

Abstract
Objective(s):
Yu-Ping-Feng-San (YPFS) is a classical traditional Chinese medicine that is widely used for treatment of the diseases in respiratory systems, including chronic obstructive pulmonary disease (COPD) recognized as chronic inflammatory disease. However, the molecular mechanism remains unclear. Here we detected the factors involved in transforming growth factor beta 1 (TGF-β1)/Smad2 signaling pathway and inflammatory cytokines, to clarify whether YPFS could attenuate inflammatory response dependent on TGF-β1/Smad2 signaling in COPD rats or cigarette smoke extract (CSE)-treated human bronchial epithelial (Beas-2B) cells.
Materials and Methods:
The COPD rat model was established by exposure to cigarette smoke and intratracheal instillation of lipopolysaccharide, YPFS was administered to the animals. The efficacy of YPFS was evaluated by comparing the severity of pulmonary pathological damage, pro-inflammation cytokines, collagen related genes and the activation of TGF-β1/Smad2 signaling pathway. Furthermore, CSE-treated cells were employed to confirm whether the effect of YPFS was dependent on the TGF-β1/Smad2 signaling via knockdown Smad2 (Si-RNA), or pretreatment with the inhibitor of TGF-β1.
Results:
Administration of YPFS effectively alleviated injury of lung, suppressed releasing of pro-inflammatory cytokines and collagen deposition in COPD animals (P<0.05), whereas exogenous TGF-β1 promoted releasing of IL-1β, IL-6, TNFα (P<0.05). Administration YPFS reduced inflammatory response significantly, also down-regulated TGF-β1/Smad2 signaling in vivo and in vitro. Unexpectedly, knockdown Smad2 or inhibition of TGF-β1 abolished anti-inflammatory effect of YPFS in CSE-treated cells.
Conclusion:
YPFS accomplished anti-inflammatory effects mainly by suppressing phosphorylation of Smad2, TGF-β1/Smad2 signaling pathway was required for YPFS-mediated anti-inflammation in COPD rats or CSE-treated Beas-2B cells.
Keywords: COPD, Pro-inflammatory, Cytokine, TGF-β1/Smad2, YPFS
Introduction
Chronic obstructive pulmonary disease (COPD) is defined as a chronic inflammatory disease (1), which is one of the leading global cause of morbidity and mortality, resulting approximately 5% of total deaths worldwide (2, 3). COPD is characterized by progressive airflow limitation irreversibly, causing by various noxious particles or gases (4). The major risk factor is cigarette smoke exposure, about 90% of deaths of COPD being attributable to smoking (5). Cigarette smoke is seen as a pro-inflammatiory stimulus leading to airway inflammation, (6). Excessive inflammation was deemed to be a typically quality for COPD (7), which is associated with an abnormal inflammatory response of the lung to mainly cigarette smoke (8). A previous study has demonstrated damage of lung pathology that in COPD animals inducing by cigarette smoke exposure, including tracheal responsiveness, oxidative stress and intemperate lung inflammation (9, 10).
Transforming growth factor-β (TGF-β) plays a key role in tissue repair and remodeling (11), which has been widely implicated in pathogenesis of COPD (12). TGF-β can promote collagen expression and fibrosis for airway remodeling (13). Genetic studies have confirmed an association of gene polymorphisms of the TGF-β with COPD (14). Ichimaru reported in 2012 that TGF-β induces perlecan deposition to aggravates the situation of airway amooth muscle cells (ASMC) from COPD (15). Activation of the TGF-β down-stream signaling pathway may cause airway remodeling and airway inflammation that is considered as the characteristic of COPD (16, 17). Previously, TGF-β has been recognized to play an essential role in the repression of inflammation; however, recent studies have confirmed the positive roles of
TGF-β in inflammatory responses (18). TGF-β plays a central role in driving inflammation by in-ducing inflammatory mediator release to accelerate the inflammatory progress, including IL-6, IL-17 and GM-CSF (15, 17, 19, 20). In addition, TGF-β overexpression may develop significant inflammation in transgenic mice (21). Thus, intervention of TGF-β signaling alleviated inflammation, which is a potentially thera-peutic strategy for COPD.
Yu-Ping-Feng-San (YPFS) is a classical Chinese medicinal formula in China. According to the Chinese Pharmacopoeia, the formula includes the following 3 herbs: Radix Astragall, the dried roots of Astragalus membranaceus, Rhizoma Atractylodis macrocephalae, the dried roots of Atractylodesmacrocephala Koidz, Radix saposhnikoviae, and the dried roots of Saposhnikovia divaricata, in the ratio of 1:2:1 on a dry weight basis, respectively, is orally administered as a decoction or in the form of granule formulations. Clinically, YPFS has been widely used in medication for treatment of respiratory systems and immune systems diseases, including anaphylactic rhinitis, asthma and COPD (22-24). However, the action mechanism of YPFS remains unclear. In this report, we demonstrated the role of YPFS on effectively alleviating injury of inflammation and deposition of collagen in COPD rats, suppression releasing of pro-inflammatory cytokines in vivo and in vitro. Furthermore, we discovered that the TGF-β1/Smad2 signaling was required for YPFS-mediated anti-inflammation in cigarette smoke extract (CSE)-treated Beas-2B cells.
Materials and Methods
Animals and treatment
Male Sprague Dawley rats were obtained from the Animal Center, Kunming Medical University, China, and used between the ages of 3-4 months. Animals were feed under a constant 12 hr light-dark cycle and were allowed to eat and drink ad libitum. All procedures were performed in accordance with the protocol outlined in the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health and approved by the Animal Care and Use Committee of Yunnan University of Traditional Chinese Medicine.
