

Traditional fermented foods represent relatively low-complexity microbial environments that can be used as model microbial communities to understand how microbes interact in natural environments. Our results illustrate the dynamic nature of kefir fermentations and microbial succession patterns therein. In the process, the link between individual species, and associated pathways, with flavor compounds is revealed and several genes that could be responsible for the purported gut health-associated benefits of consuming kefir are identified. Ultimately, in addition to providing an important fundamental insight into microbial interactions, this information can be applied to optimize the fermentation processes, flavors, and health-related attributes of this and other fermented foods.
KEYWORDS: dairy, flavor, kefir, metagenomics, microbiota
ABSTRACT
Kefir is a putatively health-promoting dairy beverage that is produced when a kefir grain, consisting of a consortium of microorganisms, is added to milk to initiate a natural fermentation. Here, a detailed analysis was carried out to determine how the microbial population, gene content, and flavor of three kefirs from distinct geographic locations change over the course of 24-h fermentations. Metagenomic sequencing revealed that Lactobacillus kefiranofaciens was the dominant bacterial species in kefir during early stages of fermentations but that Leuconostoc mesenteroides became more prevalent in later stages. This pattern is consistent with an observation that genes involved in aromatic amino acid biosynthesis were absent from L. kefiranofaciens but were present in L. mesenteroides. Additionally, these shifts in the microbial community structure, and associated pathways, corresponded to changes in the levels of volatile compounds. Specifically, Acetobacter spp. correlated with acetic acid; Lactobacillus spp. correlated with carboxylic acids, esters and ketones; Leuconostoc spp. correlated with acetic acid and 2,3-butanedione; and Saccharomyces spp. correlated with esters. The correlation data suggest a causal relationship between microbial taxa and flavor that is supported by observations that addition of L. kefiranofaciens NCFB 2797 increased the levels of esters and ketones whereas addition of L. mesenteroides 213M0 increased the levels of acetic acid and 2,3-butanedione. Finally, we detected genes associated with probiotic functionalities in the kefir microbiome. Our results illustrate the dynamic nature of kefir fermentations and microbial succession patterns therein and can be applied to optimize the fermentation processes, flavors, and health-related attributes of this and other fermented foods.
IMPORTANCE Traditional fermented foods represent relatively low-complexity microbial environments that can be used as model microbial communities to understand how microbes interact in natural environments. Our results illustrate the dynamic nature of kefir fermentations and microbial succession patterns therein. In the process, the link between individual species, and associated pathways, with flavor compounds is revealed and several genes that could be responsible for the purported gut health-associated benefits of consuming kefir are identified. Ultimately, in addition to providing an important fundamental insight into microbial interactions, this information can be applied to optimize the fermentation processes, flavors, and health-related attributes of this and other fermented foods.
Author Video: An author video summary of this article is available.
INTRODUCTION
Our knowledge of the composition of complex microbial communities from different environments has increased dramatically in recent years (1–3). However, considerably less is known about the biological interactions and other processes that drive microbial succession, or changes in the microbial population structure over time, in these environments (4). It has been proposed that microbial communities from fermented foods could provide a useful model for elucidating the determinants of microbial succession, given that they are considerably less complex than, for example, those from the gut or soil (5). Indeed, cheese rind communities have previously been used to great effect for this purpose (6).
Here, we show that kefir provides an alternative model microbial community that is less complex and provides results even more quickly. Kefir is a traditional fermented milk beverage that is typically produced by inoculating a kefir grain, a cauliflower-like exopolysaccharide (EPS) matrix containing a symbiotic community of bacteria and yeast (7), into milk and incubating it at room temperature for approximately 24 h, resulting in a beverage that has been described as having a pleasantly sour or yogurt-like taste (8). This flavor can vary, depending on the microbial composition of the grain that is used (9). High-throughput sequencing investigations have demonstrated that kefir grains are typically dominated by the bacterial genus Lactobacillus and the fungal phylum Ascomycota (9, 10). In contrast, kefir milk is dominated by the bacterial genera Lactobacillus, Lactococcus, Acetobacter, and Leuconostoc and the fungal genera Kazachstania, Kluyveromyces, Naumovozyma, and Saccharomyces (9, 11, 12).
The consumption of kefir has been associated with numerous health benefits, including anticarcinogenic, anti-inflammatory, and antipathogenic effects (13–15), as well as the alleviation of the symptoms of lactose intolerance and the reduction of cholesterol (16, 17). There is mounting evidence to suggest that the microorganisms present in kefir exert at least some of these health benefits (18–22), but there is a lack of understanding of the mechanisms by which they do so.
In this work, amplicon sequencing and whole-metagenome shotgun sequencing were combined with metabolomics and flavor analysis to highlight how the microbial composition, gene content, and flavor of kefir change over the course of 24-h fermentations. We demonstrate that the integration of multiple omics data can predict the contribution of individual microorganisms to metabolite production in a microbial environment, using flavor formation as an example, and we validate these findings through supplementation with specific microbes. To our knowledge, this is the first study to combine metagenome binning and metabolic reconstruction to determine the microbial composition, at both the species and strain levels, and the functional potential of a fermented food, respectively, at different stages of fermentation. In addition, this is the first study to combine whole-metagenome shotgun sequencing with metabolomics to link microbial species with volatile-compound production in kefir. Our findings reveal a dynamic flux from Lactobacillus kefiranofaciens domination during the early stages of fermentations to Leuconostoc mesenteroides domination during the latter stages, establish a causal relationship between microbial taxa and flavor, and highlight genes that likely contribute to kefir’s purported health-associated attributes.
RESULTS
Microbial composition of kefir.
16S rRNA and internal transcribed spacer (ITS) gene sequencing was used to determine the changes in the microbial population of kefirs over the course of 24-h fermentations initiated with three separate grains, designated Fr1, Ick, and UK3, from distinct geographic locations, namely, France, Ireland, and the United Kingdom.
Analysis of the grains showed that Lactobacillus was the dominant bacterial genus and constituted >92% of the populations of all three grains (see Fig. S1 in the supplemental material). Acetobacter was subdominant and accounted for between 1 and 2% of the population of each grain. In addition, Leuconostoc was present in all three grains, although its abundance varied from 0.2 to 1.5%. Other genera that were detected at a relative abundance of >1% were Propionibacterium, in Fr1 (4.6%) only, and Bifidobacterium, in UK3 (3.4%) only. A fungal population was detected in the grains Fr1 and Ick but not in UK3. Saccharomyces and Kazachstania were the only fungal genera present (see Fig. S1).
The bacterial (A) and fungal (B) compositions of kefir grains, as determined by amplicon sequencing. Note that we were unable to generate an ITS amplicon for the UK3 sample. Download Figure S1, PDF file, 0.09 MB (95.1KB, pdf) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Analysis of milk samples revealed that an initially relatively high bacterial diversity decreased over time, with a small number of genera becoming dominant by 8 and 24 h (see Fig. S2 in the supplemental material). On average, at 0 h, or immediately before the grains were added to the milk, the bacterial genera present at a relative abundance of ≥1% were Pseudomonas (16.9%), Anoxybacillus (7.1%), Thermus (6.5%), Acinetobacter (5%), Streptococcus (4.5%), Geobacillus (3.2%), Clostridium (2.4%), Butyrivibrio (2.2%), Serratia (2.1%), Enterobacter (1.3%), Turicibacter (1.3%), and Lactococcus (1%). A further 46.5% of the bacterial genera had a relative abundance of <1% (Fig. 1A). This microbial profile is consistent with that of pasteurized milk, as reported previously by Quigley et al. (23). We were unable to generate an ITS amplicon for the three samples collected at 0 h, and quantitative PCR (qPCR) indicated that fungal DNA was present at <2 pg/µl.
FIG 1 .

Stacked bar charts presenting the bacterial compositions of kefir samples after 0, 8, and 24 h of fermentation, as determined by 16S rRNA gene sequencing (A) and binning of metagenome sequences with Kraken (B).
Cladogram presenting the bacterial diversity of kefir samples at 0, 8, and 24 h of fermentation, as determined by 16S rRNA gene sequencing. The color intensities in the outer rings indicate the average relative abundances of the bacterial genera in Fr1, Ick, and UK3 at each stage. Download Figure S2, DOCX file, 0.6 MB (652.7KB, docx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
qPCR measurements revealed that the total bacterial and fungal levels increased after kefir grains were added to milk (see Table S1 in the supplemental material). At 8 and 24 h in Fr1, Ick, and UK3, Lactobacillus, Leuconostoc, and Acetobacter accounted for >98% of the total bacterial population, while Saccharomyces and Kazachstania accounted for >99% of the fungal population. No other bacterial or fungal genera were present at a relative abundance of >1%.
