

Abstract
Janthinobacterium and Duganella are well-known for their antifungal effects. Surprisingly, almost nothing is known on molecular aspects involved in the close bacterium-fungus interaction. To better understand this interaction, we established the genomes of 11 Janthinobacterium and Duganella isolates in combination with phylogenetic and functional analyses of all publicly available genomes. Thereby, we identified a core and pan genome of 1058 and 23,628 genes. All strains encoded secondary metabolite gene clusters and chitinases, both possibly involved in fungal growth suppression. All but one strain carried a single gene cluster involved in the biosynthesis of alpha-hydroxyketone-like autoinducer molecules, designated JAI-1. Genome-wide RNA-seq studies employing the background of two isolates and the corresponding JAI-1 deficient strains identified a set of 45 QS-regulated genes in both isolates. Most regulated genes are characterized by a conserved sequence motif within the promoter region. Among the most strongly regulated genes were secondary metabolite and type VI secretion system gene clusters. Most intriguing, co-incubation studies of J. sp. HH102 or its corresponding JAI-1 synthase deletion mutant with the plant pathogen Fusarium graminearum provided first evidence of a QS-dependent interaction with this pathogen.
Keywords: Janthinobacterium, Duganella, Fusarium graminearum, CAI-1, quorum sensing, violacein, bacterium-fungus interaction
Introduction
Janthinobacterium spp. and bacteria affiliated with the genus Duganella are Gram-negative, motile, and aerobic bacteria, which are commonly isolated from soil and aquatic samples. Janthinobacterium and Duganella spp. are grouped among 11 other genera within the family Oxalobacteraceae of the Betaproteobacteria (Lincoln et al., 1999; Baldani et al., 2014). Although most isolates have been obtained from soil or aquatic sites, Janthinobacterium can occur in marine habitats in which they can grow to remarkably high titers (Alonso-Sáez et al., 2014). While Janthinobacterium as well as Duganella appear to be non-pathogenic to humans, animals, and plants, they are well-known for their antifungal effects. For example, J. livium suppresses fungal growth on human and amphibian skin or J. agaricidamnsoum causes the soft rot disease on the mushroom Agaricus bisporus (Becker et al., 2009; Harris et al., 2009; Wiggins et al., 2011; Graupner et al., 2015; Ramsey et al., 2015). The antifungal activities within this Oxalobacteraceae family are most likely induced through a regulatory network in response to chitin or degradation products (Cretoiu et al., 2013; Kielak et al., 2013) and the involvement of the secondary metabolite violacein is hypothesized (Brucker et al., 2008; Ramsey et al., 2015).
To date, only few complete or permanent draft genomes of Janthinobacterium and Duganella are publicly available. In addition, only very few reports have analyzed the mechanisms of cell-cell communication in these genera. We recently reported, that Janthinobacterium sp. HH01 (in this study renamed as Duganella sp. HH01, Hornung et al., 2013) employs a cell-cell signaling mechanism that was previously only known for Vibrio or Legionella and is based on the synthesis of α-hydroxyketones. In V. cholerae and L. pneumophila the corresponding signaling molecule is involved in the regulation of pathogen-host interaction (Tiaden and Hilbi, 2012; Simon et al., 2015) and plays a key role in bacterial competence in V. cholerae, which could be linked to the presence of chitin (Lo Scrudato and Blokesch, 2012; Borgeaud et al., 2015). Working on HH01 we have shown that the janthinobacterial autoinducer (JAI-1) is synthesized by the autoinducer synthase JqsA. The jqsA gene is encoded within a conserved cluster together with the sensor kinase JqsS and the regulator protein JqsR. We further provided evidence in a previous study that JAI-1 affects the violacein biosynthesis in HH01 (Hornung et al., 2013). In the light of these observations we asked, whether and to which extent Janthinobacterium and Duganella have established cell-cell communication mechanisms used for intra- and inter-species communication and whether these would be important for their interaction with fungi.
To address these questions we performed comprehensive and combined genome, transcriptome, and mutational analysis of 11 strains. Furthermore, we analyzed the transcriptomes of two jqsA-knockout strains using RNA-seq and identified common and highly conserved regulatory quroum sensing (QS) circuits. Additional co-inoculation tests with the plant pathogen Fusarium graminearum indicate that the chitin degradation product N-acetyl-D-glucosamine and the QS signal JAI-1 both affect the interaction with the fungus in Janthinobacterium but not in Duganella isolates.
Materials and methods
Bacterial strains and growth
Bacterial strains and plasmids are listed in Table S1. Standard molecular cloning techniques were used (Sambrook and Russell, 2001). Environmental samples were collected from a rainwater-cistern at the Botanical Garden in Klein Flottbek (Hamburg, Germany, HH100–HH107). Samples were enriched on R2A liquid medium (Reasoner and Geldreich, 1985) and purple-pigmented bacteria were repetitively streaked on R2A plates to obtain pure cultures. The same habitat was used to exclude the influence of the isolation source on the impact of the very diverse Oxalobacteraceae family on fungi. Strain MP5059B (5059B) was isolated from fruiting bodies of Ruzenia spermoides, collected close to the Jakobi Weiher, in the Stadtwald Frankfurt (Germany). Asci were isolated with a sterile needle from perithecia and placed on standard PDA agar (Gams et al., 1998) in an attempt to cultivate the ascomycete. Bacterial strains were grown at 22°C and type strains at 28°C in R2A. Twenty-five μg/ml kanamycin was supplemented when required. 1% (w/v) starch, 4% (w/v) skimmilk or 1% (v/v) tributyrin was added for amylase, protease and lipase activity tests. Cells were grown in M9 without glucose (−G, Elbing and Brent, 2002) and shrimp shell chitin supplementation for 1 week at 22°C to test chitinoclastic activity. Violacein synthesis was measured in TY (Elbing and Brent, 2002). We included genomic data of the strains J. agaricidamnosum DSM9628 (HG322949; Lincoln et al., 1999), J. lividum strains DSM1522 (De Ley et al., 1978) PAMC 25724 (AHHB00000000; Kim et al., 2012), RIT308 (JFYR00000000; Gan et al., 2014), MTR (JRRH00000000; Valdes et al., 2015), J. sp. strains Marseille (CP000269; Audic et al., 2007), RA13 (JQNP01000001; McTaggart et al., 2015), CG3 (APFF00000000; Smith et al., 2013), KBS0711 (LBCO00000000; Shoemaker et al., 2015), Ant5-2 (LNCE00000000; Mojib et al., 2010), Pseudoduganella violaceinigra DSM15887 (AUDI00000000; Kämpfer et al., 2012), D. zoogloeoides DSM16928 (AX110134; Hiraishi et al., 1997), D. phyllosphaereae DSM23865 (Kämpfer et al., 2012), and D. sp. HH01 (AMWD00000000; Hornung et al., 2013). Genome information was derived from the Gold data base or IMG (http://www.jgi.doe.gov/): J. sp. strains OK676: 60134; 344: 57625, 551a: 57361; NFR18: 57185; B9-8: CP014222; CG23_2: CYSS00000000; and D. sp. strains CF402: 61889; OV458: 60275.
Sequencing, annotation, and bioinformatic tools
For genome sequencing, different methods were used. Strains HH101, HH105, and 5059B were sequenced by a combined approach using the 454 GS-FLX pyrosequencing system (Roche Life Science, Mannheim, Germany) and the Genome Analyzer IIx (Illumina, San Diego, CA, USA), resulting in 2 × 122 bp reads. All other genomes were sequenced on the Genome Analyzer IIx, except for strain HH102 that was sequenced employing the Illumina MiSeq system, resulting in 2 × 301 bp reads. All libraries were prepared according to the protocols of the manufacturers. The Illumina reads were quality filtered; remaining adaptor sequences and the reads with a cutoff phred-33 score of 15 were trimmed using Trimmomatic (Bolger et al., 2014). Illumina reads were assembled using SPAdes genome assembler software (Bankevich et al., 2012) and 454 reads with Newbler 2.8. For the validation of the assembly, QualiMap version 2.1 was used (García-Alcalde et al., 2012). The software tool Prokka (Seemann, 2014) was used for automatic gene prediction and automatic annotation, which was subsequently manually curated by using the IMG-ER system (Markowitz et al., 2014), the Swiss-Prot, TrEMBL, and InterPro databases (Zdobnov and Apweiler, 2001). Genomes were manually analyzed to identify homologs and orthologs of known QS systems, hydrolytic enzymes, secondary metabolites and secretion systems to bypass the bias of automated functional annotations. For Multi locus sequence analysis (MLSA) total protein sequences from the 29 genomes were extracted from the corresponding GenBank files using cds_extractor.pl v0.6 (https://github.com/aleimba/bac-genomics-scripts) and used for downstream analysis with an in-house pipeline at the Goettingen Genomics Laboratory (Poehlein et al., 2015). In detail, proteinortho version 5 (default specification: blast = blastp v2.2.24, E-value = 1e-10, alg.-conn. = 0.1, coverage = 0.5, percent_identity = 50, adaptive_similarity = 0.95, inc_pairs = 1, inc_singles = 1, selfblast = 1, unambiguous = 0, Lechner et al., 2011) was used to generate clusters of orthologs groups, inparalogs were removed, MUSCLE (Edgar, 2004) used to align the remaining sequences and poorly aligned positions were automatically filtered from the alignments using Gblocks (Castresana, 2000). A maximum-likelihood tree from 1058 orthologs groups (Table S2) was inferred with 500 bootstraps with RAxML (Stamatakis, 2014). A phylogenetic tree was inferred with neighbor joining and 1000 bootstraps. The script PO_2_MLSA.py is available at github (https://github.com/jvollme). Average nucleotide identity (ANIm) analyses were performed using pyani.py (https://github.com/widdowquinn/pyani). Briefly, nucleotide sequences were extracted from the corresponding GenBank files using seq_format-converter.pl v0.2 (https://github.com/aleimba/bac-genomics-scripts) and subsequently used to run pyani in ANIm mode (uses MUMmer, NUCmer) to align input sequences.
Mutagenesis of HH102
To obtain HH102ΔjqsA (JAB4_14950) PCR products of primers ΔjqsA102-UP-FP-BamHI, ΔjqsA102-UP-RP-XbaI, ΔjqsA102-DS-FP-XbaI and ΔjqsA102-DS-RP-EcoRI (Table S3) were cloned in the suizide plasmid pNPTS138-R6KT (Lassak et al., 2010). Strain HH102 was transformed by electroporation (Hornung et al., 2013) and single recombinant mutants carrying this construct were selected on R2A. To obtain mutants, heterogenotes were streaked on R2A agar plates supplemented with 10% (w/v) sucrose. Correctness of the deletions was verified with primers oFH106/oFH107 and oFH108/oFH109 (Table S3).