TheCOPDratmodel was established by exposure to cigarette smoke daily and intratracheal instillation of lipopolysaccharide (LPS) (25, 26). To be brief, rats were placed in the perspex chamber 30 cm (length) × 20 cm (width) × 25 cm (height), which separateed into small cigarette burn box and animals inhalation chamber. The commercial cigarettes (HongHe filter cigarette; Hong Yun Hong He Group) containing 12 mg tar and 1.2 mg nicotine per cigarette were used in this study. A lighted cigarette was placed in cigarette burn box near the small holes on the wall of the inhalation chamber, the cigarette smoke delivered into the animal inhalation chamber by a circulation fan to control gas flow at a rate of 5 min per cigarette approximately. Continuous fresh cigarette smoke was administered for 1 h/day for 6 weeks. In addition, 200 µg LPS (dissolved in saline) was administered to the airways of the rats via endotracheal intubation on days 1 and 14, after the animals were anesthetized with 10% chloral hydrate. Rats were randomly divided into 4 groups, with 8 rats in each group. Rats in normal control were bred normally for 6 weeks and administered with same dose of physiological saline. The model group (COPD group) was established with the description above. The drug intervention group was corresponded to model group, 0.5g/kg/day YPFS was administered via intragastric administration every day from day 15 until 6 weeks, the calculation of the dose equivalent was based on the application of the conversion factor recommended by the Food and Drug Administration when the animal under study is the rat (available at: http://www.fda.gov/cder/cancer/animalframe.htm). Roxithromycin 20 mg/kg was administered as the positive control.
Extract preparation of YPFS
YPFS is a traditional Chinese herbal medicine formula, which is composed of Radix Astragall, Rhizoma Atractylodes macrocephala, Radix saposhnikoviae, in the ratio of 1:2:1 on a dry weight basis, respective. All medicinal herbs were purchased from Yi Xin Tang Drugstore (Kunming, China) and identified by department of Chinese materia medica of Yunnan University of Traditional Chinese Medicine. The major identified effective phytochemical compound of YPFS is illustrated in previously published paper (27).
Extract preparation of YPFS was described previously, all medicinal herbs were decocted within a water bath for twice, the water extract was filtered and evaporated below 55°C under reduced pressure, and then the residue was freeze-dried. The residue was dissolved at the desired concentration with distilled water, sterilized by 0.22 μm filter beforeintragastric administration or adding into the medium of cells (28, 29).
Cell culture and CSE preparation
The human lung bronchial epithelial line Beas-2B cells (a kind gift of Prof. CG Zou, Yunnan University, China) were grown in RPMI-1640 medium (Invitrogen) containing L-glutamine, glucose, NaHCO3, 10% FBS and 1% penicillin-streptomycin (Invitrogen) and maintained at 37°C, 5% CO2. For experiments examining the function of exogenous TGF-β1, human recombinant TGF-β1 (R&D Systems, USA) was added at 5 ng/ml or 10 ng/ml into medium, after incubation for 24 hr, the cells were harvest for further study.
Cigarettesmokeextract (CSE) was freshly prepared, in a manner similar to that described by Tony and Kothari (30, 31). A lighted cigarette was connected to a peristaltic pump apparatus, and the smoke from one cigarette was slowly bubbled into 25 ml of sterile RPMI-1640 with 10% serum at a rate of one cigarette per min, each puff of 2 sec duration approximately, following the pH of RPMI 1640 dissolved-CSE was adjusted to 7.0 and sterile-filtered through a 0.22 μm filter. Beas-2B cells were exposed into RPMI-1640 medium with CSE for 24 hr, exogenous YPFS wasadministrated respectively. In some experiments, CSE-treated cells were preincubated for 12 hr with 30 μM TGF-β inhibitors LY2109761 (Selleck Chemicals), before administration YPFS.
RNA interference
To knockdown expression of Smad2 by RNA interference, Beas-2B cells were transfected at 60-70% confluence with 50 nM of Smad2-specific small interfering RNAs (siRNAs; GenePharma Co, Shanghai, China) in Opti-MEM medium (Invitrogen) using Lipofectamine 2000 transfection agent (Invitrogen) according to the manufacturer’s specifications. In control group, the negative control was replaced. Gene silencing efficiency was determined by Quantitative real-time PCR 72 hr post-transfection. The following siRNAs were used: Smad2, 5’-GUC CCA UGA AAA GAC UUA A-3’ (F), UUA AGU UUU CAU GGG A-3’ (R), with a control siRNA (4390846 Invitrogen)
Western blotting
The samples of lung tissues of rats were homogenized in liquid nitrogen and the homogenate was lysed into lysis buffer on ice for 30 min (Beyotime, Jiangsu, China). For detection of protein expression in Beas-2B cells, the cells were was lysed into lysis buffer on ice for 30 min. Total protein lysates were estimated using the BCA protein assay (Beyotime, Jiangsu, China). The lysates (40 μg) of total protein per well were separated using 10% SDS polyacrylamide gel, and then transferred onto immobilon-PSQ transfer PVDF membrane (Millipore, Bedford, MA). Membranes were detected of phosphorylated forms and expression of protein. Primary antibodies were anti-phospho-Smad2, anti-TGF-β1, anti-α-SMA (1: 1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), anti-Collagen I (1:3000 dilution; Abcam) and anti-Actin antibodies (1:3000 dilution; Sigma). The secondary antibody was a peroxidase-coupled anti-mouse or rabbit IgG (1:4000 dilution; Abmart). The membrane was exposed to Kodak X-OMAT film (Kodak, China), and the film was developed. Signals were quantified using Image J and normalized to controls.
Quantitative real-time PCR
Total RNA was extracted from lung tissues of rats or cells with Trizol reagent (Invitrogen, Carlsbad, CA). cDNAs were synthesized using a reverse transcription kit (Invitrogen). A real time-PCR analysis was performed on the ABI 7500 Real-Time PCR System (Applied Biosystems, Darmstadt, Germany) using SYBR®Premix-Ex Tag TM (Takara, Dalian, China). The primers used for PCR were as follows in rats: IL-1β 5’- CTG TGA CTC GTG GGA TGA TG-3’ (F), 5’-GGG ATT TTG TCG TTG CTT GT-3 ‘(R); TNFα: 5’- AGA TGT GGA ACT GGC AGA GG-3’ (F), 5’-CCC ATT TGG GAA CTT CTC CT-3’ (R); IL-6, 5’- CCG GAG AGG AGA CTT CAC AG-3’ (F), 5’-ACA GTG CAT CAT CGC TGT TC-3’ (R); TGF-β1, 5’-AGA AGT CAC CCG CGT GCT AA-3’ (F), 5’-TCC CGA ATG TCT GAC GTA TTG A- 3’ (R); β-actin: 5’-TCA TGA AGT GTG ACG TTG ACA TCC GT- 3’ (F), 5’-CCT AGA AGC ATT TGC GGT GCA CGA TG-3’ (R). The primers used for PCR were as follows in Beas-2B cells: IL-1β: 5’-GGA ACC CCA GAG CGA AAT ACA-3’ (F), 5’-CCT GAA GAA TGC CTC CTC ACA-3’ (R); IL-6: 5’-AAA TTC GGT ACA TCC TCG AC-3’ (F), 5’- CCT CTT TGC TGC TTT CAC AC-3’ (R); TNFα: 5’-GAG CAC TGA AAG CAT GAT CC-3’ (F), 5’-CGA GAA GAT GAT CTG ACT GCC-3’ (R); TGF-β1: 5’-ATT GAG GGC TTT CGC CTT AG-3’ (F), 5’-CCG GTA GTG AAC CCG TTG A-3’ (R);β-actin: 5’-ATG TTT GAG ACC TTC AAC AC-3’ (F), 5’-CAC GTC ACA CTT CAT GAT GG-3’ (R).
