

Graphical abstract

Keywords: 4-Aminoquinolines, Side chain modification, Guanidines, Antitrypanosomal agents, Brain permeability
Abstract
Current drugs against human African trypanosomiasis (HAT) suffer from several serious drawbacks. The search for novel, effective, brain permeable, safe, and inexpensive antitrypanosomal compounds is therefore an urgent need. We have recently reported that the 4-aminoquinoline derivative huprine Y, developed in our group as an anticholinesterasic agent, exhibits a submicromolar potency against Trypanosoma brucei and that its homo- and hetero-dimerization can result in to up to three-fold increased potency and selectivity. As an alternative strategy towards more potent smaller molecule anti-HAT agents, we have explored the introduction of ω-cyanoalkyl, ω-aminoalkyl, or ω-guanidinoalkyl chains at the primary amino group of huprine or the simplified 4-aminoquinoline analogue tacrine. Here, we describe the evaluation of a small in-house library and a second generation of newly synthesized derivatives, which has led to the identification of 13 side chain modified 4-aminoquinoline derivatives with submicromolar potencies against T. brucei. Among these compounds, the guanidinononyltacrine analogue 15e exhibits a 5-fold increased antitrypanosomal potency, 10-fold increased selectivity, and 100-fold decreased anticholinesterasic activity relative to the parent huprine Y. Its biological profile, lower molecular weight relative to dimeric compounds, reduced lipophilicity, and ease of synthesis, make it an interesting anti-HAT lead, amenable to further optimization to eliminate its remaining anticholinesterasic activity.
1. Introduction
Human African trypanosomiasis (HAT or sleeping sickness) is one of the 17 infectious diseases grouped under the term Neglected Tropical Diseases, which inflict a devastating effect on the health and economy of nearly 150 countries.1, 2, 3, 4 HAT is caused by two subspecies of the protozoan parasite Trypanosoma brucei, which are transmitted to humans through the bite of tsetse flies in rural areas of sub-Saharan Africa. The two subspecies of this parasite lead to distinct disease courses and display different geographical distribution. Most cases of HAT occur in western and central Africa and are due to T. brucei gambiense, which causes a chronic infection that slowly progresses from an initial hemolymphatic stage, often asymptomatic, to a late stage, in which the parasites spread into the central nervous system. This produces severe neurological pathology, including sleep disruptions, which give rise to the common name of the disease. About 2–5% of HAT cases occur in southern and eastern Africa and are caused by T. brucei rhodesiense. This leads to an acute infection that rapidly progresses from early to late stage disease. With both forms of HAT, the absence of effective treatment in the late stage inexorably leads to coma and death.5
Over the last 15 years, because of public health measures, there has been considerable success combatting HAT, with the estimated numbers of those infected falling from 300,000 to less than 20,000.6 However, the disease still occurs in 36 countries, with 65 million people at risk, and there is a constant potential for large epidemic outbreaks.
Vaccines are not a realistic option for prevention of HAT because of antigenic variation in the parasite.7 Chemotherapy is therefore of particular importance.8 Unfortunately, the few drugs that have been approved for HAT (pentamidine and suramin for early stage HAT; melarsoprol and eflornithine, alone or in combination with nifurtimox, for late stage HAT) are unsatisfactory for several reasons, which include the occurrence of major side effects, high costs associated with parenteral administration and medical supervision, lack of brain permeability (in the case of pentamidine and suramin), which precludes their use in late stage HAT, and the increasing emergence of resistance.9, 10, 11 Thus, the development of novel antitrypanosomal compounds that can overcome these issues is urgently needed.3, 12
Repurposing of known drugs is being increasingly pursued for antitrypanosomal drug discovery, particularly because this strategy should be more rapid and less expensive than the development of new chemical entities.4, 13, 14, 15 Despite this, most research efforts to replenish the antitrypanosomal pipeline remain focussed on the development of novel compounds, rationally designed or screened against one or several parasite biological targets16, 17, 18, 19 or, more often, arising from phenotypic whole cell screens of compound libraries.20, 21, 22, 23, 24, 25, 26
7-Chloro-4-aminoquinoline derivatives are within those structural classes that are being developed for the treatment of HAT.27 We recently reported that huprine Y (1, Fig. 1), a 7-chloro-4-aminoquinoline derivative with potent acetylcholinesterase (AChE) inhibitory activity, developed in our group as an anti-Alzheimer drug candidate,28 exhibited significant activity against T. brucei (IC50 = 0.61 μM, selectivity index (SI) over rat myoblast L6 cells = 13).29, 30 In 4-aminoquinoline-based antimalarials, both dimerization and side chain modification have been used to increase potency and overcome parasite resistance.31, 32, 33, 34, 35 Interestingly, we have found that homodimerization of huprine Y (as in compound 2,36 Fig. 1) and heterodimerization with the 4-aminoquinoline derivative tacrine (as in compound 3,37 Fig. 1) also results in up to 3-fold increased potency and selectivity against T. brucei.
Figure 1.
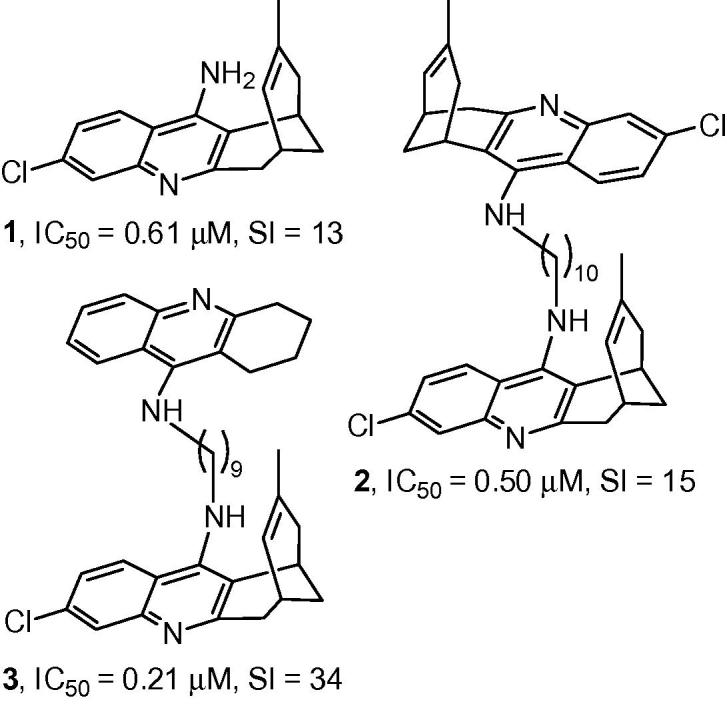
Structures, trypanocidal (T. brucei) activities, and selectivity indices of huprine Y, 1, and the oligomethylene-linked homo- and heterodimers 2 and 3.
Modification of the side chain attached to the exocyclic amino group of 7-chloro-4-aminoquinoline derivatives has also been reported to lead to increased antitrypanosomal activity.35 To explore further the structure–antitrypanosomal activity relationships around the huprine scaffold, we report here: (i) the screening of a series of huprine derivatives, substituted at the exocyclic amino group with cyanoalkyl or aminoalkyl chains of different lengths and nature (nitriles 4a–h, and amines 5a–h, Fig. 2), against cultured bloodstream forms of T. brucei, rat skeletal myoblast L6 cells, and electric eel AChE, and the evaluation of their brain permeability using an in vitro artificial membrane assay (PAMPA-BBB); (ii) the synthesis and evaluation of the antitrypanosomal, cytotoxic, and anticholinesterasic activity and brain permeability of novel huprine and structurally related tacrine derivatives with other modified side chains terminating in cyano, primary or cyclic amino, or guanidino groups.
Figure 2.
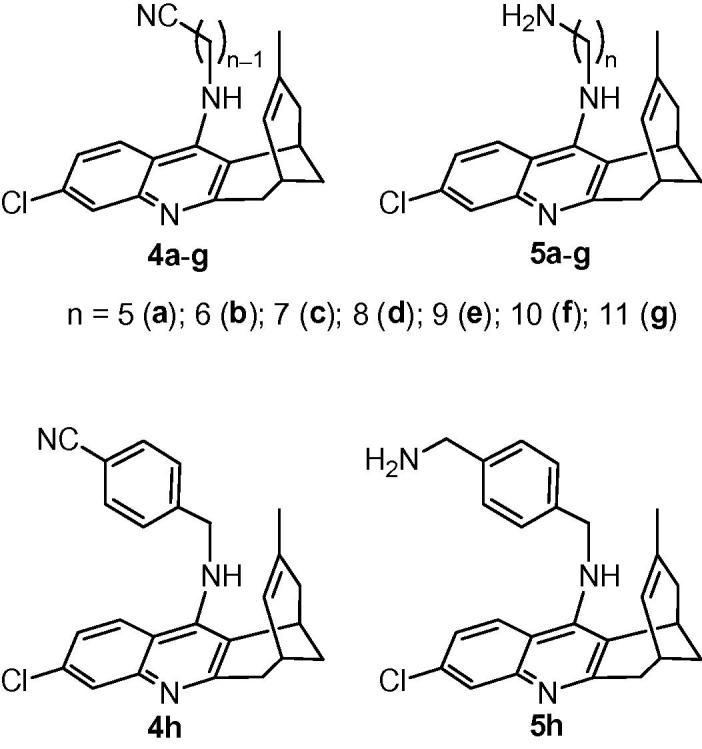
Structures of cyanoalkyl- and aminoalkyl-huprines 4a–h and 5a–h.
2. Results and discussion
2.1. Screening of the antitrypanosomal, cytotoxic, and anticholinesterasic activity and brain permeability of the ω-cyanoalkyl- and ω-aminoalkyl-huprines 4a–h and 5a–h
The ω-aminoalkyl-huprines 5a–h (Fig. 2) were recently synthesized in our group as immediate precursors of a family of huprine-based anti-Alzheimer hybrid compounds.38 We inferred that these compounds might be interesting antitrypanosomal leads based on two grounds. Firstly, aminoalkylhuprines 5a–h should be diprotonated at physiological pH, like pentamidine and other antitrypanosomal dicationic compounds,39, 40 which seemed favourable for anti-HAT activity. Secondly, we expected that the substitution of one of the lipophilic 4-aminoquinoline moieties of bis(4-aminoquinoline) dimers, like 2 and 3 (Fig. 1), by a primary amino group in aminoalkylhuprines 5a–h would result in a decreased AChE inhibitory activity, and hence, a lower risk of unwanted cholinergic side-effects. Indeed, this trend in AChE inhibitory activity has been reported for a family of dimeric tacrines,41 and can be ascribed to a less efficient interaction with a secondary binding site of the enzyme AChE, the so-called peripheral anionic site, when the second 4-aminoquinoline moiety of the dimer, bis(7)tacrine, is substituted by a simple primary amino group.
Because some nitriles have been found to display antitrypanosomal activity,9, 42 we also envisaged the biological screening of the nitriles 4a–h (Fig. 2), the synthetic precursors of amines 5a–h.38
The ω-cyanoalkyl-huprines 4a–h and the ω-aminoalkyl-huprines 5a–h were first screened against the bloodstream form of T. brucei, the clinically relevant form of the parasite,43 using nifurtimox and huprine Y as reference compounds. These side chain modified huprine derivatives displayed low micromolar to submicromolar IC50 values, with all of them being more potent antitrypanosomal agents than nifurtimox, and a few being slightly more potent than, or equipotent to, the parent huprine Y (Table 1). Somewhat unexpectedly, nitriles were found to be in general more potent than the corresponding amines, especially those featuring hepta- to nona-methylene side chains (octa- to deca-methylene side chains in the amines), which were 4–8-fold more potent than their amine counterparts. A clear trend was found in the antitrypanosomal potency of nitriles 4a–h regarding the length of the linker, with the potency increasing from n = 5 (4a) to n = 9 (4e), and then decreasing for the longer homologues 4f and 4g. For the amines, the highest potency was found for the heptamethylene-linked derivative 5c. The presence of a p-phenylene ring in the side chain does not seem to be of particular relevance for the antitrypanosomal activity, with the p-phenylene-linked nitrile 4h and amine 5h being equipotent to nitrile 4a and amine 5a with a similar side chain length.
