

Introduction
The XX male syndrome, first reported in 1964, is an unusual cause of male factor infertility. Its relative rarity coupled with the presence of unambiguous male external genitalia in most patients with this syndrome makes clinicians overlook the condition in the differential diagnosis of primary infertility. We report five genetically proven XX males, who all presented with azoospermia and primary infertility (four out of five), and discuss their phenotypic characteristics and hormonal profiles to make treating physicians aware of this diagnosis in appropriate settings.
Case Report
Case 1
A couple, married for 2 years, presented with primary infertility. The 26-year-old husband was found to have azoospermia. His height was 154.7 cm with upper segment (US)/lower segment (LS) ratio of 0.82. His arm span was 155.4 cm. He was well virilized (Tanner stage 4 pubic hair) with normal stretched penile length (SPL) (Fig. 1a). However, both the testes were 3 ml in volume and were very firm on palpation. Both the epididymes were palpable without nodularity. He also had bilateral non-tender gynaecomastia. He used to achieve orgasm with very poor ejaculatory volume.
Fig. 1.

External genitalia a and G-banded karyotype b of case 1
Hormonal investigations revealed the following:
Early morning total testosterone: 71.6 ng/dl; FSH: 37.2 mIU/ml; LH: 24.3 mIU/ml; and prolactin: 7.7 ng/ml.
Case 2
Forty-two-year-old husband of an infertile couple was evaluated for azoospermia. He was 162.5 cm tall with an arm span of 163 cm and US/LS of 0.8. Genital examination revealed Tanner stage 4 pubic hair, 2-ml firm testes and normally formed epididymis. Both the breasts were enlarged with a disc diameter of 5 cm.
Hormonal evaluation documented:
Early morning total testosterone: 64.8 ng/dl; FSH: 24.4 mIU/ml; LH: 18.2 mIU/ml; and prolactin: 5.7 ng/ml.
Case 3
Twenty-one-year-old married gentleman presented with bilateral tender gynaecomastia with low ejaculatory volume. His auxology revealed the following: height: 165 cm; arm span: 166 cm; and US/ LS: 0.75. He had Tanner stage 4 pubic hair with bilateral small testes (6 ml) with normal consistency. SPL was 9 cm.
Endocrinological evaluation showed:
Early morning total testosterone: 183 ng/dl; FSH: 44.6 mIU/ml; LH: 22 mIU/ml; and prolactin: 10.2 ng/ml.
Case 4
Fifty-two-year-old male, detected with type 2 diabetes about 5 years back, attended our clinic for glycaemic control. A detailed history revealed long standing significant hypogonadism with primary infertility. He was short (height: 162 cm), centrally obese (waist circumference: 102 cm) with US/LS of 0.79 and arm span of 163 cm. Both testes were firm and 3 ml in volume. Endocrinological work-up documented hypergonadotropic hypogonadism as evidenced by low testosterone and elevated gonadotrophin.
Case 5
Clinical examination in a 17-year-old unmarried male, presenting with bilateral gynaecomastia, revealed the following:
Height: 170.2 cm; arm span: 172 cm; and US/LS of 0.75
Pubic hair: Tanner stage 4 (Fig. 2a); testicular volume: 4 ml bilateral; both testes were firm; and bilateral tender gynaecomastia with a disc diameter of 4 cm.
Fig. 2.

External genitalia a and G-banded karyotype b of case 5
Biochemical examination revealed: testosterone: 254 ng/dl; FSH: 46.6 mIU/ml; and LH: 20.2 mIU/ml.
Seminal fluid examination in all five cases showed azoospermia with low ejaculatory volume. All the five patients had identical presentation with eunuchoidism, less than 5-cm difference between arm span and height, gynaecomastia, unambiguous male genitalia with adequate virilization, bilateral small testes, normal epididymis and hypergonadotropic hypogonadism. The working diagnosis in each of the above five cases was Klinefelter’s syndrome (KFS) or its variant, and G-banded karyotype from the peripheral blood lymphocytes was asked for in all of them.