MTT assay
The MTT assay was performed in a 96-well plate, 1 × 104 cells/100 μl/well were cultured with YPFS at the concentrations of 0.01 mg-0.5 mg/ml. After the 12 hr and 24 hr incubation period, 20 μl of MTT reagent was added to each well, and incubated for 1 hr in a 5% CO2 incubator at 37°C. Afterwards the supernatant was discarded and 150 μl of DMSO per well was added to solubilize formazan crystals for 10 min on a shaker. The absorbance was measured using a microplate reader at a wavelength of 490 nm. (Thermo Scientific, Finland)
Pathohistology analysis
All the groups of animals (including model group, YPFS administration group and the control) were killed, the lung tissue of right lower lobes were collected. The tissues were dipped in 4% formalin, dehydrated in graded ethylic alcohol, embedded in paraffin, stained with hematoxilin/eosin (HE) or Masson’s trichrome staining. Inflammation was determined using a semiquantitative pathohisto-logical score as described previously. The level of airway inflammation was classified into the categories: almost not visible (0-5); slight (6-20); moderate (21-40); severe/profound inflammation (41-60) (32).
Statistical analysis
Data from all the experiments are expressed as mean ± SEM. A significant difference was determined by a one-way ANOVA test followed by a Student-Newman-Keuls test. P-values < 0.05 were considered significant.
Results
YPFS represses inflammatory response in the model of COPD rat
To investigate the role of YPFS in the inflamma-tory response, the COPD animals were administrated with 0.5 g/kg/day YPFS, through intragastric admi-nistration from 15 day until 6 weeks. Then lung tissues were examined using hematoxylin and eosin (HE) staining. At the endpoint of 6 weeks, airway inflammatory infiltrates were measured by a semiquantitative histology score. Administration with YPFS inhibited lymphocyte influx and led to an approximately 2-fold decrease in histology score comparing to the COPD group (P<0.05) (Figure 1A and 1B). As inflammation is a characteristic for COPD, IL-1β, IL-6 and TNFα are the key pro-inflammatory cytokines playing critical roles in the process of inflammation, so we assessed the expression of aforesaid pro-inflammatory cytokines in alveolar tissues. The mRNA expressions of IL-1β, IL-6 and TNFα were significantly elevated in COPD group (P<0.05) (Figure. 1C-1E), however, all of them were crippled after administration YPFS (P<0.05) (Figure. 1C-1E). These results revealed that administration YPFS alleviated injury of airway inflammation and repressed pro-inflammatory cytokines in alveolar tissues of COPD animals.
Figure 1.
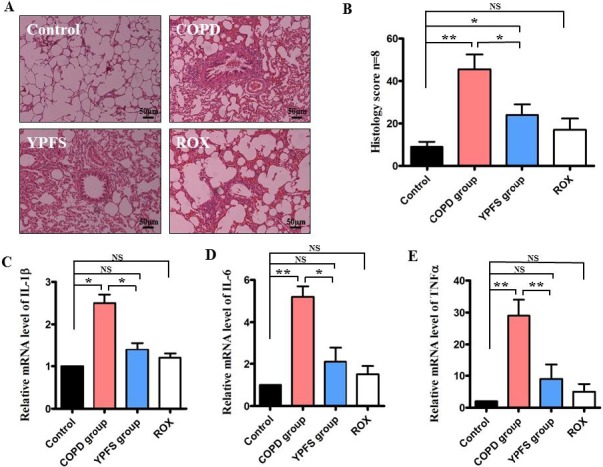
The effects of YPFS on suppression airway inflammation in COPD rats. After YPFS was administrated via intragastric administration at 0.5 g/kg/day from 15 day until 6 weeks in COPD rats, roxithromycin (ROX) 20 mg/kg was administrated as positivecontrol. (A) Alveolar tissues were collected and stained by hematoxylin and eosin, magnification x100. (B) The airway inflammation classified by the histology score was measured. (C), (D) and (E) Quantitative PCR analysis was used to study the mRNA levels of IL-1β, IL-6 and TNFα in the lung tissue. Data are expressed as mean ± SEM of three independent experiments.*P< 0.05 **P< 0.01 NS, not significantly versus control
Effect of YPFS on inhibition of inflammatory cytokines releases in CSE-treated human lung bronchial epithelial line Beas-2B cells
Smoking is contribute to serious inflammation of bronchial epithelial cells, which is a risk factor of development of COPD (33). To investigate the effect of YPFS in vitro, human lung bronchial epithelial line Beas-2B cells were employed by cigarette smoke extract (CSE) then to test the activity of YPFS. Firstly, we determined the cytotoxicity of YPFS from 0.01 mg/ml to 0.5 mg/ml in Beas-2B cells using the MTT assay, the result showed that 0.5 g/ml of YPFS significantly inhibited cell proliferation (P<0.05) (Figure 2A). Next, we tested the mRNA levels of IL-1β, IL-6 and TNFα. The results indicated that administration YPFS reduced significantly levels of inflammatory cytokines in CSE-treated cells (P<0.05) (Figure 2B), also attenuated CSE-triggered Beas-2B cells death. The proportion of living cells is more than 93% in CSE-triggered Beas-2B cells, compared to the model group without administration YPFS (P<0.05) (Figure 2C). These results suggest that the YPFS repress pro-inflammatory cytokine release to against CSE-triggered apoptosis.
Figure 2.