Absolute abundances of bacteria and fungi in kefir samples after 0, 8, and 24 h of fermentation, as determined by qPCR measurements. Download Table S1, PDF file, 0.06 MB (62KB, pdf) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Although there were some differences in their compositions at each time point, the bacterial communities of the three kefirs all followed the same pattern of succession (Fig. 1A). Between 0 and 8 h, there was an increase in the relative abundances of Lactobacillus, Leuconostoc, and Acetobacter. Lactobacillus was the dominant genus at 8 h. However, between 8 and 24 h, the relative abundance of Lactobacillus decreased. Concurrently, the relative abundances of Leuconostoc and Acetobacter increased. On average, Leuconostoc accounted for approximately one-third of the bacterial population at 24 h. In contrast to the bacterial communities of the three kefirs, the respective fungal communities displayed various patterns of succession (see Fig. S3A in the supplemental material).
Stacked bar charts presenting the fungal compositions of kefir samples after 0, 8, and 24 h of fermentation, as determined by ITS gene sequencing (A), and the microbial composition of kefir samples after 0, 8, and 24 h of fermentation (B), as determined by MetaPhlan2. Download Figure S3, TIF file, 0.4 MB (405.4KB, tif) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
16S rRNA and ITS compositional data were supplemented by composition-based analysis of shotgun metagenomic data. Kraken (24) was used to determine the bacterial composition of kefir after 0, 8, and 24 h of fermentation and yielded results that corresponded well to amplicon sequencing results at the genus level but which could be further assigned to the species level. It was established that the kefir milk was dominated by L. kefiranofaciens at 8 h (Fig. 1B). However, between 8 and 24 h, the relative abundance of L. kefiranofaciens decreased, whereas the relative abundance of Leuconostoc mesenteroides increased. During the same period, there were also increases in the relative abundances of Acetobacter pasteurianus, Lactobacillus helveticus, Leuconostoc citreum, Leuconostoc gelidum, and Leuconostoc kimchii. These results were generally consistent with those generated by MetaPhlan2 (25) (see Fig. S3B in the supplemental material), except that MetaPhlan2 did not detect some of the species present in lower abundance (i.e., A. pasteurianus, L. citreum, L. gelidum, or L. kimchii). MetaPhlan2 predicted that Saccharomyces cerevisiae was the dominant fungal species and that it accounted for 0.9% and 0.2% of the microbiota in kefir at 8 and 24 h of fermentation, respectively. However, it did not detect Kazachstania species.
In addition, PanPhlAn (26) was used to provide strain level characterization of the most dominant bacterial species identified by Kraken and MetaPhlan2. The results indicated that, across all of the kefirs tested, the strains present were most closely related to L. kefiranofaciens DSM 10550, L. mesenteroides ATCC 8293, and L. helveticus MTCC 5463 (see Fig. S5 in the supplemental material). Despite this relative homogeneity, it was still apparent that the strains in a particular kefir were more closely related to each other than they were to strains from other kefirs (see Fig. S4 in the supplemental material).
Hierarchically clustered heat maps showing the relatedness of L. kefiranofaciens (A), L. mesenteroides (B), and L. helveticus (C) from Fr1, Ick, and UK3 to reference strain genomes, as determined by PanPhlAn. Clustering was done with hclust2 (https://bitbucket.org/nsegata/hclust2). Download Figure S4, TIF file, 0.9 MB (942.1KB, tif) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
APLSR plot (principal components 1 and 2) of sensory acceptance and RDA data for spiked and unspiked kefir samples. Download Figure S5, PDF file, 0.08 MB (87.8KB, pdf) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Gene content of kefir.
Whole-metagenome shotgun sequencing was used to characterize the functional potential of the kefir microbiome at different stages of fermentation, and the HUMAnN2 pipeline (https://bitbucket.org/biobakery/humann2) was used for metagenomic metabolic reconstruction. The default HUMAnN2 pathway abundance table was regrouped by using a custom mapping file to assign individual MetaCyc pathways (27) to a hierarchy of 534 gene product categories to achieve an overview of the kefir microbiome (Fig. 2). The statistical tool LEfSe (28) was used to identify changes in the abundances of genetic pathways over the course of fermentation. Notably, we observed that pathways involved in carbohydrate metabolism, carboxylate degradation, and unsaturated fatty acid biosynthesis were most prevalent at 8 h, whereas those involved in amino acid metabolism and 2,3-butanediol degradation were most prevalent at 24 h (Fig. 2). Inspection of the default pathway abundance table revealed that pathways involved in fatty acid beta oxidation were present in kefirs. The pathways mentioned here are of particular interest because they are potentially involved in the production of volatile compounds (Table 1).
FIG 2 .

Cladogram presenting a hierarchical overview of the MetaCyc pathways detected in the kefir microbiome with HUMAnN2. Central nodes represent general pathway category functions, like carbohydrate catabolism, and their descendant nodes represent more specific pathway category functions, like sucrose degradation. The colors of the clades indicate the times at which pathways of particular interest were most prevalent, as determined by LEfSe. The outer rings indicate the presence/absence of pathways in L. kefiranofaciens (blue), L. mesenteroides (orange), L. helveticus (red), and S. cerevisiae (maroon).
TABLE 1 .
Volatile compounds detected in kefir by GC-MS
| Compound | LRIa | Ref LRIb | Odor descriptor(s) | Source |
|---|---|---|---|---|
| Carboxylic acids | ||||
| Acetic acid | 692 | 629 | Vinegar, peppers, green, fruity, floral, sour | Carbohydrate metabolism |
| Hexanoic acid | 968 | 983 | Sweaty, cheesy, sharp, goaty, bad breath, acidic | Lipid metabolism |
| Octanoic acid | 1,163 | 1,160 | Cheesy, rancid, pungent, sweat, soapy, goaty | Lipid metabolism |
| Nonanoic acid | 1,254 | 1,276 | Fatty, soapy, waxy, green, goat | Lipid metabolism |
| n-Decanoic acid | 1,355 | 1,379 | Soapy, waxy, stale, buttery, fruity, grassy, cheesy, milky | Lipid metabolism |
| Alcohols | ||||
| 2-Methyl-1-butanol | 733 | 755 | Penetrating, alcohol, wine-like, plastic | Amino acid metabolism |
| 2-Ethyl-1-hexanol | 1,025 | 1,031 | Animal, cardboard | Lipid metabolism |
| Ethanol | 468 | 426 | Dry, dust | Carbohydrate metabolism |
| 2-Butanol | 601 | 596 | Fruity | Carbohydrate metabolism |
| 2-Methyl-1-propanol | 621 | 647 | Malty | Amino acid metabolism |
| 3-Methyl-butanol | 730 | 768 | Fresh cheese, breathtaking, alcoholic, fruity, grainy, solvent-like, floral, malty | Amino acid metabolism |
| Phenylethyl alcohol | 1,119 | 1,112 | Unclean, rose, violet-like, honey, floral, spicy | Amino acid metabolism |
| 1-Pentanol | 730 | 768 | Fruity, alcoholic, green, balsamic, fusel oil, woody | Lipid metabolism |
| Aldehydes | ||||
| 3-Methyl-butanal | 649 | 654 | Malty, cheesy, green, dark chocolate, cocoa | Amino acid metabolism |
| 2-Methyl-butanal | 658 | 662 | Malty, dark chocolate, almond, cocoa, coffee | Amino acid metabolism |
| Octanal | 1,002 | 1,004 | Green, fatty, soapy, fruity, orange peel | Lipid metabolism |
| Nonanal | 1,103 | 1,106 | Green, citrus, fatty, floral | Lipid metabolism |
| Pentanal | 694 | 697 | Pungent, almond-like, chemical, malty, apple, green | Lipid metabolism |
| Hexanal | 798 | 801 | Green, slightly fruity, lemon, herbal, grassy, tallow | Lipid metabolism |
| Heptanal | 900 | 901 | Slightly fruity (balsam), fatty, oily, green, woody | Lipid metabolism |
| Esters | ||||
| Ethyl acetate | 609 | 614 | Solvent, pineapple, fruity, apples | Carbohydrate metabolism |
| Ethyl butanoate | 802 | 800 | Ripe fruit, buttery, green, apple, pineapple, banana, sweet | Carbohydrate metabolism |
| Ethyl hexanoate | 995 | 1,002 | Fruity, malty, young cheese, moldy, apple, green, orange, pineapple, banana | Carbohydrate metabolism |
| Ethyl octanoate | 1,190 | 1,198 | Fruity, apple, green, fatty, orange, winey, pineapple, apricot | Carbohydrate metabolism |
| Ethyl decanoate | 1,388 | 1,396 | Fruity, grape, cognac | Carbohydrate metabolism |
| 3-Methyl-1-butanol, acetate | 874 | 879 | Fruity, banana, candy, sweet, apple peel | Unknown |
| Ketones | ||||
| 2,3-Butanedione | 589 | 596 | Buttery, strong | Carbohydrate metabolism |
| 2,3-Pentanedione | 694 | 693 | Creamy, cheesy, oily, sweet buttery, caramellic | Carbohydrate metabolism |
| 2,3-Hexanedione | 781 | 788 | Sweet, creamy, caramellic, buttery | Carbohydrate metabolism |
| 2-Heptanone | 887 | 891 | Blue cheese, spicy, Roquefort | Lipid metabolism |
| 2-Undecanone | 1,288 | 1,294 | Floral, fruity, green, musty, tallow | Lipid metabolism |
| 2-Pentanone | 679 | 687 | Orange peel, sweet, fruity | Lipid metabolism |
| 2-Nonanone | 1,088 | 1,094 | Malty, fruity, hot milk, smoked cheese | Lipid metabolism |
| Acetone | 494 | 496 | Earthy, fruity, wood pulp, hay | Lipid metabolism |
| 2-Butanone | 598 | 593 | Buttery, sour milk, etheric | Carbohydrate metabolism |
| Sulfur compounds | ||||
| Dimethyl sulfone | 920 | 926 | Sulfurous, hot milk, burned | Amino acid metabolism |
| Carbon disulfide | 537 | 568 | Sweet, ethereal | Amino acid metabolism |
LRI, linear retention index.