RNA-Seq analyses—HH01, HH01ΔjqsA, HH102, and HH102ΔjqsA
Cells were cultivated in 20 ml R2A for 11 h (HH102, HH102ΔjqsA) or 13 h (HH01, HH01ΔjqsA) and extracted with hot phenol (Aiba et al., 1981). Briefly, cultures were mixed with 25 ml ice-cold killing buffer (20 mM Tris-HCl, pH 7.5, 5 mM MgCl2), centrifuged (10 min, 4°C) and shock frozen. Cells were resuspended in 125 μl ice-cold 300 mM sucrose/10 mM sodium acetate (pH 5.2), mixed with 125 μl 2% (w/v) SDS/10 mM sodium acetate (pH 5.2) and incubated (90 s, 65°C). Four hundred microliter hot phenol was added, incubated (3 min, 65°C), frozen and spun down (10 min, RT). Step was repeated. Four hundred microliter phenol:chloroform:isoamylalcohol (50:48:2 v/v) was added twice to the supernatant and spun down. Four hundred microliter chloroform:isoamylalcohol (96:2 v/v) was added and centrifuged (2 min, RT). For RNA precipitation overnight (−20°C) 40 μl 3 M sodium acetate (pH 5.2) and 1 ml 100% (v/v) ethanol were added. RNA was spun down (20 min, 4°C), washed and solved in diethylpyrocarbonate-treated H2O. Remaining genomic DNA was removed by digesting with DNAse I (Fermentas, St. Leon-Rot, Germany). The Ribo-Zero magnetic kit (Epicentre Biotechnologies, Madison, WI, USA) was used to reduce the amont of rRNA derived sequences. For sequencing, the strand-specific cDNA libraries were constructed with a NEBNext Ultra directional RNA library preparation kit for Illumina (New England BioLabs, Frankfurt am Main, Germany) and sequenced by using a GAIIx or MiSeq instrument (Illumina Inc., San Diego, CA, USA) in the paired-end mode and running 2 × 75 cycles. Between 28,738,406 and 56,507,330 raw reads were generated for the samples. For quality filtering and removing of remaining adaptor sequences, Trimmomatic (Bolger et al., 2014) and a cutoff phred-33 score of 15 were used. The mapping of the remaining sequences was performed with the Bowtie (version 2) program (Langmead and Salzberg, 2012) using the implemented end-to-end mode, which requires the entire read align from one end to the other. Differential expression analyses were performed with the baySeq program (Mortazavi et al., 2008). Genes with a fold change in expression of ≥2.0, a likelihood value of ≥0.9, and an adjusted P-value of ≤ 0.05 (the P-value was corrected by the false discovery rate [FDR] on the basis of the Benjamini-Hochberg procedure) were considered differentially expressed. Data set jan4t3 was excluded from calculation. Transcriptomic data were verified with qRT-PCR. Therefore, primer efficiency was tested with a dilution series of genomic DNA using the CFX96 Touch™ Real-Time PCR Detection System (Bio Rad, München, Germany), SYBR Green SuperMix (Quanta Biosciences, Gaithersburg, MD, USA), processed with the Bio-Rad CFX Manager 3.1 software (Bio Rad, München, Germany) and included in the normalized expression (ΔΔCq). Two microgram of DNase treated RNA (RTS DNase™, Mo Bio Laboratories, Inc., Carlsbad, CA, USA) was transcribed in cDNA (SuperScript® VILO cDNA Synthesis Kit and Master Mix, Thermo Scientific, Hampshire, United Kingdom). The genes rpoD (HH01, HH102), dnaG (HH01), and dnaB (HH102) were used as house keeping genes (Table S3). The relative quantity of the three biological samples composed of triplicates are shown in a graph with data relative to control (Figure S1).
Promoter-fusion studies
Plasmids and primers are listed in Tables S1, S3. Promoter sequence of vioA (JAB9_09370, Pvio107+JAI-FP-BamHI, and Pvio107+JAI-RP-EcoRI) was cloned in pBBR1MCS-2::mCherry (mCherry-FP-EcoRI, mCherry-RP-HindIII with pK18mobII_pKOScvm as template) to obtain pBBR1MCS-2::pvio107+JAI::mCherry. Amplification with Pvio107-JAI-FP-XhoI and mCherry-RP-HindIII led to the exclusion of the JAI-1 motif (pBBR1MCS-2::pvio107-JAI::mCherry). Bacterial strains were electroporated as described previously (Hornung et al., 2013).
Inhibition assay of Janthinobacterium, Duganella, and Fusarium graminearum
Mycelial plugs of the strain F. graminearum 8/1 (Bönnighausen et al., 2015) with a constitutively expressed GFP (Jansen et al., 2005) were taken of a 3-day-old colony grown on complete medium (Leach et al., 1982), placed on R2A plates with 1 × 105 bacterial cells/ml and incubated for 6 days at 22°C. For the liquid assay, 1 × 103 bacterial cells/μl grown in R2A lacking glucose (−G) were mixed with 400 fungal conidia grown on SNA (Nirenberg, 1981) for 1.5–2 weeks, rinsed with ice-cold distilled H2O and separated with 100 μm filter. To test the QS effect, 1 × 109 bacterial cells/ml grown in R2A −G supplemented with 0.05% (w/v) G, 10 mM D-glucosamine (DG) or 10 mM N-acetyl-D-gluocosamine (NADG) were filtered, mixed with 400 conidia to a volume of 200 μl in R2A −G and incubated for 72 h at 22°C prior to GFP detection.
Microscopic analysis of Janthinobacterium—F. graminearum interactions
1 × 104 bacterial cells harboring pBBR1MCS-2::P-mCherry were incubated with 400 conidia in R2A –G supplemented with 10 mM NADG for 48 h at 28°C. Subcloning of the promoterless pBBR1MCS-2::mCherry construct using SacI and XhoI endonucleases created the pBBR1MCS-2::P-mCherry plasmid to constitutively express mCherry (Table S1). Micrographs and Z series were captured with the confocal laser-scanning microscope LSM 780 (Zeiss, Germany) by using a Zeiss EC Plan-Neofluar 10 × objective for overview micrographs and a Zeiss C-Aprochromat 63 × water-immersion objective for Z series. First, GFP was excited and detected as described (Ellinger et al., 2013); second, mCherry was excited at 561 nm by using a diode-pumped solid-state (DPSS) laser. Emission filtering was done with a 567–618 nm bandpass filter. Brightfield micrographs were gathered with a transmitted light detector (T-PMT). For image processing the ZEN 2010 (Zeiss) operating software was used.
Sequence deposition and accession numbers
The whole genome DNA sequences have been deposited at GenBank under the accession numbers LROM00000000 (D. phyllosphaerae DSM23865), LRON00000000 (HH101), LRHV00000000 (HH105), J. lividum DSM1522 (LRHW00000000), MP5059B (LRHX00000000), HH100 (LRHY00000000), HH102 (LRHZ00000000), HH103 (LRIA00000000), HH104 (LRIB00000000), HH106 (LRIC00000000), and HH107 (LRID00000000). RNA-seq data have been deposited in the SRA archive under the accession number SRP073418 (HH102) and SRP073365 (HH01).
Results
Janthinobacterium and Duganella species suppress growth of the plant pathogen F. graminearum in standardized growth assays under laboratory conditions
Because of previous reports on antifungal activities of Janthinobacterium, we asked the question, whether these inhibitory effects are a common phenomenon of the Janthinobacterium and Duganella genera within the family Oxalobacteraceae and whether this response is linked to QS related processes or the isolation source. To address these questions, we first isolated six Janthinobacterium and two Duganella species (HH100–HH107) from the same habitat to exclude isolation source-dependent factors and broaden the genetic spectrum of Oxalobacteraceae. We included the strains HH01, MP5059B, and the type strains J. livdium, J. agaricidamnosum, D. phyllosphaerae, and D. zoogloeoides to include diverse environmental habitats, as these strains are derived from aquatic sources, soils, and plant surfaces. The antifungal capacity of all isolates was tested on the well-known plant pathogen F. graminearum (Goswami and Kistler, 2004). We established two different assays to assess the antifungal activities. Firstly, we used a growth assay on R2A agar plates and co-incubated fungal and bacterial cells together on these plates (Figure 1A). Secondly, we employed a gfp-tagged F. graminearum 8/1 variant (Bönnighausen et al., 2015) and monitored its growth based on the fluorescence of the GFP protein in microtiter plates when co-incubated with bacteria (Figure 1B). Since most of the above-named bacterial isolates produce violacein, we also coincubated F. graminearum with E. coli harboring the violacein synthesis genes vioABCDE on a self-replicable plasmid (derived from Hornung et al., 2013, Table S1). This liquid assay allowed a time-dependent monitoring of the fungal response to bacterial supernatants. Both tests implied that all strains affected growth of F. graminearum but to different extents (Figures 1A,B). A more than 50% reduction of fungal growth was observed for nearly all isolates (Figure 1B). Further, we observed that the overall fungal growth inhibition was independent of the differing amounts of violacein produced by the different isolates: D. phyllosphaerae and D. zoogloeoides produce no violacein, but display antifungal activities (Table 1, Figures 1A,B). Additionally, we observed that supernatants of E. coli clones expressing the vioABCDE genes had no major impact on F. graminearum growth. These findings indicated that violacein per-se was not the primary cause of the fungal growth inhibition. Together with this, we observed that the majority of the strains displayed hydrolytic and chitinoclastic activities, which are possibly involved in fungal defense (Table 1, Schuster et al., 2003; Wagner et al., 2003; Goo et al., 2010).
Figure 1.

Inhibition of F. graminearum 8/1 by isolates and the E. coli vioABCDE strain. The E. coli strain harbors the violacein vioABCDE genes on a pDrive plasmid (Hornung et al., 2013). (A) Plate assay to determine the inhibition of F. graminearum 8/1 by HH01, HH102, or E. coli. Co-incubation studies were performed with 1 × 109 bacterial cells per ml and hyphae of F. graminearum 8/1 on solid R2A. The plates were incubated for 6 days at 22°C. Experiments were performed thrice. (B) Results of liquid assays to test the F. graminearum 8/1 inhibition by the isolates HH100, HH101, HH102, HH103, HH104, HH105, HH106, HH107, and 5059B, the four type strains J. agaricidamnosum, J. lividum, D. phyllosphaerae and D. zoogloeoides, and E. coli vioABCDE. Liquid tests were performed with 180 μl filtered supernatant of 1 × 109 bacterial cells per ml grown in R2A –G and 400 conidia from F. graminearium 8/1, solved in 20 μl medium. The assay was incubated for 72 h at 28°C. One out of three independent experiments is shown and each experiment contained four replicates. The error lines indicate standard deviations.
Table 1.