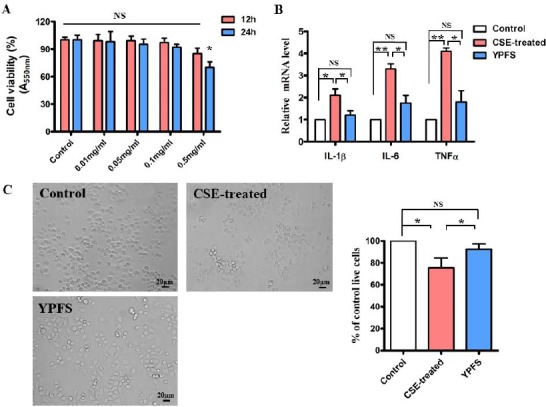
YPFS inhibited inflammatory cytokine releasing in CSE-treated Beas-2B cells. (A) The effect of YPFS on cell viability at the concentration of 0.01 mg/ml-0.5 mg/ml in human bronchial epithelial cells Beas-2B for 12 hr and 24 hr. Cell viability was performed with MTT assay. *P<0.05 NS, not significantly, versus control (normal saline). (B) After Beas-2B cells were exposed into RPMI-1640 medium with CSE for 24 hr, then 0.1 mg/ml YPFS was administrated for 24 hr, Quantitative PCR analysis was used to study the mRNA levels of IL-1β, IL-6 and TNFα. Data are expressed as mean±SEM of three independent experiments. *P<0.05 **P<0.01 NS, not significantly versus control. (C) Phase-contrast images of the CSE-treated Beas-2B cells were administrated with 0.1 mg/ml YPFS or normal saline for 24 hr, magnification x100. The right panel shows cell viability was determined using MTT assay. Data are expressed as mean±SEM of three independent experiments. *P< 0.05 NS, not significantly versus control
YPFS decreases TGF-β1/Smad-2 signaling in the lung tissue of the COPD rats
TGF-β1 is known as a risk element of idiopathic pulmonary fibrosis and airway remodeling (34). There is a close link between smoking and expression of TGF-β1 in small airway epithelium of tobacco smokers (35). The results above prompted us to determine activity of TGF-β1 signal in COPD rats, so TGF-β1 was measured. Both the mRNA level and the protein level of TGF-β1 were significantly increased in COPD animals as compared with the normal rat, whereas the expression of TGF-β1 were reversed dramatically in COPD animals after administration YPFS (P<0.05) (Figure 3A and 3B). To further determine whether YPFS decreased TGF-β1/smad-2 signaling, the phosphorylation levels of Smad-2 were detected. The result showed that the phosphorylation levels of Smad-2 were elevated in COPD rats and CSE-treated cells (P<0.05) (Figure 3C). However, phosphorylation levels of Smad-2 were overturned dramatically in YPFS group (P<0.05) (Figure 3C). Clinical trials showed COPD patients often couple with collagen depositionin the lung (36), hence we further analyzed deposition of collagen in alveolar tissues and terminal bronchiole. Masson’s trichrome staining revealed more pronounced fibrosis in COPD animals as compared with YPFS group (Figure 3D). Moreover, the expression of collagen I and α-SMA in the lung tissue of rats was detected by Western blotting. As expected, we observed a dramatic decrease in the protein levels of collagen I and α-SMA in the lung tissue of COPD animals, after administration with YPFS (P<0.05) (Figure 3E). Taken together with the data, it was evident that TGF-β1/smad2 signaling was characteristically elevated in COPD animals, and YPFS played a key role in inhibition TGF-β1/smad2 signaling and collagen deposition.
Figure 3.
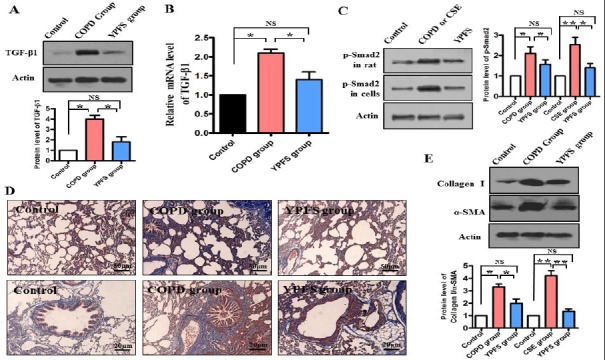
YPFS decreased TGF-β1/Smad-2 signaling. After YPFS 0.5 g/kg/day or normal saline was administrate from 15 day until 6 weeks in COPD rats. (A) The protein levels of TGF-β1 were determined by Western blotting. The blot is typical of three experiments. The lower panel shows quantification of the ratio of TGF-β1 to actin. Data are expressed as mean±SEM of three independent experiments. *P< 0.05 NS, not significantly versus control. (B) Quantitative PCR analysis was used to study the mRNA levels of TGF-β1. *P<0.05 NS, not significantly versus control. (C) After administration YPFS or normal saline in COPD rats (The upper band) or 0.1mg/ml YPFS wasadministrated for 24 hr in CSE-treated Beas-2B cells (The middle band), the protein levels of phosphorylation Smad2 were determined by Western blotting. The blot is typical of three experiments. The right panel shows quantification of the ratio of phosphorylation of Smad2 to actin. Data are expressed as mean ± SEM of three independent experiments. *P<0.05 **P<0.01 NS, not significantly versus control. (D) Effects of YPFS on pulmonary structural remodeling of COPD rats assessed by Masson Trichrome staining for collagen in blue, magnification x100 and x400 (lower panel). (E) The protein levels of collagen I and α-SMA were determined by Western blotting. The blot is typical of three experiments. The low panel shows quantification of the ratio of collagen I or α-SMA to actin. Data are expressed as mean ± SEM of three independent experiments. *P<0.05 **P<0.01 NS, not significantly versus control
TGF-β1 induces phosphorylation of Smad2 and promotes the expression of pro-inflammatory cytokines in Beas-2B cells
TGF-β1 overexpression may develop significant inflammation in transgenic mice (21), to investigate whether TGF-β1 is a major component of inducing inflammation in vitro, we further analyzed levels of pro-inflammatory cytokines in vitro. After supplementation ofexogenous TGF-β1 (5 or 10 ng/ml) in Beas-2B cells, the amount of phosphorylated Smad2 was induced in Beas-2B cells in 24 hr (P<0.05) (Figure 4A). We also found that the mRNA levels of IL-6, IL-1β and TNFα were significantly increased in different extent for 24 hr (P<0.05) (Figure 4B).
Figure 4.