The karyotype revealed the following:
Case 1 46, XX (Fig. 1b)
Case 2 46, XX
Case 3 46, XX
Case 4 46, XX
Case 5 46, X, der(X)t(X;Y) (p22.1; p11.2) (Fig. 2b)
(Small chromosomal material on p arm of one of the X chromosomes and the extrachromosomal material is likely a part of p arm of the Y chromosome)
A subsequent fluorescent in situ hybridization (FISH) using sex-determining region Y (SRY) gene probe revealed the presence of SRY (red signal) on one of the X chromosomes in each of them (blue signal) (Fig. 3).
Fig. 3.
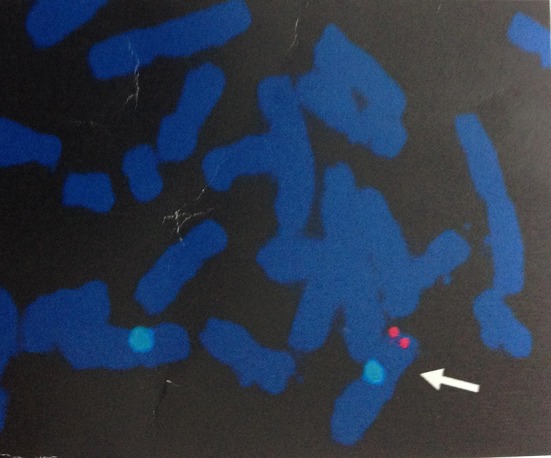
FISH showing presence of SRY (red signal) on one of the X chromosomes in one of the sample (blue signal)
Discussions
Primary infertility, affecting about 15 % couples in reproductive age group, is defined as failure to conceive after at least 1 year of regular unprotected intercourse. Male factor infertility, due to varying forms and severity of impaired spermatogenesis, constitutes about half of these cases. Chromosomal abnormalities constitute <10 % of these infertile males; however, the proportion increases in males with azoospermia and severe oligospermia. The most common genetic cause of male factor infertility is microdeletions involving the long arm of Y chromosome (Yq), and the most frequent karyotype abnormality encountered in them is 47, XXY KFS.
46, XX karyotype is rarely encountered in patients getting investigated for primary infertility. The incidence varies from less than 1 % to about 9 %. Based on clinical presentations, sex-reversed 46, XX individuals fall into following three categories: (1) Classic XX males (also known as 46, XX testicular disorder of sex development), who are otherwise unambiguous males having normally differentiated male internal and external genitalia, usually present with primary infertility, hypogonadism and gynaecomastia, (2) XX males with ambiguous external genitalia, usually detected at birth, and (3) XX true hermaphrodites or XX ovotesticular disorder of sex development, who usually demonstrate ambiguous external and internal genitalia and have ovary and/or testes and/or ovotestes as the gonads [1]. The 46, XX male is considered a rare syndrome with an incidence of about 1:20,000 in newborn males; however, as most of them usually go unnoticed because of normal appearing male external genitalia, the true incidence perhaps is much higher than this.
The SRY, situated in the short arm of Y chromosome (Yp), is considered the major testes-determining factor, and its presence drives the bipotential primitive gonad towards testes development. About 90 % of the 46, XX males are known to carry variable amount of Y chromosome material containing the SRY region due to an X/Y chromosomal interchange during paternal meiosis, leading to the differentiation of primitive gonad into testes. Genes related to spermatogenesis are known to be harboured in the three azoospermia factor regions (AZFa, AZFb and AZFc) located in Yq. Absence of Yq explains the universal presence of azoospermia, despite testes being the gonad present in these patients. Testicular histology usually reveals complete Sertoli cell-only syndrome and hyperplasia of the Leydig cells.
The mean height of our patients was 162.88 cm. In a study involving 11 genetically proven XX male patients, the heights were between 160 and 165 cm with a mean of 161 cm, which matches the auxological findings of our patients. The appearance of the external genitalia of our patients are also identical to that particular cohort with normal masculinization and small testes [2].
