/infdis/jir327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moser M., Leo O. Key concepts in immunology. Vaccine. 2010;28:2–13. doi: 10.1016/j.vaccine.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Siegrist C.-A. Vaccines. Elsevier; 2013. Vaccine immunology; pp. 14–32. [Google Scholar]
- 6.Fine P., Eames K., Heymann D.L. “Herd immunity”: a rough guide. Clin. Infect. Dis. 2011;52:911–916. doi: 10.1093/cid/cir007. [DOI] [PubMed] [Google Scholar]
- 7.Rashid H., Khandaker G., Booy R. Vaccination and herd immunity. Curr. Opin. Infect. Dis. 2012;25:243–249. doi: 10.1097/QCO.0b013e328352f727. [DOI] [PubMed] [Google Scholar]
- 8.Fine P. Science and society: vaccines and public health. Public Health. 2014;128:686–692. doi: 10.1016/j.puhe.2014.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Smith K.A. Edward Jenner and the small pox vaccine. Front. Immunol. 2011;2:1–6. doi: 10.3389/fimmu.2011.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plotkin S.A., Plotkin S.L. The development of vaccines: how the past led to the future. Nat. Rev. Microbiol. 2011;9:889–893. doi: 10.1038/nrmicro2668. [DOI] [PubMed] [Google Scholar]
- 11.Berche P. Louis Pasteur, from crystals of life to vaccination. Clin. Microbiol. Infect. 2012;18:1–6. doi: 10.1111/j.1469-0691.2012.03945.x. [DOI] [PubMed] [Google Scholar]
- 12.Ehrlich P. Croonian Lecture: on immunity with special reference to cell life. Proc. R. Soc. Lond. 1899;66:424–448. [Google Scholar]
- 13.Glenny A.T., Hopkins B.E. Diphtheria toxoid as an immunising agent. Br. J. Exp. Pathol. 1923;4(5):283–288. [Google Scholar]
- 14.Fitzgerald J.G. Diphtheria toxoid as an immunizing agent. Can. Med. Assoc. J. 1927;17:524–529. [PMC free article] [PubMed] [Google Scholar]
- 15.Hayflick L., Moorhead P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 16.Witkowski J.A. Dr. Carrel’s immortal cells. Med. Hist. 1980;24:129–142. doi: 10.1017/s0025727300040126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebeling A.H. A ten year old strain of fibroblasts. J. Exp. Med. 1922;35:755–759. doi: 10.1084/jem.35.6.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weller T.H., Enders J.F. Production of hemagglutinin by mumps and influenza A viruses in suspended cell tissue cultures. Exp. Biol. Med. 1948;69:124–128. doi: 10.3181/00379727-69-16638. [DOI] [PubMed] [Google Scholar]
- 19.Enders J.F., Weller T.H., Robbins F.C. Cultivation of the Lansing Strain of poliomyelitis virus in cultures of various human embryonic tissues. Science. 1949;109:85–87. doi: 10.1126/science.109.2822.85. [DOI] [PubMed] [Google Scholar]
- 20.Trevelyan B., Smallman-Raynor M., Cliff A.D. The spatial dynamics of poliomyelitis in the United States: from epidemic emergence to vaccine-induced retreat, 1910–1971. Ann. Assoc. Am. Geogr. 2005;95:269–293. doi: 10.1111/j.1467-8306.2005.00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.John T.J. The Golden Jubilee of vaccination against poliomyelitis. Indian J. Med. Res. 2004;119:1–17. [PubMed] [Google Scholar]
- 22.Salk J.E. Studies in human subjects on active immunization against poliomyelitis. I. A preliminary report of experiments in progress. J. Am. Med. Assoc. 1953;151:1081–1098. [PubMed] [Google Scholar]
- 23.Salk J.E., Krech U., Youngner J.S., Bennett B.L., Lewis L.J., Bazeley P.L. Formaldehyde treatment and safety testing of experimental poliomyelitis vaccines. Am. J. Public Health. 1954;44:563–570. doi: 10.2105/ajph.44.5.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strebel P.M., Sutter R.W., Cochi S.L., Biellik R.J., Brink E.W., Kew O.M. Epidemiology of poliomyelitis in the United States one decade after the last reported case of indigenous wild virus-associated disease. Clin. Infect. Dis. 1992;14:568–579. doi: 10.1093/clinids/14.2.568. [DOI] [PubMed] [Google Scholar]
- 25.Sabin A.B., Hennessen W.A., Winsser J. Studies on variants of poliomyelitis virus. J. Exp. Med. 1954;99:551–576. doi: 10.1084/jem.99.6.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabin A.B. Oral poliovirus vaccine: history of its development and use and current challenge to eliminate poliomyelitis from the world. J. Infect. Dis. 1985;151:420–436. doi: 10.1093/infdis/151.3.420. [DOI] [PubMed] [Google Scholar]
- 27.Schonberger L.B., Kaplan J., Kim-Farley R., Moore M., Eddins D.L., Hatch M. Control of paralytic poliomyelitis in the United States. Rev. Infect. Dis. 2015;6(Suppl. 2):S424–S426. doi: 10.1093/clinids/6.supplement_2.s424. [DOI] [PubMed] [Google Scholar]
- 28.Johns R.B., Farnsworth S., Thompson H., Brady F. Two voluntary mass immunization programs using Sabin oral vaccine. JAMA. 1963;183:171–175. doi: 10.1001/jama.1963.03700030047010. [DOI] [PubMed] [Google Scholar]
- 29.Sabin A.B. Paralytic poliomyelitis: old dogmas and new perspectives. Rev. Infect. Dis. 1981;3:543–564. doi: 10.1093/clinids/3.3.543. [DOI] [PubMed] [Google Scholar]
- 30.Pliaka V., Kyriakopoulou Z., Markoulatos P. Risks associated with the use of live-attenuated vaccine poliovirus strains and the strategies for control and eradication of paralytic poliomyelitis. Expert Rev. Vaccines. 2012;11:609–628. doi: 10.1586/erv.12.28. [DOI] [PubMed] [Google Scholar]
- 31.Immunization Systems Management Group of the Global Polio Eradication Initiative Introduction of inactivated poliovirus vaccine and switch from trivalent to bivalent oral poliovirus vaccine – worldwide, 2013–2016. MMWR Morb. Mortal. Wkly Rep. 2015;64:699–702. [PMC free article] [PubMed] [Google Scholar]
- 32.Orenstein W.A., Papania M.J., Wharton M.E. Measles elimination in the United States. J. Infect. Dis. 2004;189(Suppl):S1–S3. doi: 10.1086/377693. [DOI] [PubMed] [Google Scholar]
- 33.Hilleman M.R. Past, present, and future of measles, mumps, and rubella virus vaccines. Pediatrics. 1992;90:149–153. [PubMed] [Google Scholar]