Physiological features of the sequenced Oxalobacteraceae strains and related species*.
| Feature | Janthinobacterium | Duganella | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HH100 | HH102 | HH103 | HH104 | HH106 | HH107 | 5059 | 1522 | 9628 | HH101 | HH105 | 23865 | 16928 | HH01 | |
| Violacein synthesis (μg/ml) | 1.27 ± 0.12 | 1.54 ± 0.4 | 1.19 ± 0.02 | 1.83 ± 0.08 | 0.15 ± 0.07 | 0.93 ± 0.26 | 0.37 ± 0.02 | 0.03 ± 0.01 | − | 6.33 ± 0.15 | 3.36 ± 0.76 | − | − | 1.98 ± 0.02 |
| Amylolytic activity | − | − | − | − | − | − | − | − | + | + | + | + | + | + |
| Lipolytic activity | + | + | + | + | + | + | + | ++ | + | +++ | +++ | +++ | + | +++ |
| Proteolytic activity | ++ | ++ | ++ | ++ | ++ | ++ | ++ | − | + | (+) | (+) | − | − | (+) |
| Chitinoclastic activity | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | + | + | − | − | + |
Strains sequenced in this work are indicated by gray shadings. 9628 refers to J. agaricidamnosum, 1522 to J. lividum, 23865 to D. phyllosphaerae and 16928 to D. zoogloeoides. Violacein (μg/ml) was extracted after growth for 24 h in R2A. Amylolytic and proteolytic activities were measured after 72 h of growth on plates and lipolytic activities after 144 h of growth on solid media containing starch, skim milk, or tributyrin. Activities were recorded as diameter of clearing zones around individual colonies. Chitinolytic activity was determined after 1 week of growth in M9 lacking glucose and supplemented with chitin. +, enzymatic was observed; ++, strong activity; +++, very strong activity; (+), weak activity; –, no activity observed, data are mean values of at least three independent biological test; ± indicates the standard deviations.
Key traits of the sequenced strains and phylogenetic relationship
To unravel potential molecular mechanisms involved in bacterium-fungus interaction, we established the genome sequences of the isolated strains and the type strains J. lividum and D. phyllosphaerae to broaden the genetic spectrum of the Oxalobacteraceae family. First, rRNA analysis of the isolates, type strains and Nitrosomonas europaeae ATCC25976 (HE862405) classified the strains as phylogenetically associated within the genera Janthinobacterium and Duganella (data not shown). We included the genomic data of the J. lividum strains PAMC 25724 and RIT308 and J. sp. strains Marseille, RA13, CG3, OK676, 344, and 551a (Table 2). The genomes ranged from 5.5 to 7.4 Mb and the G+C content from 62.4 to 65.6%. The strains harbored 5467 to 6535 predicted protein-encoding genes, 6 to 32 rRNA genes, and 57 to 82 tRNA genes. The pan- and core-genome of altogether 29 publicly available genome data of Janthinobacterium, Duganella, and Pseudoduganella genomes comprised of 23,628 and 1058 genes, respectively. Based on the core genome analysis, we constructed a phylogenetic tree using a multilocus sequence analysis (MLSA, Figure 2) as well as the average nucleotide identity (ANIm, Figure S2) to focus on the phylogenetical distribution of the genera Janthinboacterium and Duganella. Considering these two methods, the isolates HH100, HH102, HH103, HH104, HH106, HH107, and 5059B cluster in proximity to J. lividum 1522. Especially HH104, RIT308, and NFR18 are closely related, as well as 5059B, MTR, and J. lividum 1522. Based on these findings, the isolates HH100, HH102, HH103, HH104, HH106, HH107, and 5059B are most likely affiliated with the genus Janthinobacterium. We designated this cluster as OxaI and HH102 as representative. The strains HH101, HH105, and HH01 cluster within the Duganella branch. HH01, HH101, and HH105 are most likely affiliated with the genus Duganella and we designated this cluster OxaII. HH01 was selected as representative. Comparative genome analyses indicated that relatively high levels of synteny exist between all analyzed Janthinobacterium genomes. Only few regions were identified in the OxaI cluster and J. lividum strain that showed relatively high levels of genome divergence (Figure S3A). The analyzed Duganella genomes, however, showed higher levels of divergence (Figure S3B). The highest levels of synteny were observed between the Duganella strains HH01, HH101, and HH105.
Table 2.
General genomic features of the strains sequenced in this work and other closely related and sequenced strains*.
| Genome traits | Janthinobacterium | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HH100 | HH102 | HH103 | HH104 | HH106 | HH107 | 5059 | 1522 | 9628 | Mars | 25724 | |
| Size (Mbp) | 6.7 | 6.7 | 6.6 | 6.4 | 6.3 | 5.5 | 6.4 | 6.7 | 5.9 | 4.1 | 4.98 |
| G+C content (%) | 65.6 | 62.4 | 62.5 | 62.6 | 62.9 | 63.0 | 62.8 | 62.4 | 61.0 | 54.23 | 60.6 |
| rRNAs | 9 | 8 | 8 | 6 | 8 | 8 | 32 | 11 | 7 | 6 | 21 |
| tRNAs | 64 | 69 | 73 | 61 | 64 | 66 | 82 | 72 | 73 | 46 | 80 |
| Other RNA genes | 22 | 23 | 22 | 11 | 11 | 14 | 12 | 17 | 0 | 14 | 13 |
| Coding genes | 5970 | 5987 | 5873 | 5657 | 5604 | 5467 | 5645 | 5820 | 5493 | 3697 | 4432 |
| With function | 4757 | 4776 | 4673 | 4647 | 4575 | 4481 | 4596 | 4760 | 4634 | 2813 | 3540 |
| %Secondary metabolites | 1.04 | 1.19 | 1.11 | 1.39 | 1.24 | 1.34 | 1.41 | 3.59 | 8.1 | n.d. | n.d. |
| Scaffolds | 150 | 121 | 141 | 65 | 73 | 116 | 100 | 127 | 1 | 1 | 48 |
| Genome traits | Janthinobacterium | Duganella | |||||||||
| RA13 | CG3 | OK676 | 344 | 551a | RIT308 | HH101 | HH105 | 23865 | 16928 | HH01 | |
| Size (Mbp) | 6.4 | 6.3 | 6.3 | 6.4 | 6.5 | 6.2 | 7.4 | 7.4 | 6.2 | 6.3 | 7.1 |
| G+C content (%) | 62.5 | 65.5 | 62.8 | 63.7 | 63.6 | 62.8 | 64.4 | 64.1 | 63.9 | 63.6 | 64.2 |
| rRNAs | 25 | 15 | 15 | 13 | 9 | 10 | 10 | 24 | 8 | 14 | 20 |
| tRNAs | 92 | 81 | 65 | 68 | 70 | 83 | 57 | 71 | 58 | 69 | 84 |
| Other RNA genes | 9 | 13 | 14 | 12 | 12 | − | 14 | 12 | 14 | 13 | 0 |
| Coding genes | 5650 | 5426 | 5502 | 5526 | 5541 | 5431 | 6535 | 6277 | 5390 | 5342 | 5996 |
| With function | 4612 | 4369 | 4594 | 4572 | 4594 | 4668 | 5340 | 5140 | 4375 | 4431 | 4323 |
| %Secondary metabolites | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 4.21 | 6.78 | 1.46 | 2.33 | 6.14 |
| Scaffolds | 1 | 7 | 31 | 27 | 28 | 44 | 223 | 89 | 158 | 25 | 2 |
We included genomic data of the strains J. agaricidamnosum (9628), J. lividum strains (1522), PAMC 25724 (25724), RIT308, J. sp. strains Marseille (Mars), RA13, CG3, D. zoogloeoides (16928), D. phyllosphaereae (23865), and HH01. Genome information were derived from the Gold data base or IMG (http://www.jgi.doe.gov/): J. sp. strains OK676, 344, and 551a. HH100–HH107 and MP5059B (5059) refers to the isolates of our lab. Strains sequenced in this work are indicated by gray shadings. “n.d.” refers to not determined and “−“ to not present. Data collection: January 2016.
Figure 2.

Multilocus sequence alignment (MLSA) tree. For Multi locus sequence analysis (MLSA) total protein sequences from the 29 genomes were extracted from the corresponding GenBank files using cds_extractor.pl v0.6 (https://github.com/aleimba/bac-genomics-scripts) and used for downstream analysis with an in-house pipeline at the Goettingen Genomics Laboratory. A maximum-likelihood tree from 1058 orthologs groups (Table S2) was inferred with 500 bootstraps with RAxML (Stamatakis, 2014). A phylogenetic tree was inferred with neighbor joining and 1000 bootstraps. The script PO_2_MLSA.py is available at github (https://github.com/jvollme). Strains sequenced in this study are written in black. The two OxaI (HH102) and OxaII (HH01) representatives are marked with an arrow.
Secondary metabolite encoding potential does not correlate with fungal growth repression
All strains sequenced in this study encoded a variable number of secondary metabolite gene clusters ranging from 1 to 6.8% of the genome (Table 2) with the higher percentages being comparable to well-known secondary metabolite producers like Streptomyces or Myxobacteria (Bode and Müller, 2005). The identified gene clusters were mostly linked to the biosynthesis of secondary metabolites, including non-ribosomal peptide synthetases (NRPS), and NRPS-polyketide synthases (PKS) hybrid clusters (Medema et al., 2011). Besides NRPS and NRPS/PKS hybrids (HH01, HH101, HH105, J. agaricidamnosum, and J. lividum), clusters encoding for proteinaceous toxins were observed in all strains. Further, clusters synthesizing terpene class of organic compounds (all strains), a siderophore (D. phyllosphaerae), butyrolactone (HH105), hserlactone (J. lividum), and a cluster to synthesize the peptides linaridin and thiopeptide-lantipeptides (J. agaricidamnosum) were found. Gene clusters coding for compounds linked to the synthesis of pigments (e.g., violacein and aryl-polyenes) were identified. Violacein was synthesized by all strains except for D. phyllosphaerae and D. zoogloeoides (Table 1), excluding violacein as fungal growth reducing factor. Interestingly, the overall number of genes and ORFs involved in secondary metabolite biosynthesis did not correlate with the observed antifungal effects. However, the presence of proteinaceous toxins and terpenes in all strains indicated a possible involvement in reducing fungal growth.