Table 1.
Antitrypanosomal, cytotoxic, and anticholinesterasic activity and BBB permeability of cyanoalkylhuprines 4a–h and aminoalkylhuprines 5a–h and reference compounds 1 and nifurtimoxa
| Compd | T. brucei IC50 (μM) | T. brucei IC90 (μM) | L6 cells IC50 (μM) | SITbb | EeAChE IC50 (nM) | Pe (10−6 cm s−1)c (prediction) |
|---|---|---|---|---|---|---|
| 4a | 2.13 ± 0.49 | 4.04 ± 0.06 | 20.3 ± 0.2 | 9.7 | 26.9 ± 1.1 | d |
| 4b | 1.60 ± 0.13 | 3.21 ± 0.32 | 19.0 ± 0.3 | 11.9 | 31.1 ± 3.1 | 12.4 ± 0.6 (CNS+) |
| 4c | 0.86 ± 0.05 | 1.22 ± 0.03 | 11.6 ± 3.0 | 13.5 | d | d |
| 4d | 0.62 ± 0.02 | 0.85 ± 0.01 | 7.65 ± 0.41 | 12.3 | d | d |
| 4e | 0.32 ± 0.01 | 0.42 ± 0.01 | 8.04 ± 0.53 | 25.1 | d | d |
| 4f | 0.46 ± 0.01 | 0.61 ± 0.01 | 4.79 ± 0.25 | 10.4 | 9.67 ± 0.89 | 11.3 ± 1.2 (CNS+) |
| 4g | 1.37 ± 0.06 | 1.91 ± 0.02 | 4.98 ± 0.21 | 3.6 | d | 16.2 ± 1.3 (CNS+) |
| 4h | 1.86 ± 0.08 | 3.39 ± 0.15 | 10.0 ± 0.8 | 5.4 | 158 ± 21 | 19.3 ± 1.2 (CNS+) |
| 5a | 0.92 ± 0.08 | 2.92 ± 0.43 | 3.82 ± 0.11 | 4.2 | d | d |
| 5b | 2.03 ± 0.10 | 3.09 ± 0.21 | 4.78 ± 0.16 | 2.4 | d | d |
| 5c | 0.68 ± 0.20 | 1.15 ± 0.05 | 4.33 ± 0.08 | 6.4 | 36.0 ± 3.5 | 7.6 ± 0.7 (CNS+) |
| 5d | 2.28 ± 0.29 | 4.28 ± 0.46 | 12.5 ± 3.3 | 5.5 | 17.3 ± 1.2 | 9.8 ± 0.6 (CNS+) |
| 5e | 2.61 ± 0.16 | 4.58 ± 0.31 | 13.6 ± 3.4 | 5.2 | 16.4 ± 1.8 | 7.1 ± 0.7 (CNS+) |
| 5f | 3.33 ± 0.10 | 4.33 ± 0.18 | 7.55 ± 0.19 | 2.3 | 20.3 ± 2.5 | 7.0 ± 0.3 (CNS+) |
| 5g | 1.79 ± 0.03 | 2.24 ± 0.04 | 8.06 ± 0.63 | 4.5 | 35.3 ± 3.9 | 4.0 ± 0.2 (CNS±) |
| 5h | 0.92 ± 0.03 | 1.44 ± 0.15 | 4.27 ± 0.13 | 4.6 | d | 11.7 ± 1.1 (CNS+) |
| 1e | 0.61 ± 0.03 | 2.94 ± 0.20 | 7.80 ± 0.47 | 13 | 0.30 ± 0.01 | 23.8 ± 2.7 (CNS+)f |
| Nifurtimox | 4.4 ± 0.7g | 32.0 ± 1.1 | 7.3 | d | d |
In vitro activity against bloodstream form of T. brucei (pH 7.4), rat myoblast L6 cells, and Electrophorus electricus AChE, expressed as the concentration that inhibited growth or enzyme activity by 50% (IC50) and 90% (IC90, for T. brucei). Data are the mean of triplicate experiments ± SEM.
SITb: selectivity index as the ratio of cytotoxic to anti-T. brucei IC50 values.
Permeability values from the PAMPA-BBB assay. Values are expressed as the mean ± SD of three independent experiments.
Not determined.
Trypanocidal and cytotoxicity activity values taken from Ref. 29.
Taken from Ref. 38.
Taken from Ref. 44.
Thus, the most interesting side chain modified huprine derivative was nitrile 4e, which, with an IC50 value against T. brucei of 320 nM (and an IC90 value of 420 nM), was 14-fold more potent than nifurtimox and 2-fold more potent than huprine Y (7-fold more potent than huprine Y in terms of the IC90 values), in agreement with the expected increase in antitrypanosomal potency upon modification of the side chain at the exocyclic amino group.
Interestingly, nitriles 4a–h and amines 5a–h turned out to be less toxic to rat skeletal myoblast L6 cells than to T. brucei, with nitriles 4a–h being less cytotoxic than the corresponding amines 5a–h (Table 1), especially nitrile 4e, which displayed a selectivity index of 25, i.e. 2- and 3.5-fold more than that of the parent huprine Y and nifurtimox, respectively.
As expected, the introduction of the ω-cyanoalkyl and ω-aminoalkyl chains at the primary amino group of huprine Y led to a clear decrease in AChE inhibitory activity (up to 500-fold). Notwithstanding the lower AChE inhibitory potency relative to the parent huprine Y, nitriles 4a–h and amines 5a–h were more potent AChE inhibitors than would be desirable in antitrypanosomal agents, with nanomolar IC50 values for electric eel AChE inhibition (Table 1).
Because good brain penetration is necessary for the treatment of late-stage HAT, the brain permeability of nitriles 4a–h and amines 5a–h was assessed in vitro through the well-established parallel artificial membrane permeability assay (PAMPA-BBB).45 As with the parent huprine Y, the permeabilities of most of these modified analogues, through the porcine brain lipid extract used as an artificial blood–brain barrier (BBB) model, were found to be above the threshold established for high BBB permeation (CNS+, Pe (10−6 cm s−1) > 5.17, Table 1). Therefore, these compounds are predicted to be able to cross the BBB, with nitriles 4a–h being more permeable than the corresponding amines (Table 1), probably due to the dicationic character of the latter at physiological pH.
Overall, the screening of this small in-house compound library pointed to neutral cyano or basic primary amino groups at the end of a side chain of 9 or 7 carbon atoms, respectively, as being favourable substitution patterns for potent, selective, and brain permeable antitrypanosomal agents.
2.2. Synthesis of novel side chain modified 4-aminoquinoline derivatives
Even though the ω-aminoalkyl-huprines 5a–h turned out to be less potent antitrypanosomal agents than the corresponding nitriles 4a–h, they still displayed a submicromolar antitrypanosomal potency in some cases, as well as some selectivity and brain permeability. Indeed, other classes of compounds, such as bis-guanidines and bis-amidines, are diprotonated at physiological pH, like aminoalkylhuprines 5a–h, and exhibit potent antitrypanosomal activity,40, 46, 47, 48, 49 and brain permeability.40 To further extend the SAR around side chain modified huprine derivatives, we undertook the synthesis and biological profiling of novel dibasic huprine derivatives featuring a terminal guanidine (6b and 6e, Scheme 1), piperidine (7, Scheme 2) or morpholine (8, Scheme 2) moiety, together with different side chain lengths.
Scheme 1.
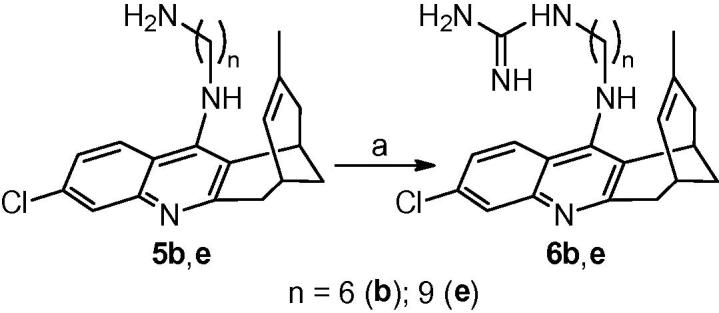
Reagents and conditions: (a) 1H-pyrazole-1-carboxamidine hydrochloride, Et3N, CH3CN, reflux, overnight.
Scheme 2.
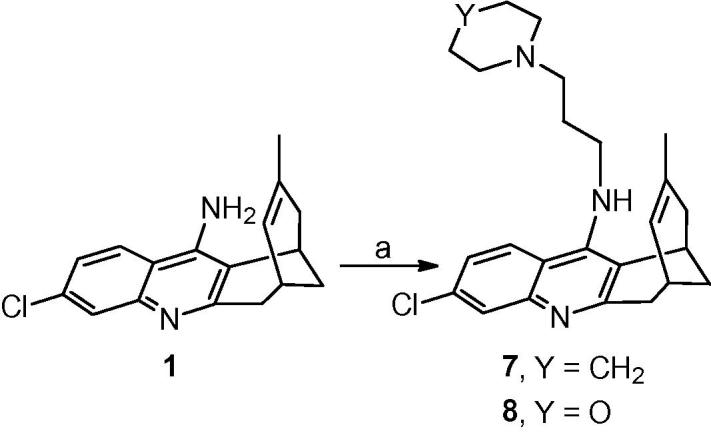
Reagents and conditions: (a) KOH, 4 Å molecular sieves, DMSO, rt, 2 h; then, 1-(3-chloropropyl)piperidine hydrochloride or 3-morpholinopropyl methanesulfonate, DMSO, rt, overnight.
In addition, to assess the role of the unsaturated methyl-substituted three-carbon bridge of the huprine moiety, we also synthesised a series of ω-cyanoalkyl-, ω-aminoalkyl-, and ω-guanidinoalkyl derivatives (11c, 11e, 12c–e, 13c, 13e, 14e, 15c, 15e, 16e, Scheme 3), in which the huprine core was substituted by the simpler, less lipophilic (by around 2 log P units), and easier-to-synthesize tricyclic core of the 4-aminoquinoline derivatives tacrine and 6-chlorotacrine (9 and 10, respectively, Scheme 3). These featured oligomethylene chains of lengths in the range that was found optimal for antitrypanosomal activity in the huprine series (n = 7–9).
Scheme 3.
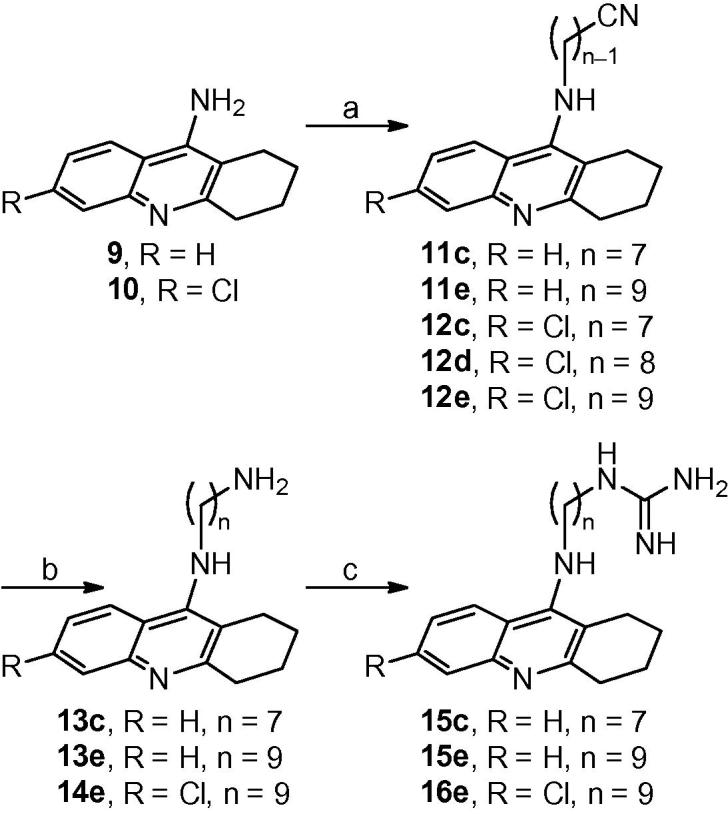
Reagents and conditions: (a) KOH, 4 Å molecular sieves, DMSO, rt, 2 h; then, ω-bromoalkanenitrile, DMSO, rt, overnight; (b) LiAlH4, Et2O, rt, overnight; (c) 1H-pyrazole-1-carboxamidine hydrochloride, Et3N, CH3CN, reflux, overnight.