- 34.Lievano F., Galea S.A., Thornton M., Wiedmann R.T., Manoff S.B., Tran T.N. Measles, mumps, and rubella virus vaccine (M-M-R™II): a review of 32 years of clinical and postmarketing experience. Vaccine. 2012;30:6918–6926. doi: 10.1016/j.vaccine.2012.08.057. [DOI] [PubMed] [Google Scholar]
- 35.Berg P., Mertz J.E. Personal reflections on the origins and emergence of recombinant DNA technology. Genetics. 2010;184:9–17. doi: 10.1534/genetics.109.112144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moyle P.M., Toth I. Modern subunit vaccines: development, components, and research opportunities. ChemMedChem. 2013;8:360–376. doi: 10.1002/cmdc.201200487. [DOI] [PubMed] [Google Scholar]
- 37.Purcell A.W., McCluskey J., Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007;6:404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 38.García A., De Sanctis J.B. An overview of adjuvant formulations and delivery systems. APMIS. 2014;122:257–267. doi: 10.1111/apm.12143. [DOI] [PubMed] [Google Scholar]
- 39.Brito L.A., Malyala P., O’Hagan D.T. Vaccine adjuvant formulations: a pharmaceutical perspective. Semin. Immunol. 2013;25:130–145. doi: 10.1016/j.smim.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Sesardic D., Dobbelaer R. European Union regulatory developments for new vaccine adjuvants and delivery systems. Vaccine. 2004;22:2452–2456. doi: 10.1016/j.vaccine.2003.11.071. [DOI] [PubMed] [Google Scholar]
- 41.Glenny A.T., Pope C.G., Waddington H., Wallace U. Immunological notes. XVII–XXIV. J. Pathol. Bacteriol. 1926;29:31–40. [Google Scholar]
- 42.Kool M., Fierens K., Lambrecht B.N. Alum adjuvant: some of the tricks of the oldest adjuvant. J. Med. Microbiol. 2012;61:927–934. doi: 10.1099/jmm.0.038943-0. [DOI] [PubMed] [Google Scholar]
- 43.Oleszycka E., Lavelle E.C. Immunomodulatory properties of the vaccine adjuvant alum. Curr. Opin. Immunol. 2014;28:1–5. doi: 10.1016/j.coi.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 44.Freund J., Bonanto M.V. The effect of paraffin oil, lanolin-like substances and killed tubercle bacilli on immunization with diphtheric toxoid and bact. typhosum. J. Immunol. 1944;48:325–334. [Google Scholar]
- 45.Fox C.B., Haensler J. An update on safety and immunogenicity of vaccines containing emulsion-based adjuvants. Expert Rev. Vaccines. 2013;12:747–758. doi: 10.1586/14760584.2013.811188. [DOI] [PubMed] [Google Scholar]
- 46.Hawken J., Troy S.B. Adjuvants and inactivated polio vaccine: a systematic review. Vaccine. 2012;30:6971–6979. doi: 10.1016/j.vaccine.2012.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Hagan D.T., Ott G.S., De Gregorio E., Seubert A. The mechanism of action of MF59 – an innately attractive adjuvant formulation. Vaccine. 2012;30:4341–4348. doi: 10.1016/j.vaccine.2011.09.061. [DOI] [PubMed] [Google Scholar]
- 48.Heinemann L., Woodfield L., Amer M., Hibma M. Effective induction of type 1 helper IgG2a and cytotoxic T-cell responses in mice following immunization with human papillomavirus type 16 E2 in MF59. Viral Immunol. 2008;21:225–233. doi: 10.1089/vim.2007.0101. [DOI] [PubMed] [Google Scholar]
- 49.Bohannon J.K., Hernandez A., Enkhbaatar P., Adams W.L., Sherwood E.R. The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants. Shock. 2013;40:451–462. doi: 10.1097/SHK.0000000000000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan Y., Kagan J.C. A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol. Cell. 2014;54:212–223. doi: 10.1016/j.molcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang Z., Mak T.W., Sen G., Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kagan J.C., Su T., Horng T., Chow A., Akira S., Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deutschman C.S., Tracey K.J. Sepsis: current dogma and new perspectives. Immunity. 2014;40:463–475. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 54.Schneerson R., Fattom A., Szu S.C., Bryla D., Ulrich J.T., Rudbach J.A. Evaluation of monophosphoryl lipid A (MPL) as an adjuvant. Enhancement of the serum antibody response in mice to polysaccharide-protein conjugates by concurrent injection with MPL. J. Immunol. 1991;147:2136–2140. [PubMed] [Google Scholar]
- 55.Mata-Haro V., Cekic C., Martin M., Chilton P.M., Casella C.R., Mitchell T.C. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 56.Moore A., McCarthy L., Mills K.H.G. The adjuvant combination monophosphoryl lipid A and QS21 switches T cell responses induced with a soluble recombinant HIV protein from Th2 to Th1. Vaccine. 1999;17:2517–2527. doi: 10.1016/s0264-410x(99)00062-6. [DOI] [PubMed] [Google Scholar]
- 57.Thoelen S., Van Damme P., Mathei C., Leroux-Roelst G., Desomberet I. Safety and immunogenicity of a hepatitis B vaccine formulated with a novel adjuvant system. Vaccine. 1998;16:708–714. doi: 10.1016/s0264-410x(97)00254-5. [DOI] [PubMed] [Google Scholar]
- 58.Astiz M.E., Rackow E.C., Still J.G., Howell S.T., Cato A., Von Eschen K.B. Pretreatment of normal humans with monophosphoryl lipid A induces tolerance to endotoxin: a prospective, double-blind, randomized, controlled trial. Crit. Care Med. 1995;23:9–17. doi: 10.1097/00003246-199501000-00006. [DOI] [PubMed] [Google Scholar]
- 59.Samatey F.A., Imada K., Nagashima S., Vonderviszt F., Kumasaka T., Yamamoto M. Structure of the bacterial flagellar protofilament and implications for a switch for supercoiling. Nature. 2001;410:331–337. doi: 10.1038/35066504. [DOI] [PubMed] [Google Scholar]
- 60.Donnelly M.A., Steiner T.S. Two nonadjacent regions in enteroaggregative Escherichia coli flagellin are required for activation of toll-like receptor 5. J. Biol. Chem. 2002;277:40456–40461. doi: 10.1074/jbc.M206851200. [DOI] [PubMed] [Google Scholar]
- 61.Eaves-Pyles T.D., Wong H.R., Odoms K., Pyles R.B. Salmonella flagellin-dependent proinflammatory responses are localized to the conserved amino and carboxyl regions of the protein. J. Immunol. 2001;167:7009–7016. doi: 10.4049/jimmunol.167.12.7009. [DOI] [PubMed] [Google Scholar]