Oxalobacteraceae harbor the Vibrio cholerae- and Legionella pneumophila-like QS systems in a conserved cluster as part of the core genome
To better understand, how these bacteria use intra- and inter-species communication to communicate and for the expression and secretion of hydrolytic enzymes and secondary metabolites potentially involved in the effect on F. graminearum, the sequenced Oxalobacteraceae family affiliated isolates were examined for QS systems present in their genomes. Interestingly, only the Janthinobacterium strains 1522, RA13 and CG3 strains harbored luxI/luxR homologs genes, potentially involved in the synthesis of N-acyl-homoserine lactones (N-AHL, JALI_51420/JALI_51430, FG13DRAFT_1957/FG13DRAFT_1956, and JANGC3DRAFT_0717/JANGC3DRAFT_0712). Strain 1522 codes for one additional luxI homolog (JALI_55550). Several potential luxR solos were detected in these strains. It is noteworthy that none of the analyzed strains coded for a luxS homolog, necessary for the synthesis of AI-2 (Bassler et al., 1993; Surette et al., 1999). All but one strain addressed in this study code for the JQS system, formerly described in V. cholerae and L. pneumophila (Tiaden and Hilbi, 2012). The JQS system of HH01 consists of the autoinducer synthase gene jqsA (Jab_2c24330), the sensor kinase/phosphatase gene jqsS (Jab_2c24340) and the response regulator jqsR gene (Jab_2c24350) with the same organization in all strains, but differences in the intergenic region flanking the jqsA and jqsS genes. This region varied from 42 to 172 bp.
To further examine the importance of the jqsA gene for QS-dependent regulation in the family Oxalobacteraceae, we constructed a deletion mutant of the jqsA gene of strain HH102 (HH102ΔjqsA) as OxaI representative and used the strain HH01ΔjqsA as OxaII representative (Hornung et al., 2013). Mutants were verified by DNA sequencing and complemented with functional AI synthases (Figure S4), whereby HH01ΔjqsA had previously been complemented (Hornung et al., 2013). A deletion of the jqsA gene resulted in HH102 in a strong reduction of the violacein production. This was most pronounced when grown in liquid R2A or on solid TY (Figure 3B). A further phenotype associated with the jqsA mutation included an altered violacein production in response to chitin embedment for strain HH102 (Figure 3B). Thereby, planctonic cells and chitin-attached cells of the wild type HH102 produced violacein. Interestingly, HH102ΔjqsA cells synthesized violacein only when attached to chitin, indicating a possible regulatory role of chitin. This effect was not observed for HH01 or its jqsA deletion mutant. Additionally, in HH102ΔjqsA the proteolytic activity was attenuated compared to the parent strain (Figure 3C).
Figure 3.

jqsA gene deletion of HH01 and HH102, associated phenotypes and influence of the conserved JAI-1 motif. (A) Physical map of the of the JQS cluster encoding the deleted jqsA gene and the jqsS and jqsR genes, including the schematic location of the jqsA deletion. (B) Growth of HH01, HH01ΔjqsA, HH102, and HH102ΔjqsA on solid TY media (72 h at 22°C), in liquid R2A media (72 h at 22°C), and in liquid M9 –G, supplemented with chitin (growth for 1 week at 22°C). (C) HH102 and HH102ΔjqsA on R2A agar plates supplemented with 4% (w/v) skim milk (144 h at 22°C). (D) Predicted JAI-1 motif determined by sequence analysis of 28 bp 5′upstream region of the chiA, jqsA, and vioA genes summarized in Table S5. Sequences extending 14 bp were excluded from analysis. The logo was created using http://skylign.org/ (Wheeler et al., 2014). (E) Influence of the JAI-1 motif as part of a 49 bp deletion on the JAI-1 induced mCherry expression. The RFU of the strains HH01, HH102, and their corresponding jqsA deletion mutants HH01ΔjqsA and HH102ΔjqsA, harboring the plasmids pBBR1MCS-2::PvioHH107+JAI::mCherry (dark gray) and pBBR1MCS-2::PvioHH107-JAI::mCherry (bright gray), were determined by measuring the fluorescence at 587/610 nm per OD600 = 1. In pBBR1MCS-2::PvioHH107-JAI::mCherry a 49 bp region was deleted containing the complete JAI-1 motif. The cells were grown for 24 h at 22°C in R2A media and RFUs measured. Error bars indicate the standard deviations. Data are values of a minimum of three tests. Bars carrying the same letters of HH01/HH01ΔjqsA or HH102/HH102ΔjqsA are statistically not significantly different using P-values of < 0.05 (students t-test).
RNA-Seq identifies few QS-regulated genes in HH01 and HH102 and a conserved JAI-1 motif in the promoter region of QS regulated genes
Based on the results, we analyzed the global gene expression patterns of the strains HH01 and HH102 and their corresponding JAI-1 synthase mutants at the transition from exponential to stationary growth phase. We chose the early time point, because at the onset of the stationary growth phase many of QS-dependent processes are turned on. For each sample a minimum of 19.6 million cDNA reads could be uniquely mapped to the reference genomes. In the comparative analysis of RNA-seq data we considered genes with a log fold-change of ≥ 2.0, a likelihood value of ≥ 0.9, and a FDR-value of ≤ 0.05 as statistically significant. Expression analysis by qRT-PCR technology was used to confirm part of the RNA-seq data (Table 3). In HH01 a total set of 31 genes was differentially expressed and JAI-1 affects the expression of 14 in HH102 (Table 3). The HH01 QS-regulated genes were mainly organized in gene clusters that could be linked to four functional classes: secondary metabolite biosynthesis, lipopolysaccharide biosynthesis, a short chain dehydrogenase cluster and a cluster coding for a novel transport system. In HH102 two gene clusters and five single genes were differentially regulated. This included one out of two encoded type VI secretion system (T6SS) clusters and one cluster necessary to assemble a Flp pilus. With the exception of the secondary metabolite violacein no shared homologs genes of HH01 and HH102 were QS regulated, despite the core genome affiliation (Table 3).
Table 3.
QS-dependent genes identified in HH01 and HH102 using RNA-seq and qRT-PCR*.
| Locus tag | Possible function | Log-fold change | Phylogenetic classification | JAI-1 motif | qRT-PCR (fold change) |
|---|---|---|---|---|---|
| HH01 | |||||
| Transport System | |||||
| Jab_2c10580* | Outer membrane protein, oprM | −1.71 | I + II + J | y | |
| Jab_2c10590 | HlyD family secretion protein | −2.28 | I + II + J | −1.77 | |
| Jab_2c10600* | ATPase | −1.09 | I + II + J | ||
| Jab_2c10610 | Inner membrane transport permease, yhhJ | −2.37 | I + II + J | ||
| Lipopolysaccharide Synthesis | |||||
| Jab_2c07990 | Lipopolysaccharide biosynthesis protein | −2.14 | II + J | y | −3.92 |
| Jab_2c08000 | Putative tyrosine-protein kinase | −2 | II + J | ||
| Jab_2c08010* | Hypothetical protein | −1.97 | II + J | ||
| Jab_2c08020* | UDP-N-acetylglucosamine 2-epimerase, wecB | −1.95 | |||
| Jab_2c08030 | Polysaccharide deacetylase | −2.21 | II + J | ||
| Jab_2c08040 | Hypothetical protein | −2.09 | II + J | ||
| Jab_2c08050* | Glycosyl transferase | −1.97 | II + J | ||
| Jab_2c08060* | Eight transmembrane protein, epsH | −1.94 | II + J | ||
| Jab_2c08070 | Possible glycosyl transferase | −2.42 | II + J | ||
| Jab_2c08080* | Possible asparagine synthetase | −1.9 | II | ||
| Jab_2c08090* | Possible glycosyltransferase | −1.69 | II + J | ||
| Jab_2c08100* | Putative capsular polysaccharide biosynthesis protein | −1.94 | II + J | ||
| Secondary Metabolites | |||||
| Jab_2c08810 | L-tryptophan oxidase, vioA | −2.61 | I + II + J | y | −5.97 |
| Jab_2c08820 | Violacein biosynthesis protein, vioB | −2.41 | I + II + J | ||
| Jab_2c08830 | Monooxygenase, vioC | −2.32 | I + II + J | ||
| Jab_2c08840 | Tryptophan hydroxylase, vioD | −2.34 | I + II + J | ||
| Jab_2c08850 | Violacein biosynthesis protein, vioE | −2.45 | I + II + J | ||
| Jab_2c08860 | Predicted arabinose efflux permease | −2.54 | I + II + J | ||
| Jab_2c16610 | Kynureninase, kynU | +2.67 | I + II − HH105 | y | +5.4 |
| Jab_2c16620 | Tryptophan 2,3-dioxygenase, kynA | +2.4 | I + II − HH101 | ||
| Jab_2c35330* | Hypothetical protein | −1.18 | U | n.a. | |
| Jab_2c35360 | Possible polyketide synthase | −2.39 | U | ||
| Jab_2c35370 | Possible homoserine O-succinyltransferase, metA | −3.68 | U | ||
| Jab_2c35380 | Non-ribosomal peptide synthetase/amino acid adenylation protein | −3.34 | U | ||
| Jab_2c35390 | Predicted glycine/serine hydroxymethyltransferase, glyA | −3.04 | U | ||
| Jab_2c35400 | Non-ribosomal peptide synthetase | −3.49 | U | −7.76 | |
| Jab_2c35410 | Non-ribosomal peptide synthetase | −3.68 | U | ||
| Sdr Cluster | |||||
| Jab_2c26560 | Predicted aldo/keto reductase | −7.77 | U | y | |
| Jab_2c26580 | Predicted short-chain dehydrogenase/reductase | −8.77 | II | −353.35 | |
| Jab_2c26590 | Predicted transcriptional regulator AraC family | −3.96 | II | ||
| Jab_2c26600* | Hypothetical protein | −1.14 | II | ||
| Other | |||||
| Jab_1c10070 | Putative serine/threonine protein kinase | −2.48 | II | n | |
| Jab_1c13730 | Predicted porin | −2.24 | I + II − HH104 | n | |
| Jab_1c18190 | Putative polysaccharide deacetylase | +2.3 | II | y | |
| Jab_1c09990 | Tetracycline resistance protein | −3.18 | I + II − HH104 | y | |
| Jab_2c08460 | Predicted histidine kinase | −2.17 | I + II + D +J | y | |
| Jab_2c08470 | Predicted diguanylat cyclase | −2.21 | I + II + D +J | ||
| Jab_2c19720 | TIR domain containing protein | −2.29 | C − Marseille | y | |
| HH102 | |||||
| Type VI Secretion System | |||||
| Jab4_02740* | Type VI secretion system protein, impL | −3.91 | I, II, D, J-1522 | y | |
| Jab4_02750* | Type VI secretion system protein, impM | −3.29 | C −M-Pd | ||
| Jab4_02760* | OmpA-OmpF porin, OOP family | −4.22 | C −M-Pd | ||
| Jab4_02770* | Type VI secretion system protein, impK | −2.53 | I, II, D-16928, J | y | |