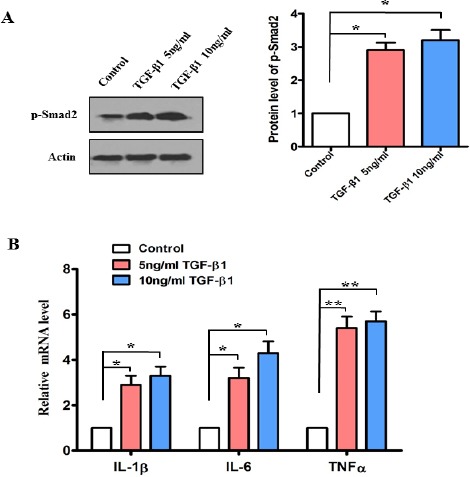
Exogenous TGF-β1 promoted the expression of pro-inflammatory cytokines in Beas-2B cells. After exogenous TGF-β1 was added into Beas-2B cells at the concentration of 5 ng/ml or 10 ng/ml for 24 hr. (A) The protein level of phosphorylation Smad2 was determined by Western blotting. The blot is typical of three experiments. The right panel shows quantification of the ratio of phosphorylation Smad2 to actin. Data are expressed as mean ± SEM of three independent experiments. *P<0.05. (B) Quantitative PCR analysis was used to study the mRNA levels of IL-1β, IL-6 and TNFα Data are expressed as mean±SEM of three independent experiments.*P<0.05 **P<0.01
Inhibition TGF-β1/Smad2 signaling abolishes anti-
inflammatory effect of YPFS in CSE-treated cells
As show in Figure 1-4, the results have demonstrated that YPFS repressed inflammatory response and attenuated releasing of TGF-β1 in vivo and in vitro. In order to further verify whether Smad2 functions is upstream of anti-inflammatory response, endogenous Smad2 expression was significantly ablated by siRNA in Beas-2B cells (P<0.05) (Figure 5A). However, knockdown of Smad-2 by siRNA, the mRNA levels of these pro-inflammatory cytokines IL-1β, IL-6 and TNF-α had no significant change even so administration with YPFS in CSE-treated Beas-2B cells (Figure 5B-5D). We also tested a specific TGF-β inhibitors LY2109761 (30 μM), and found the inhibitors failed to suppress expression of IL-1β, IL-6 and TNFα after administration with YPFS in CSE-treated Beas-2B cells (Figure 5E). These results suggest that the anti-inflammation activation of YPFS is dependent on TGF-β1/Smad2 signaling, devitali-zation of Smad2 impaired the anti-inflammatory effect of YPFS.
Figure 5.
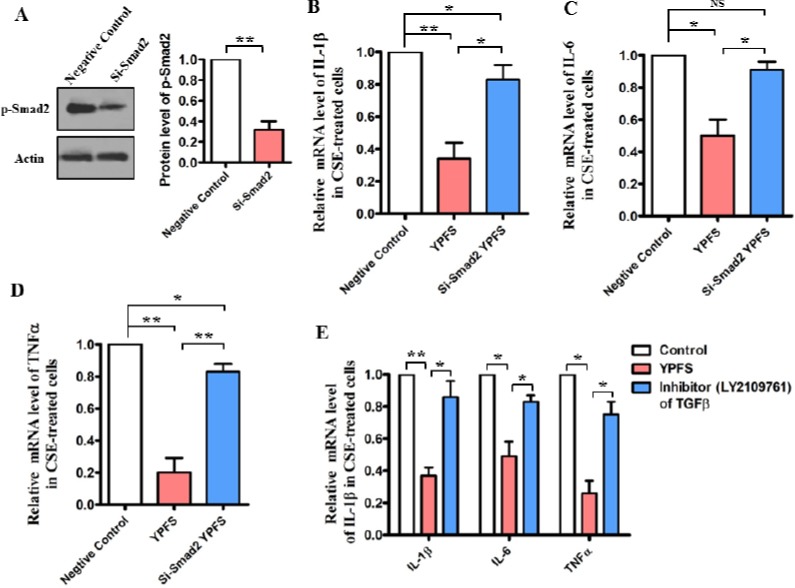
Smad-2 is required for anti-inflammatory effects of YPFS in CSE-treated cells. (A) After Smad2 was knockdown by siRNA in Beas-2B cells, the protein levels of phosphorylation Smad2 were determined by Western blotting. The blot is typical of three experiments. The right panel shows quantification of the ratio of phosphorylation Smad2 to actin. Data are expressed as mean±SEM of three independent experiments. *P<0.05 versus negative control (NC). (B), (C) and (D) Knockdown of endogenous Smad2 expression by siRNA in CSE-treated Beas-2B cells, 0.1mg/ml YPFS wasadministrated for 24 h, Quantitative PCR analysis was used to study the mRNA levels of IL-1β,IL-6 and TNFα. *P<0.05 **P<0.01 NS, not significantly versus control. (E) After pre-incubated with 30 μM TGF-β inhibitors (LY2109761) for 12 hr in CSE-treated Beas-2B cells, 0.1 mg/ml YPFS wasadministrated for 24 hr, Quantitative PCR analysis was used to study the mRNA levels of IL-1β,IL-6 and TNFα. *P<0.05 **P<0.01
Discussion
YPFS is a widely used immunomodulatory herbal medication used in traditional Chinese medicine to cure the diseases of respiratory systems and immune systems (22). YPFS markedly decreases IL-4, IL-17 levels, also inhibits Th2 cell that mediate allergic contact dermatitis and ovalbumin (OVA)-induced allergic asthma(37, 38). Recently, Du et al report in 2015 that YPFS induces gene expression of anti-viral proteins by interferon signaling and inhibiting neuraminidase activity (39). Natural products were seen as the sources of new drug to treat COPD, the group of Mohammad et al reported a series of effects of Nigella sativa, Zataria multiflora and the constituent on immune response and animal model of COPD (40-42). Here, we provide the first evidence that YPFS decreased inflammatory injury of pulmonary and relieved collagen deposition causing by cigarette smoke-induced COPD rats, also suppressed the releasing of pro-inflammatory cytokines, including IL-1β, IL-6 and TNFα in CSE-treated human bronchial epithelial Beas-2B cells. Moreover, our results demonstrated that in both the COPD rats and the CSE-treated Beas-2B cells, YPFS showed good effects of anti-inflammation via the TGF-β1/Smad2 signaling pathway. The molecular mechanism by which the TGF-β1/Smad2 signaling pathway promotes anti-inflammation involved the dephosphorylation of Smad2. However, knockdown of Smad2 by RNAi seriously obstructs anti-inflammation of YPFS CSE-treated cell. Our data clearly demonstrated that the anti-inflammatory effect of YPFS is associated with the suppression of the TGF-β1/Smad2 signaling pathway. Forallthis, the mechanism by how YPFS suppress phosphory-lation of smad-2 remains unclear, and need to be investigated further.