Reference LRI.
In addition, the default HUMAnN2 gene family table was regrouped to Gene Ontology (GO) terms (gene product categories [29]) and, in total, we detected 1,288, 1,006, and 947 GO terms associated with carbohydrate, amino acid, and lipid metabolism in the kefir microbiome. Interestingly, pathways involved in aromatic amino acids and proline biosynthesis were assigned to L. mesenteroides but not to L. kefiranofaciens. Similarly, pathways involved in arabinose, maltose, pentose, sucrose, xylose, and xylulose metabolism were present in L. mesenteroides but not in L. kefiranofaciens.
Finally, the HUMANnN2 gene family table was inspected for genes associated with probiotic functionalities to better understand the basis of the health benefits of kefir. We observed that L. kefiranofaciens in Fr1, Ick, and UK3 contained genes encoding EPS synthesis proteins (UniRef50_W5XGS2, UniRef50_F6CC46, and UniRef50_F0TGY1), bile salt transporter proteins (UniRef50_Q74LX5 and UniRef50_F6CE74), adhesion proteins (UniRef50_F6CFB4 and UniRef50_Q040W2), mucus binding proteins (UniRef50_F6CE70, UniRef50_F6CE69, UniRef50_F6CDG7, and UniRef50_F6CBX6), and the type III bacteriocins/bacteriolysins helveticin J (UniRef50_D5GYX2) and enterolysin A (UniRef50_D5GXY3 and UniRef50_F6CAP6). On the basis of these findings, we downloaded publicly available metagenome sequences from cheeses and kimchi (Table 2) todetermine the prevalence of similar genes in other fermented foods. HUMAnN2 indicated that genes encoding EPS synthesis proteins, adhesion proteins, mucus binding proteins, bile salt hydrolases, bile salt symporters, and bacteriocins/prebacteriocins were widespread in the 14 cheese varieties investigated (Fig. 3). In addition, we observed several instances where multiple genes were assigned to individual species (see Table S2 in the supplemental material). We identified similar genes in kimchi (Fig. 5), although HUMAnN2 was unable to assign them to individual species because of the lower sequencing depth of those samples.
TABLE 2 .
Accession numbers of the cheese and kimchi metagenomes analyzed in this study
| Origin | Accession no. | Repository | Sample description | Reference |
|---|---|---|---|---|
| Cheese | 4524483.3 | MG-Rast | Washed unpasteurized cow cheese | 6 |
| Cheese | 4524484.3 | MG-Rast | Bloomy unpasteurized goat cheese | 6 |
| Cheese | 4524488.3 | MG-Rast | Natural unpasteurized cow cheese | 6 |
| Cheese | 4524489.3 | MG-Rast | Bloomy unpasteurized goat cheese | 6 |
| Cheese | 4524490.3 | MG-Rast | Natural unpasteurized cow cheese | 6 |
| Cheese | 4524493.3 | MG-Rast | Natural unpasteurized cow cheese | 6 |
| Cheese | 4524494.3 | MG-Rast | Washed pasteurized cow cheese | 6 |
| Cheese | 4524495.3 | MG-Rast | Washed unpasteurized cow cheese | 6 |
| Cheese | 4524496.3 | MG-Rast | Washed unpasteurized cow cheese | 6 |
| Cheese | 4524497.3 | MG-Rast | Natural pasteurized cow cheese | 6 |
| Cheese | 4524499.3 | MG-Rast | Washed unpasteurized cow cheese | 6 |
| Cheese | 4524500.3 | MG-Rast | Washed unpasteurized cow cheese | 6 |
| Cheese | 4524505.3 | MG-Rast | Washed unpasteurized cow cheese | 6 |
| Kimchi | SRX072929 | Sequence Read Archive | Kimchi fermentation, day 1 | 41 |
| Kimchi | SRX072930 | Sequence Read Archive | Kimchi fermentation, day 7 | 41 |
| Kimchi | SRX072931 | Sequence Read Archive | Kimchi fermentation, day 13 | 41 |
| Kimchi | SRX072932 | Sequence Read Archive | Kimchi fermentation, day 16 | 41 |
| Kimchi | SRX072933 | Sequence Read Archive | Kimchi fermentation, day 18 | 41 |
| Kimchi | SRX072934 | Sequence Read Archive | Kimchi fermentation, day 21 | 41 |
| Kimchi | SRX072935 | Sequence Read Archive | Kimchi fermentation, day 23 | 41 |
| Kimchi | SRX072936 | Sequence Read Archive | Kimchi fermentation, day 25 | 41 |
| Kimchi | SRX072937 | Sequence Read Archive | Kimchi fermentation, day 27 | 41 |
| Kimchi | SRX072938 | Sequence Read Archive | Kimchi fermentation, day 29 | 41 |
| Cheese | SAMEA3232870 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232871 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232872 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232873 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232874 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232875 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232876 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232877 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232878 | European Nucleotide Archive | Continental-type cheese | 23 |
| Cheese | SAMEA3232879 | European Nucleotide Archive | Continental-type cheese | 23 |
FIG 3 .

Hierarchically clustered binary heat map showing the presence/absence of genes associated with probiotic action in cheese and kimchi metagenomes, as determined by HUMAnN2. Clustering was performed with the hclust function in the R package gplots.
FIG 5 .

Hierarchically clustered heat map showing correlations between the relative abundances of microbial species and the levels of volatile compounds in kefir samples. Clustering was performed with the hclust function in the R package gplots. The color of each tile of the heat map indicates the type/strength of the correlation for a given species-compound combination, as indicated by the color key.
Microbial species from cheese samples that contain two or more genes associated with probiotic action, as determined by HUMAnN2. Download Table S2, XLSX file, 0.01 MB (13KB, xlsx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Volatile-compound profiling and sensory analysis of kefir milk.
Gas chromatography-mass spectrometry (GC-MS) was used to determine the volatile-compound profile of kefir milk after 0, 8, and 24 h of fermentation. Thirty-nine volatile compounds that could contribute to flavor were identified and semiquantified in kefir milks produced with each of the three kefir grains. These consisted of nine ketones, seven aldehydes, six esters, eight alcohols, five carboxylic acids, and two sulfur compounds (Table 1). The results of the volatile-compound analysis are presented in Fig. 4. The levels of all of the compounds detected increased after 0 h, apart from 1-pentanol, pentanal, hexanal, heptanal, heptanol, acetone, and 2-butanone (Fig. 4).