Guanidinoalkyl huprines 6b and 6e were readily synthesized in moderate yield (30% and 59%, respectively) from the corresponding amines 5b and 5e upon reaction with 1H-pyrazole-1-carboxamidine hydrochloride in dry CH3CN in the presence of Et3N (Scheme 1).
Likewise, the synthesis of the piperidinopropyl- and morpholinopropyl-huprines 7 and 8 only required one step, i.e. the alkylation of huprine Y, 1, with the commercial 1-(3-chloropropyl)piperidine hydrochloride or the readily available 3-morpholinopropyl methanesulfonate,50 after deprotonation of the primary amino group with KOH (Scheme 2).
For the synthesis of the novel cyanoalkyl tacrines 11c, 11e, and 12c–e, the known aminoalkyl tacrines 13c,41 13e,51 and 14e,52 and the novel guanidinoalkyl tacrines 15c, 15e, and 16e, we followed the same three-step protocol that we had used in the huprine series, based on the initial alkylation of tacrine or chlorotacrine with the corresponding ω-bromoalkanenitrile, followed by LiAlH4 reduction of the cyano to a primary amino group, and final conversion of the amines into the guanidines upon treatment with 1H-pyrazole-1-carboxamidine hydrochloride (Scheme 3).
All the target compounds were transformed into the corresponding hydrochloride or dihydrochloride salts, with which their chemical and biological characterization was performed.
2.3. Biological profiling of the novel side chain modified 4-aminoquinoline derivatives
The therapeutic potential of the second generation side chain modified huprine and tacrine derivatives was assessed by evaluation of their antitrypanosomal activity against cultured bloodstream forms of T. brucei, and by their brain permeability. Additionally, their potential toxicity was assessed by measuring their effect on the viability of rat L6 cells as a model of normal mammalian cells and by their AChE inhibitory activity.
We found several compounds that exhibited nanomolar antitrypanosomal IC50 and IC90 values, favourable selectivity indices, and brain permeability (Table 2).
Table 2.
Antitrypanosomal, cytotoxic, and anticholinesterase activity and BBB permeability of the N-cyanoalkyl, N-aminoalkyl, and N-guanidinoalkyl 4-aminoquinoline derivatives 6b,e, 7, 8, 11c,e, 12c–e, 13c,e, 14e, 15c,e, and 16e and reference compounds 1 and nifurtimoxa
| Compd | T. brucei IC50 (μM) | T. brucei IC90 (μM) | L6 cells IC50 (μM) | SITbb | EeAChE IC50 (nM) | Pe (10−6 cm s−1)c (prediction) |
|---|---|---|---|---|---|---|
| 6b | 1.37 ± 0.02 | 1.54 ± 0.01 | 21.0 ± 1.6 | 15.3 | 11.8 ± 1.3 | 2.9 ± 0.4 (CNS±) |
| 6e | 0.33 ± 0.06 | 0.70 ± 0.09 | 10.9 ± 0.9 | 33 | 10.6 ± 0.8 | 5.4 ± 0.3 (CNS+) |
| 7 | 0.83 ± 0.04 | 1.70 ± 0.21 | 2.40 ± 0.19 | 2.9 | 48.4 ± 2.7 | 5.9 ± 0.4 (CNS+) |
| 8 | 1.75 ± 0.19 | 4.88 ± 0.27 | 14.3 ± 0.5 | 8.2 | 13.3 ± 0.8 | 15.0 ± 1.0 (CNS+) |
| 11c | 3.81 ± 1.13 | 10.3 ± 2.7 | 34.8 ± 1.6 | 9.1 | 53.9 ± 3.0 | 11.2 ± 0.2 (CNS+) |
| 11e | 0.98 ± 0.05 | 1.38 ± 0.09 | 5.73 ± 0.16 | 4.2 | 44.9 ± 1.6 | 7.6 ± 0.35 (CNS+) |
| 12c | 7.92 ± 0.17 | 12.2 ± 0.2 | 34.7 ± 0.6 | 4.4 | d | 16.7 ± 0.9 (CNS+) |
| 12d | 6.89 ± 0.13 | 8.75 ± 0.24 | 27.8 ± 1.6 | 4.0 | 46.0 ± 6.0 | 14.7 ± 0.8 (CNS+) |
| 12e | 1.57 ± 0.17 | 2.56 ± 0.07 | 8.00 ± 0.44 | 5.1 | 24.3 ± 4.5 | 5.4 ± 0.4 (CNS+) |
| 13c | 2.13 ± 0.05 | 3.12 ± 0.14 | 7.28 ± 0.86 | 3.4 | 23.9 ± 2.6 | 7.4 ± 0.1 (CNS+) |
| 13e | 4.07 ± 0.12 | 5.55 ± 0.10 | 16.6 ± 0.6 | 4.1 | 15.6 ± 1.5 | 6.9 ± 0.5 (CNS+) |
| 14e | 2.31 ± 0.36 | 4.88 ± 0.18 | 7.62 ± 0.81 | 3.3 | 14.5 ± 1.6 | 5.8 ± 0.3 (CNS+) |
| 15c | 0.85 ± 0.09 | 1.30 ± 0.20 | <1.20 | <1.4 | 23.9 ± 2.6 | 5.9 ± 0.3 (CNS+) |
| 15e | 0.12 ± 0.01 | 0.25 ± 0.04 | 15.9 ± 0.8 | 133 | 30.5 ± 1.9 | 6.5 ± 0.3 (CNS+) |
| 16e | 0.63 ± 0.08 | 0.96 ± 0.02 | 11.9 ± 1.2 | 19 | 16.4 ± 1.5 | 6.7 ± 0.4 (CNS+) |
| 1e | 0.61 ± 0.03 | 2.94 ± 0.20 | 7.80 ± 0.47 | 13 | 0.30 ± 0.01 | 23.8 ± 2.7 (CNS+)f |
| Nifurtimox | 4.4 ± 0.7g | 32.0 ± 1.1 | 7.3 | d | d |
In vitro activity against bloodstream form of T. brucei (pH 7.4), rat myoblast L6 cells, and Electrophorus electricus AChE, expressed as the concentration that inhibited growth or enzyme activity by 50% (IC50) and 90% (IC90, for T. brucei). Data are the mean of triplicate experiments ± SEM.
SITb: selectivity index as the ratio of cytotoxic to anti-T. brucei IC50 values.
Permeability values from the PAMPA-BBB assay. Values are expressed as the mean ± SD of three independent experiments.
Not determined.
Trypanocidal and cytotoxicity activity values taken from Ref. 29.
Taken from Ref. 38.
Taken from Ref. 44.
With the huprine derivatives, we found that the introduction of a guanidine at the end of the side chain led to increased antitrypanosomal potencies relative to the corresponding primary amine counterparts, and to roughly equipotent activity relative to the nitriles. Thus, the novel guanidine 6e, with a total of 9 carbon atoms in the side chain, displayed the same antitrypanosomal potency as the most active compound of the initially screened series, nitrile 4e (IC50 0.3 μM) (Table 2). The introduction of a piperidino or a morpholino substituent at the side chain of the huprine derivatives 7 and 8 did not seem to confer any particular contribution to the antitrypanosomal properties, with these compounds displaying similar activities to several of the aminoalkylhuprines.
The same trend observed with the huprine derivatives in regard of the terminal functionality and length of the side chain was found in the tacrine derivatives. The order of antitrypanosomal potencies was guanidines > nitriles > amines, with higher potencies for the guanidines and nitriles that have a total number of 9 carbon atoms in the side chain. Higher antitrypanosomal potencies were also found for those derivatives with an unsubstituted tacrine moiety. Thus, the nonamethylene-linked guanidine 15e (IC50 120 nM) was found to be the most potent side chain modified tacrine derivative (Table 2).
With regard to the 4-aminoquinoline core, replacement of the huprine with the less complex tacrine moiety led, in general, to a drop in antitrypanosomal activity, with ω-aminoalkyl- and ω-cyanoalkyl (6-chloro)tacrines being 1–3-fold and 3–9-fold, respectively, less potent than the corresponding ω-aminoalkyl- and ω-cyanoalkyl huprine derivatives. A notable exception was the ω-guanidinononyl tacrine 15e, which was 3-fold more potent than the ω-guanidinononyl huprine 6e. Overall this was the most potent side chain modified 4-aminoquinoline derivative.
Interestingly, all the second generation side chain modified huprine and tacrine derivatives could be inferred to be capable of entering the central nervous system (with the sole exception of guanidine 6b), on the basis of their measured PAMPA-BBB permeabilities. All of them, with the exception of 15c, also turned out to be less toxic to mammalian L6 cells than to T. brucei, with selectivity indices ranging from 3 to 133 (Table 2). Unfortunately, as with the first generation of screened compounds, despite inhibiting AChE 35–180-fold less potently than the parent compound huprine Y, the anticholinesterasic activity of the second generation side chain modified huprine and tacrine derivatives was still not ideal. Their anticholinesterasic activity was 1–2 orders of magnitude higher than their antitrypanosomal potencies, with the guanidinononyl tacrine 15e being the best balanced compound (with the anticholinesterasic activity only 4-fold greater than that against typanosomes).
3. Conclusion
We recently found that both homodimerization and heterodimerization of the 4-aminoquinoline derivative huprine Y results in increased potency against T. brucei and improved selectivity over mammalian cells relative to the parent compound,36, 37 albeit at the expense of increasing lipophilicity and molecular weight. In this current paper, we have explored the effect on antitrypanosomal activity of the introduction of a side chain, featuring a terminal cyano, amino, or guanidino group, at the primary amino group of huprine Y or the simpler structurally related tacrine or 6-chlorotacrine, as an alternative approach to improve the antitrypanosomal profile. We found that the introduction of a guanidino or a cyano group at the end of a chain of nine carbon atoms was the best type of substitution to produce good antitrypanosomal activity in both the huprine and the tacrine series. The presence of the tetracyclic huprine core leads to higher antitrypanosomal potency in the cyanoalkyl derivatives, whereas the opposite trend was found for guanidinoalkyl derivatives, with the guanidinononyltacrine analogue 15e being the most promising compound of the side chain modified 4-aminoquinoline derivatives. Overall, 13 out of the 31 side chain modified 4-aminoquinoline derivatives displayed submicromolar potencies against cultured bloodstream form T. brucei and good brain permeability. The antitrypanosomal potency of these compounds is greater than their toxicity to mammalian cells, but lower than their anticholinesterasic activity, which might result in undesirable cholinergic side effects.
Guanidinononyltacrine 15e emerges as the most interesting antitrypanosomal lead of this class. It is endowed with potent (IC50 = 120 nM) and selective (SI = 133) activity against T. brucei, should be brain permeable, and has the least unfavourable antitrypanosomal/anticholinergic activity ratio. This compound has 5-fold increased antitrypanosomal potency, 10-fold increased selectivity index, and 100-fold decreased anticholinesterasic activity compared with the parent huprine Y, together with lower lipophilicity and molecular weight relative to huprine-based homo- and hetero-dimeric compounds. Our findings confirm that introduction of a conveniently functionalized side chain at the primary amino group of 4-aminoquinoline derivatives may be superior to homo- and hetero-dimerization as a strategy to improve the potential anti-HAT therapeutic profile of this structural class.