| Jab4_02780* | Type VI secretion system protein, impJ | −2.09 | C −M | ||
| Jab4_02790* | Type VI secretion system protein, vasD | −2.12 | I, 01, J-9628 | ||
| Jab4_02800* | Hypothetical protein | −3.73 | C −M | y | |
| Jab4_02810* | Type VI secretion system protein, impB | −3.4 | C −M,-9628 | ||
| Jab4_02820 | Type VI secretion system protein, impC | −3.64 | C −M | ||
| Jab4_02830* | Type VI secretion system secreted protein, hcp | −3.99 | C −M | ||
| Jab4_02840 | Type VI secretion system protein, impF | −3.44 | C −M | ||
| Jab4_02850 | Type VI secretion system protein, vasG | −3.44 | C −M, J-9628 | −13.86 | |
| Jab4_02860* | Type VI secretion system protein, impA | −3.35 | C −M | ||
| Flp Pilus Assembly | |||||
| Jab4_35690 | Flp pilus assembly protein, flp/pilA | −2.58 | I, J | y | −5.07 |
| Jab4_35700* | Prepilin peptidase, cpaA | −2.5 | C −M | ||
| Jab4_35710* | Hypothetical protein | −3.07 | C −M | ||
| Jab4_35720 | Flp pilus assembly protein, cpaB | −3.21 | C −M | ||
| Jab4_35730* | Flp pilus assembly protein, cpaC | −3.01 | C −M. −CG23 | ||
| Jab4_35740* | Hypothetical protein | −3.13 | I, J | ||
| Jab4_35750* | Tight adherence protein, tadG | −3.02 | C −M | ||
| Jab4_35760* | Tight adherence protein, tadE-like | −3.58 | I, J | ||
| Jab4_35770 | Tight adherence protein, tadE-like | −3.56 | C −M | ||
| Jab4_35780* | Flp pilus assembly protein, cpaE | −3.35 | I −5059, II, D, J-1522 | ||
| Jab4_35790 | Flp pilus assembly protein, cpaF | −3.6 | C | ||
| Jab4_35800* | Tight adherence protein, tadB | −3.49 | C −M | −10.53 | |
| Jab4_35810* | Tight adherence protein, tadC | −3.37 | C −M | ||
| Jab4_35820* | TPR repeat-containing protein | −3.22 | I, J | ||
| Secondary Metabolites | |||||
| Jab4_20480* | Inner membrane transport protein YdhP | −1.76 | I + II + J | y | |
| Jab4_20490* | Hypothetical protein | −1.7 | I + II + J | ||
| Jab4_20500* | Tryptophan hydroxylase, vioD | −1.79 | I + II + J | ||
| Jab4_20510* | Monooxygenase, vioC | −1.84 | I + II + J | ||
| Jab4_20520 | Violacein biosynthesis protein, vioB | −2.09 | I + II + J | ||
| Jab4_20530 | L-tryptophan oxidase, vioA | −2.22 | I + II + J | −6.48 | |
| Others | |||||
| JAB4_03960* | Predicted phospholipase | −2.9 | I—5059 | n | |
| JAB4_13490 | Hypothetical protein | −2.6 | I, J | y | |
| JAB4_13500* | Hypothetical protein | −3.22 | I, J | ||
| JAB4_16300 | PRC-barrel domain protein | −2.67 | I, II, J, D | y | −6.25 |
| JAB4_23200 | Hemerythrin-like metal-binding domain protein | −2.58 | I, II, J-9628 | y | |
| JAB4_30510 | Response regulator receiver domain-containing protein | −2.52 | I, J-9628 | y | |
| Jab4_42080 | Predicted aminoglycoside 3-N-acetyltransferase | −2.89 | I, J-9628 | y | −3.57 |
| Jab4_42090* | Hypothetical protein | −3.21 | I, J-9628 | ||
| Jab4_42320 | Hypothetical protein | −2.81 | I, J | n | |
| Jab4_42330* | Hypothetical protein | −2.83 | I, J | ||
| JAB4_54620* | Predicted soluble aldose sugar dehydrogenase | −3.04 | I, J-9628 | y | |
We considered genes with a fold-change of ≥ 2.0, a likelihood value ≥ 0.9 and a FDR-value of ≤ 0.05 as statistically significant. Genes that did not match these criteria but were part of an operon and confirmed by qRT-PCR are indicated with*. y refers to the presence of the JAI-1 motif, n to no JAI-1 motif identified; n.a., not available as the promoter lies within a contig border. For phylogenetic classification, isolates are classified according to the OxaI (I) or OxaII (II) affiliation and type strains were abbreviated with J (Janthinobacterium) and D (Duganella). 9628 refers to J. agaricidamnosum, 1522 to J. lividum, 23865 to D. phyllosphaerae, 16928 to D. zoogloeoides, 5059 to J. sp. MP5059B, M to J. sp. Marseille and PD to P. violaceinigra. − indicates the lack of this gene within the genome of the following strain, C refers to the core genome and U to the unique presence within this strain. Genes/ORFs being part of the same gene cluster or operon as indicated by consecutive locus tags are either light or dark gray shaded.
Intrigued by these observations, we asked, whether a common regulatory motif would be located upstream of all the QS regulated genes. Comparing the 5′ region of the differentially expressed genes in HH01 and HH102, we identified a conserved palindromic sequence in the promoter region of almost all regulated genes and operons (Table 3, Table S4). This conserved sequence consisted of 14/15 nucleotides with the sequence TTGAN6/7TCAA (Figure 3D) and was located 243 bp upstream of the presumed vioA translational start site. This motif was also observed upstream of all vioA, jqsA, and chiA genes of the strains sequenced and analyzed in this work (Table S5). Analyzing the motif position of the JAI-1 regulated genes (Table S4), the position ranged from 80 to 339 bp upstream of the translational start sites. To further verify the importance of this motif we constructed a promoter fusion carrying the vioA promoter within a 280 bp fragment fused to a mCherry reporter gene on a self-replicable plasmid (Table S1). This fusion was mobilized into HH01 and HH102 and the corresponding jqsA mutants. As expected, it was active in the parent strains and two-fold less active in the jqsA mutants. Deletion of the TTGAN6/7TCAA motif as part of a 49 bp deletion resulted in a 75–90% reduction of promoter activity compared to the strains carrying the complete vioA promoter (Figure 3E). These observations indicate a regulatory role of this conserved sequence with respect to the JAI-1-dependent gene expression. Thus, we designated this TTGAN6/7TCAA sequence JAI-1 motif. However, next to JAI-1 the chitin derivate DG and NADG affect the vioA promoter controlled mCherry expression in dependency on the JAI-1 motif as well (data not shown). This suggests a complex regulatory circuit between QS and chitin metabolism and might explain why a discrepancy between the wild type strains harboring the construct lacking the JAI-1 motif and the corresponding gene deletion mutants with the JAI-1 motif was observed.
HH102ΔjqsA is attenuated in its virulence against the plant pathogen F. graminearum
In the light of these observations we asked, whether the JAI-1 dependent gene expression and signaling circuit would affect the interaction of HH01 or HH102 with F. graminearum. To address this question we assayed the inhibition of fungal growth caused through culture supernatants of both bacterial strains and their corresponding jqsA mutants (Figures 4A,B). We chose culture supernatants, because we hypothesized that the bacteria would release growth inhibitory compounds into the surrounding medium in a QS-dependent manner. Therefore, filtered bacterial supernatants were co-incubated with F. graminearum for 72 h in microtiter plates as described in “Material and Methods” and fungal growth was recorded as relative fluorescence (RFU). Further, we preincubated the bacterial cells with either glucose (G), D-glucosamine (DG) or N-acetyl-D-glucosamine (NADG) to test whether these chitin degradation products have impact on the overall bacterial response. As expected culture supernatants obtained from the parent strains of HH102 and HH01 inhibited fungal growth (Figures 4A,B). Interestingly, HH102 and HH01 strongly inhibited fungal growth when supernatants of HH102 or HH01 were employed that were derived from cultures grown in the presence of DG. The lack of the jqsA gene, however, did not significantly affect growth inhibition in the mutants compared to the parent strains, when supernatants of cells were employed that had been grown in the presence of DG. When HH102 or HH102ΔjqsA were grown in the presence of 10 mM NADG, the growth inhibition of the fungus differed strongly between wild type and the corresponding mutant. Under these conditions HH102ΔjqsA was 2.5 ± 0.3-fold less active against F. graminearum (Figure 4A). Interestingly, this difference in fungal growth inhibition was only observed in the presence of culture supernatants obtained from cells that had been grown in the presence of NADG but not DG or G (Figure 4A). These inhibition data correlated well with microscopic analyses of the interaction between F. graminearum and the bacterial strains HH102 or HH102ΔjqsA (Figure 5). In general, the presence of both bacterial strains caused a reduced germination rate of the fungal conidia and twisted growth of hyphae with irregular lateral branches (Figure 5A, four lower panels and Figure 6A). In contrast and in the absence of bacteria, the hyphae grew straight and uni-directional (Figure 5A, two upper panels). In these tests HH102 formed dense biofilms or cell assemblages around hyphae of F. graminearum (Figure 5A, mid section panels). The hyphae were often completely covered and enclosed by the bacteria. Interestingly, the presence of NADG appeared to promote a direct attachment of HH102 cells on hyphae of F. graminearum (Figures 5B,C). Furthermore, the HH102ΔjqsA strain formed less dense and looser biofilms around the hyphae (Figure 5A, four lower panels). This observation was supported by images obtained using scanning electron microcopy. Thereby, several intriguing observations could be made. First, wild type strain HH102 secreted outer membrane vesicles (OMVs, Figure 6B, white arrows) and second, HH102 attached tightly to fungal hyphae by forming netlike structures around the bacterial cells. Interestingly, OMVs were less frequently observed in mutant cells. Further, the netlike structures attaching mutant cells to hyphae surfaces appeared different and less dense (Figure 6C). Altogether these data imply that the JAI-1-dependent signaling is involved in the interaction with the fungus and that it attenuates the growth suppression in the presence of NADG. Our data further suggest that DG and NADG interfere with this rather complex regulatory circuit.
Figure 4.