KFS is undoubtedly the commonest cause of hypergonadotropic hypogonadism with oligozoospermia and is manifested by relatively tall stature with long-leggedness (the difference between height and arm span is less than 5 cm) and firm, small testes. Male hypogonadism, manifested during childhood, results in increased body height caused by retardation of sex steroid-induced closure of epiphyses. In most of the patients of KFS, the onset of hypogonadism is peri-pubertal and the long-leggedness and relatively taller height is due to SHOX (short stature homeobox) gene overdosage, located in the pseudoautosomal region 1 (PAR1) of the short arm of X and Y chromosomes.
XX males, who usually have normal for-age testosterone levels during puberty and higher frequency of hypergonadotropic hypogonadism in adulthood, can be differentiated phenotypically from KFS by shorter height and increased incidence of cryptorchidism and gynaecomastia [1]. XX males are also shorter than healthy men, suggesting the role of genes on Y chromosome that control height [3]. The potential Y chromosomal growth-control gene is putatively located on the long arm of the Y chromosome.
To conclude, the diagnosis of XX male should be considered in all infertile males with azoospermia who are short and clinically resembles KFS otherwise. It also carries an immense therapeutic implication because these patients are at increased risk of breast carcinoma [4] and testicular sperm extraction as a modality of assisted reproduction is going to be futile in these patients.
Acknowledgments
Funding
The study was not funded by anybody. The patients themselves bore the cost of suggested investigations.
Authors contribution
PPC, RB, SM and SC were involved in evaluation and management of first four patients. AR was involved in evaluation and management of case 5. PPC and RB were involved in the literature search and writing of the manuscript. SM and SC checked and finalized the manuscript.
Partha P. Chakraborty
holds the following qualification: MBBS (Hons); MD (Medicine); DM (Endocrinology); DNB (Endocrinology). He is a consultant endocrinologist of repute. He has more than 50 publications in various national and international journals to his credit. He is in West Bengal Medical Education Service since 2006, and currently he holds the post of Assistant Professor, Department of Medicine, Midnapore Medical College & Hospital.
Conflicts of interest
Disclosure of potential conflicts of interest if any: None. All the authors (PPC, RB, AR, SM and SC) declare that they have no conflict of interest.
Ethical Approval
This article contains case series only. The patients were evaluated according to their presentations. They were not included in any study protocol by any of the authors. The Institute approves publication of case series.
Informed Consent
Informed consents were obtained from all individual patients.
Footnotes
Partha P. Chakraborty is an Assistant Professor at the Department of Medicine, Midnapore Medical College & Hospital, Midnapore, West Bengal, India; Rana Bhattacharjee is a RMO-cum-clinical tutor; Satinath Mukhopadhyay is a Professor; Subhankar Chowdhury is a Professor at the Department of Endocrinology & Metabolism, IPGME&R/SSKM Hospital, Kolkata, West Bengal, India; Ajitesh Roy is a Consultant Endocrinologist at the Ruby General Hospital, Kolkata.
References
- 1.Vorona E, Zitzmann M, Gromoll J, et al. Clinical, endocrinological, and epigenetic features of the 46, XX male syndrome, compared with 47, XXY Klinefelter patients. J Clin Endocrinol Metab. 2007;92:3458–3465. doi: 10.1210/jc.2007-0447. [DOI] [PubMed] [Google Scholar]
- 2.Gao X, Chen G, Huang J, et al. Clinical, cytogenetic, and molecular analysis with 46, XX male sex reversal syndrome: case reports. J Assist Reprod Genet. 2013;30(3):431–435. doi: 10.1007/s10815-013-9939-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Chapelle A. The etiology of maleness in XX men. Hum Genet. 1981;58:105–116. doi: 10.1007/BF00284157. [DOI] [PubMed] [Google Scholar]
- 4.Hado HS, Helmy SW, Klemm K, et al. XX male: a rare cause of short stature, infertility, gynaecomastia and carcinoma of the breast. Int J Clin Pract. 2003;57(9):844–845. [PubMed] [Google Scholar]