4. Experimental
4.1. Chemistry. General methods
Melting points were determined in open capillary tubes with a MFB 595010M Gallenkamp melting point apparatus. 400 MHz 1H/100.6 MHz 13C NMR spectra were recorded on a Varian Mercury 400 spectrometer at the Centres Científics i Tecnològics of the University of Barcelona (CCiTUB). The chemical shifts are reported in ppm (δ scale) relative to solvent signals (CD3OD at 3.31 and 49.0 ppm in the 1H and 13C NMR spectra, respectively), and coupling constants are reported in Hertz (Hz). The syn (anti) notation of the protons at position 13 of the huprine moiety of compounds 6b,e, 7, and 8 means that the corresponding proton at position 13 is on the same (different) side of the quinoline moiety with respect to the cyclohexene ring. IR spectra were run on a Perkin-Elmer Spectrum RX I spectrophotometer. Absorption values are expressed as wavenumbers (cm−1); only significant absorption bands are given. Column chromatography was performed on silica gel 60 AC.C (35–70 mesh, SDS, ref 2000027). Thin-layer chromatography was performed with aluminium-backed sheets with silica gel 60 F254 (Merck, ref. 1.05554), and spots were visualized with UV light and 1% aqueous solution of KMnO4. High resolution mass spectra were carried out at the CCiTUB with a LC/MSD TOF Agilent Technologies spectrometer. The analytical samples of all of the compounds that were subjected to pharmacological evaluation were dried at 65 °C/2 Torr for at least 2 days (standard conditions). Nitriles 11c and 12c are protected in a patent of our group,53 where, however, no NMR spectra data were given. A more consistent chemical characterization of these compounds is included in this section.
4.1.1. 1-{6-[(3-Chloro-6,7,10,11-tetrahydro-9-methyl-7,11-methanocycloocta[b]quinolin-12-yl)amino]hexyl}guanidine (6b)
To a solution of amine 5b (462 mg, 1.20 mmol) in dry CH3CN (5 mL), anhydrous Et3N (0.5 mL, 363 mg, 3.59 mmol) and 1H-pyrazole-1-carboxamidine hydrochloride (172 mg, 1.17 mmol) were added. The reaction mixture was stirred under reflux overnight. The resulting precipitated solid was taken in CH2Cl2 (25 mL) and treated with 2 N NaOH (20 mL). The organic phase was washed with H2O (3 × 20 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure to afford guanidine 6b (150 mg, 30% yield), as a brownish solid.
A solution of 6b (150 mg, 0.39 mmol) in CH2Cl2 (10 mL) was treated with a methanolic solution of HCl (0.75 N, 4.2 mL, 3.15 mmol) and concentrated under reduced pressure. The resulting solid was taken in MeOH (0.25 mL) and precipitated by addition of EtOAc (1.4 mL). The precipitate was washed with pentane (3 × 2 mL) to give, after drying under standard conditions, 6b·2HCl (37 mg) as a brownish hygroscopic solid; mp 166–168 °C; IR (ATR) ν 3500–2500 (max at 3260, 3134, 2929, 2865, +NH, NH, CH st), 1659, 1631, 1582, 1567, 1514 (C N, Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.42–1.54 (complex signal, 4H, 3-H2, 4-H2), 1.58 (s, 3H, 9′-CH3), superimposed in part 1.62 (tt, J = J′ = 6.8 Hz, 2H, 2-H2), superimposed in part 1.91 (tt, J = J′ = 7.2 Hz, 2H, 5-H2), 1.93 (br d, J = 17.2 Hz, 1H, 10′-Hendo), superimposed in part 1.94 (br d, J = 12.8 Hz, 1H, 13′-Hsyn), 2.09 (dm, J = 12.8 Hz, 1H, 13′-Hanti), 2.56 (dd, J = 17.2 Hz, J′ = 4.4 Hz, 1H, 10′-Hexo), 2.77 (m, 1H, 7′-H), 2.86 (br d, J = 17.6 Hz, 1H, 6′-Hendo), 3.19 (t, J = 6.8 Hz, 2H, 1-H2), superimposed in part 3.20 (dd, J = 17.6 Hz, J′ = 5.2 Hz, 1H, 6′-Hexo), 3.46 (m, 1H, 11′-H), 4.00 (t, J = 7.2 Hz, 2H, 6-H2), 4.85 (s, +NH, +NH2, NH, NH2), 5.59 (br d, J = 4.8 Hz, 1H, 8′-H), 7.57 (dd, J = 9.2 Hz, J′ = 2.0 Hz, 1H, 2′-H), 7.77 (d, J = 2.0 Hz, 1H, 4′-H), 8.41 (d, J = 9.2 Hz, 1H, 1′-H); 13C NMR (100.6 MHz, CD3OD) δ 23.5 (CH3, 9′-CH3), 27.26 (CH, C11′), 27.29 (CH2), 27.4 (CH2) (C3, C4), 27.8 (CH, C7′), 29.3 (CH2, C13′), 29.8 (CH2, C2), 31.2 (CH2, C5), 36.0 (CH2, C6′), 36.1 (CH2, C10′), 42.4 (CH2, C1), 49.5 (CH2, C6), 115.6 (C, C12a′), 117.6 (C, C11a′), 119.1 (CH, C4′), 125.1 (CH, C8′), 126.7 (CH, C2′), 129.5 (CH, C1′), 134.6 (C, C9′), 140.2 (C, C3′), 141.0 (C, C4a′), 151.3 (C, C5a′), 156.8 (C, C12′), 158.6 (C, guanidine C N); HRMS (ESI), calcd for (C24H3235ClN5+H+) 426.2419, found 426.2414.
4.1.2. 1-{9-[(3-Chloro-6,7,10,11-tetrahydro-9-methyl-7,11-methanocycloocta[b]quinolin-12-yl)amino]nonyl}guanidine (6e)
It was prepared as described for 6b. From amine 5e (50 mg, 0.12 mmol) and 1H-pyrazole-1-carboxamidine hydrochloride (21 mg, 0.14 mmol), a brownish solid residue (39 mg) was obtained. This crude product was taken in MeOH (2 mL) and eluted through a Dowex™ Marathon™ A OH– anion exchange resin (5 g) using MeOH (500 mL) as the eluent, to provide 6e (33 mg, 59% yield) as a brownish oil.
6e·2HCl: brownish solid; mp 210–211 °C; IR (ATR) ν 3500–2500 (max at 3250, 3137, 2923, 2852, +NH, NH, CH st), 1630, 1577, 1513 (C N, Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.34–1.48 (complex signal, 10H, 3-H2, 4-H2, 5-H2, 6-H2, 7-H2), superimposed in part 1.56 (m, 2H, 2-H2), 1.59 (s, 3H, 9′-CH3), 1.86 (tt, J = J′ = 7.2 Hz, 2H, 8-H2), 1.93 (br d, J = 17.6 Hz, 1H, 10′-Hendo), superimposed in part 1.94 (dm, J = 12.4 Hz, 1H, 13′-Hsyn), 2.09 (dm, J = 12.4 Hz, 1H, 13′-Hanti), 2.55 (dd, J = 17.6 Hz, J′ = 4.4 Hz, 1H, 10′-Hexo), 2.77 (m, 1H, 7′-H), 2.86 (br d, J = 18.0 Hz, 1H, 6′-Hendo), 3.16 (t, J = 7.2 Hz, 2H, 1-H2), 3.21 (dd, J = 18.0 Hz, J′ = 5.6 Hz, 1H, 6′-Hexo), 3.45 (m, 1H, 11′-H), 3.98 (t, J = 7.2 Hz, 2H, 9-H2), 4.85 (s, +NH, +NH2, NH, NH2), 5.59 (br d, J = 4.4 Hz, 1H, 8′-H), 7.56 (dd, J = 9.2 Hz, J′ = 2.4 Hz, 1H, 2′-H), 7.77 (d, J = 2.4 Hz, 1H, 4′-H), 8.40 (d, J = 9.2 Hz, 1H, 1′-H); 13C NMR (100.6 MHz, CD3OD) δ 23.5 (CH3, 9′-CH3), 27.3 (CH, C11′), 27.7 (CH2), 27.8 [(CH, C7′) + CH2] (C3, C7), 29.3 (CH2, C13′), 29.9 (CH2, C2), 30.3 (2CH2), 30.6 (CH2) (C4, C5, C6), 31.3 (CH2, C8), 36.0 (CH2, C6′), 36.1 (CH2, C10′), 42.5 (CH2, C1), 49.6 (CH2, C9), 115.7 (C, C12a′), 117.6 (C, C11a′), 119.1 (CH, C4′), 125.1 (CH, C8′), 126.6 (CH, C2′), 129.5 (CH, C1′), 134.5 (C, C9′), 140.3 (C, C3′), 141.0 (C, C4a′), 151.2 (C, C5a′), 156.9 (C, C12′), 158.6 (C, guanidine C N); HRMS (ESI), calcd for (C27H3835ClN5+H+) 468.2889, found 468.2903.
4.1.3. 3-Chloro-6,7,10,11-tetrahydro-9-methyl-12-[(3-piperidinopropyl)amino]-7,11-methanocycloocta[b]quinoline (7)
A mixture of huprine Y, 1 (500 g, 1.76 mmol), finely powdered KOH (85% purity, 346 mg, 5.24 mmol), and 4 Å molecular sieves in anhydrous DMSO (4 mL) was stirred, heating every 10 min approximately with a heat gun for 1 h and at rt for an additional hour, and then treated with a solution of 1-(3-chloropropyl)piperidine hydrochloride (429 mg, 2.17 mmol) in anhydrous DMSO (2.5 mL). The reaction mixture was stirred at room temperature overnight, diluted with H2O (50 mL) and extracted with CH2Cl2 (3 × 25 mL). The combined organic extracts were washed with H2O (3 × 100 mL), dried over anhydrous Na2SO4, and evaporated at reduced pressure to give a yellowish solid (413 mg), which was purified by column chromatography (40–60 μm silica gel, CH2Cl2/MeOH/50% aq NH4OH mixtures, gradient elution). On elution with CH2Cl2/MeOH/50% aq NH4OH 99:1:0.4, piperidinopropylhuprine 7 (35 mg, 5% yield) was isolated as a yellowish oil; Rf 0.38 (CH2Cl2/MeOH/50% aq NH4OH 9:1:0.1).