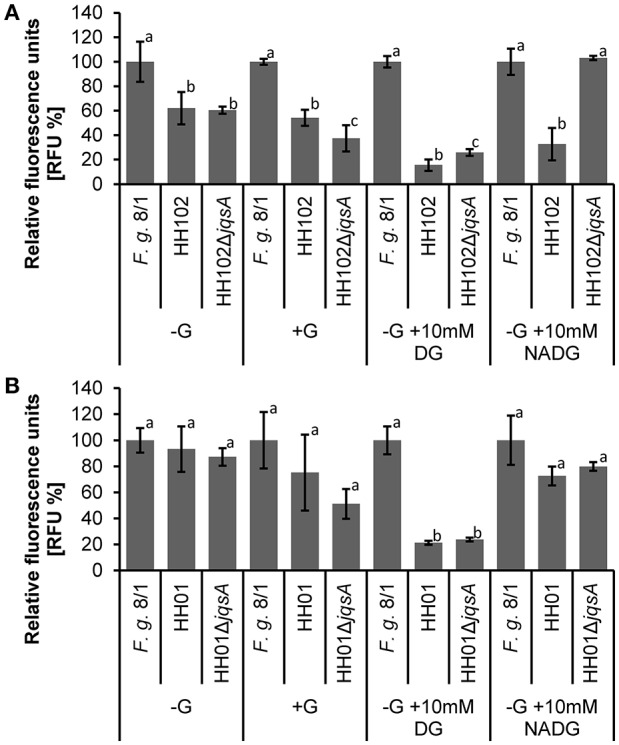
Bacterial inhibition of F. graminearum growth in a microtiter plate assay. The HH102, HH01, HH102ΔjqsA, and HH01ΔjqsA cells were grown in R2A −G. Media were supplemented with 0.05% (w/v) glucose (G), 10 mM D-glucosamine (DG), or 10 mM N-acetyl-D-glucosamine (NADG) and cells were incubated at 22°C for 24 h. One-hundred and eighty microliter of filtered supernatant (1 × 109 cells per ml) and 400 fungal conidia were incubated in a volume of 200 μl for 72 h at 28°C, shaking. Expression of chromosomal integrated GFP in F. graminerarum 8/1 was detected at 485/20; 528/20 nm. GFP expression of F. graminearum 8/1 in different media was based on F. graminearum 8/1 grown in R2A −G. This is set as 100%. Herein, fold changes are calculated setting each F. graminearum media control as 100% and calculating the relative fluorescence of co-incubated F. graminearum with bacteria for the respective medium. Experiments were performed at least four-times with four replicates. Bars carrying the same letters within each condition are statistically not significantly different using P-values of < 0.05 (students t-test). One individual experiment of (A) HH102/HH102ΔjqsA and (B) HH01/HH01ΔjqsA is shown.
Figure 5.
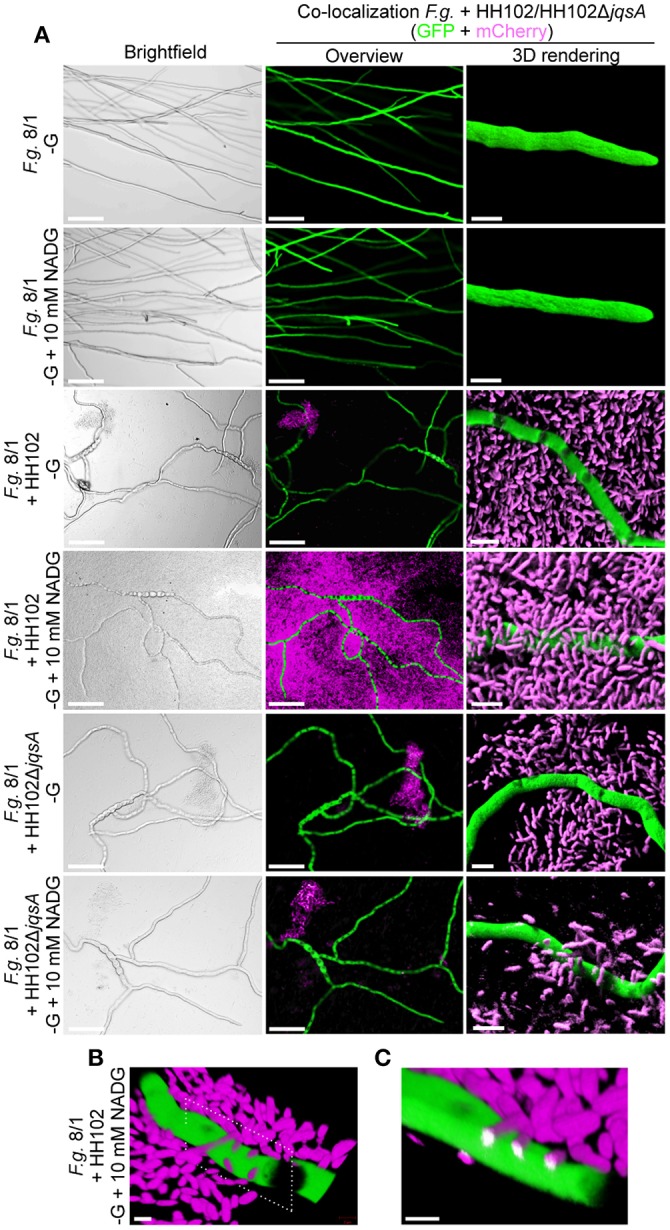
Microscopic images reflecting the interaction between HH102 or HH102ΔjqsA and F. graminearum. (A) Co-localization of mCherry-tagged HH102 and HH102ΔjqsA cells and GFP-tagged F. graminearum in co-cultures. Left panels, bright field overview images; middle panels, same section image as on left side panel but using fluorescence detection for GFP and mCherry; and right panels, 3D rendering of selected regions. Scale bars: overview (left and middle panels) micrographs = 100 μm; 3D-rendered micrographs = 5 μm (right panels). No mCherry-based fluorescence was visible in control cultures without bacteria. (B) 3D-projection of HH102 cells attached to hyphae of F. graminearum in cultures supplemented with NADG. Dotted frame indicates plane of in silico cross section in (C). Scale bar = 2 μm. (C) In silico cross section at sites of bacterial attachment on F. graminearum hyphae. Bacterial interaction and penetration into fungal hypha is indicated by white color. Scale bar = 2 μm. The HH102 and HH102ΔjqsA cells constitutively expressing mCherry were co-incubated with F. graminearum expressing GFP in 200 μl R2A −G supplemented with or without 10 mM N-acetyl-D-glucosamine (NADG) at 28°C for 72 h. Micrographs were taken by confocal laser-scanning microcopy. Green color shows GFP-, magenta color shows mCherry-emitted fluorescence.
Figure 6.

Scanning electron microscopic images reflecting the interaction between HH102 or HH102ΔjqsA and F. graminearum. (A) Uninfected hyphae growing linear; (B) HH102 colonizing fungal hypae OMVs are indicated with white arrows in the left hand panel and netlike structures attaching the cells to the fungal hyphae are indicated in the right hand panel with blue arrows; (C) HH102ΔjqsA colonizing fungal hypae with less dense and differentially structured netlike structures and lacking OMVs. Maginfications used are indicated in the individual images. The HH102 and HH102ΔjqsA cells were pretreated for 24 h at 22°C with 10 mM N-acetyl-D-glucosamine (NADG) and co-incubated with F. graminearum expressing GFP in 200 μl R2A −G supplemented with 10 mM NADG at 28°C for 72 h.
Discussion
Today our knowledge on bacterium-fungus interactions is sparse and only now the first reports are appearing that give insight into this important form of microbe-microbe interaction (Kobayashi and Crouch, 2009; Frey-Klett et al., 2011; Lackner et al., 2011; Stopnisek et al., 2016). Within these settings, we have established the genomes of 11 bacteria affiliated with the genera of Janthinobacterium and Duganella to get insight into the bacterial mechanism employed for cell-cell communication and the interaction with the fungus F. graminearum. We produced, by far, the largest and most comprehensive dataset for this bacterial group. Using these data we constructed a detailed MLSA tree (Figure 2). This MLS analysis indicated that Janthinobacterium and Duganella strains form a polyphyletic group within the familiy Oxalobacteraceae. The majority of the analyzed genomes cluster into two distinct phylogenetic groups, which we designated OxaI and OxaII. The core genome of the 29 Oxalobacteraceae strains consisted of 1058 genes and the pan genome of 23,628 genes, indicating that there is a high degree of variation within this family. This reflects the ability of the species to adapt to a wide range of environmental niches. Even though complete and draft genomes were used for core/pan genome analysis, the use of draft genomes had no effect on the size of the core and pan genome. All draft genomes contained contigs larger than 0.5 kb and gaps in the draft genomes represent repetitive regions such as rRNA clusters, transposases, or phage regions which are mainly covered by small contigs (0.5–5 kb).
Within this study, we provide evidence that all tested isolates code for antifungal properties, regardless of the phylogenetic affiliation to the genus Janthinobacterium or Duganella. These are most likely secondary metabolites, bacterial hydrolases and other “factors” secreted to attack the fungus (Tables 1, 2) and as suggested for the Oxalobacteraceae genus Collimonas (Song et al., 2015). The occurrence of secondary metabolite biosynthesis gene clusters appears to be a common trait of Janthinobacterium and Duganella species (Table 2). However, the overall number of genes coding for secondary metabolites is highly variable within both genera and ranges from 1 to 8% of all predicted protein-encoding genes (Table 2). It is noteworthy that almost all strains with the exception of the two Duganella type strains code for the biosynthesis of violacein. However, we further provide strong evidence that this antifungal effect is not primarily linked to the synthesis of violacein per-se, but is also affiliated to other not yet identified proteinaceous factors (Figures 1A,B, Table 1), as the fungal growth reducing effect is evened out by heating bacterial supernatants (data not shown). Interestingly, while the violacein biosynthesis genes are organized in the same order and in a conserved operon as reported for Chromobacterium violaceum, the expression regulation of the vioABCDE genes appears to be different. Instead of a N-AHL-dependent regulatory circuit of C. violaceum the violacein biosynthesis in Janthinobacterium and Duganella is partly regulated through a CAI-1/LAI-1-like molecule (Morohoshi et al., 2010; Stauff and Bassler, 2011; Hornung et al., 2013). This signaling pathway has been identified in V. cholera and uses a (S)-3-hydroxytridecan-4-one autoinducer molecule (CAI-1) for communication. In V. cholera, CAI-1 is involved in the repression of virulence and plays an important role in biofilm dissolution (Zhu and Mekalanos, 2003; Higgins et al., 2007). It has also been shown that CAI-1 plays a major role in natural competence (Lo Scrudato and Blokesch, 2013), as well as the structurally similar L. pneumophila LAI-1 molecule (3-hydroxypentadecan-4-one). LAI-1 promotes pathogen-host cell interactions, the production of virulence factors and controls the change of the L. pneumophila life cycle from the replicative to the transmissive growth phase. Interestingly, LAI-1 is directly involved in inter-kingdom signaling, as a lack of LAI-1 modulates migration of eukaryotic cells (Spirig et al., 2008; Tiaden et al., 2010; Simon et al., 2015; Schell et al., 2016). Moreover, V. harveyi uses amongst others (Z)-3-aminoundec-2-en-4-one as CAI-1 molecule to regulate luminescence, for example (Henke and Bassler, 2004). Most remarkably, the genes linked to the synthesis of α-hydroxyketone-like AI molecules were identified in all analyzed Janthinobacterium and Duganella isolates, but J. sp. Marseille. The JQS system consists of the designated AI synthase jqsA, adjacent to the cognate sensor kinase jqsS and the regulator jqsR (Figure 3A).