FIG 4 .
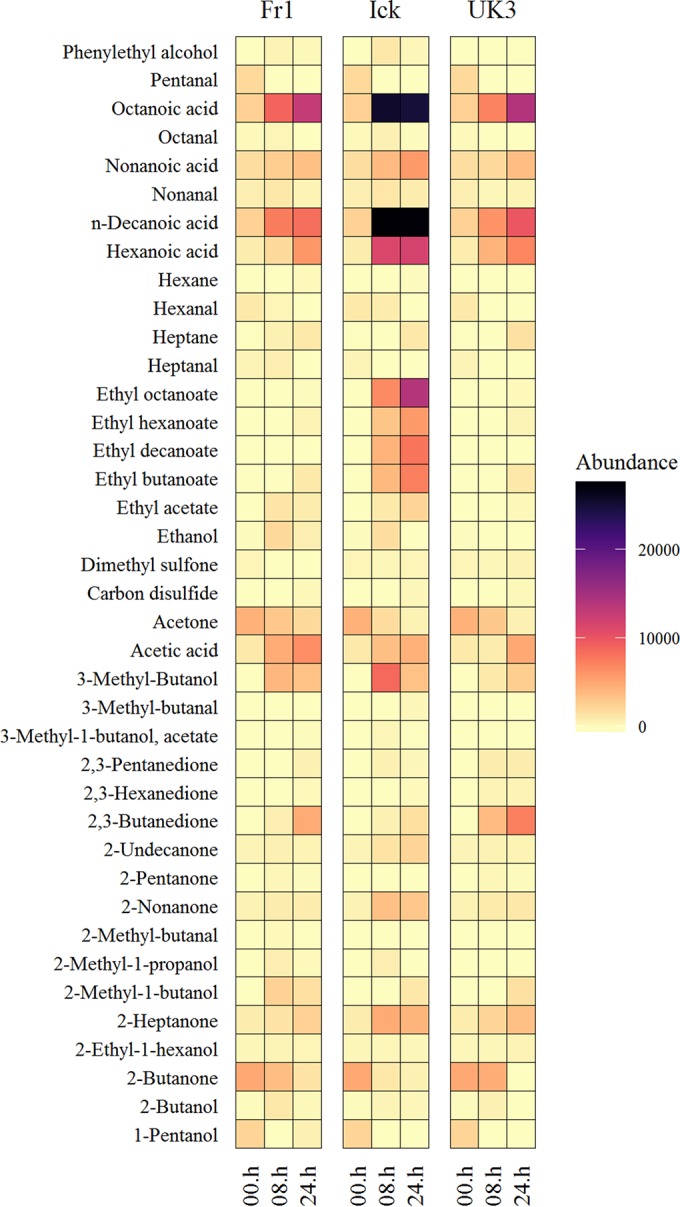
Faceted heat map showing changes in the volatile-compound profiles of Fr1, Ick, and UK3. Note that the 00.h column in each section refers to the volatile-compound profile of milk immediately before fermentation initiation. The same starting milk was used for Fr1, Ick, and UK3.
Sensory acceptance evaluation and ranking descriptive analysis (RDA) of the Fr1 and Ick kefir milks were performed after 24-h fermentations. These revealed perceptible differences between the milks. Specifically, Fr1 samples had a more likeable, buttery flavor whereas Ick samples had a less likeable but fruity flavor (see Fig. S5 in the supplemental material). These results confirm that the volatile-compound profile data are consistent with subsequent flavor.
Correlations between microbial taxa and volatile compounds.
The Spearman rank correlation test was used to identify correlations between the levels of individual taxa and flavor compounds. At the genus level, based on amplicon sequencing results, there were strong correlations between Lactobacillus and carboxylic acids, esters, and 3-methyl-1-butanol; between Saccharomyces and carboxylic acids and esters; between Acetobacter and acetic acid, 2-methyl-1-butanol, and 2,3-butanedione; between Leuconostoc and 2,3-butanedione; and between Kazachstania and acetic acid, 2-methyl-1-butanol, 2,3-butanedione, 2,3-pentanedione, and 2,3-hexanedione (see Table S3 in the supplemental material). At the bacterial species level, on the basis of Kraken results, there were strong correlations between L. kefiranofaciens and carboxylic acids and esters; between A. pasteurianus and carboxylic acids and 2,3-butanedione; and between L. mesenteroides and 2,3-butanedione. At the fungal species level, on the basis of MetaPhlan2 results, there were strong correlations between S. cerevisiae and alcohols and esters (Table 3; Fig. 5). In summary, correlations were found between compounds associated with vinegary flavors and A. pasteurianus, those associated with cheesy flavors and L. kefiranofaciens, those associated with buttery flavors and L. mesenteroides, and those associated with fruity flavors and L. kefiranofaciens and S. cerevisiae.
TABLE 3 .
Summary of strong positive correlations identified between the relative abundances of species and the levels of metabolites in kefir
| Species | Compound(s) | R value |
P value |
|
|---|---|---|---|---|
| Unadjusted | FDR adjusteda | |||
| Leuconostoc mesenteroides | 2,3-Butanedione | 0.79 | 0.0005 | 0.011 |
| Lactobacillus kefiranofaciens | 2-Nonanone | 0.79 | 0.0005 | 0.011 |
| Lactobacillus helveticus | Acetic acid | 0.75 | 0.0013 | 0.017 |
| Leuconostoc mesenteroides | 2-Methyl-1-butanol | 0.74 | 0.0015 | 0.017 |
| Lactobacillus kefiranofaciens | Hexanoic acid | 0.71 | 0.0033 | 0.024 |
| Lactobacillus kefiranofaciens | 2-heptanone | 0.71 | 0.0033 | 0.024 |
| Lactobacillus kefiranofaciens | Octanoic acid | 0.70 | 0.0040 | 0.024 |
| Lactobacillus kefiranofaciens | Acids | 0.68 | 0.0049 | 0.024 |
| Lactobacillus kefiranofaciens | n-Decanoic acid | 0.68 | 0.0049 | 0.024 |
| Saccharomyces cerevisiae | Nonanal | 0.66 | 0.0080 | 0.035 |
| Lactobacillus kefiranofaciens | Ethyl decanoate | 0.65 | 0.0089 | 0.035 |
| Lactobacillus kefiranofaciens | Esters | 0.64 | 0.0099 | 0.035 |
| Saccharomyces cerevisiae | Ethyl acetate | 0.63 | 0.0110 | 0.035 |
| Saccharomyces cerevisiae | 3-Methyl-butanol | 0.63 | 0.0118 | 0.035 |
| Acetobacter pasteurianus | 2-Methyl-1-butanol | 0.63 | 0.0126 | 0.035 |
| Saccharomyces cerevisiae | Phenylethyl alcohol | 0.62 | 0.0134 | 0.035 |
| Saccharomyces cerevisiae | Octanal | 0.62 | 0.0141 | 0.035 |
| Acetobacter pasteurianus | Nonanoic acid | 0.60 | 0.0169 | 0.040 |
| Lactobacillus kefiranofaciens | Phenylethyl alcohol | 0.60 | 0.0179 | 0.040 |
| Saccharomyces cerevisiae | Alcohols | 0.59 | 0.0203 | 0.040 |
| Acetobacter pasteurianus | Acetic acid | 0.59 | 0.0206 | 0.040 |
| Lactobacillus helveticus | 2,3-Butanedione | 0.57 | 0.0255 | 0.040 |
| Acetobacter pasteurianus | Ethyl butanoate | 0.57 | 0.0268 | 0.040 |
| Lactobacillus kefiranofaciens | Ethyl hexanoate | 0.57 | 0.0279 | 0.040 |
| Lactobacillus helveticus | 3-Methyl-butanol | 0.56 | 0.0283 | 0.040 |
| Saccharomyces cerevisiae | Ethyl decanoate | 0.56 | 0.0283 | 0.040 |
| Lactobacillus kefiranofaciens | Ethyl butanoate | 0.56 | 0.0292 | 0.040 |
| Acetobacter pasteurianus | Ethyl hexanoate | 0.56 | 0.0314 | 0.040 |
| Lactobacillus kefiranofaciens | Nonanoic acid | 0.56 | 0.0315 | 0.040 |
| Lactobacillus kefiranofaciens | 2-Undecanone | 0.56 | 0.0315 | 0.040 |
| Lactobacillus kefiranofaciens | Ethyl octanoate | 0.55 | 0.0321 | 0.040 |
| Lactobacillus kefiranofaciens | 3-Methyl-butanol | 0.55 | 0.0321 | 0.040 |
| Acetobacter pasteurianus | Acids | 0.55 | 0.0324 | 0.040 |
| Acetobacter pasteurianus | Hexanoic acid | 0.54 | 0.0368 | 0.044 |
| Acetobacter pasteurianus | 2,3-Butanedione | 0.54 | 0.0375 | 0.044 |
| Acetobacter pasteurianus | Ethyl acetate | 0.54 | 0.0381 | 0.044 |
| Saccharomyces cerevisiae | Esters | 0.53 | 0.0413 | 0.046 |
| Leuconostoc mesenteroides | Acetic acid | 0.53 | 0.0419 | 0.046 |
| Lactobacillus helveticus | 2-Methyl-1-butanol | 0.53 | 0.0433 | 0.046 |
| Saccharomyces cerevisiae | 2-Undecanone | 0.53 | 0.0435 | 0.046 |
| Lactobacillus kefiranofaciens | Ethyl acetate | 0.52 | 0.0478 | 0.048 |
| Acetobacter pasteurianus | Octanoic acid | 0.52 | 0.0478 | 0.048 |
FDR, false discovery rate.