Inflammation is seen as a double-edged sword, the most important role is a fundamental protective response to infection, irritation, or other injury (43). However, excessive inflammatory response has been suggested as a risk factor for the development ofmany diseases, including atherosclerosis, rheumatoid arthritis, inflammatory bowel disease, asthma and COPD (3, 44). Inflammation intensity is a key inducement in COPD, which is an enhanced or abnormal inflammatory immune response (45). Frequently exposure to noxious particles and smoking, can active a series of inflammatory res-ponse in the small airways and lung parenchyma (46). Progress of inflammation involves several different cell types such as macrophages, lympho-cytes, neutrophils and inflammatory mediators including proteinases cytokines, growth factors, and chemokines (3). Clinical studies have demonstrated that patients with COPD release greater amounts of IL-6, IL-8, IL-32, TNFα, GM-CSF and CXCL8 compared to those healthy subjects (47-49). So suppression of the inflammatory response is a logical approach to the treatment of COPD (50). For instance, to decrease production of inflammatory cytokines in inflamma-tory bowel disease patients is known as a strategy for attenuation clinical activity in Crohn’s disease patients (51, 52). Furthermore, adipose-derived stromal cell therapy decrease tracheal hyperresponsiveness and lung inflammation to relieve symptom in COPD animals (53). In this paper, YPFS inhibited pro-inflammatory cytokines (including, IL-1β, IL-6 and TNFα) and attenuated the destruction of inducing by smoking in COPD rats or CSE-treated cells.
TGF-β/Smad2 signaling link inflammation to fibrogenesis (54). The relationship between TGF-β/Smad-2 signaling and airway inflammation has been extensively studied in pulmonary fibrosis model animals (55). The expression of TGF-β was detected in multiple cells, such as bronchial epithelial cells, infiltrating eosinophils, mast cells, alveolar macrophages and myofibroblasts (55). Activation of TGF-β induces the phosphorylation of Smad2 and Smad3, forms a complex with the Smad4 to translocate into nucleus to bind and regulate down-stream targets, while the whole process is essential role in the pathogenesis of fibrosis (56). Smad7 is an inhibitor of Smad2 and Smad3, and it had confirmed the role of Smad7 and NF-κB crosstalk pathway in renal inflammation; In contrast, overexpression of Smad7 can inhibit the activation of NF-κB (57). Indeed transgenic mice of Smad7 develop more severe inflammation (58). Except Smad7, in the current study, Smad2 also is the key mediator fibrosis and inflammation, for instance, activation of TGF-β/Smad2 signaling is induced deposition of extracellular matrix (ECM) components and airway remodeling in asthma, whereas overexpression of Smad-2 can specifically alter airway hyperreactivity and remodeling (54, 59). In the murine model of airway inflammation inducing by ovalbumin, phosphorylated Smad-2 is dramatically increased in multiple cells (eg. infiltrating inflammatory cells and bronchial epithelial cells) (55). Our results demonstrated that YPFS reduced the levels of phosphorylated Smad-2 in vivo and in vitro, while dephosphorylation of Smad2 was inhibited collagen deposition in the lung tissue of the COPD rats. Nevertheless, knockdown of Smad-2 by RNAi seriously obstructed anti-inflammation of YPFS CSE-treated cell, thus Smad-2 actually played a mediatory role in anti-inflammation of YPFS. Thus, our study indicated potential mechanism of YPFS on therapy COPD.
Conclusion
In the present study we provided evidence of the protective role of YPFS in suppression inflammation of COPD animals and CSE-treated human bronchial epithelial line Beas-2B cells. YPFS accomplished anti-inflammatory effects mainly by suppressing the TGF-β1/Smad2 signaling pathway which might contribute to inhibit inflammatory mediators, also attenuation collagen deposition. The possible mechanisms may involve dephosphorylation of Smad2, which may be responsible for the observed effects of YPFS on alleviating inflammation in vivo and in vitro.
Acknowledgment
This work was supported by National Science Foundation of China (No. 81460684 and No. 81503323) and a grant form Yunnan Department of Science and Technology (No. 2013FA041 and No. 2015FD035).