A solution of 7 (74 mg, 0.18 mmol) in CH2Cl2 (3 mL) was filtered through a 0.2 μm NYL filter, treated with HCl/Et2O (2.36 N, 0.16 mL, 0.38 mmol), and evaporated under reduced pressure. The resulting solid was washed with pentane (3 × 2 mL) to give, after drying under standard conditions, 7·2HCl (81 mg) as a pale yellow solid; mp 189–190 °C (dec.); IR (ATR) ν 3500–2500 (max at 3370, 3261, 3070, 2932, +NH, NH, CH st), 1630, 1580, 1513 (Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.59 (s, 3H, 9-CH3), superimposed 1.86–1.96 [complex signal, 6H, piperidine 3(5)-H2, piperidine 4-H2], 1.93 (br d, J = 17.2 Hz, 1H, 10-Hendo), superimposed in part 1.97 (dm, J = 12.4 Hz, 1H, 13-Hsyn), 2.10 (dm, J = 12.4 Hz, 1H, 13-Hanti), 2.37 (tt, J = 7.6 Hz, J′ = 7.2 Hz, 2H, 2′-H2), 2.59 (dd, J = 17.2 Hz, J′ = 4.8 Hz, 1H, 10-Hexo), 2.79 (m, 1H, 7-H), 2.89 (br d, J = 17.6 Hz, 1H, 6-Hendo), 2.90–3.06 [br signal, 2H, piperidine 2(6)-Hax], 3.23 (dd, J = 17.6 Hz, J′ = 5.6 Hz, 1H, 6-Hexo), 3.25 (t, J = 7.6 Hz, 2H, 3′-H2), 3.53 (m, 1H, 11-H), 3.50–3.62 [br signal, 2H, piperidine 2(6)-Heq], 4.08 (t, J = 7.2 Hz, 2H, 1′-H2), 4.85 (s, +NH, NH), 5.59 (br d, J = 4.8 Hz, 1H, 8-H), 7.61 (dd, J = 9.2 Hz, J′ = 2.0 Hz, 1H, 2-H), 7.79 (d, J = 2.0 Hz, 1H, 4-H), 8.41 (d, J = 9.2 Hz, 1H, 1-H); 13C NMR (100.6 MHz, CD3OD) δ 22.7 (CH2, piperidine C4), 23.4 (CH3, 9-CH3), 24.2 [2CH2, piperidine C3(5)], 26.0 (CH2, C2′), 27.3 (CH, C11), 27.8 (CH, C7), 29.2 (CH2, C13), 36.1 (CH2, C6), 36.3 (CH2, C10), 46.5 (CH2, C1′), 54.4 [2CH2, piperidine C2(6)], 55.3 (CH2, C3′), 115.8 (C, C12a), 118.3 (C, C11a), 119.2 (CH, C4), 125.1 (CH, C8), 127.1 (CH, C2), 129.2 (CH, C1), 134.6 (C, C9), 140.3 (C, C3), 140.9 (C, C4a), 151.8 (C, C5a), 156.8 (C, C12); HRMS (ESI), calcd for (C25H3235ClN3+H+) 410.2358, found 410.2365.
4.1.4. (±)-3-Chloro-6,7,10,11-tetrahydro-9-methyl-12-[(3-morpholinopropyl)amino]-7,11-methanocycloocta[b]quinoline (8)
It was prepared as described for 7. From 1 (500 mg, 1.76 mmol) and 3-morpholinopropyl methanesulfonate (470 mg, 2.10 mmol), a brownish oily residue (543 mg) was obtained and purified by column chromatography (40–60 μm silica gel, CH2Cl2/50% aq NH4OH 100:0.4), to give morpholinopropylhuprine 8 (121 mg, 17% yield) as a yellow oil; Rf 0.42 (CH2Cl2/MeOH/50% aq NH4OH 9:1:0.1).
8·2HCl: pale yellow solid; mp 173–174 °C (dec.); IR (ATR) ν 3500–2500 (max at 3384, 3225, 3109, 3048, 2922, 2790, 2728, 2683, 2610, +NH, NH, CH st), 1631, 1582, 1563, 1512 (Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.59 (s, 3H), 1.93 (br d, J = 17.2 Hz, 1H, 10-Hendo), superimposed in part 1.97 (dm, J = 12.8 Hz, 1H, 13-Hsyn), 2.10 (dm, J = 12.8 Hz, 1H, 13-Hanti), 2.39 (tt, J = J′ = 7.2 Hz, 2H, 2′-H2), 2.59 (dd, J = 17.2 Hz, J′ = 4.8 Hz, 1H, 10-Hexo), 2.78 (m, 1H, 7-H), 2.89 (br d, J = 18.0 Hz, 1H, 6-Hendo), superimposed 3.12–3.22 (m, 2H, 3′-H2), 3.23 (dd, J = 18.0 Hz, J′ = 5.2 Hz, 1H, 6-Hexo), superimposed with the CD3OD signal 3.28–3.36 [br signal, 2H, morpholine 3(5)-Hax], 3.53 (m, 1H, 11-H), superimposed 3.46–3.58 (br signal, 2H, morpholine 3(5)-Heq], 3.82–3.93 [br signal, 2H, morpholine 2(6)-Hax], 4.02–4.08 [br signal, 2H, morpholine 2(6)-Heq], 4.10 (t, J = 7.2 Hz, 2H, 1′-H2), 4.85 (s, +NH, NH), 5.59 (br d, J = 4.8 Hz, 1H, 8-H), 7.61 (dd, J = 9.6 Hz, J′ = 2.0 Hz, 1H, 2-H), 7.79 (d, J = 2.0 Hz, 1H, 4-H), 8.41 (d, J = 9.6 Hz, 1H, 1-H); 13C NMR (100.6 MHz, CD3OD) δ 23.5 (CH3, 9-CH3), 25.7 (CH2, C2′), 27.3 (CH, C11), 27.9 (CH, C7), 29.2 (CH2, C13), 36.1 (CH2, C6), 36.4 (CH2, C10), 46.5 (CH2, C1′), 53.3 [2CH2, morpholine C3(5)], 55.6 (CH2, C3′), 65.1 [2CH2, morpholine C2(6)], 115.8 (C, C12a), 118.3 (C, C11a), 119.3 (CH, C4), 125.1 (CH, C8), 127.2 (CH, C2), 129.3 (CH, C1), 134.7 (C, C9), 140.3 (C, C3), 140.9 (C, C4a), 151.8 (C, C5a), 156.9 (C, C12); HRMS (ESI), calcd for (C24H3035ClN3O+H+) 412.2150, found 412.2164.
4.1.5. 7-[(1,2,3,4-Tetrahydroacridin-9-yl)amino]heptanenitrile (11c)53
A suspension of tacrine, 9 (1.70 g, 8.57 mmol) and finely powdered KOH (85% purity, 0.97 g, 14.7 mmol), and 4 Å molecular sieves in anhydrous DMSO (20 mL) was stirred, heating every 10 min approximately with a heat gun for 1 h and at room temperature one additional hour, and then treated with a solution of 7-bromoheptanenitrile (90% purity, 1.55 mL, 1.96 g, 9.28 mmol) in anhydrous DMSO (12 mL) dropwise during 30 min. The reaction mixture was stirred at room temperature overnight, diluted with 5 N NaOH (50 mL) and extracted with EtOAc (3 × 150 mL). The combined organic extracts were washed with H2O (4 × 100 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure to give a crude product (2.53 g). Purification of this residue by column chromatography (40–60 μm silica gel, CH2Cl2/50% aq NH4OH 100:0.2) afforded a 85:15 mixture of the dialkylated byproduct and nitrile 11c (190 mg) and pure nitrile 11c (1.75 g, 66% isolated yield) as a yellow oil; Rf 0.77 (CH2Cl2/MeOH/50% aq NH4OH 9:1:0.05).
A solution of 11c (79 mg, 0.26 mmol) in CH2Cl2 (15 mL) was filtered through a 0.2 μm NYL filter, treated with methanolic HCl (0.53 N, 2.15 mL, 1.14 mmol) and evaporated under reduced pressure. The resulting solid was taken in MeOH (0.20 mL) and precipitated by addition of EtOAc (0.60 mL). The precipitate was washed with pentane (3 × 2 mL) to give, after drying under standard conditions, 11c·HCl (39 mg) as a pale brown sticky solid; IR (ATR) ν 3500–2500 (max at 3237, 2932, 2860, 2770, +NH, NH, CH st), 2242 (CN st), 1633, 1586, 1573, 1523 (Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.46–1.54 (complex signal, 4H, 4-H2, 5-H2), 1.66 (tt, J = 7.2 Hz, J′ = 6.8 Hz, 2H, 3-H2), 1.86 (tt, J = 7.2 Hz, J′ = 6.8 Hz, 2H, 6-H2), 1.94–2.02 (complex signal, 4H, 2′-H2, 3′-H2), 2.44 (t, J = 6.8 Hz, 2H, 2-H2), 2.71 (br t, J = 5.6 Hz, 2H, 1′-H2), 3.02 (br t, J = 6.0 Hz, 2H, 4′-H2), 3.97 (t, J = 7.2 Hz, 2H, 7-H2), 4.85 (s, +NH, NH), 7.59 (dd, J = 8.4 Hz, J′ = 7.6 Hz, 1H, 7′-H), 7.77 (d, J = 8.8 Hz, 1H, 5′-H), 7.85 (dd, J = 8.8 Hz, J′ = 7.6 Hz, 1H, 6′-H), 8.40 (d, J = 8.4 Hz, 1H, 8′-H); 13C NMR (100.6 MHz, CD3OD) δ 17.2 (CH2, C2), 21.8 (CH2, C3′), 23.0 (CH2, C2′), 24.9 (CH2, C1′), 26.3 (CH2, C3), 26.9 (CH2, C5), 29.30 (CH2), 29.33 (CH2) (C4, C4′), 31.2 (CH2, C6), 49.0 (CH2, C7), 112.9 (C, C9a′), 117.1 (C, C8a′), 120.1 (CH, C5′), 121.1 (C, C1), 126.3 (CH, C7′), 126.5 (CH, C8′), 134.1 (CH, C6′), 139.8 (C, C10a′), 151.7 (C, C4a′), 158.0 (C, C9′); HRMS (ESI), calcd for (C20H25N3+H+) 308.2121, found 308.2117.
4.1.6. 9-[(1,2,3,4-Tetrahydroacridin-9-yl)amino]nonanenitrile (11e)
It was prepared as described for 11c. From 9 (500 mg, 2.52 mmol) and 9-bromononanenitrile (659 mg, 3.02 mmol), a yellowish oily residue (984 mg) was obtained and purified by column chromatography (40–60 μm silica gel, CH2Cl2/MeOH/50% aq NH4OH mixtures, gradient elution). On elution with CH2Cl2/MeOH/50% aq NH4OH 99.9:0.1:0.4 to 99.7:0.3:0.4, nitrile 11e (272 mg, 32% yield) was isolated as a yellow oil; Rf 0.51 (CH2Cl2/MeOH/50% aq NH4OH 9:1:0.1).
11e·HCl: yellow sticky solid; IR (ATR) ν 3500–2500 (max at 3268, 2931, 2848, 2756, 2700, 2667, +NH, NH, CH st), 2241 (CN st), 1630, 1590, 1572, 1522 (Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.34–1.48 (complex signal, 8H, 4-H2, 5-H2, 6-H2, 7-H2), 1.62 (tt, J = J′ = 7.2 Hz, 2H, 3-H2), 1.84 (tt, J = J′ = 7.2 Hz, 2H, 8-H2), 1.94–2.00 (complex signal, 4H, 2′-H2, 3′-H2), 2.42 (t, J = 7.2 Hz, 2H, 2-H2), 2.71 (br t, J = 5.6 Hz, 2H, 1′-H2), 3.02 (br t, J = 6.0 Hz, 2H, 4′-H2), 3.96 (t, J = 7.2 Hz, 2H, 9-H2), 4.85 (s, +NH, NH), 7.59 (ddd, J = 8.4 Hz, J′ = 7.2 Hz, J″ = 1.2 Hz, 1H, 7′-H), 7.75 (br d, J = 7.6 Hz, 1H, 5′-H), 7.85 (ddd, J = 7.6 Hz, J′ = 7.2 Hz, J″ = 1.2 Hz, 1H, 6′-H), 8.40 (br d, J = 8.4 Hz, 1H, 8′-H); 13C NMR (100.6 MHz, CD3OD) δ 17.3 (CH2, C2), 21.8 (CH2, C3′), 23.0 (CH2, C2′), 24.8 (CH2, C1′), 26.3 (CH2, C3), 27.5 (CH2, C7), 29.3 (CH2, C4′), 29.5 (CH2), 29.6 (CH2), 29.9 (CH2) (C4, C5, C6), 31.4 (CH2, C8), 49.1 (CH2, C9), 112.8 (C, C9a′), 117.0 (C, C8a′), 120.1 (CH, C5′), 121.3 (C, C1), 126.3 (CH, C7′), 126.5 (CH, C8′), 134.1 (CH, C6′), 139.8 (C, C10a′), 151.7 (C, C4a′), 158.0 (C, C9′); HRMS (ESI), calcd for (C22H29N3+H+) 336.2434, found 336.2436.