Correlations between the relative abundances of microbial genera and the levels of volatile compounds. Download Table S3, DOCX file, 0.01 MB (14.8KB, docx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Impact of supplementing kefir with kefir isolates.
The consequences of adding L. kefiranofaciens NCFB 2797 to Fr1, a kefir with a low indigenous L. kefiranofaciens population level, was investigated. GC-MS revealed that this addition caused increases in the levels of the esters ethenyl acetate (by 59.15%), ethyl acetate (100%), methyl-3-butyrate (26.83%), and 2-methylbutyl-acetate (11.44%) and the ketone 2-heptanone (65.86%). In contrast, the addition of L. mesenteroides 213M0 to Ick, a kefir with a low indigenous L. mesenteroides population level, resulted in increases in the levels of acetic acid (168.28%) and 2,3-butanediol (14.91%), a precursor to 2,3-butanedione (see Table S4 in the supplemental material). Despite changes in the volatile-compound profile, there were no perceptible changes in flavor (see Fig. S5 in the supplemental material).
Changes in the volatile-compound profiles of kefirs supplemented with L. kefiranofaciens 484 NCFB 2797 (A) and L. mesenteroides 213M0 (B). Download Table S4, XLSX file, 0.01 MB (12.5KB, xlsx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Many traditional fermented foods have been reported to have health benefits (30, 31). These foods are often produced on a small-scale, artisanal basis. However, the increased demand for health-promoting foods among the public presents an opportunity to bring traditional fermented foods to a wider audience and serves as an incentive to optimize starter cultures for the mass production of fermented foods with enhanced sensory qualities (32). In recent years, genetic characterization has been increasingly employed to guide starter culture development for numerous fermented foods, including wines, beers, cocoa, and meats (33–36). Similarly, integrated molecular omics approaches (37) have emerged as powerful methods of investigating the microbial dynamics of food fermentations with the aim of optimizing processes like flavor production (38). In this study, we combined compositional and shotgun DNA sequencing with GC-MS and flavor analysis to predict microbes involved in the production of different flavor compounds in kefir.
We identified significant correlations between the abundances of particular microbial genera and species and the levels of different volatile compounds and showed that the microbes in kefir had genes necessary for the production of these compounds. Specifically, Acetobacter pasteurianus correlated with acetic acid, which is associated with vinegary flavors; L. kefiranofaciens correlated with carboxylic acids and ketones associated with cheesy flavors and with esters associated with fruity flavors; L. mesenteroides correlated with 2,3-butanedione, which is associated with buttery flavors, and with acetic acid; and S. cerevisiae correlated with esters. Sensory analysis revealed that Fr1, a kefir high in L. mesenteroides, had a likeable buttery flavor, whereas Ick, a kefir high in L. kefiranofaciens, had a less likeable but fruity flavor. Thus, our data suggested a causal relationship between specific taxa and flavor characteristics that was subsequently supported by experimentally manipulating the kefir community. In line with predictions, adding L. kefiranofaciens NCFB 2797 to Fr1 resulted in increases in the levels of 2-heptanone and esters, whereas the addition of L. mesenteroides 213M0 to Ick resulted in increases in the levels of acetic acid and 2,3-butanediol, a precursor of 2,3-butanedione. However, sensory analysis indicated that these changes were imperceptible and therefore higher inoculum levels might be necessary to change flavor.
On the basis of these results, we predict that the final flavor of kefir can be manipulated by altering the ratio of microbes in the grain. Unfortunately, to date, it has not been possible to artificially reconstruct kefir grains in the laboratory and this might hamper the practical application of our findings. However, we propose that the approach outlined here can be used to accelerate the development of superior multistrain starter cultures to improve the flavor of a variety of fermented foods.
From a systems biology perspective, our work confirms that kefir is suitable as a model microbial community. There are two advantages to using the kefir model, rather than other fermented foods, in this way. First, kefir contains fewer species and so is a simpler environment in which to investigate how microbial communities are formed. Second, kefir is quick and easy to produce, with the fermentation taking just 24 h when it is incubated at room temperature. In addition, others have demonstrated that kefir is a highly culturable system and, indeed, all of the species that were detected at a relative abundance of >1% at 8 and 24 h across the kefirs examined have been isolated previously (39).
Ultimately, Kraken and MetaPhlAn2 showed that the microbial population of kefir was dominated by L. kefiranofaciens at 8 h of fermentation. However, between 8 and 24 h, there was a fall in the relative abundance of L. kefiranofaciens and L. mesenteroides superseded it as the dominant species. The shift from L. kefiranofaciens to L. mesenteroides is similar to patterns of microbial succession seen in other fermented foods (40, 41). We propose that kefir could be a particularly appropriate model community in which to determine the driving forces behind microbial succession. Early colonizing bacteria in other fermentations have been reported to modify the environment in such a way as to make it more suitable for the growth of other bacteria, thus driving succession (5), and this could explain the observed shift that occurs during kefir fermentation. Our HUMAnN2 results revealed that genes involved in aromatic amino acid biosynthesis were assigned to L. mesenteroides but not to L. kefiranofaciens. This may be significant because free amino acid analysis showed that there was a significant decrease in the levels of tyrosine in kefir between 8 and 24 h (see Text S1 in the supplemental material). It is possible that its ability to synthesize tyrosine underlies the increased prevalence of L. mesenteroides, relative to L. kefiranofaciens, in the latter stages of fermentation. Future work will focus on investigating the effect of modifying the levels of tyrosine on the microbiota and volatile-compound profile of kefir. Thus, a “kefir model” has the potential to yield insights into the effects of nutrient availability on microbial succession and metabolite production in other, more complicated, environments.
Supplemental materials and methods and results. Download Text S1, DOCX file, 0.02 MB (22.3KB, docx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Finally, we showed that L. kefiranofaciens has genes that encode proteins that are considered to be important for probiotic action, including EPS synthesis proteins, bile salt transporters, mucus binding proteins, and bacteriolysins (42, 43). The presence of these genes suggests that the L. kefiranofaciens strains present in these kefirs have the potential to survive gastric transit, colonize the gut, and inhibit the growth of pathogens. Indeed, previous studies using mice have shown that L. kefiranofaciens protects against enterohemorrhagic Escherichia coli infection (44). Further analysis of shotgun metagenomic data from cheese and kimchi indicated that similar genes are present in other fermented foods. Our findings are consistent with previous observations that fermented food-borne microbes can colonize the gut (45) and support designating some fermented foods, like kimchi, “probiotic foods” (31).
In summary, in this study, it has been demonstrated that a combined metagenomic and metabolomic approach can potentially be used to identify the microbes from a particular environment that are responsible for the production of certain metabolites, using the production of flavor compounds during kefir fermentation as a model. Furthermore, we have provided additional evidence of the use of microbial fermentations to provide valuable insights into the dynamics of microbial succession and, in the process, identified genes in L. kefiranofaciens that potentially confer important probiotic traits. To conclude, our analyses confirm the value of using kefir as a model microbial community, while also providing a valuable insight into the microbiology of this natural health-promoting beverage.