References
- 1.Hagstad S, Bjerg A, Ekerljung L, Backman H, Lindberg A, Rönmark E, et al. Passive smoking exposure is associated with increased risk of COPD in never smokers. Chest. 2014;145:1298–1304. doi: 10.1378/chest.13-1349. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB. Total and state-specific medical and absenteeism costs of COPD among adults aged≥ 18 years in the United States for 2010 and projections through 2020. Chest. 2015;147:31–45. doi: 10.1378/chest.14-0972. [DOI] [PubMed] [Google Scholar]
- 3.Sethi S, Mahler DA, Marcus P, Owen CA, Yawn B, Rennard S. Inflammation in COPD: implications for management. Am J Med. 2012;125:1162–1270. doi: 10.1016/j.amjmed.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 4.Chung K, Adcock I. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–1356. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- 5.Jiménez-Ruiz CA, Andreas S, Lewis KE, Tonnesen P, van Schayck C, Hajek P, et al. Statement on smoking cessation in COPD and other pulmonary diseases and in smokers with comorbidities who find it difficult to quit. Eur Respir J. 2015;46:61–79. doi: 10.1183/09031936.00092614. [DOI] [PubMed] [Google Scholar]
- 6.Shah S, Gangan N, Bechtol R, Vaidya V. Smoking In Chronic Obstructive Pulmonary Disease (Copd) Patients: Socio-Demographic Factors Associated With Smoking Cessation. Value Health. 2013;16:A240. [Google Scholar]
- 7.Rabe KF, Bateman ED, O’Donnell D, Witte S, Bredenbröker D, Bethke TD. Roflumilast-an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2005;366:563–571. doi: 10.1016/S0140-6736(05)67100-0. [DOI] [PubMed] [Google Scholar]
- 8.Günter S, John-Schuster G, Conlon TM, Hager K, Amarie OV, Eickelberg O, et al. Immunoaging augments sensitivity to cigarette smoke-induced COPD. Eur Respir J. 2014;44:3248. [Google Scholar]
- 9.Leila GM, Mohammad Hossein B, Mohammad RN. The effect of Zataria multiflora and its constituent, carvacrol, on tracheal responsiveness and lung pathology in guinea pig model of COPD. Phytother Res. 2015;29:730–736. doi: 10.1002/ptr.5309. [DOI] [PubMed] [Google Scholar]
- 10.Mohammad Hossein B, Leila GM. Lung inflammation changes and oxidative stress induced by cigarette smoke exposure in guinea pigs affected by Zataria multiflora and its constituent, carvacrol. BMC Complement Altern Med. 2015;15:1–10. doi: 10.1186/s12906-015-0574-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandsma C-A, Timens W, Jonker MR, Rutgers B, Noordhoek JA, Postma DS. Differential effects of fluticasone on extracellular matrix production by airway and parenchymal fibroblasts in severe COPD. Am J Physiol Lung Cell Mol Physiol. 2013;305:582–589. doi: 10.1152/ajplung.00152.2013. [DOI] [PubMed] [Google Scholar]
- 12.Königshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, Jahn A, et al. WNT1-inducible signaling protein–1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119:772–787. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, et al. Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol. 2012;23:252–265. doi: 10.1681/ASN.2011010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su Zg, Wen Fq, Feng Yl, Xiao M, Wu Xl. Transforming growth factor-β1 gene polymorphisms associated with chronic obstructive pulmonary disease in Chinese population. Acta Pharmacol Sin. 2005;26:714–720. [PubMed] [Google Scholar]
- 15.Ichimaru Y, Krimmer DI, Burgess JK, Black JL, Oliver BG. TGF-β enhances deposition of perlecan from COPD airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2012;302:325–333. doi: 10.1152/ajplung.00453.2010. [DOI] [PubMed] [Google Scholar]
- 16.Warburton D. Developmental responses to lung injury: repair or fibrosis. Fibrogenesis Tissue Repair. 2012;5:S2. doi: 10.1186/1755-1536-5-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitamura H, Cambier S, Somanath S, Barker T, Minagawa S, Markovics J, et al. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8–mediated activation of TGF-β. J Clin Invest. 2011;121:2863–2875. doi: 10.1172/JCI45589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-β. J Biochem. 2010;147:781–792. doi: 10.1093/jb/mvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharkey DJ, Tremellen KP, Jasper MJ, Gemzell-Danielsson K, Robertson SA. Seminal fluid induces leukocyte recruitment and cytokine and chemokine mRNA expression in the human cervix after coitus. J Immunol. 2012;188:2445–2454. doi: 10.4049/jimmunol.1102736. [DOI] [PubMed] [Google Scholar]
- 20.Hovhannisyan L, Mkrtchyan G, Boyajyan A, Avetyan D, Tadevosyan MY SS. Inflammatory markers in post-traumatic stress disorder. Cytokines Inflammation. 2012;11:42–45. [Google Scholar]
- 21.Han G, Li F, Singh TP, Wolf P, Wang XJ. The pro-inflammatory role of TGFβ1: a paradox? Int J Biol Sci. 2012;8:228–235. doi: 10.7150/ijbs.8.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li T, Wang Y, Wang Y, Liang R, Zhang D, Zhang H, et al. Development of an SPE–HPLC–MS method for simultaneous determination and pharmacokinetic study of bioactive constituents of Yu Ping Feng San in rat plasma after oral administration. J Ethnopharmacol. 2013;145:784–92. doi: 10.1016/j.jep.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao Yl, An W, Zhang W. Yu ping feng san auxiliary treatment of 30 cases of chronic obstructive pulmonary disease. Herald Med. 2011;30:63–66. [Google Scholar]
- 24.Ma YT, Wang DH. Yu Ping Feng San stabilization treatment of chronic obstructive pulmonary disease pulmonary qi deficiency syndrome curative effect observation. Modern Traditional Chinese Med. 2009:6–8. [Google Scholar]
- 25.Wang CM, Jiang M, Wang HJ. Effect of NF-κB inhibitor on high-mobility group protein B1 expression in a COPD rat model. Mol Med Rep. 2013;7:499–502. doi: 10.3892/mmr.2012.1181. [DOI] [PubMed] [Google Scholar]
- 26.Zheng H, Liu Y, Tian H, Fang Z, Li G, He S. Development and characterization of a rat model of chronic obstructive pulmonary disease (COPD) induced by sidestream cigarette smoke. Toxicol Lett. 2009;189:225–234. doi: 10.1016/j.toxlet.2009.06.850. [DOI] [PubMed] [Google Scholar]
- 27.Du CY, Choi RC, Zheng KY, Dong TT, Lau DT, Tsim KW. Yu Ping Feng San, an ancient Chinese herbal decoction containing Astragali radix Atractylodis macrocephalae Rhizoma and Saposhnikoviae radix regulates the release of cytokines in murine macrophages. PLoS One. 2013;8:e78622. doi: 10.1371/journal.pone.0078622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaw LH, Lin LC, Tsai TH. HPLC–MS/MS analysis of a traditional Chinese medical formulation of Bu-Yang-Huan-Wu-Tang and its pharmacokinetics after oral administration to rats. PLoS One. 2012;7:e43848. doi: 10.1371/journal.pone.0043848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao HM, Huang XY, Zhou F, Tong WT, Wan PT, Huang MF, et al. Si Shen Wan inhibits mRNA expression of apoptosis-related molecules in p38 MAPK signal pathway in mice with colitis. Evid Based Complement Alternat Med. 2013;2013:432097. doi: 10.1155/2013/432097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tony P, Sarah LP, Daniel P, Monique LVH. Cigarette smoke extract induces differential expression levels of beta-defensin peptides in human alveolar epithelial cells. Tob Induc Dis. 2013;11:10. doi: 10.1186/1617-9625-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kothari HPS. Immune-oxidative alterations in cultured human lymphocytes by cigarette smoke. Global J Multidisciplinary study. Global J of Multidisciplinary Studies. 2015;4:281–290. [Google Scholar]