- 62.Murthy K.G.K., Deb A., Goonesekera S., Szabó C., Salzman A.L. Identification of conserved domains in Salmonella muenchen flagellin that are essential for its ability to activate TLR5 and to induce an inflammatory response in vitro. J. Biol. Chem. 2004;279:5667–5675. doi: 10.1074/jbc.M307759200. [DOI] [PubMed] [Google Scholar]
- 63.Zhao Y., Shao F. The NAIP-NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol. Rev. 2015;265:85–102. doi: 10.1111/imr.12293. [DOI] [PubMed] [Google Scholar]
- 64.Mizel S.B., Bates J.T. Flagellin as an adjuvant: cellular mechanisms and potential. J. Immunol. 2010;185:5677–5682. doi: 10.4049/jimmunol.1002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta S.K., Bajwa P., Deb R., Chellappa M.M., Dey S. Flagellin a toll-like receptor 5 agonist as an adjuvant in chicken vaccines. Clin. Vaccine Immunol. 2014;21:261–270. doi: 10.1128/CVI.00669-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gewirtz A.T., Navas T.A., Lyons S., Godowski P.J., Madara J.L. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 67.Honko A.N., Mizel S.B. Mucosal administration of flagellin induces innate immunity in the mouse lung. Infect. Immun. 2004;72:6676–6679. doi: 10.1128/IAI.72.11.6676-6679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsujimoto H., Uchida T., Efron P.A., Scumpia P.O., Verma A., Matsumoto T. Flagellin enhances NK cell proliferation and activation directly and through dendritic cell-NK cell interactions. J. Leukoc. Biol. 2005;78:888–897. doi: 10.1189/jlb.0105051. [DOI] [PubMed] [Google Scholar]
- 69.Vicente-Suarez I., Brayer J., Villagra A., Cheng F., Sotomayor E.M. TLR5 ligation by flagellin converts tolerogenic dendritic cells into activating antigen-presenting cells that preferentially induce T-helper 1 responses. Immunol. Lett. 2009;125:114–118. doi: 10.1016/j.imlet.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Didierlaurent A., Ferrero I., Otten L.A., Dubois B., Reinhardt M., Carlsen H. Flagellin promotes myeloid differentiation factor 88-dependent development of Th2-type response. J. Immunol. 2004;172:6922–6930. doi: 10.4049/jimmunol.172.11.6922. [DOI] [PubMed] [Google Scholar]
- 71.Means T.K., Hayashi F., Smith K.D., Aderem A., Luster A.D. The toll-like receptor 5 stimulus bacterial flagellin induces maturation and chemokine production in human dendritic cells. J. Immunol. 2003;170:5165–5175. doi: 10.4049/jimmunol.170.10.5165. [DOI] [PubMed] [Google Scholar]
- 72.Braga C.J.M., Massis L.M., Sbrogio-Almeida M.E., Alencar B.C.G., Bargieri D.Y., Boscardin S.B. CD8+ T cell adjuvant effects of Salmonella FliCd flagellin in live vaccine vectors or as purified protein. Vaccine. 2010;28:1373–1382. doi: 10.1016/j.vaccine.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 73.Cunningham A.F., Khan M., Ball J., Toellner K.M., Serre K., Mohr E. Responses to the soluble flagellar protein FliC are Th2, while those to FliC on Salmonella are Th1. Eur. J. Immunol. 2004;34:2986–2995. doi: 10.1002/eji.200425403. [DOI] [PubMed] [Google Scholar]
- 74.Nguyen C.T., Hong S.H., Sin J.I., Vu H.V.D., Jeong K., Cho K.O. Flagellin enhances tumor-specific CD8+ T cell immune responses through TLR5 stimulation in a therapeutic cancer vaccine model. Vaccine. 2013;31:3879–3887. doi: 10.1016/j.vaccine.2013.06.054. [DOI] [PubMed] [Google Scholar]
- 75.Bates J.T., Graff A.H., Phipps J.P., Grayson J.M., Mizel S.B. Enhanced antigen processing of flagellin fusion proteins promotes the antigen-specific CD8+ T cell response independently of TLR5 and MyD88. J. Immunol. 2011;186:6255–6262. doi: 10.4049/jimmunol.1001855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huleatt J.W., Jacobs A.R., Tang J., Desai P., Kopp E.B., Huang Y. Vaccination with recombinant fusion proteins incorporating Toll-like receptor ligands induces rapid cellular and humoral immunity. Vaccine. 2007;25:763–775. doi: 10.1016/j.vaccine.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 77.Lightfield K.L., Persson J., Brubaker S.W., Witte C.E., von Moltke J., Dunipace E.A. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 2008;9:1171–1178. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Halff E.F., Diebolder C.A., Versteeg M., Schouten A., Brondijk T.H.C., Huizinga E.G. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 2012;287:38460–38472. doi: 10.1074/jbc.M112.393512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumagai Y., Takeuchi O., Akira S. TLR9 as a key receptor for the recognition of DNA. Adv. Drug Deliv. Rev. 2008;60:795–804. doi: 10.1016/j.addr.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 80.Scheiermann J., Klinman D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine. 2014;32:6377–6389. doi: 10.1016/j.vaccine.2014.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bode C., Zhao G., Steinhagen F., Kinjo T., Klinman D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines. 2011;10:499–511. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shirota H., Klinman D.M. Recent progress concerning CpG DNA and its use as a vaccine adjuvant. Expert Rev. Vaccines. 2014;13:299–312. doi: 10.1586/14760584.2014.863715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cervantes J.L., Weinerman B., Basole C., Salazar J.C. TLR8: the forgotten relative revindicated. Cell. Mol. Immunol. 2012;9:434–438. doi: 10.1038/cmi.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ohto U., Tanji H., Shimizu T. Structure and function of Toll-like receptor 8. Microbes Infect. 2014;16:273–282. doi: 10.1016/j.micinf.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 85.Vasilakos J.P., Tomai M.A. The use of Toll-like receptor 7/8 agonists as vaccine adjuvants. Expert Rev. Vaccines. 2013;12:809–819. doi: 10.1586/14760584.2013.811208. [DOI] [PubMed] [Google Scholar]