MATERIALS AND METHODS
Kefir fermentations.
Three kefir grains, Fr1, Ick, and UK3, from distinct geographic locations, France, Ireland, and the United Kingdom, respectively, were used for kefir fermentations. The grains were weighed and inoculated in full-fat pasteurized milk at a concentration of 2% (wt/vol) in separate fermentation vessels. The milk was incubated at 25°C for 24 h. A 20-ml volume of milk was collected after 0, 8, or 24 h. In total, there were 15 2% (wt/vol) kefir milk samples: three 0-h samples that were collected immediately before the addition of Fr1, Ick, or UK3; three 8-h samples (one each from Fr1, Ick, and UK3); and nine 24-h samples (one from each of the three replicate fermentations with Fr1, Ick, or UK3). The samples were stored at −20°C until DNA extraction and volatile-compound analysis. Kefir grains were washed with sterile deionized water between fermentations.
Additional fermentations were performed in which milk inoculated with specific kefir grains was supplemented with kefir isolates to assess the consequences of increased levels of these taxa on volatile-compound levels and flavor. Specifically, L. kefiranofaciens NCFB 2797 and L. mesenteroides 213M0 were grown overnight in 10 ml of de Man, Rogosa, and Sharpe broth; pelleted at 5,444 × g; and resuspended in 5 ml of pasteurized milk. L. kefiranofaciens NCFB 2797 cells were added to Fr1 milk, and L. mesenteroides 213M0 cells were added to Ick milk. Unspiked Fr1 and Ick served as negative controls. As described above, milk was incubated at 25°C for 24 h and the fermentations were carried out in triplicate. A 5-ml volume of milk was collected for volatile-compound analysis, and the samples were stored at −20°C. A 400-ml volume of milk was collected for sensory evaluation, and the samples were stored at −80°C.
Volatile-compound profiling of kefir by GC-MS.
For volatile-compound analysis of kefir, 1 g of the sample was added to a 20-ml screw-cap solid-phase microextraction (SPME) vial with a silicone/polytetrafluoroethylene septum (Apex Scientific, Maynooth, Ireland) and equilibrated to 75°C for 5 min with pulsed agitation for 5 s at 400 rpm with a GC Sampler 80 (Agilent Technologies Ltd., Little Island, Cork, Ireland). A single 50/30-µm Carboxen-divinylbenzene-polydimethylsiloxane SPME fiber (Agilent Technologies Ltd., Ireland) was used; it was exposed to the headspace above the samples for 20 min at a depth of 1 cm at 75°C. The fiber was retracted and injected into the GC inlet and desorbed for 2 min at 250°C. After injection, the fiber was heated in a bakeout station for 3 min at 270°C to cleanse the fiber. The samples were analyzed in triplicate. Injections were made on an Agilent 7890A GC apparatus with an Agilent DB-5 column (60 m by 0.25 mm by 0.25 µm) with a multipurpose injector with a Merlin microseal (Agilent Technologies Ltd., Ireland). The temperature of the column oven was set at 35°C, held for 0.5 min, increased at 6.5°C·min−1 to 230°C, and then increased at 15°C·min−1 to 325°C, yielding at total run time of 36.8 min. The carrier gas was helium held at a constant pressure of 23 lb/in2. The detector was an Agilent 5975C MSD single-quadrupole mass spectrometer detector (Agilent Technologies Ltd., Ireland). The ion source temperature was 230°C, the interface temperature was set at 280°C, and the MS mode was electronic ionization (−70 V) with the mass range scanned between 35 and 250 atomic mass units. Compounds were identified by mass spectrum comparisons to the National Institute of Standards and Technology 2011 mass spectral library, the automated mass spectral deconvolution and identification system, and an in-house library created in TargetView software (Markes International, Llantrisant, United Kingdom) with target and qualifier ions and linear retention indices for each compound. Autotuning of the GC-MS system was carried out prior to the analysis to ensure optimal GC-MS performance. A set of external standards was also run at the start and end of the sample set, and abundances were compared to known amounts to ensure that both the SPME extraction and MS detection were performing within specifications.
Volatile-compound profiling of spiked and unspiked kefir samples was done by a slightly modified GC-MS protocol (see Text S1 in the supplemental material).
Sensory analysis of spiked and nonspiked kefir.
Twenty-five naive assessors were recruited for sensory acceptance evaluation, and 10 trained assessors were recruited for RDA. Analysis of variance–partial least-squares regression (APLSR) was used to process the results of the sensory acceptance evaluation test and RDA with Unscrambler software version 10.3. See Text S1 in the supplemental material for a more in-depth description of the sensory analysis methods used.
Total DNA extraction from kefir (milks and grains).
DNA was extracted from 15 ml of kefir milk as follows. Milk was centrifuged at 5,444 × g for 30 min at 4°C to pellet the microbial cells in the liquid. The cell pellet was resuspended in 200 µl of PowerBead solution from the PowerSoil DNA Isolation kit (Cambio, Cambridge, United Kingdom). The resuspended cells were transferred to a PowerBead tube (Cambio, Cambridge, United Kingdom). A 90-µl volume of 50 mg/ml lysozyme (Sigma-Aldrich, Dublin, Ireland) and 50 µl of 100 U/ml mutanolysin (Sigma-Aldrich, Dublin, Ireland) were added, and the sample was incubated at 60°C for 15 min. A 28-µl volume of proteinase K (Sigma-Aldrich, Dublin, Ireland) was added, and the sample was incubated at 60°C for a further 15 min. DNA was then purified from the sample by the standard PowerSoil DNA Isolation kit protocol (Cambio, Cambridge, United Kingdom). Total DNA was also extracted from each of the three grains. Fragments of 50 mg were removed from different sites on each of the grains and added to separate PowerBead tubes (Cambio, Cambridge, United Kingdom). The grain fragments were homogenized by shaking the PowerBead tube on the TissueLyser II (Qiagen, West Sussex, United Kingdom) at 20 Hz for 10 min. Following homogenization, DNA was purified from the sample by the method outlined above. Total DNA was initially quantified and qualified by gel electrophoresis and the NanoDrop 1000 (BioSciences, Dublin, Ireland) before more accurate quantification with the Qubit High Sensitivity DNA assay (BioSciences, Dublin, Ireland). Bacterial and fungal abundances were determined by qPCR by the protocol described by Fouhy et al. (46) and the Femto Fungal DNA Quantification kit (Cambridge Biosciences, United Kingdom), respectively.
Amplicon sequencing.
16S rRNA gene libraries were prepared from extracted DNA by the 16S Metagenomic Sequencing Library Preparation protocol from Illumina (47). ITS gene libraries were prepared for the samples with a modified version of the 16S rRNA gene extraction protocol. Briefly, the initial genomic DNA amplification was performed with primers specific to the ITS1-ITS2 region of the ITS gene (48), but they were modified to incorporate the Illumina overhang adaptor (i.e., ITSF1 primer 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCTTGGTCATTTAGAGGAAGTAA 3′ and ITS2 primer 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGCTGCGTTCTTCATCGATGC 3′). After amplification of the ITS1-ITS2 region, PCR products were treated as described in the Illumina protocol. Samples were sequenced on the Illumina MiSeq in the Teagasc sequencing facility, with a 2 × 250 cycle V2 kit, in accordance with standard Illumina sequencing protocols.
Whole-metagenome shotgun sequencing.
Whole-metagenome shotgun libraries were prepared in accordance with the Nextera XT DNA Library Preparation Guide from Illumina (47). Samples were sequenced on the Illumina MiSeq sequencing platform in the Teagasc sequencing facility, with a 2 × 300 cycle V3 kit, in accordance with standard Illumina sequencing protocols.
Bioinformatic analysis.
16S rRNA gene sequencing data were processed with the pipeline described by Fouhy et al. (49). Briefly, sequences were quality checked, clustered into operational taxonomic units, and aligned and diversity (both alpha and beta) was calculated with a combination of the Qiime (1.8.0) (50) and USearch (v7-64bit) (51) pipelines. Taxonomy was assigned with a BLAST search (52) against SILVA SSURef database release 1 (53). ITS gene sequencing data were processed with a slightly modified pipeline. Taxonomy was assigned by using a BLAST search against the ITSoneDB database (54). Raw reads from whole-metagenome shotgun sequencing were filtered on the basis of quality and quantity and trimmed to 200 bp with a combination of Picardtools (https://github.com/broadinstitute/picard) and SAMtools (55). Subsequently, function was assigned to reads with the HUMAnN2 suite of tools (56), which assigned function based on the ChocoPhlan databases and genes based on UniRef (57). The HUMAnN2 gene abundance table was regrouped by a mapping of MetaCyc pathways and a mapping of GO terms for amino acid, carbohydrate, and lipid metabolism. MetaPhlAn2 and Kraken were used to profile changes in the microbial composition of kefir milk at the species level (24, 25).