4.1.7. 7-[(6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino]heptanenitrile (12c)53
It was prepared as described for 11c. From 6-chlorotacrine, 10 (2.00 g, 8.59 mmol), and 7-bromoheptanenitrile (90% purity, 1.55 mL, 1.96 g, 9.28 mmol), a yellow oily residue (3.06 g) was obtained. Purification of this residue by column chromatography (40–60 μm silica gel, CH2Cl2/50% aq NH4OH 100:0.2) afforded a 87:13 mixture of dialkylated byproduct and nitrile 12c (789 mg) and pure nitrile 12c (2.06 g, 70% isolated yield) as a yellow oil; Rf 0.92 (CH2Cl2/MeOH/50% aq NH4OH 9:1:0.05).
12c·HCl: yellowish solid; mp 86–87 °C; IR (ATR) ν 3500–2500 (max at 3347, 3138, 3059, 2949, 2928, 2858, 2744, +NH, NH, CH st), 2245 (CN st), 1639, 1605, 1573, 1524 (Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.46–1.54 (complex signal, 4H, 4-H2, 5-H2), 1.66 (tt, J = 7.2 Hz, J′ = 6.8 Hz, 2H, 3-H2), 1.87 (tt, J = J′ = 7.2 Hz, 2H, 6-H2), 1.92–2.02 (complex signal, 4H, 2′-H2, 3′-H2), 2.45 (t, J = 7.2 Hz, 2H, 2-H2), 2.69 (br t, J = 6.0 Hz, 2H, 1′-H2), 3.01 (br t, J = 5.6 Hz, 2H, 4′-H2), 3.96 (t, J = 7.2 Hz, 2H, 7-H2), 4.84 (s, +NH, NH), 7.56 (dd, J = 9.2 Hz, J′ = 2.4 Hz, 1H, 7′-H), 7.79 (d, J = 2.4 Hz, 1H, 5′-H), 8.39 (d, J = 9.2 Hz, 1H, 8′-H); 13C NMR (100.6 MHz, CD3OD) δ 17.2 (CH2, C2), 21.7 (CH2, C3′), 22.9 (CH2, C2′), 24.8 (CH2, C1′), 26.3 (CH2, C3), 26.9 (CH2, C5), 29.29 (CH2), 29.34 (CH2) (C4, C4′), 31.1 (CH2, C6), 49.1 (CH2, C7), 113.4 (C), 115.4 (C) (C8a′, C9a′), 119.1 (CH, C5′), 121.2 (C, C1), 126.8 (CH, C7′), 128.8 (CH, C8′), 140.1 (CH, C6′), 140.5 (C, C10a′), 152.2 (C, C4a′), 157.8 (C, C9′); HRMS (ESI), calcd for (C20H2435ClN3+H+) 342.1732, found 342.1737.
4.1.8. 8-[(6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino]octanenitrile (12d)
It was prepared as described for 11c. From 6-chlorotacrine, 10 (1.00 g, 4.30 mmol), and 8-bromooctanenitrile (1.01 g, 4.95 mmol), a yellow oily residue (1.73 g) was obtained. Purification of this crude by column chromatography (40–60 μm silica gel, CH2Cl2/50% aq NH4OH 100:0.4), afforded nitrile 12d (411 mg, 27% yield) as a yellow oil; Rf 0.80 (CH2Cl2/MeOH/50% aq NH4OH 9:1:0.1).
12d·HCl: yellowish solid; mp 210–213 °C; IR (ATR) ν 3500–2500 (max at 3251, 3052, 2934, 2853, 2711, +NH, NH, CH st), 2246 (CN st), 1633, 1616, 1588, 1567, 1542, 1517 (Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.40–1.52 (complex signal, 6H, 4-H2, 5-H2, 6-H2), 1.63 (tt, J = J′ = 7.2 Hz, 2H, 3-H2), 1.85 (tt, J = J′ = 7.2 Hz, 2H, 7-H2), 1.92–2.00 (complex signal, 4H, 2′-H2, 3′-H2), 2.43 (t, J = 7.2 Hz, 2H, 2-H2), 2.68 (br t, J = 6.0 Hz, 2H, 1′-H2), 3.00 (br t, J = 6.0 Hz, 2H, 4′-H2), 3.95 (t, J = 7.2 Hz, 2H, 8-H2), 4.85 (s, +NH, NH), 7.57 (dd, J = 9.2 Hz, J′ = 2.0 Hz, 1H, 7′-H), 7.77 (d, J = 2.0 Hz, 1H, 5′-H), 8.39 (d, J = 9.2 Hz, 1H, 8′-H); 13C NMR (100.6 MHz, CD3OD) δ 17.2 (CH2, C2), 21.8 (CH2, C3′), 22.9 (CH2, C2′), 24.7 (CH2, C1′), 26.3 (CH2, C3), 27.4 (CH2, C6), 29.36 (CH2), 29.40 (CH2) (C4, C4′), 29.5 (CH2, C5), 31.2 (CH2, C7), 49.2 (CH2, C8), 113.4 (C), 115.5 (C) (C8a′, C9a′), 119.2 (CH, C5′), 121.2 (C, C1), 126.8 (CH, C7′), 128.8 (CH, C8′), 140.1 (CH, C6′), 140.6 (C, C10a′), 152.2 (C, C4a′), 157.8 (C, C9′); HRMS (ESI), calcd for (C21H2635ClN3+H+) 356.1888, found 356.1878.
4.1.9. 9-[(6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino]nonanenitrile (12e)
It was prepared as described for 11c. From 6-chlorotacrine, 10 (500 mg, 2.15 mmol), and 9-bromononanenitrile (561 mg, 2.57 mmol), a yellow oily residue (849 mg) was obtained and purified by column chromatography (40–60 μm silica gel, CH2Cl2/MeOH/50% aq NH4OH mixtures, gradient elution). On elution with CH2Cl2/MeOH/50% aq NH4OH 100:0:0.4 to 99.8:0.2:0.4, nitrile 12e (380 mg, 48% yield) was isolated as a yellow oil; Rf 0.82 (CH2Cl2/MeOH/50% aq NH4OH 9:1:0.1).
12e·HCl: yellowish solid; mp 176–177 °C; IR (ATR) ν 3500–2500 (max at 3248, 3048, 2931, 2852, 2714, +NH, NH, CH st), 2246 (CN st), 1632, 1589, 1566, 1523 (Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.36–1.50 (complex signal, 8H, 4-H2, 5-H2, 6-H2, 7-H2), 1.62 (tt, J = J′ = 7.2 Hz, 2H, 3-H2), 1.84 (tt, J = J′ = 7.2 Hz, 2H, 8-H2), 1.92–2.02 (complex signal, 4H, 2′-H2, 3′-H2), 2.43 (t, J = 7.2 Hz, 2H, 2-H2), 2.68 (br t, J = 6.0 Hz, 2H, 1′-H2), 3.00 (br t, J = 6.4 Hz, 2H, 4′-H2), 3.95 (t, J = 7.2 Hz, 2H, 9-H2), 4.85 (s, +NH, NH), 7.56 (dd, J = 9.2 Hz, J′ = 2.0 Hz, 1H, 7′-H), 7.78 (d, J = 2.0 Hz, 1H, 5′-H), 8.39 (d, J = 9.2 Hz, 1H, 8′-H); 13C NMR (100.6 MHz, CD3OD) δ 17.3 (CH2, C2), 21.8 (CH2, C3′), 22.9 (CH2, C2′), 24.7 (CH2, C1′), 26.4 (CH2, C3), 27.6 (CH2, C7), 29.3 (CH2, C4′), 29.6 (CH2), 29.7 (CH2), 30.0 (CH2) (C4, C5, C6), 31.3 (CH2, C8), 49.2 (CH2, C9), 113.4 (C), 115.5 (C) (C8a′, C9a′), 119.2 (CH, C5′), 121.2 (C, C1), 126.8 (CH, C7′), 128.8 (CH, C8′), 140.1 (CH, C6′), 140.5 (C, C10a′), 152.1 (C, C4a′), 157.9 (C, C9’); HRMS (ESI), calcd for (C22H2835ClN3+H+) 370.2045, found 370.2037.
4.1.10. 1-{7-[(1,2,3,4-Tetrahydroacridin-9-yl)amino]heptyl}guanidine (15c)
It was prepared as described for 6b. From amine 13c (100 mg, 0.32 mmol) and 1H-pyrazole-1-carboxamidine hydrochloride (57 mg, 0.39 mmol), a brownish solid residue (87 mg) was obtained. This crude product was taken in MeOH (2 mL) and eluted through a Dowex™ Marathon™ A OH– anion exchange resin (5 g) using MeOH (500 mL) as the eluent, to provide guanidine 15c (81 mg, 72% yield) as a brownish oil.
15c·2HCl: brownish sticky solid; IR (ATR) ν 3500–2500 (max at 3257, 3132, 2930, 2858, +NH, NH, CH st), 1631, 1574, 1520 (C N, Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.38–1.50 (complex signal, 6H, 3-H2, 4-H2, 5-H2), 1.59 (tt, J = J′ = 6.8 Hz, 2H, 2-H2), 1.86 (tt, J = J′ = 7.2 Hz, 2H, 6-H2), 1.92–2.02 (complex signal, 4H, 2′-H2, 3′-H2), 2.71 (br t, J = 5.2 Hz, 2H, 1′-H2), 3.02 (t, J = 5.6 Hz, 2H, 4′-H2), 3.16 (t, J = 6.8 Hz, 2H, 1-H2), 3.96 (t, J = 7.2 Hz, 2H, 7-H2), 4.85 (s, +NH, +NH2, NH, NH2), 7.59 (ddd, J = 8.4 Hz, J′ = 6.8 Hz, J″ = 1.6 Hz, 1H, 7′-H), 7.76 (dd, J = 8.4 Hz, J′ = 1.6 Hz, 1H, 5′-H), 7.86 (ddd, J = 8.4 Hz, J′ = 6.8 Hz, J″ = 1.2 Hz, 1H, 6′-H), 8.40 (br d, J = 8.4 Hz, 1H, 8′-H); 13C NMR (100.6 MHz, CD3OD) δ 21.8 (CH2, C3′), 23.0 (CH2, C2′), 24.9 (CH2, C1′), 27.57 (CH2), 27.63 (CH2) (C3, C5), 29.3 (CH2, C4′), 29.8 (CH2), 29.9 (CH2) (C2, C4), 31.5 (CH2, C6), 42.4 (CH2, C1), 49.1 (CH2, C7), 112.8 (C, C9a′), 117.0 (C, C8a′), 120.1 (CH, C5′), 126.3 (CH, C7′), 126.5 (CH, C8′), 134.1 (CH, C6′), 139.8 (C, C10a′), 151.7 (C, C4a′), 158.0 (C, C9′), 158.5 (C, guanidine C N); HRMS (ESI), calcd for (C21H31N5+H+) 354.2652, found 354.2665.
4.1.11. 1-{9-[(1,2,3,4-Tetrahydroacridin-9-yl)amino]nonyl}guanidine (15e)
It was prepared as described for 6b. From 13e·HCl (47 mg, 0.12 mmol) and 1H-pyrazole-1-carboxamidine hydrochloride (21 mg, 0.14 mmol), a brownish solid residue (46 mg) was obtained. This crude product was taken in MeOH (2 mL) and eluted through a Dowex™ Marathon™ A OH– anion exchange resin (5 g) using MeOH (500 mL) as the eluent, to provide guanidine 15e (35 mg, 76% yield) as a brownish oil.