- 86.Bagnoli F., Fontana M.R., Soldaini E., Mishra R.P.N., Fiaschi L., Cartocci E. Vaccine composition formulated with a novel TLR7-dependent adjuvant induces high and broad protection against Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 2015;112:3680–3685. doi: 10.1073/pnas.1424924112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prince A.M. An antigen detected in the blood during the incubation period of serum hepatitis. Proc. Natl. Acad. Sci. U.S.A. 1968;60:814–821. doi: 10.1073/pnas.60.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blumberg B.S., Alter H.J., Visnich S. A “new” antigen in leukemia sera. JAMA. 1965;191:541–546. doi: 10.1001/jama.1965.03080070025007. [DOI] [PubMed] [Google Scholar]
- 89.Pan C.Q., Zhang J.X. Natural history and clinical consequences of hepatitis B virus infection. Int. J. Med. Sci. 2005;2:36–40. doi: 10.7150/ijms.2.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kushnir N., Streatfield S.J., Yusibov V. Virus-like particles as a highly efficient vaccine platform: diversity of targets and production systems and advances in clinical development. Vaccine. 2012;31:58–83. doi: 10.1016/j.vaccine.2012.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cabral G.A., Marciano-Cabral F., Funk G.A., Sanchez Y., Hollinger F.B., Melnick J.L. Cellular and humoral immunity in guinea pigs to two major polypeptides derived from hepatitis B surface antigen. J. Gen. Virol. 1978;38:339–350. doi: 10.1099/0022-1317-38-2-339. [DOI] [PubMed] [Google Scholar]
- 92.Maupas P., Goudeau A., Coursaget P., Drucker J., Bagros P. Immunisation against hepatitis B in man. Lancet. 1976;307:1367–1370. doi: 10.1016/s0140-6736(76)93023-3. [DOI] [PubMed] [Google Scholar]
- 93.Szmuness W., Stevens C.E., Harley E.J., Zang E.A., Oleszko W.R., William D.C. Hepatitis B vaccine: demonstration of efficacy in a controlled clinical trial in a high-risk population in the United States. N. Engl. J. Med. 1980;303:833–841. doi: 10.1056/NEJM198010093031501. [DOI] [PubMed] [Google Scholar]
- 94.Krugman S., Giles J.P., Hammond J. Viral hepatitis, type B (MS-2 strain). Studies on active immunization. JAMA. 1971;217:41–45. [PubMed] [Google Scholar]
- 95.Purcell R.H., Gerin J.L. Hepatitis B subunit vaccine: a preliminary report of safety and efficacy tests in chimpanzees. Am. J. Med. Sci. 1975;270:395–399. [PubMed] [Google Scholar]
- 96.Hilleman M.R., Buynak E.B., Roehm R.R., Tytell A.A., Bertland A.U., Lampson G.P. Purified and inactivated human hepatitis B vaccine. Am. J. Med. Sci. 1975;270:401–404. doi: 10.1097/00000441-197509000-00025. [DOI] [PubMed] [Google Scholar]
- 97.Michel M.-L., Tiollais P. Hepatitis B vaccines: protective efficacy and therapeutic potential. Pathol. Biol. (Paris) 2010;58:288–295. doi: 10.1016/j.patbio.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 98.MacKay P., Pasek M., Magazin M., Kovacic R.T., Allet B., Stahl S. Production of immunologically active surface antigens of hepatitis B virus by Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1981;78:4510–4514. doi: 10.1073/pnas.78.7.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Burrell C.J., Mackay P., Greenaway P.J., Hofschneider P.H., Murray K. Expression in Escherichia coli of hepatitis B virus DNA sequences cloned in plasmid pBR322. Nature. 1979;279:43–47. doi: 10.1038/279043a0. [DOI] [PubMed] [Google Scholar]
- 100.Edman J.C., Hallewell R.A., Valenzuela P., Goodman H.M., Rutter W.J. Synthesis of hepatitis B surface and core antigens in E. coli. Nature. 1981;291:503–506. doi: 10.1038/291503a0. [DOI] [PubMed] [Google Scholar]
- 101.Valenzuela P., Medina A., Rutter W. Synthesis and assembly of hepatitis B virus surface antigen particles in yeast. Nature. 1982;298:347–350. doi: 10.1038/298347a0. [DOI] [PubMed] [Google Scholar]
- 102.Zajac B.A., West D.J., Mcaleer W.J., Scolnick E.M. Overview of clinical studies with hepatitis B vaccine made by recombinant DNA B. J. Infect. 1986;3:39–45. doi: 10.1016/s0163-4453(86)92668-x. [DOI] [PubMed] [Google Scholar]
- 103.Dandolos E., Roumeliotou-Karayannis A., Richardson S.C., Papaevangelou G. Safety and immunogenicity of a recombinant hepatitis B vaccine. J. Med. Virol. 1985;17:57–62. doi: 10.1002/jmv.1890170109. [DOI] [PubMed] [Google Scholar]
- 104.Jilg W., Schmidt M., Zoulek G., Lorbeer B., Wilske B., Deinhardt F. Clinical evaluation of a recombinant hepatitis B vaccine. Lancet. 1984;324:1174–1175. doi: 10.1016/s0140-6736(84)92740-5. [DOI] [PubMed] [Google Scholar]
- 105.Malik H., Khan F.H., Ahsan H. Human papillomavirus: current status and issues of vaccination. Arch. Virol. 2014;159:199–205. doi: 10.1007/s00705-013-1827-z. [DOI] [PubMed] [Google Scholar]
- 106.Rutgers T., Gordon D., Gathoye A.M., Hollingdale M., Hockmeyer W., Rosenberg M. Hepatitis B surface antigen as carrier matrix for the repetitive epitope of the circumsporozoite protein of Plasmodium falciparum. Nat. Biotechnol. 1988;6:1065–1070. [Google Scholar]
- 107.Baschong W., Hasler L., Häner M., Kistler J., Aebi U. Repetitive versus monomeric antigen presentation: direct visualization of antibody affinity and specificity. J. Struct. Biol. 2003;143:258–262. doi: 10.1016/j.jsb.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 108.Chackerian B. Virus-like particles: flexible platforms for vaccine development. Expert Rev. Vaccines. 2007;6:381–390. doi: 10.1586/14760584.6.3.381. [DOI] [PubMed] [Google Scholar]
- 109.Grgacic E.V.L., Anderson D.A. Virus-like particles: passport to immune recognition. Methods. 2006;40:60–65. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Feynman R.P. There’ s plenty of room at the bottom. J. Microelectromech. Syst. 1992;1:60–66. [Google Scholar]
- 111.Drexler K.E. Molecular engineering: an approach to the development of general capabilities for molecular manipulation. Proc. Natl. Acad. Sci. U.S.A. 1981;78:5275–5278. doi: 10.1073/pnas.78.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Santamaria A. Historical overview of nanotechnology and nanotoxicology. Methods Mol. Biol. 2012;926:1–12. doi: 10.1007/978-1-62703-002-1_1. [DOI] [PubMed] [Google Scholar]
- 113.What is nanotechnology? <http://www.nano.gov/nanotech-101/what/definition>, n.d. (accessed August 21, 2015).