Statistical analysis of metagenomic and metabolomic data.
Statistical analysis was done with R-3.2.2 (58) and LEfSe (28). The R packages ggplot2 and gplots and the cladogram generator Graphlan (59) were used for data visualization.
Accession number(s).
Sequence data have been deposited in the European Nucleotide Archive (ENA) under the project accession number PRJEB15432.
ACKNOWLEDGMENTS
We thank Ben Bourrie and Mairéad Coakley for providing us with the strains used in this study. In addition, we thank Eric Fransoza for providing us with mapping files to regroup the HUMAnN2 gene abundance table.
This research was funded by Science Foundation Ireland in the form of a center grant (APC Microbiome Institute grant no. SFI/12/RC/2273). Research in the Cotter laboratory is also funded by Science Foundation Ireland through the PI award “Obesibiotics” (11/PI/1137). Orla O’Sullivan is funded by Science Foundation Ireland through a Starting Investigator Research Grant award (13/SIRG/2160).
Funding Statement
This research was funded by Science Foundation Ireland in the form of a centre grant (APC Microbiome Institute Grant Number SFI/12/RC/2273). Research in the Cotter laboratory is funded by SFI through the PI award "Obesibiotics" (11/PI/1137). Orla O'Sullivan is funded by Science Foundation Ireland through a Starting Investigator Research Grant award (13/SIRG/2160).
REFERENCES
- 1.Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL, Owens S, Gilbert JA, Wall DH, Caporaso JG. 2012. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci U S A 109:21390–21395. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Human Microbiome Project Consortium 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci U S A 103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faust K, Raes J. 2012. Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 5.Wolfe BE, Dutton RJ. 2015. Fermented foods as experimentally tractable microbial ecosystems. Cell 161:49–55. doi: 10.1016/j.cell.2015.02.034. [DOI] [PubMed] [Google Scholar]
- 6.Wolfe BE, Button JE, Santarelli M, Dutton RJ. 2014. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 158:422–433. doi: 10.1016/j.cell.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen B, Gürakan GC, Ünlü G. 2014. Kefir: a multifaceted fermented dairy product. Probiotics Antimicrob Proteins 6:123–135. doi: 10.1007/s12602-014-9168-0. [DOI] [PubMed] [Google Scholar]
- 8.Güzel-Seydim ZB, Seydim AC, Greene AK, Bodine AB. 2000. Determination of organic acids and volatile flavor substances in kefir during fermentation. J Food Compost Anal 13:35–43. doi: 10.1006/jfca.1999.0842. [DOI] [Google Scholar]
- 9.Marsh AJ, O’Sullivan O, Hill C, Ross RP, Cotter PD. 2013. Sequencing-based analysis of the bacterial and fungal composition of kefir grains and milks from multiple sources. PLoS One 8:e69371. doi: 10.1371/journal.pone.0069371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nalbantoglu U, Cakar A, Dogan H, Abaci N, Ustek D, Sayood K, Can H. 2014. Metagenomic analysis of the microbial community in kefir grains. Food Microbiol 41:42–51. doi: 10.1016/j.fm.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Garofalo C, Osimani A, Milanović V, Aquilanti L, De Filippis F, Stellato G, Di Mauro S, Turchetti B, Buzzini P, Ercolini D, Clementi F. 2015. Bacteria and yeast microbiota in milk kefir grains from different Italian regions. Food Microbiol 49:123–133. doi: 10.1016/j.fm.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Korsak N, Taminiau B, Leclercq M, Nezer C, Crevecoeur S, Ferauche C, Detry E, Delcenserie V, Daube G. 2015. Short communication: evaluation of the microbiota of kefir samples using metagenetic analysis targeting the 16S and 26S ribosomal DNA fragments. J Dairy Sci 98:3684–3689. doi: 10.3168/jds.2014-9065. [DOI] [PubMed] [Google Scholar]
- 13.de Moreno de Leblanc A, Matar C, Farnworth E, Perdigón G. 2007. Study of immune cells involved in the antitumor effect of kefir in a murine breast cancer model J Dairy Sci 90:1920–1928. doi: 10.3168/jds.2006-079. [DOI] [PubMed] [Google Scholar]
- 14.Lee M-Y, Ahn K-S, Kwon O-K, Kim M-J, Kim M-K, Lee I-Y, Oh S-R, Lee H-K. 2007. Anti-inflammatory and anti-allergic effects of kefir in a mouse asthma model. Immunobiology 212:647–654. doi: 10.1016/j.imbio.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Rodrigues KL, Caputo LR, Carvalho JC, Evangelista J, Schneedorf JM. 2005. Antimicrobial and healing activity of kefir and kefiran extract. Int J Antimicrob Agents 25:404–408. doi: 10.1016/j.ijantimicag.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 16.Hertzler SR, Clancy SM. 2003. Kefir improves lactose digestion and tolerance in adults with lactose maldigestion. J Am Diet Assoc 103:582–587. doi: 10.1053/jada.2003.50111. [DOI] [PubMed] [Google Scholar]
- 17.Liu J-R, Wang S-Y, Chen M-J, Chen H-L, Yueh P-Y, Lin C-W. 2006. Hypocholesterolaemic effects of milk-kefir and soyamilk-kefir in cholesterol-fed hamsters. Br J Nutr 95:939–946. doi: 10.1079/BJN20061752. [DOI] [PubMed] [Google Scholar]
- 18.Bolla PA, Abraham AG, Pérez PF, de los Angeles Serradell M. 27 October 2015. Kefir-isolated bacteria and yeasts inhibit Shigella flexneri invasion and modulate pro-inflammatory response on intestinal epithelial cells. Benef Microbes. [DOI] [PubMed] [Google Scholar]
- 19.Leite AM, Miguel MA, Peixoto RS, Ruas-Madiedo P, Paschoalin VM, Mayo B, Delgado S. 2015. Probiotic potential of selected lactic acid bacteria strains isolated from Brazilian kefir grains. J Dairy Sci 98:3622–3632. doi: 10.3168/jds.2014-9265. [DOI] [PubMed] [Google Scholar]
- 20.Carasi P, Racedo SM, Jacquot C, Romanin DE, Serradell MA, Urdaci MC. 2015. Impact of kefir derived Lactobacillus kefiri on the mucosal immune response and gut microbiota. J Immunol Res 2015:361604. doi: 10.1155/2015/361604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Montijo-Prieto S, Moreno E, Bergillos-Meca T, Lasserrot A, Ruiz-López M-D, Ruiz-Bravo A, Jiménez-Valera M. 2015. A Lactobacillus plantarum strain isolated from kefir protects against intestinal infection with Yersinia enterocolitica O9 and modulates immunity in mice. Res Microbiol 166:626–632. doi: 10.1016/j.resmic.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Bolla PA, Carasi P, Bolla Mde L, De Antoni GL, Serradell Mde L. 2013. Protective effect of a mixture of kefir-isolated lactic acid bacteria and yeasts in a hamster model of Clostridium difficile infection. Anaerobe 21:28–33. doi: 10.1016/j.anaerobe.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 23.Quigley L, McCarthy R, O’Sullivan O, Beresford TP, Fitzgerald GF, Ross RP, Stanton C, Cotter PD. 2013. The microbial content of raw and pasteurized cow milk as determined by molecular approaches. J Dairy Sci 96:4928–4937. doi: 10.3168/jds.2013-6688. [DOI] [PubMed] [Google Scholar]
- 24.Wood DE, Salzberg SL. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. 2012. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, Segata N. 2016. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat Methods 13:435–438. doi: 10.1038/nmeth.3802. [DOI] [PubMed] [Google Scholar]
- 27.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Weerasinghe D, Zhang P, Karp PD. 2014. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 42:D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: tool for the unification of biology. Nat Genet 25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsh AJ, Hill C, Ross RP, Cotter PD. 2014. Fermented beverages with health-promoting potential: past and future perspectives. Trends Food Sci Technol 38:113–124. doi: 10.1016/j.tifs.2014.05.002. [DOI] [Google Scholar]
- 31.Park K-Y, Jeong J-K, Lee Y-E, Daily JW III. 2014. Health benefits of kimchi (Korean fermented vegetables) as a probiotic food. J Med Food 17:6–20. doi: 10.1089/jmf.2013.3083. [DOI] [PubMed] [Google Scholar]