15e·2HCl: brownish sticky solid; IR (ATR) ν 3500–2500 (max at 3256, 3132, 2926, 2852, +NH, NH, CH st), 1661, 1631, 1575, 1521 (C N, Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.34–1.48 (complex signal, 10H, 3-H2, 4-H2, 5-H2, 6-H2, 7-H2), 1.58 (tt, J = J′ = 6.8 Hz, 2H, 2-H2), 1.84 (tt, J = J′ = 7.2 Hz, 2H, 8-H2), 1.94–2.00 (complex signal, 4H, 2′-H2, 3′-H2), 2.70 (br t, J = 6.0 Hz, 2H, 1′-H2), 3.02 (br t, J = 6.4 Hz, 2H, 4′-H2), 3.16 (t, J = 6.8 Hz, 2H, 1-H2), 3.95 (t, J = 7.2 Hz, 2H, 9-H2), 4.85 (s, +NH, +NH2, NH, NH2), 7.59 (ddd, J = 8.8 Hz, J′ = 6.8 Hz, J″ = 1.2 Hz, 1H, 7′-H), 7.76 (dd, J = 8.4 Hz, J′ = 1.2 Hz, 1H, 5′-H), 7.85 (ddd, J = 8.4 Hz, J′ = 6.8 Hz, J″ = 1.2 Hz, 1H, 6′-H), 8.40 (br d, J = 8.8 Hz, 1H, 8′-H); 13C NMR (100.6 MHz, CD3OD) δ 21.8 (CH2, C3′), 22.9 (CH2, C2′), 24.9 (CH2, C1′), 27.6 (2CH2, C3, C7), 29.3 (CH2, C4′), 29.8 (CH2, C2), 30.1 (2CH2), 30.4 (CH2) (C4, C5, C6), 31.5 (CH2, C8), 42.4 (CH2, C1), 49.1 (CH2, C9), 112.8 (C, C9a′), 116.9 (C, C8a′), 120.1 (CH, C5′), 126.3 (CH, C7′), 126.4 (CH, C8′), 134.1 (CH, C6′), 139.7 (C, C10a′), 151.6 (C, C4a′), 157.9 (C, C9′), 158.5 (C, guanidine C N); HRMS (ESI), calcd for (C23H35N5+H+) 382.2965, found 382.2975.
4.1.12. 1-{9-[(6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino]nonyl}guanidine (16e)
It was prepared as described for 6b. From 14e·HCl (90 mg, 0.22 mmol) and 1H-pyrazole-1-carboxamidine hydrochloride (39 mg, 0.27 mmol), a brownish solid residue (81 mg) was obtained. This crude product was taken in MeOH (2 mL) and eluted through a Dowex™ Marathon™ A OH− anion exchange resin (5 g) using MeOH (500 mL) as the eluent, to provide guanidine 16e (61 mg, 67% yield) as a brownish oil.
16e·2HCl: brownish solid; mp 209–210 °C; IR (ATR) ν 3500–2500 (max at 3253, 3126, 2928, 2855, +NH, NH, CH st), 1631, 1572, 1514 (C N, Ar–C–C, Ar–C–N st) cm−1; 1H NMR (400 MHz, CD3OD) δ 1.38–1.52 (complex signal, 10H, 3-H2, 4-H2, 5-H2, 6-H2, 7-H2), 1.60 (tt, J = J′ = 6.8 Hz, 2H, 2-H2), 1.86 (tt, J = J′ = 7.2 Hz, 2H, 8-H2), 1.92–2.02 (complex signal, 4H, 2′-H2, 3′-H2), 2.68 (br t, J = 6.0 Hz, 2H, 1′-H2), 3.00 (br t, J = 6.0 Hz, 2H, 4′-H2), 3.17 (t, J = 6.8 Hz, 2H, 1-H2), 3.95 (t, J = 7.2 Hz, 2H, 9-H2), 4.85 (s, +NH, +NH2, NH, NH2), 7.57 (dd, J = 9.2 Hz, J′ = 2.4 Hz, 1H, 7′-H), 7.79 (d, J = 2.4 Hz, 1H, 5′-H), 8.40 (d, J = 9.2 Hz, 1H, 8′-H); 13C NMR (100.6 MHz, CD3OD) δ 21.8 (CH2, C3′), 22.9 (CH2, C2′), 24.8 (CH2, C1′), 27.5 (CH2), 27.6 (CH2) (C3, C7), 29.3 (CH2, C4′), 29.7 (2CH2), 29.8 (2CH2) (C2, C4, C5, C6), 31.3 (CH2, C8), 42.4 (CH2, C1), 49.2 (CH2, C9), 113.3 (C), 115.4 (C) (C8a′, C9a′), 119.1 (CH, C5′), 126.8 (CH, C7′), 128.8 (CH, C8′), 140.0 (CH, C6′), 140.4 (C, C10a′), 152.1 (C, C4a′), 157.7 (C, C9′), 158.6 (C, guanidine C N); HRMS (ESI), calcd for (C23H3435ClN5+H+) 416.2576, found 416.2589.
4.2. Biological assays
4.2.1. T. brucei culturing and evaluation of antitrypanosomal activity
Bloodstream form T. brucei (strain 221) was cultured at 37 °C in modified Iscove’s medium.54 Trypanocidal activity was assessed by growing parasites in the presence of various concentrations of the novel compounds and determining the levels which inhibited growth by 50% (IC50) and 90% (IC90). T. brucei in the logarithmic phase of growth were diluted back to 2.5 × 104 mL−1 and aliquoted into 96-well plates. The compounds were then added at a range of concentrations and the plates incubated at 37 °C. Each drug concentration was tested in triplicate. Resazurin was added after 48 h and the plates incubated for a further 16 h and the plates then read in a Spectramax plate reader. Results were analysed using GraphPad Prism.
4.2.2. Cytotoxic activity against rat skeletal myoblast L6 cells
Cytotoxicity against mammalian cells was assessed using microtitre plates following a described procedure.55 Briefly, rat skeletal muscle L6 cells were seeded at 1 × 104 mL−1 in 200 μL of growth medium containing different compound concentrations. The plates were incubated for 6 days at 37 °C and 20 μL resazurin was then added to each well. After a further 8 h incubation, the fluorescence was determined using a Spectramax plate reader.
4.2.3. Acetylcholinesterase inhibitory activity
The inhibitory activity against Electrophorus electricus (Ee) AChE (Sigma–Aldrich) was evaluated spectrophotometrically by the method of Ellman et al.56 The reactions took place in a final volume of 300 μL of 0.1 M phosphate-buffered solution pH 8.0, containing EeAChE (0.03 U/mL) and 333 μM 5,5′-dithiobis(2-nitrobenzoic) acid (DTNB; Sigma–Aldrich) solution used to produce the yellow anion of 5-thio-2-nitrobenzoic acid. Inhibition curves were performed in duplicates using at least 10 increasing concentrations of inhibitors and preincubated for 20 min at 37 °C before adding the substrate. One duplicate sample without inhibitor was always present to yield 100% of AChE activity. Then substrate, acetylthiocholine iodide (450 μM; Sigma–Aldrich), was added and the reaction was developed for 5 min at 37 °C. The colour production was measured at 414 nm using a labsystems Multiskan spectrophotometer.
Data from concentration−inhibition experiments of the inhibitors were calculated by non-linear regression analysis, using the GraphPad Prism program package (GraphPad Software; San Diego, USA), which gave estimates of the IC50 (concentration of drug producing 50% of enzyme activity inhibition). Results are expressed as mean ± SEM of at least 4 experiments performed in duplicate.
4.2.4. Determination of brain permeability: PAMPA-BBB assay
The in vitro permeability (Pe) of the novel compounds and fourteen commercial drugs through lipid extract of porcine brain membrane was determined by using a parallel artificial membrane permeation assay.45 Commercial drugs and the target compounds were tested using a mixture of PBS/EtOH 70:30. Assay validation was made by comparing experimental and described permeability values of the commercial drugs (Table 3), which showed a good correlation: Pe (exp) = 1.6079 Pe (lit) − 1.2585 (R2 = 0.9217). From this equation and the limits established by Di et al. for BBB permeation, three ranges of permeability were established: compounds of high BBB permeation (CNS+): Pe (10−6 cm s−1) > 5.17; compounds of low BBB permeation (CNS−): Pe (10−6 cm s−1) < 1.95; and compounds of uncertain BBB permeation (CNS±): 5.17 > Pe (10−6 cm s−1) > 1.95.
Table 3.
Reported and experimental permeabilities (Pe 10−6 cm s−1) of 14 commercial drugs used for the PAMPA-BBB assay validation
| Compd | Literature valuea | Experimental valueb |
|---|---|---|
| Cimetidine | 0.0 | 0.7 ± 0.1 |
| Lomefloxacin | 1.1 | 0.7 ± 0.1 |
| Norfloxazin | 0.1 | 0.9 ± 0.1 |
| Ofloxazin | 0.8 | 1.0 ± 0.1 |
| Hydrocortisone | 1.9 | 1.4 ± 0.1 |
| Piroxicam | 2.5 | 2.2 ± 0.1 |
| Clonidine | 5.3 | 6.5 ± 0.1 |
| Corticosterone | 5.1 | 6.7 ± 0.1 |
| Imipramine | 13.0 | 12.3 ± 0.1 |
| Promazine | 8.8 | 13.8 ± 0.3 |
| Progesterone | 9.3 | 16.8 ± 0.3 |
| Desipramine | 12.0 | 17.8 ± 0.1 |
| Testosterone | 17.0 | 26.5 ± 0.4 |
| Verapamil | 16.0 | 28.4 ± 0.5 |
Taken from Ref. 45.
Values are expressed as the mean ± SD of three independent experiments.
Acknowledgments
This work was supported by Ministerio de Economía y Competitividad (MINECO) (SAF2014-57094-R) and Generalitat de Catalunya (GC) (2014SGR52). J.M.K. acknowledges funding support from the Wellcome Trust (Grant number WT092573MA). Fellowships from GC to I.S., F.J.P.-A., and E.V. are gratefully acknowledged.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2016.08.036.
Supplementary data
Spectral data.
References and notes
- 1.Centers for Disease Control and Prevention, Neglected Tropical Diseases, http://www.cdc.gov/globalhealth/ntd/. Accessed June 15th 2016.
- 2.Hotez P.J., Fenwick A., Savioli L., Molyneux D.H. Lancet. 2009;373:1570. doi: 10.1016/S0140-6736(09)60233-6. [DOI] [PubMed] [Google Scholar]
- 3.Chibale K. Bioorg. Med. Chem. 2015;23:5085. doi: 10.1016/j.bmc.2015.04.077. [DOI] [PubMed] [Google Scholar]
- 4.Njoroge M., Njuguna N.M., Mutai P., Ongarora D.S.B., Smith P.W., Chibale K. Chem. Rev. 2014;114:11138. doi: 10.1021/cr500098f. [DOI] [PubMed] [Google Scholar]
- 5.Woodland A., Grimaldi R., Luksch T., Cleghorn L.A.T., Ojo K.K., Van Voorhis W.C., Brenk R., Frearson J.A., Gilbert I.H., Wyatt P.G. ChemMedChem. 2013;8:1127. doi: 10.1002/cmdc.201300072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization, Trypanosomiasis, human African (sleeping sickness), http://www.who.int/mediacentre/factsheets/fs259/en/, accessed on July 5th 2016.