- 114.Doll T.A.P.F., Raman S., Dey R., Burkhard P. Nanoscale assemblies and their biomedical applications. J. R. Soc. Interface. 2013;10 doi: 10.1098/rsif.2012.0740. 20120740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao L., Seth A., Wibowo N., Zhao C.X., Mitter N., Yu C. Nanoparticle vaccines. Vaccine. 2014;32:327–337. doi: 10.1016/j.vaccine.2013.11.069. [DOI] [PubMed] [Google Scholar]
- 116.Lu J.-M., Wang X., Marin-Muller C., Wang H., Lin P.H., Yao Q. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev. Mol. Diagn. 2009;9:325–341. doi: 10.1586/erm.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Danhier F., Ansorena E., Silva J.M., Coco R., Le Breton A., Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J. Control. Release. 2012;161:505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 118.Lutsiak M.E.C., Kwon G.S., Samuel J. Biodegradable nanoparticle delivery of a Th2-biased peptide for induction of Th1 immune responses. J. Pharm. Pharmacol. 2006;58:739–747. doi: 10.1211/jpp.58.6.0004. [DOI] [PubMed] [Google Scholar]
- 119.Manish M., Rahi A., Kaur M., Bhatnagar R., Singh S. A single-dose PLGA encapsulated protective antigen domain 4 nanoformulation protects mice against Bacillus anthracis spore challenge. PLoS One. 2013;8 doi: 10.1371/journal.pone.0061885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Silva A.L., Rosalia R.A., Sazak A., Carstens M.G., Ossendorp F., Oostendorp J. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: low-burst release is crucial for efficient CD8+ T cell activation. Eur. J. Pharm. Biopharm. 2013;83:338–345. doi: 10.1016/j.ejpb.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 121.Demento S.L., Cui W., Criscione J.M., Stern E., Tulipan J., Kaech S.M. Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials. 2012;33:4957–4964. doi: 10.1016/j.biomaterials.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vila A., Sánchez A., Évora C., Soriano I., Vila Jato J.L., Alonso M.J. PEG-PLA nanoparticles as carriers for nasal vaccine delivery. J. Aerosol Med. 2004;17:174–185. doi: 10.1089/0894268041457183. [DOI] [PubMed] [Google Scholar]
- 123.Kim S.Y., Doh H.J., Jang M.H., Ha Y.J., Chung S.I., Park H.J. Oral immunization with Helicobacter pylori-loaded poly(d, l-lactide-co-glycolide) nanoparticles. Helicobacter. 1999;4:33–39. doi: 10.1046/j.1523-5378.1999.09046.x. [DOI] [PubMed] [Google Scholar]
- 124.Thomas C., Rawat A., Hope-Weeks L., Ahsan F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol. Pharm. 2011;8:405–415. doi: 10.1021/mp100255c. [DOI] [PubMed] [Google Scholar]
- 125.Moon J.J., Suh H., Polhemus M.E., Ockenhouse C.F., Yadava A., Irvine D.J. Antigen-displaying lipid-enveloped PLGA nanoparticles as delivery agents for a Plasmodium vivax malaria vaccine. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ilyinskii P.O., Roy C.J., O’Neil C.P., Browning E.A., Pittet L.A., Altreuter D.H. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine. 2014;32:2882–2895. doi: 10.1016/j.vaccine.2014.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang L., Chan J.M., Gu F.X., Rhee J., Wang A.Z., Radovic-moreno A.F. Self-assembled lipid polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2:1696–1702. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Scheerlinck J.P.Y., Gloster S., Gamvrellis A., Mottram P.L., Plebanski M. Systemic immune responses in sheep, induced by a novel nano-bead adjuvant. Vaccine. 2006;24:1124–1131. doi: 10.1016/j.vaccine.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 129.Johrden L., Tenbusch M., Lietz R., Bonsmann M.S.G., Niezold T., Wildner O. Comparison of polystyrene nanoparticles and UV-inactivated antigen-displaying adenovirus for vaccine delivery in mice. Virol. J. 2013;10:108. doi: 10.1186/1743-422X-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kalkanidis M., Pietersz G.A., Xiang S.D., Mottram P.L., Crimeen-Irwin B., Ardipradja K. Methods for nano-particle based vaccine formulation and evaluation of their immunogenicity. Methods. 2006;40:20–29. doi: 10.1016/j.ymeth.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 131.Mohamud R., Xiang S.D., Selomulya C., Rolland J.M., O’Hehir R.E., Hardy C.L. The effects of engineered nanoparticles on pulmonary immune homeostasis. Drug Metab. Rev. 2013;2532:1–15. doi: 10.3109/03602532.2013.859688. [DOI] [PubMed] [Google Scholar]
- 132.Kumar D., Saini N., Jain N., Sareen R., Pandit V. Gold nanoparticles: an era in bionanotechnology. Expert Opin. Drug Deliv. 2013;10:397–409. doi: 10.1517/17425247.2013.749854. [DOI] [PubMed] [Google Scholar]
- 133.Cao-Milán R., Liz-Marzán L.M. Gold nanoparticle conjugates: recent advances toward clinical applications. Expert Opin. Drug Deliv. 2014;11:741–752. doi: 10.1517/17425247.2014.891582. [DOI] [PubMed] [Google Scholar]
- 134.Hadrup N., Sharma A.K., Poulsen M., Nielsen E. Toxicological risk assessment of elemental gold following oral exposure to sheets and nanoparticles – a review. Regul. Toxicol. Pharmacol. 2015;72:216–221. doi: 10.1016/j.yrtph.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 135.Niikura K., Matsunaga T., Suzuki T., Kobayashi S., Yamaguchi H. Gold nanoparticles as a vaccine platform: influence of size and shape on immunological responses in vitro and in vivo. 2013:3926–3938. doi: 10.1021/nn3057005. [DOI] [PubMed] [Google Scholar]