- 32.Bachmann H, Pronk JT, Kleerebezem M, Teusink B. 2015. Evolutionary engineering to enhance starter culture performance in food fermentations. Curr Opin Biotechnol 32:1–7. doi: 10.1016/j.copbio.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Bellon JR, Yang F, Day MP, Inglis DL, Chambers PJ. 2015. Designing and creating Saccharomyces interspecific hybrids for improved, industry relevant, phenotypes. Appl Microbiol Biotechnol 99:8597–8609. doi: 10.1007/s00253-015-6737-4. [DOI] [PubMed] [Google Scholar]
- 34.Steensels J, Snoek T, Meersman E, Picca Nicolino M, Voordeckers K, Verstrepen KJ. 2014. Improving industrial yeast strains: exploiting natural and artificial diversity. FEMS Microbiol Rev 38:947–995. doi: 10.1111/1574-6976.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meersman E, Steensels J, Paulus T, Struyf N, Saels V, Mathawan M, Koffi J, Vrancken G, Verstrepen KJ. 2015. Breeding strategy to generate robust yeast starter cultures for cocoa pulp fermentations. Appl Environ Microbiol 81:6166–6176. doi: 10.1128/AEM.00133-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flores M, Corral S, Cano-García L, Salvador A, Belloch C. 2015. Yeast strains as potential aroma enhancers in dry fermented sausages. Int J Food Microbiol 212:16–24. doi: 10.1016/j.ijfoodmicro.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 37.Franzosa EA, Hsu T, Sirota-Madi A, Shafquat A, Abu-Ali G, Morgan XC, Huttenhower C. 2015. Sequencing and beyond: integrating molecular ’omics’ for microbial community profiling. Nat Rev Microbiol 13:360–372. doi: 10.1038/nrmicro3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z-M, Lu Z-M, Shi J-S, Xu Z-H. 2016. Exploring flavour-producing core microbiota in multispecies solid-state fermentation of traditional Chinese vinegar. Sci Rep 6:26818. doi: 10.1038/srep26818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bourrie BC, Willing BP, Cotter PD. 2016. The microbiota and health promoting characteristics of the fermented beverage kefir. Front Microbiol 7:647. doi: 10.3389/fmicb.2016.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong SH, Jung JY, Lee SH, Jin HM, Jeon CO. 2013. Microbial succession and metabolite changes during fermentation of dongchimi, traditional Korean watery kimchi. Int J Food Microbiol 164:46–53. doi: 10.1016/j.ijfoodmicro.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 41.Jung JY, Lee SH, Lee HJ, Jeon CO. 2013. Microbial succession and metabolite changes during fermentation of saeu-jeot: traditional Korean salted seafood. Food Microbiol 34:360–368. doi: 10.1016/j.fm.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 42.Bermudez-Brito M, Plaza-Díaz J, Muñoz-Quezada S, Gómez-Llorente C, Gil A. 2012. Probiotic mechanisms of action. Ann Nutr Metab 61:160–174. doi: 10.1159/000342079. [DOI] [PubMed] [Google Scholar]
- 43.Ventura M, O’Flaherty S, Claesson MJ, Turroni F, Klaenhammer TR, van Sinderen D, O’Toole PW. 2009. Genome-scale analyses of health-promoting bacteria: probiogenomics. Nat Rev Microbiol 7:61–71. doi: 10.1038/nrmicro2047. [DOI] [PubMed] [Google Scholar]
- 44.Chen YP, Lee TY, Hong WS, Hsieh HH, Chen MJ. 2013. Effects of Lactobacillus kefiranofaciens M1 isolated from kefir grains on enterohemorrhagic Escherichia coli infection using mouse and intestinal cell models. J Dairy Sci 96:7467–7477. doi: 10.3168/jds.2013-7015. [DOI] [PubMed] [Google Scholar]
- 45.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fouhy F, Guinane CM, Hussey S, Wall R, Ryan CA, Dempsey EM, Murphy B, Ross RP, Fitzgerald GF, Stanton C, Cotter PD. 2012. High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob Agents Chemother 56:5811–5820. doi: 10.1128/AAC.00789-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clooney AG, Fouhy F, Sleator RD, O’ Driscoll A, Stanton C, Cotter PD, Claesson MJ. 2016. Comparing apples and oranges?: next generation sequencing and its impact on Microbiome analysis. PLoS One 11:e0148028. doi: 10.1371/journal.pone.0148028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orgiazzi A, Lumini E, Nilsson RH, Girlanda M, Vizzini A, Bonfante P, Bianciotto V. 2012. Unravelling soil fungal communities from different Mediterranean land-use backgrounds. PLoS One 7:e34847. doi: 10.1371/journal.pone.0034847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fouhy F, Deane J, Rea MC, O’Sullivan Ó, Ross RP, O’Callaghan G, Plant BJ, Stanton C. 2015. The effects of freezing on faecal microbiota as determined using MiSeq sequencing and culture-based investigations. PLoS One 10:e0119355. doi: 10.1371/journal.pone.0119355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 52.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 53.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santamaria M, Fosso B, Consiglio A, De Caro G, Grillo G, Licciulli F, Liuni S, Marzano M, Alonso-Alemany D, Valiente G. 2012. Reference databases for taxonomic assignment in metagenomics. Brief Bioinform 13:682–695. [DOI] [PubMed] [Google Scholar]
- 55.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methé B, Schloss PD, Gevers D, Mitreva M, Huttenhower C. 2012. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8:e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. 2007. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 58.R Core Team 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 59.Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. 2015. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3:e1029. doi: 10.7717/peerj.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The bacterial (A) and fungal (B) compositions of kefir grains, as determined by amplicon sequencing. Note that we were unable to generate an ITS amplicon for the UK3 sample. Download Figure S1, PDF file, 0.09 MB (95.1KB, pdf) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Cladogram presenting the bacterial diversity of kefir samples at 0, 8, and 24 h of fermentation, as determined by 16S rRNA gene sequencing. The color intensities in the outer rings indicate the average relative abundances of the bacterial genera in Fr1, Ick, and UK3 at each stage. Download Figure S2, DOCX file, 0.6 MB (652.7KB, docx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Absolute abundances of bacteria and fungi in kefir samples after 0, 8, and 24 h of fermentation, as determined by qPCR measurements. Download Table S1, PDF file, 0.06 MB (62KB, pdf) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Stacked bar charts presenting the fungal compositions of kefir samples after 0, 8, and 24 h of fermentation, as determined by ITS gene sequencing (A), and the microbial composition of kefir samples after 0, 8, and 24 h of fermentation (B), as determined by MetaPhlan2. Download Figure S3, TIF file, 0.4 MB (405.4KB, tif) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Hierarchically clustered heat maps showing the relatedness of L. kefiranofaciens (A), L. mesenteroides (B), and L. helveticus (C) from Fr1, Ick, and UK3 to reference strain genomes, as determined by PanPhlAn. Clustering was done with hclust2 (https://bitbucket.org/nsegata/hclust2). Download Figure S4, TIF file, 0.9 MB (942.1KB, tif) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
APLSR plot (principal components 1 and 2) of sensory acceptance and RDA data for spiked and unspiked kefir samples. Download Figure S5, PDF file, 0.08 MB (87.8KB, pdf) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Microbial species from cheese samples that contain two or more genes associated with probiotic action, as determined by HUMAnN2. Download Table S2, XLSX file, 0.01 MB (13KB, xlsx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Correlations between the relative abundances of microbial genera and the levels of volatile compounds. Download Table S3, DOCX file, 0.01 MB (14.8KB, docx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Changes in the volatile-compound profiles of kefirs supplemented with L. kefiranofaciens 484 NCFB 2797 (A) and L. mesenteroides 213M0 (B). Download Table S4, XLSX file, 0.01 MB (12.5KB, xlsx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Supplemental materials and methods and results. Download Text S1, DOCX file, 0.02 MB (22.3KB, docx) .
Copyright © 2016 Walsh et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.