In our study, we provided experimental evidence that JAI-1-like signaling affects the violacein biosynthesis in HH01 and HH102 and influences protease secretion in HH102 (Figure 3C). Furthermore, RNA-seq analyses implied that 31 genes are controlled through JAI-1-dependent signaling in HH01 and 14 in HH102 (Table 3). Most of the JAI-1-dependent genes were organized in clusters or putative operons. Interestingly, the majority of the QS-regulated genes carried a highly conserved motif in the 5′ direction of the transcriptional start site (Figure 3D). The frequent occurrence of this motif suggests a common regulatory principle for all these genes. We named this TTGAN6/7TCAA sequence JAI-1 motif. Analyzing the RNA-seq data, the regulation of genes for the biosynthesis of secondary metabolites, T6SS, and Flp-Pilus assembly are of special interest. Within this framework the biosynthesis of secondary metabolites of Collimonas was suggested to affect fungal growth (Song et al., 2015), the T6SS of V. cholerae acts together with chitin and CAI-1 as competence pheromone to function as predatory killing device (Borgeaud et al., 2015) and the T4P of V. vulnificus was implied to be necessary for the attachment to chitin (Williams et al., 2015). Testing the wild type strain HH102 and the jqsA gene deletion mutant HH102ΔjqsA in a BFI with F. graminearum, one of the most intriguing observations in this study is that the lack of the jqsA gene resulted in a decreased fungal growth inhibition (Figure 4A). The observation is remarkable, because this α-hydroxyketone-synthesizing QS system has previously not been reported to be involved in bacterial-fungal interactions. Further, the observation that the JAI-1 dependent fungal growth inhibition also depends on the presence of the chitin degradation product NADG, implies a role of chitin in the Janthinobacterium-specific QS regulatory circuit. NADG is a chitin degradation product mostly occurring in aquatic microorganisms (Hillman et al., 1989; Gooday, 1990). As the expression of chitinases is induced by chitin degradation products (Beier and Bertilsson, 2011), we suggest that the bacteria mostly attached to fungal hyphae express and secrete chitinases in response to NADG. Therefore, we conclude that JQS and NADG are involved in reducing fungal growth. This includes the JAI-1-regulated T6SS and T4P expression, both interfering with the chitin metabolism and fungal growth suppression (Borgeaud et al., 2015; Williams et al., 2015) and the QS- and NADG-induced exopolysaccharide expression. Further contributing factors might be the QS-independent expressed secondary metabolites. Altogether, our observation is in line with work reported for V. cholerae, which induces natural competence in the presence of chitin and CAI-1 and with work describing an enhanced chitin degrading activity in a pathogen suppressive environment (Lo Scrudato and Blokesch, 2013; Dalia et al., 2014). Thus, the observation that the lack of the JAI-1 signaling molecule affects fungal growth inhibition implies an involvement of the janthinobacterial AI in the interaction with eucaryotic organisms. Future work has to unravel this complex regulatory circuit.
Author contributions
Conceived and designed the experiments: FH, CK, and WRS. Performed the experiments: FH, CK, AP, and CV. Analyzed the data: FH, AP, and WRS. Contributed reagents/materials/analysis tools: RD, WS, WRS, CV, and MP. Wrote the paper: FH and WRS. Edited the paper: FH, WRS, CV, RD, WS, HB, and MP.
Funding
This work was supported in part by the BMBF grants 0315586F and 0315587A to WRS.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01668/full#supplementary-material
References
- Aiba H., Adhya S., de Crombrugghe B. (1981). Evidence for two functional gal promoters in intact Escherichia coli cells. J. Biol. Chem. 256, 11905–11910. [PubMed] [Google Scholar]
- Alikhan N. F., Petty N. K., Ben Zakour N. L., Beatson S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. 10.1186/1471-2164-12-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Sáez L., Zeder M., Harding T., Pernthaler J., Lovejoy C., Bertilsson S., et al. (2014). Winter bloom of a rare betaproteobacterium in the Arctic Ocean. Front. Microbiol. 5:425. 10.3389/fmicb.2014.00425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic S., Robert C., Campagna B., Parinello H., Claverie J. M., Raoult D., et al. (2007). Genome analysis of Minibacterium massiliensis highlights the convergent evolution of water-living bacteria. PLoS Genet. 3:e138 10.1371/journal.pgen.0030138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldani J. I., Rouws L., Cruz L. M., Olivares F. L., Schmid M., Hartmann A. (2014). The family Oxalobacteraceae, in The Prokaryotes - Alphaproteobacteria and Betaproteobacteria, eds Rosenverg E., DeLong E. F., Lory S., Stackebrandt E., Thompson F. (Berlin; Heidelberg: Springer-Verlag; ), 919–974. 10.1007/978-3-642-30197-1_291 [DOI] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler B. L., Wright M., Showalter R. E., Silverman M. R. (1993). Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol. Microbiol. 9, 773–786. 10.1111/j.1365-2958.1993.tb01737.x [DOI] [PubMed] [Google Scholar]
- Becker M. H., Brucker R. M., Schwantes C. R., Harris R. N., Minbiole K. P. (2009). The bacterially produced metabolite violacein is associated with survival of amphibians infected with a lethal fungus. Appl. Environ. Microbiol. 75, 6635–6638. 10.1128/AEM.01294-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier S., Bertilsson S. (2011). Uncoupling of chitinase activity and uptake of hydrolysis products in freshwater bacterioplankton. Limnol. Oceanogr. 56, 1179–1188. 10.4319/lo.2011.56.4.1179 [DOI] [Google Scholar]
- Bode H. B., Müller R. (2005). The impact of bacterial genomics on natural product research. Angew. Chem. Int. Ed. Engl. 44, 6828–6846. 10.1002/anie.200501080 [DOI] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönnighausen J., Gebhard D., Kröger C., Hadeler B., Tumforde T., Lieberei R., et al. (2015). Disruption of the GABA shunt affects mitochondrial respiration and virulence in the cereal pathogen Fusarium graminearum. Mol. Microbiol. 98, 1115–1132. 10.1111/mmi.13203 [DOI] [PubMed] [Google Scholar]
- Borgeaud S., Metzger L. C., Scrignari T., Blokesch M. (2015). The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science 347, 63–67. 10.1126/science.1260064 [DOI] [PubMed] [Google Scholar]
- Brucker R. M., Harris R. N., Schwantes C. R., Gallaher T. N., Flaherty D. C., Lam B. A., et al. (2008). Amphibian chemical defense: antifungal metabolites of the microsymbiont Janthinobacterium lividum on the salamander Plethodon cinereus. J. Chem. Ecol. 34, 1422–1429. 10.1007/s10886-008-9555-7 [DOI] [PubMed] [Google Scholar]
- Castresana J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- Cretoiu M. S., Korthals G. W., Visser J. H. M., van Elsas J. D. (2013). Chitin amendment increases soil suppressiveness toward plant pathogens and modulates the actinobacterial and oxalobacteraceal communities in an experimental agricultural field. Appl. Environ. Microbiol. 79, 5291–5301. 10.1128/AEM.01361-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalia A. B., Lazinski D. W., Camilli A. (2014). Identification of a membrane-bound transcriptional regulator that links chitin and natural competence in Vibrio cholerae. Mbio 5, e01028–e01013. 10.1128/mBio.01028-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ley J., Segers P., Gillis M. (1978). Intrageneric and intergeneric similarities of Chromobacterium and Janthinobacterium ribosomal ribonucleic-acid cistrons. Int. J. Syst. Bacteriol. 28:154 10.1099/00207713-28-2-154 [DOI] [Google Scholar]
- Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbing K., Brent R. (2002). Media preparation and baceriological tools. Curr. Protoc. Mol. Biol. 59 Chapter I:Unit 1.1, 1.1.1–1.1.7. 10.1002/0471142727.mb0101s59 [DOI] [PubMed] [Google Scholar]
- Ellinger D., Naumann M., Falter C., Zwikowics C., Jamrow T., Manisseri C., et al. (2013). Elevated early callose deposition results in complete penetration resistance to powdery mildew in Arabidopsis. Plant Physiol. 161, 1433–1444. 10.1104/pp.112.211011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey-Klett P., Burlinson P., Deveau A., Barret M., Tarkka M., Sarniguet A. (2011). Bacterial-fungal interactions: hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol. Mol. Biol. Rev. 75, 583–609. 10.1128/MMBR.00020-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gams W., Hoekstra E. S., Aptroot A. (1998). CBS Course of Mycology. Delft: Centraalbureau vorr Schimmelcultures Baarn. [Google Scholar]
- Gan H. Y., Gan H. M., Savka M. A., Triassi A. J., Wheatley M. S., Smart L. B., et al. (2014). Whole-genome sequences of 13 endophytic bacteria isolated from shrub willow (salix) grown in Geneva, New York. Genome Announc. 2:e00288–14. 10.1128/genomeA.00288-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alcalde F., Okonechnikov K., Carbonell J., Cruz L. M., Götz S., Tarazona S., et al. (2012). Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679. 10.1093/bioinformatics/bts503 [DOI] [PubMed] [Google Scholar]
- Goo E., Kang Y., Kim H., Hwang I. (2010). Proteomic analysis of quorum sensing-dependent proteins in Burkholderia glumae. J. Proteome Res. 9, 3184–3199. 10.1021/pr100045n [DOI] [PubMed] [Google Scholar]
- Gooday G. W. (1990). The ecology of chitin degradation. Adv. Microb. Ecol. 11, 387–430. 10.1007/978-1-4684-7612-5_10 [DOI] [Google Scholar]
- Goswami R. S., Kistler H. C. (2004). Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 5, 515–525. 10.1111/j.1364-3703.2004.00252.x [DOI] [PubMed] [Google Scholar]
- Graupner K., Lackner G., Hertweck C. (2015). Genome sequence of mushroom soft-rot pathogen Janthinobacterium agaricidamnosum. Genome Announc. 3:e00277–15. 10.1128/genomeA.00277-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R. N., Brucker R. M., Walke J. B., Becker M. H., Schwantes C. R., Flaherty D. C., et al. (2009). Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 3, 818–824. 10.1038/ismej.2009.27 [DOI] [PubMed] [Google Scholar]
- Henke J. M., Bassler B. L. (2004). Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J. Bacteriol. 186, 6902–6914. 10.1128/JB.186.20.6902-6914.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D. A., Pomianek M. E., Kraml C. M., Taylor R. K., Semmelhack M. F., Bassler B. L. (2007). The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature 450, 883–886. 10.1038/nature06284 [DOI] [PubMed] [Google Scholar]
- Hillman K., Gooday G. W., Prosser J. I. (1989). The mineralization of chitin in the sediments of the ythan-estuary, Aberdeenshire, Scotland. Estuar. Coast. Shelf Sci. 29, 601–612. 10.1016/0272-7714(89)90013-9 [DOI] [Google Scholar]