- 136.Gregory A.E., Williamson E.D., Prior J.L., Butcher W.A., Thompson I.J., Shaw A.M. Conjugation of Y. pestis F1-antigen to gold nanoparticles improves immunogenicity. Vaccine. 2012;30:6777–6782. doi: 10.1016/j.vaccine.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tao W., Gill H.S. M2e-immobilized gold nanoparticles as influenza A vaccine: role of soluble M2e and longevity of protection. Vaccine. 2015;33:2307–2315. doi: 10.1016/j.vaccine.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Biswas S., Medina S.H., Barchi J.J. Synthesis and cell-selective antitumor properties of amino acid conjugated tumor-associated carbohydrate antigen-coated gold nanoparticles. Carbohydr. Res. 2015;405:93–101. doi: 10.1016/j.carres.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Almeida J.P.M., Lin A.Y., Figueroa E.R., Foster A.E., Drezek R.A. In vivo gold nanoparticle delivery of peptide vaccine induces anti-tumor immune response in prophylactic and therapeutic tumor models. Small. 2015;11:1453–1459. doi: 10.1002/smll.201402179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fadel T.R., Fahmy T.M. Immunotherapy applications of carbon nanotubes: from design to safe applications. Trends Biotechnol. 2014;32:198–209. doi: 10.1016/j.tibtech.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 141.Palomäki J., Välimäki E., Sund J., Vippola M., Clausen P.A., Jensen K.A. Long, needle-like carbon nanotubes and asbestos activate the NLRP3 inflammasome through a similar mechanism. ACS Nano. 2011;5:6861–6870. doi: 10.1021/nn200595c. [DOI] [PubMed] [Google Scholar]
- 142.Katwa P., Wang X., Urankar R.N., Podila R., Hilderbrand S.C., Fick R.B. A carbon nanotube toxicity paradigm driven by mast cells and the IL-33/ST2 axis. Small. 2012;8:2904–2912. doi: 10.1002/smll.201200873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kagan V.E., Tyurina Y.Y., Tyurin V.A., Konduru N.V., Potapovich A.I., Osipov A.N. Direct and indirect effects of single walled carbon nanotubes on RAW 264.7 macrophages: role of iron. Toxicol. Lett. 2006;165:88–100. doi: 10.1016/j.toxlet.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 144.Smith J.D., Morton L.D., Ulery B.D. Nanoparticles as synthetic vaccines. Curr. Opin. Biotechnol. 2015;34:217–224. doi: 10.1016/j.copbio.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 145.Fadeel B., Garcia-Bennett A.E. Better safe than sorry: understanding the toxicological properties of inorganic nanoparticles manufactured for biomedical applications. Adv. Drug Deliv. Rev. 2010;62:362–374. doi: 10.1016/j.addr.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 146.Bangham A.D., Standish M.M., Watkins J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965;13:238–252. doi: 10.1016/s0022-2836(65)80093-6. [DOI] [PubMed] [Google Scholar]
- 147.Allison A.C., Gregoriadis G. Liposomes as immunological adjuvants. Nature. 1974;252 doi: 10.1038/252252a0. 252-252. [DOI] [PubMed] [Google Scholar]
- 148.Giddam A.K., Giddam A.K., Zaman M., Skwarczynski M., Toth I. Liposome-based delivery system for vaccine candidates: constructing an effective formulation. Nanomedicine (Lond). 2012;7:1877–1893. doi: 10.2217/nnm.12.157. [DOI] [PubMed] [Google Scholar]
- 149.Smith Korsholm K., Agger E.M., Foged C., Christensen D., Dietrich J., Andersen C.S. The adjuvant mechanism of cationic dimethyldioctadecylammonium liposomes. Immunology. 2007;121:216–226. doi: 10.1111/j.1365-2567.2007.02560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nakanishi T., Kunisawa J., Hayashi A., Tsutsumi Y., Kubo K., Nakagawa S. Positively charged liposome functions as an efficient immunoadjuvant in inducing cell-mediated immune response to soluble proteins. J. Control. Release. 1999;61:233–240. doi: 10.1016/s0168-3659(99)00097-8. [DOI] [PubMed] [Google Scholar]
- 151.Foged C., Arigita C., Sundblad A., Jiskoot W., Storm G., Frokjaer S. Interaction of dendritic cells with antigen-containing liposomes: effect of bilayer composition. Vaccine. 2004;22:1903–1913. doi: 10.1016/j.vaccine.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 152.Nordly P., Agger E.M., Andersen P., Nielsen H.M., Foged C. Incorporation of the TLR4 agonist monophosphoryl lipid a into the bilayer of DDA/TDB liposomes: physico-chemical characterization and induction of CD8+ T-cell responses in vivo. Pharm. Res. 2011;28:553–562. doi: 10.1007/s11095-010-0301-9. [DOI] [PubMed] [Google Scholar]
- 153.Faham A., Altin J.G. Antigen-containing liposomes engrafted with flagellin-related peptides are effective vaccines that can induce potent antitumor immunity and immunotherapeutic effect. J. Immunol. 2010;185:1744–1754. doi: 10.4049/jimmunol.1000027. [DOI] [PubMed] [Google Scholar]
- 154.Huckriede A., Bungener L., Stegmann T., Daemen T., Medema J., Palache A.M. The virosome concept for influenza vaccines. Vaccine. 2005;23:26–38. doi: 10.1016/j.vaccine.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 155.Moser C., Amacker M., Zurbriggen R. Influenza virosomes as a vaccine adjuvant and carrier system. Expert Rev. Vaccines. 2011;10:437–446. doi: 10.1586/erv.11.15. [DOI] [PubMed] [Google Scholar]
- 156.Herzog C., Hartmann K., Künzi V., Kürsteiner O., Mischler R., Lazar H. Eleven years of Inflexal® V-a virosomal adjuvanted influenza vaccine. Vaccine. 2009;27:4381–4387. doi: 10.1016/j.vaccine.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 157.Bovier P.A. Epaxal: a virosomal vaccine to prevent hepatitis A infection. Expert Rev. Vaccines. 2008;7:1141–1150. doi: 10.1586/14760584.7.8.1141. [DOI] [PubMed] [Google Scholar]