- 32.Munder A, Wölbeling F, Kerber-Momot T, Wedekind D, Baumann U, Gulbins E, et al. Acute intratracheal Pseudomonas aeruginosa infection in cystic fibrosis mice is age-independent. Respir Res. 2011;12:148. doi: 10.1186/1465-9921-12-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Persson C. Simvastatin inhibits smoke-induced airway epithelial injury: implications for COPD therapy. Eur Respir J. 2014;43:1208–1211. doi: 10.1183/09031936.00099913. [DOI] [PubMed] [Google Scholar]
- 34.Gorowiec MR, Borthwick LA, Parker SM, Kirby JA, Saretzki GC, Fisher AJ. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-β1-dependent mechanism. Free Radic Biol Med. 2012;52:1024–1032. doi: 10.1016/j.freeradbiomed.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 35.Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, et al. Increased expression of transforming growth factor-β 1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD) Am J Respir Criti Care Med. 2001;163:1476–1483. doi: 10.1164/ajrccm.163.6.9908135. [DOI] [PubMed] [Google Scholar]
- 36.Kranenburg AR, Willems-Widyastuti A, Mooi WJ, Sterk PJ, Alagappan VK, de Boer WI, et al. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol. 2006;126:725–735. doi: 10.1309/jc477fael1ykv54w. [DOI] [PubMed] [Google Scholar]
- 37.Gu j, Jiang J, Shen C, Lu L, Dai Q. Effects of “Jude-Screen Powder” on Th1/Th2 balance in mice model of systemic allergic airway disease. ShangHai J Traditional Chinese Med. 2005:50–52. [Google Scholar]
- 38.Shen D, Xie X, Zhu Z, Yu X, Liu H, Wang H, et al. Screening active components from Yu-Ping-Feng-San for regulating initiative key factors in allergic sensitization. PLoS One. 2014;9:e107279. doi: 10.1371/journal.pone.0107279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du CYQ, Zheng KYZ, Bi CWC, Dong TTX, Lin HL, Tsim KWK. Yu Ping Feng San, an Ancient Chinese herbal decoction, Induces gene expression of anti-viral proteins and inhibits neuraminidase activity. Phytother Res. 2015;29:656–661. doi: 10.1002/ptr.5290. [DOI] [PubMed] [Google Scholar]
- 40.Mohammad Hossein B, Leila GM. Effect of the Zataria multiflora on systemic inflammation of experimental animals model of COPD. BioMed Res Int. 2014;2014:802189. doi: 10.1155/2014/802189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gholamnezhad Z, Mohammad Hossein B, Hosseini M. Effect of Nigella sativa on immune response in treadmill exercised rat. BMC Complement Altern Med. 2014;14:437. doi: 10.1186/1472-6882-14-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leila GM, Feizpour A, Kianmehr M, Soukhtanloo M, Mohammad-Hossein B. The effect of carvacrol on systemic inflammation in guinea pigs model of COPD induced by cigarette smoke exposure. Pharmacol Rep. 2015;67:140–145. doi: 10.1016/j.pharep.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 43.Gu S, Yin N, Pei J, Lai L. Understanding traditional Chinese medicine anti-inflammatory herbal formulae by simulating their regulatory functions in the human arachidonic acid metabolic network. Mol Biosyst. 2013;9:1931–1938. doi: 10.1039/c3mb25605g. [DOI] [PubMed] [Google Scholar]
- 44.Kuzubova N, Gichkin A, Surkova E, Titova O. Left ventricular dysfunction in patients with COPD: Inflammation and endothelial dysfunction. Eur Respir J. 2013;42:178. [Google Scholar]
- 45.Yanbaeva DG, Dentener MA, Creutzberg EC, Wouters EF. Systemic inflammation in COPD: is genetic susceptibility a key factor? COPD. 2006;3:51–61. doi: 10.1080/15412550500493436. [DOI] [PubMed] [Google Scholar]
- 46.Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8:183–192. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- 47.Calabrese F, Baraldo S, Bazzan E, Lunardi F, Rea F, Maestrelli P, et al. IL-32, a novel proinflammatory cytokine in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178:894–901. doi: 10.1164/rccm.200804-646OC. [DOI] [PubMed] [Google Scholar]
- 48.Profita M, Chiappara G, Mirabella F, Di Giorgi R, Chimenti L, Costanzo G, et al. Effect of cilomilast (Ariflo) on TNF-α, IL-8, and GM-CSF release by airway cells of patients with COPD. Thorax. 2003;58:573–579. doi: 10.1136/thorax.58.7.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Leary L, Tildy B, Papazoglou E, Adcock I, Chung K, Perry M. S50 Airway smooth muscle inflammation is controlled by microrna-145 targeting of smad3 in COPD. Thorax. 2014;69:A28. [Google Scholar]
- 50.Cazzola M, Calzetta L, Bettoncelli G, Cricelli C, Romeo F, Matera MG, et al. Cardiovascular disease in asthma and COPD: a population-based retrospective cross-sectional study. Respir Med. 2012;106:249–256. doi: 10.1016/j.rmed.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 51.Sanchez-Muñoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–4288. doi: 10.3748/wjg.14.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 53.Feizpour A, Mohammad Hossein B, Ghorbani A. Adipose-derived stromal cell therapy affects lung inflammation and tracheal responsiveness in guinea pig model of COPD. PLoS One. 2014;9:e108974. doi: 10.1371/journal.pone.0108974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, et al. Progressive transforming growth factor β1–induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am J Respir Crit Care Med. 2005;171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- 55.Rosendahl A, Checchin D, Fehniger TE, ten Dijke P, Heldin C-H, Sideras P. Activation of the TGF-β/activin-Smad2 pathway during allergic airway inflammation. Am J Respir Cell Mol Biol. 2001;25:60–68. doi: 10.1165/ajrcmb.25.1.4396. [DOI] [PubMed] [Google Scholar]
- 56.Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth factors. 2011;29:196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lan HY. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7:1056–1067. doi: 10.7150/ijbs.7.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chung AC, Huang XR, Zhou L, Heuchel R, Lai KN, Lan HY. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol Dial Transplant. 2009;24:1443–1454. doi: 10.1093/ndt/gfn699. [DOI] [PubMed] [Google Scholar]
- 59.Gregory LG, Mathie SA, Walker SA, Pegorier S, Jones CP, Lloyd CM. Overexpression of Smad2 drives house dust mite–mediated airway remodeling and airway hyperresponsiveness via activin and IL-25. Am J Respir Crit Care Med. 2010;182:143–54. doi: 10.1164/rccm.200905-0725OC. [DOI] [PMC free article] [PubMed] [Google Scholar]