- 7.Horn D. Mol. Biochem. Parasitol. 2014;195:123. doi: 10.1016/j.molbiopara.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrett M.P., Croft S.L. Br. Med. Bull. 2012;104:175. doi: 10.1093/bmb/lds031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mallari J.P., Shelat A.A., Obrien T., Caffrey C.R., Kosinski A., Connelly M., Harbut M., Greenbaum D., McKerrow J.H., Guy R.K. J. Med. Chem. 2008;51:545. doi: 10.1021/jm070760l. [DOI] [PubMed] [Google Scholar]
- 10.Clark R.L., Clements C.J., Barret M.P., Mackay S.P., Rathman R.P., Owusu-Dapaah G., Spencer J., Huggan J.K. Biooorg. Med. Chem. 2012;20:6019. doi: 10.1016/j.bmc.2012.08.049. [DOI] [PubMed] [Google Scholar]
- 11.Mäser P., Wittlin S., Rottmann M., Wenzler T., Kaiser M., Brun R. Curr. Opin. Pharmacol. 2012;12:562. doi: 10.1016/j.coph.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Shi P.-Y., Smith P.W., Diagana T.T. ACS Infect. Dis. 2015;1:76. doi: 10.1021/id500043r. [DOI] [PubMed] [Google Scholar]
- 13.Carrillo A.K., Guiguemde W.A., Guy R.K. Bioorg. Med. Chem. 2015;23:5151. doi: 10.1016/j.bmc.2014.12.066. [DOI] [PubMed] [Google Scholar]
- 14.Amata E., Bland N.D., Hoyt C.T., Settimo L., Campbell R.K., Pollastri M.P. Bioorg. Med. Chem. Lett. 2014;24:4084. doi: 10.1016/j.bmcl.2014.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagle A.S., Khare S., Babu Kumar A., Supek F., Buchynskyy A., Mathison C.J.N., Kumar Chennamaneni N., Pendem N., Buckner F.S., Gelb M.H., Molteni V. Chem. Rev. 2014;114:11305. doi: 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prati F., Uliassi E., Bolognesi M.L. Med. Chem. Commun. 2014;5:853. [Google Scholar]
- 17.Lizzi F., Veronesi G., Belluti F., Bergamini C., López-Sánchez A., Kaiser M., Brun R., Krauth-Siegel R.L., Hall D.G., Rivas L., Bolognesi M.L. J. Med. Chem. 2012;55:10490. doi: 10.1021/jm301112z. [DOI] [PubMed] [Google Scholar]
- 18.Urich R., Grimaldi R., Luksch T., Frearson J.A., Brenk R., Wyatt P.G. J. Med. Chem. 2014;57:7536. doi: 10.1021/jm500239b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fairlamb A.H. Nature. 2012;482:167. doi: 10.1038/482167a. [DOI] [PubMed] [Google Scholar]
- 20.Patrick D.A., Wenzler T., Yang S., Weiser P.T., Wang M.Z., Brun R., Tidwell R.R. Bioorg. Med. Chem. 2016;24:2451. doi: 10.1016/j.bmc.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cleghorn L.A.T., Albrecht S., Stojanovski L., Simeons F.R.J., Norval S., Kime R., Collie I.T., De Rycker M., Campbell L., Hallyburton I., Frearson J.A., Wyatt P.G., Read K.D., Gilbert I.H. J. Med. Chem. 2015;58:7695. doi: 10.1021/acs.jmedchem.5b00596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woodland A., Thompson S., Cleghorn L.A.T., Norcross N., De Rycker M., Grimaldi R., Hallyburton I., Rao B., Norval S., Stojanovski L., Brun R., Kaiser M., Frearson J.A., Gray D.W., Wyatt P.G., Read K.D., Gilbert I.H. ChemMedChem. 2015;10:1809. doi: 10.1002/cmdc.201500300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llurba-Montesino N., Kaiser M., Brun R., Schmidt T.J. Molecules. 2015;20:14118. doi: 10.3390/molecules200814118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Annang F., Pérez-Moreno G., García-Hernández R., Cordon-Obras C., Martín J., Tormo J.R., Rodríguez L., de Pedro N., Gómez-Pérez V., Valente M., Reyes F., Genilloud O., Vicente F., Castanys S., Ruiz-Pérez L.M., Navarro M., Gamarro F., González-Pacanowska D. J. Biomol. Screening. 2015;20:82. doi: 10.1177/1087057114555846. [DOI] [PubMed] [Google Scholar]
- 25.Tatipaka H.B., Gillespie J.R., Chatterjee A.K., Norcross N.R., Hulverson M.A., Ranade R.M., Nagendar P., Creason S.A., McQueen J., Duster N.A., Nagle A., Supek F., Molteni V., Wenzler T., Brun R., Glynne R., Buckner F.S., Gelb M.H. J. Med. Chem. 2014;57:828. doi: 10.1021/jm401178t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavalli A., Lizzi F., Bongarzone S., Belluti F., Piazzi L., Bolognesi M.L. FEMS Immunol. Med. Microbiol. 2010;58:51. doi: 10.1111/j.1574-695X.2009.00615.x. [DOI] [PubMed] [Google Scholar]
- 27.Singh Grewal A., Pandita D., Bhardwaj S., Lather V. Curr. Top. Med. Chem. 2016;16:2245. doi: 10.2174/1568026616666160413125335. [DOI] [PubMed] [Google Scholar]
- 28.Camps P., El Achab R., Morral J., Muñoz-Torrero D., Badia A., Baños J.E., Vivas N.M., Barril X., Orozco M., Luque F.J. J. Med. Chem. 2000;43:4657. doi: 10.1021/jm000980y. [DOI] [PubMed] [Google Scholar]
- 29.Defaux J., Sala M., Formosa X., Galdeano C., Taylor M.C., Alobaid W.A.A., Kelly J.M., Wright C.W., Camps P., Muñoz-Torrero D. Bioorg. Med. Chem. 2011;19:1702. doi: 10.1016/j.bmc.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oluwafemi A.J., Okanla E.O., Camps P., Muñoz-Torrero D., Mackey Z.B., Chiang P.K., Seville S., Wright C.W. Nat. Prod. Commun. 2009;4:193. [PubMed] [Google Scholar]
- 31.Kaur K., Jain M., Reddy R.P., Jain R. Eur. J. Med. Chem. 2010;45:3245. doi: 10.1016/j.ejmech.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 32.Gomes A., Pérez B., Alburquerque I., Machado M., Prudêncio M., Nogueira F., Teixeira C., Gomes P. ChemMedChem. 2014;9:305. doi: 10.1002/cmdc.201300459. [DOI] [PubMed] [Google Scholar]
- 33.Ryckebusch A., Debreu-Fontaine M.-A., Mouray E., Grellier P., Sergheraert C., Melnyk P. Bioorg. Med. Chem. Lett. 2005;15:297. doi: 10.1016/j.bmcl.2004.10.080. [DOI] [PubMed] [Google Scholar]
- 34.Ekoue-Kovi K., Yearick K., Iwaniuk D.P., Natarajan J.K., Alumasa J., de Dios A.C., Roepe P.D., Wolf C. Bioorg. Med. Chem. 2009;17:270. doi: 10.1016/j.bmc.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musonda C.C., Gut J., Rosenthal P.J., Yardley V., Carvalho de Souza R.C., Chibale K. Bioorg. Med. Chem. 2006;14:5605. doi: 10.1016/j.bmc.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 36.Sola I., Artigas A., Taylor M.C., Gbedema S.Y., Pérez B., Clos M.V., Wright C.W., Kelly J.M., Muñoz-Torrero D. Bioorg. Med. Chem. Lett. 2014;24:5435. doi: 10.1016/j.bmcl.2014.10.025. [DOI] [PubMed] [Google Scholar]
- 37.Sola I., Castellà S., Viayna E., Galdeano C., Taylor M.C., Gbedema S.Y., Pérez B., Clos M.V., Jones D.C., Fairlamb A.H., Wright C.W., Kelly J.M., Muñoz-Torrero D. Bioorg. Med. Chem. 2015;23:5156. doi: 10.1016/j.bmc.2015.01.031. [DOI] [PubMed] [Google Scholar]
- 38.Viayna E., Sola I., Bartolini M., De Simone A., Tapia-Rojas C., Serrano F.G., Sabaté R., Juárez-Jiménez J., Pérez B., Luque F.J., Andrisano V., Clos M.V., Inestrosa N.C., Muñoz-Torrero D. J. Med. Chem. 2014;57:2549. doi: 10.1021/jm401824w. [DOI] [PubMed] [Google Scholar]
- 39.Patrick D.A., Bakunov S.A., Bakunova S.M., Kumar E.V.K.S., Chen H., Kilgore Jones S., Wenzler T., Barzcz T., Werbovetz K.A., Brun R., Tidwell R.R. Eur. J. Med. Chem. 2009;44:3543. doi: 10.1016/j.ejmech.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 40.Wenzler T., Yang S., Patrick D.A., Braissant O., Ismail M.A., Tidwell R.R., Boykin D.W., Wang M.Z., Brun R. Antimicrob. Agents Chemother. 2014;58:4452. doi: 10.1128/AAC.02309-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlier P.R., Chow E.S.-H., Han Y., Liu J., El Yazal J., Pang Y.-P. J. Med. Chem. 1999;42:4225. doi: 10.1021/jm990224w. [DOI] [PubMed] [Google Scholar]
- 42.Löser R., Schilling K., Dimmig E., Gütschow M. J. Med. Chem. 2005;48:7688. doi: 10.1021/jm050686b. [DOI] [PubMed] [Google Scholar]
- 43.Sykes M.L., Avery V.M. J. Med. Chem. 2013;56:7727. doi: 10.1021/jm4004279. [DOI] [PubMed] [Google Scholar]
- 44.Havrylyuk D., Zimenkovsky B., Karpenko O., Grellier P., Lesyk R. Eur. J. Med. Chem. 2014;85:245. doi: 10.1016/j.ejmech.2014.07.103. [DOI] [PubMed] [Google Scholar]
- 45.Di L., Kerns E.H., Fan K., McConnell O.J., Carter G.T. Eur. J. Med. Chem. 2003;38:223. doi: 10.1016/s0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 46.Reid C.M., Ebikeme C., Barret M.P., Patzewitz E.-M., Müller S., Robins D.J., Sutherland A. Biooog. Med. Chem. Lett. 2008;18:5399. doi: 10.1016/j.bmcl.2008.09.047. [DOI] [PubMed] [Google Scholar]
- 47.McKeever C., Kaiser M., Rozas I. J. Med. Chem. 2013;56:700. doi: 10.1021/jm301614w. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez J.L., Stephens C.E., Wenzler T., Brun R., Tanious F.A., Wilson W.D., Barszcz T., Werbovetz K.A., Boykin D.W. Eur. J. Med. Chem. 2007;42:552. doi: 10.1016/j.ejmech.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 49.Arafa R.K., Wenzler T., Brun R., Chai Y., Wilson W.D. Eur. J. Med. Chem. 2011;46:5852. doi: 10.1016/j.ejmech.2011.09.047. [DOI] [PubMed] [Google Scholar]
- 50.Anandan S.K., Webb H.K., Do Z.N., Gless R.D. Bioorg. Med. Chem. Lett. 2009;19:4259. doi: 10.1016/j.bmcl.2009.05.102. [DOI] [PubMed] [Google Scholar]
- 51.Muñoz-Ruiz P., Rubio L., García-Palomero E., Dorronsoro I., del Monte-Millán M., Valenzuela R., Usán P., de Austria C., Bartolini M., Andrisano V., Bidon-Chanal A., Orozco M., Luque F.J., Medina M., Martínez A. J. Med. Chem. 2005;48:7223. doi: 10.1021/jm0503289. [DOI] [PubMed] [Google Scholar]
- 52.Carlier P.R., Du D.-M., Han Y., Liu J., Pang Y.-P. Bioorg. Med. Chem. Lett. 1999;9:2335. doi: 10.1016/s0960-894x(99)00396-0. [DOI] [PubMed] [Google Scholar]
- 53.Muñoz-Torrero, D.; Ferrer, I.; Sola, I.; Aso, E. EP 2818467 A1; WO 2014/206877 A1.
- 54.Wilkinson S.R., Prathalingam S.R., Taylor M.C., Ahmed A., Horn D., Kelly J.M. Free Radical Biol. Med. 2006;40:198. doi: 10.1016/j.freeradbiomed.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 55.Bot C., Hall B.S., Bashir N., Taylor M.C., Helsby N.A., Wilkinson S.R. Antimicrob. Agents Chemother. 2010;54:4246. doi: 10.1128/AAC.00800-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellman G.L., Courtney K.D., Andres V., Jr., Featherstone R.M. Biochem. Pharmacol. 1961;7:88. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectral data.