- 158.Trovato M. Novel antigen delivery systems. World J. Virol. 2015;4:156. doi: 10.5501/wjv.v4.i3.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.He D., Marles-Wright J. Ferritin family proteins and their use in bionanotechnology. N. Biotechnol. 2015:1–7. doi: 10.1016/j.nbt.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lawson D.M., Artymiuk P.J., Yewdall S.J., Smith J.M., Livingstone J.C., Treffry A. Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature. 1991;349:541–544. doi: 10.1038/349541a0. [DOI] [PubMed] [Google Scholar]
- 161.Grant R.A., Filman D.J., Finkel S.E., Kolter R., Hogle J.M. The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat. Struct. Biol. 1998;5:294–303. doi: 10.1038/nsb0498-294. [DOI] [PubMed] [Google Scholar]
- 162.Kanekiyo M., Wei C.-J., Yassine H.M., McTamney P.M., Boyington J.C., Whittle J.R.R. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature. 2013;499:102–106. doi: 10.1038/nature12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yassine H.M., Boyington J.C., Mctamney P.M., Wei C., Kanekiyo M., Kong W. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 2015 doi: 10.1038/nm.3927. [DOI] [PubMed] [Google Scholar]
- 164.Berger W., Steiner E., Grusch M., Elbling L., Micksche M. Vaults and the major vault protein: novel roles in signal pathway regulation and immunity. Cell. Mol. Life Sci. 2009;66:43–61. doi: 10.1007/s00018-008-8364-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rome L.H., Kickhoefer V.A. Development of the vault particle as a platform technology. ACS Nano. 2013;7:889–902. doi: 10.1021/nn3052082. [DOI] [PubMed] [Google Scholar]
- 166.Kedersha N.L., Rome L.H. Isolation and characterization of a novel ribonucleoprotein particle: large structures contain a single species of small RNA. J. Cell Biol. 1986;103:699–709. doi: 10.1083/jcb.103.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kar U.K., Jiang J., Champion C.I., Salehi S., Srivastava M., Sharma S. Vault nanocapsules as adjuvants favor cell-mediated over antibody-mediated immune responses following immunization of mice. PLoS One. 2012;7:1–14. doi: 10.1371/journal.pone.0038553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Champion C.I., Kickhoefer V.A., Liu G., Moniz R.J., Freed A.S., Bergmann L.L. A vault nanoparticle vaccine induces protective mucosal immunity. PLoS One. 2009;4 doi: 10.1371/journal.pone.0005409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Papapostolou D., Howorka S. Engineering and exploiting protein assemblies in synthetic biology. Mol. BioSyst. 2009;5:723–732. doi: 10.1039/b902440a. [DOI] [PubMed] [Google Scholar]
- 170.Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 171.Howorka S. Rationally engineering natural protein assemblies in nanobiotechnology. Curr. Opin. Biotechnol. 2011;22:485–491. doi: 10.1016/j.copbio.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 172.Bromley E.H.C., Channon K., Moutevelis E., Woolfson D.N. Peptide and protein building blocks for synthetic biology: from programming biomolecules to Self-organized biomolecular systems. ACS Chem. Biol. 2008;3:38–50. doi: 10.1021/cb700249v. [DOI] [PubMed] [Google Scholar]
- 173.Powell T.J., Palath N., DeRome M.E., Tang J., Jacobs A., Boyd J.G. Synthetic nanoparticle vaccines produced by layer-by-layer assembly of artificial biofilms induce potent protective T-cell and antibody responses in vivo. Vaccine. 2011;29:558–569. doi: 10.1016/j.vaccine.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 174.Jorquera P., Oakley K., Powell T., Palath N., Boyd J., Tripp R. Layer-by-layer nanoparticle vaccines carrying the G protein CX3C motif protect against RSV infection and disease. Vaccines. 2015;3:829–849. doi: 10.3390/vaccines3040829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Powell T.J., Tang J., DeRome M.E., Mitchell R.A., Jacobs A., Deng Y. Plasmodium falciparum synthetic LbL microparticle vaccine elicits protective neutralizing antibody and parasite-specific cellular immune responses. Vaccine. 2013;31:1898–1904. doi: 10.1016/j.vaccine.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Collier J.H., Messersmith P.B. Enzymatic modification of self-assembled peptide structures with tissue transglutaminase. Bioconjug. Chem. 2003;14:748–755. doi: 10.1021/bc034017t. [DOI] [PubMed] [Google Scholar]
- 177.Jung J.P., Nagaraj A.K., Fox E.K., Rudra J.S., Devgun J.M., Collier J.H. Co-assembling peptides as defined matrices for endothelial cells. Biomaterials. 2009;30:2400–2410. doi: 10.1016/j.biomaterials.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rudra J.S., Tian Y.F., Jung J.P., Collier J.H. A self-assembling peptide acting as an immune adjuvant. Proc. Natl. Acad. Sci. U.S.A. 2010;107:622–627. doi: 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chesson C.B., Huelsmann E.J., Lacek A.T., Kohlhapp F.J., Webb M.F., Nabatiyan A. Antigenic peptide nanofibers elicit adjuvant-free CD8+ T cell responses. Vaccine. 2014;32:1174–1180. doi: 10.1016/j.vaccine.2013.11.047. [DOI] [PubMed] [Google Scholar]