- Hiraishi A., Shin Y. K., Sugiyama J. (1997). Proposal to reclassify Zoogloea ramigera IAM 12670 (P. R. Dugan 115) as Duganella zoogloeoides gen. nov., sp. nov. Int. J. Syst. Bacteriol. 47, 1249–1252. 10.1099/00207713-47-4-1249 [DOI] [PubMed] [Google Scholar]
- Hornung C., Poehlein A., Haack F. S., Schmidt M., Dierking K., Pohlen A., et al. (2013). The Janthinobacterium sp. HH01 genome encodes a homologue of the V. cholerae CqsA and L. pneumophila LqsA autoinducer synthases. PLoS ONE 8:e55045. 10.1371/journal.pone.0055045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen C., von Wettstein D., Schäfer W., Kogel K. H., Felk A., Maier F. J. (2005). Infection patterns in barley and wheat spikes inoculated with wild-type and trichodiene synthase gene disrupted Fusarium graminearum. Proc. Natl. Acad. Sci. U.S.A. 102, 16892–16897. 10.1073/pnas.0508467102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kämpfer P., Wellner S., Lohse K., Martin K., Lodders N. (2012). Duganella phyllosphaerae sp. nov., isolated from the leaf surface of Trifolium repens and proposal to reclassify Duganella violaceinigra into a novel genus as Pseudoduganella violceinigra gen. nov., comb. nov. Syst. Appl. Microbiol. 35, 278–278. 10.1016/j.syapm.2012.02.001 [DOI] [PubMed] [Google Scholar]
- Kielak A. M., Cretoiu M. S., Semenov A. V., Sørensen S. J., van Elsas J. D. (2013). Bacterial chitinolytic communities respond to chitin and pH alteration in soil. Appl. Environ. Microbiol. 79, 263–272. 10.1128/AEM.02546-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. J., Shin S. C., Hong S. G., Lee Y. M., Lee H., Lee J., et al. (2012). Genome sequence of Janthinobacterium sp. strain PAMC 25724, isolated from alpine glacier cryoconite. J. Bacteriol. 194, 2096. 10.1128/JB.00096-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi D. Y., Crouch J. A. (2009). Bacterial/fungal interactions: from pathogens to mutualistic endosymbionts. Annu. Rev. Phytopathol. 47, 63–82. 10.1146/annurev-phyto-080508-081729 [DOI] [PubMed] [Google Scholar]
- Lackner G., Moebius N., Hertweck C. (2011). Endofungal bacterium controls its host by an hrp type III secretion system. ISME J. 5, 252–261. 10.1038/ismej.2010.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassak J., Henche A.-L., Binnenkade L., Thormann K. M. (2010). ArcS, the cognate sensor kinase in an atypical Arc system of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 76, 3263–3274. 10.1128/AEM.00512-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach J., Lang B. R., Yoder O. C. (1982). Methods for selection of mutants and in vitro culture of Cochliobolus heterostrophus. J. Gen. Microbiol. 128, 1719–1729. 10.1099/00221287-128-8-1719 [DOI] [Google Scholar]
- Lechner M., Findeiss S., Steiner L., Marz M., Stadler P. F., Prohaska S. J. (2011). Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 12:124. 10.1186/1471-2105-12-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln S. P., Fermor T. R., Tindall B. J. (1999). Janthinobacterium agaricidamnosum sp. nov., a soft rot pathogen of Agaricus bisporus. Int. J. Syst. Bacteriol. 49(Pt 4), 1577–1589. 10.1099/00207713-49-4-1577 [DOI] [PubMed] [Google Scholar]
- Lo Scrudato M., Blokesch M. (2012). The regulatory network of natural competence and ransformation of Vibrio cholerae. PLoS Genet. 8:e1002778. 10.1371/journal.pgen.1002778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Scrudato M., Blokesch M. (2013). A transcriptional regulator linking quorum sensing and chitin induction to render Vibrio cholerae naturally transformable. Nucleic Acids Res. 41, 3644–3658. 10.1093/nar/gkt041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz V. M., Chen I.-M. A., Palaniappan K., Chu K., Szeto E., Pillay M., et al. (2014). IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42, D560–D567. 10.1093/nar/gkt963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTaggart T. L., Shapiro N., Woyke T., Chistoserdova L. (2015). Draft genome of Janthinobacterium sp. RA13 isolated from lake Washington sediment. Genome Announc. 3:e01588–14. 10.1128/genomeA.01588-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M. H., Blin K., Cimermancic P., de Jager V., Zakrzewski P., Fischbach M. A., et al. (2011). antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346. 10.1093/nar/gkr466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojib N., Philpott R., Huang J. P., Niederweis M., Bej A. K. (2010). Antimycobacterial activity in vitro of pigments isolated from Antarctic bacteria. Antonie van Leeuwenhoek, 98, 531–540. 10.1007/s10482-010-9470-0 [DOI] [PubMed] [Google Scholar]
- Morohoshi T., Fukamachi K., Kato M., Kato N., Ikeda T. (2010). Regulation of the violacein biosynthetic gene cluster by acylhomoserine lactone-mediated quorum sensing in Chromobacterium violaceum ATCC 12472. Biosci. Biotechnol. Biochem. 74, 2116–2119. 10.1271/bbb.100385 [DOI] [PubMed] [Google Scholar]
- Mortazavi A., Williams B. A., McCue K., Schaeffer L., Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- Nirenberg H. I. (1981). A simplified method for identifying Fusarium spp. occurring on wheat. Can. J. Bot. 59, 1599–1609. 10.1139/b81-217 [DOI] [Google Scholar]
- Poehlein A., Riegel K., König S. M., Leimbach A., Daniel R., Dürre P. (2015). Genome sequence of Clostridium sporogenes DSM 795(T), an amino acid-degrading, nontoxic surrogate of neurotoxin-producing Clostridium botulinum. Stand. Genomic Sci. 10, 40. 10.1186/s40793-015-0016-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey J. P., Mercurio A., Holland J. A., Harris R. N., Minbiole K. P. (2015). The cutaneous bacterium Janthinobacterium lividum inhibits the growth of Trichophyton rubrum in vitro. Int. J. Dermatol. 54, 156–159. 10.1111/ijd.12217 [DOI] [PubMed] [Google Scholar]
- Reasoner D. J., Geldreich E. E. (1985). A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 49, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J. A., Russell D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd Edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Schell U., Simon S., Sahr T., Hager D., Albers M. F., Kessler A., et al. (2016). The α-hydroxyketone LAI-1 regulates motility, Lqs-dependent phosphorylation signalling and gene expression of Legionella pneumophila. Mol. Microbiol. 99, 778–793. 10.1111/mmi.13265 [DOI] [PubMed] [Google Scholar]
- Schuster M., Lostroh C. P., Ogi T., Greenberg E. P. (2003). Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 185, 2066–2079. 10.1128/JB.185.7.2066-2079.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Shoemaker W. R., Muscarella M. E., Lennon J. T. (2015). Genome sequence of the soil bacterium Janthinobacterium sp. KBS0711. Genome Announc. 3:e00689–15. 10.1128/genomeA.00689-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon S., Schell U., Heuer N., Hager D., Albers M. F., Matthias J., et al. (2015). Inter-kingdom signaling by the Legionella quorum sensing molecule LAI-1 modulates cell migration through an IQGAP1-Cdc42-ARHGEF9-dependent pathway. PLoS Pathogens 11:e1005307. 10.1371/journal.ppat.1005307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith H., Akiyama T., Foreman C., Franklin M., Woyke T., Teshima H., et al. (2013). Draft genome sequence and description of Janthinobacterium sp. strain CG3, a psychrotolerant Antarctic supraglacial stream bacterium. Genome Announc. 1:e00960–13. 10.1128/genomeA.00960-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C., Schmidt R., de Jager V., Krzyzanowska D., Jongedijk E., Cankar K., et al. (2015). Exploring the genomic traits of fungus-feeding bacterial genus Collimonas. BMC Genomics 16:1103. 10.1186/s12864-015-2289-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirig T., Tiaden A., Kiefer P., Buchrieser C., Vorholt J. A., Hilbi H. (2008). The Legionella autoinducer synthase LqsA produces an α-hydroxyketone signaling molecule. J. Biol. Chem. 283, 18113–18123. 10.1074/jbc.M801929200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauff D. L., Bassler B. L. (2011). Quorum sensing in Chromobacterium violaceum: DNA recognition and gene regulation by the CviR receptor. J. Bacteriol. 193, 3871–3878. 10.1128/JB.05125-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopnisek N., Zühlke D., Carlier A., Barberán A., Fierer N., Becher D., et al. (2016). Molecular mechanisms underlying the close association between soil Burkholderia and fungi. ISME J. 10, 253–264. 10.1038/ismej.2015.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surette M. G., Miller M. B., Bassler B. L. (1999). Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: a new family of genes responsible for autoinducer production. Proc. Natl. Acad. Sci. U.S.A. 96, 1639–1644. 10.1073/pnas.96.4.1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiaden A., Hilbi H. (2012). α-hydroxyketone synthesis and sensing by Legionella and Vibrio. Sensors (Basel) 12, 2899–2919. 10.3390/s120302899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiaden A., Spirig T., Sahr T., Wälti M. A., Boucke K., Buchrieser C., et al. (2010). The autoinducer synthase LqsA and putative sensor kinase LqsS regulate phagocyte interactions, extracellular filaments and a genomic island of Legionella pneumophila. Environ. Microbiol. 12, 1243–1259. 10.1111/j.1462-2920.2010.02167.x [DOI] [PubMed] [Google Scholar]
- Valdes N., Soto P., Cottet L., Alarcon P., Gonzalez A., Castillo A., et al. (2015). Draft genome sequence of Janthinobacterium lividum strain MTR reveals its mechanism of capnophilic behavior. Stand. Genomic Sci. 10, 110. 10.1186/s40793-015-0104-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner V. E., Bushnell D., Passador L., Brooks A. I., Iglewski B. H. (2003). Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J. Bacteriol. 185, 2080–2095. 10.1128/JB.185.7.2080-2095.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. J., Clements J., Finn R. D. (2014). Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics 15:7. 10.1186/1471-2105-15-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins P. J., Smith J. M., Harris R. N., Minbiole K. P. C. (2011). Gut of red-backed salamanders (Plethodon cinereus) may serve as a reservoir for an antifungal cutaneous bacterium. J. Herpetol. 45, 329–332. 10.1670/10-231.1 [DOI] [Google Scholar]
- Williams T. C., Ayrapetyan M., Oliver J. D. (2015). Molecular and physical factors that influence attachment of Vibrio vulnificus to chitin. Appl. Environ. Microbiol. 81, 6158–6165. 10.1128/AEM.00753-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov E. M., Apweiler R. (2001). InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. 10.1093/bioinformatics/17.9.847 [DOI] [PubMed] [Google Scholar]
- Zhu J., Mekalanos J. J. (2003). Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev. Cell 5, 647–656. 10.1016/S1534-5807(03)00295-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.