- 180.Rudra J.S., Mishra S., Chong A.S., Mitchell R.A., Nardin E.H., Nussenzweig V. Self-assembled peptide nanofibers raising durable antibody responses against a malaria epitope. Biomaterials. 2012;33:6476–6484. doi: 10.1016/j.biomaterials.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pompano R.R., Chen J., Verbus E.A., Han H., Fridman A., McNeely T. Titrating T-cell epitopes within self-assembled vaccines optimizes CD4+ helper T cell and antibody outputs. Adv. Healthc. Mater. 2014;3:1898–1908. doi: 10.1002/adhm.201400137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Azmi F., Ahmad Fuaad A.A.H., Giddam A.K., Batzloff M.R., Good M.F., Skwarczynski M. Self-adjuvanting vaccine against group A streptococcus: application of fibrillized peptide and immunostimulatory lipid as adjuvant. Bioorg. Med. Chem. 2014;22:6401–6408. doi: 10.1016/j.bmc.2014.09.042. [DOI] [PubMed] [Google Scholar]
- 183.Chen J., Pompano R.R., Santiago F.W., Maillat L., Sciammas R., Sun T. The use of self-adjuvanting nanofiber vaccines to elicit high-affinity B cell responses to peptide antigens without inflammation. Biomaterials. 2013;34:8776–8785. doi: 10.1016/j.biomaterials.2013.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Burkhard P., Stetefeld J., Strelkov S.V. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 185.Tamborrini M., Geib N., Marrero-Nodarse A., Jud M., Hauser J., Aho C. A synthetic virus-like particle streptococcal vaccine candidate using B-cell epitopes from the proline-rich region of pneumococcal surface protein A. Vaccines. 2015;3:850–874. doi: 10.3390/vaccines3040850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ghasparian A., Riedel T., Koomullil J., Moehle K., Gorba C., Svergun D.I. Engineered synthetic virus-like particles and their use in vaccine delivery. ChemBioChem. 2011;12:100–109. doi: 10.1002/cbic.201000536. [DOI] [PubMed] [Google Scholar]
- 187.Raman S., Machaidze G., Lustig A., Olivieri V., Aebi U., Burkhard P. Design of peptide nanoparticles using simple protein oligomerization domains. Open Nanomed. J. 2009;2:15–26. [Google Scholar]
- 188.Yang Y., Ringler P., Müller S.A., Burkhard P. Optimizing the refolding conditions of self-assembling polypeptide nanoparticles that serve as repetitive antigen display systems. J. Struct. Biol. 2012;177:168–176. doi: 10.1016/j.jsb.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Indelicato G., Wahome N., Ringler P., Müller S.A., Nieh M.-P., Burkhard P. Principles governing the self-assembly of coiled-coil protein nanoparticles. Biophys. J. 2016;110:646–660. doi: 10.1016/j.bpj.2015.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Raman S., Machaidze G., Lustig A., Aebi U., Burkhard P. Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomedicine. 2006;2:95–102. doi: 10.1016/j.nano.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 191.Apostolovic B., Danial M., Klok H.-A. Coiled coils: attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials. Chem. Soc. Rev. 2010;39:3541–3575. doi: 10.1039/b914339b. [DOI] [PubMed] [Google Scholar]
- 192.Augustin T., Cehlar O., Skrabana R., Majerova P., Hanes J. Unravelling viral camouflage: approaches to the study and characterization of conformational epitopes. Acta Virol. 2015;59:103–116. doi: 10.4149/av_2015_02_103. [DOI] [PubMed] [Google Scholar]
- 193.Kaba S.A., McCoy M.E., Doll T.A.P.F., Brando C., Guo Q., Dasgupta D. Protective antibody and CD8(+) T-cell responses to the Plasmodium falciparum circumsporozoite protein induced by a nanoparticle vaccine. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Babapoor S., Neef T., Mittelholzer C., Girshick T., Garmendia A., Shang H. A novel vaccine using nanoparticle platform to present immunogenic M2e against avian influenza infection. Influenza Res. Treat. 2011;2011 doi: 10.1155/2011/126794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pimentel T.A.P.F., Yan Z., Jeffers S.A., Holmes K.V., Hodges R.S., Burkhard P. Peptide nanoparticles as novel immunogens: design and analysis of a prototypic severe acute respiratory syndrome vaccine. Chem. Biol. Drug Des. 2009;73:53–61. doi: 10.1111/j.1747-0285.2008.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wahome N., Pfeiffer T., Ambiel I., Yang Y., Keppler O.T., Bosch V. Conformation-specific display of 4E10 and 2F5 epitopes on self-assembling protein nanoparticles as a potential HIV vaccine. Chem. Biol. Drug Des. 2012;80:349–357. doi: 10.1111/j.1747-0285.2012.01423.x. [DOI] [PubMed] [Google Scholar]
- 197.Mutapi F., Billingsley P.F., Secor W.E. Infection and treatment immunizations for successful parasite vaccines. Trends Parasitol. 2013;29:135–141. doi: 10.1016/j.pt.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kaba S.A., Brando C., Guo Q., Mittelholzer C., Raman S., Tropel D. A nonadjuvanted polypeptide nanoparticle vaccine confers long-lasting protection against rodent malaria. J. Immunol. 2009;183:7268–7277. doi: 10.4049/jimmunol.0901957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.McCoy M.E., Golden H.E., Doll T.A.P.F., Yang Y., Kaba S.A., Zou X. Mechanisms of protective immune responses induced by the Plasmodium falciparum circumsporozoite protein-based, self-assembling protein nanoparticle vaccine. Malar. J. 2013;12:136. doi: 10.1186/1475-2875-12-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.El Bissati K., Zhou Y., Dasgupta D., Cobb D., Dubey J.P., Burkhard P. Effectiveness of a novel immunogenic nanoparticle platform for Toxoplasma peptide vaccine in HLA transgenic mice. Vaccine. 2014;32:3243–3248. doi: 10.1016/j.vaccine.2014.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]