

Abstract
Bacterial communities of rhizospheric soils play an important role in the tolerance and uptake of metal-tolerant/hyperaccumulating plants to metals, e.g. the Cu-tolerant Elsholtzia splendens native to China. In this work, pyrosequencing of the bacterial 16S rRNA gene was firstly applied to investigate the rhizospheric bacterial community of E. splendens grown at Cu contaminated sites. The 47 phyla including 11 dominant phyla (>1%) in E. splendens rhizosphere were presented. The effects of Cu and other environmental factors (total organic carbon, total nitrogen and pH) on the rhizospheric bacterial community were studied comprehensively. The phyla abundances were affected by the environmental factors to different extent, and we found pH, instead of Cu concentration, influenced UniFrac distance significantly and was identified as the most important environmental factor affecting bacterial community. In addition, the influence of environmental factors on gene profiles was explored according to the predicted metagenomes obtained by PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states). Our study illustrates a view about Cu-tolerant E. splendens rhizospheric bacterial communities (composition, diversity and gene profiles) and their influencing factors, giving a hand for the understanding on bacterial community is formed and affected in rhizosphere.
Soil contamination by metals has been of a great concern on a global scale because of their threats to the environment, food security and human health. Cu is one of these metals, although it is an essential micronutrient for normal plant growth and development, which can impair and damage plant growth when exceeding the optimal concentration1,2. Various physical and chemical technologies have been developed to remediate soils contaminated with metals, and phytoremediation is becoming increasingly popular because of its economic, energy-efficient, and environmentally friendly features3. Elsholtzia splendens, native to China, has gained special attention due to its high tolerance to Cu4. Studies have revealed the capability of E. splendens to enhance metal solubility via a root-bacteria interaction, absorb metals with metal transporters, and detoxify metals by chelating and distributing them to the apoplast5,6,7.
Rhizospheric bacteria are crucial for the success of phytoremediation. Normally, the low solubility and availability of metals in most soils has limited the performance of phytoremediation. Rhizospheric bacteria can improve phytoremediation through altering metal bioavailability by changing the environmental pH, producing chelating substances and altering redox potentials8. Additionally, bacteria can increase metal removal efficiency via promoting plant growth by synthetizing plant hormones, secreting enzymes, producing siderophores and solubilizing phosphate in soils9. Research on E. splendens rhizospheric bacteria reveals that Cu-tolerant bacteria help in enhancing soil Cu solubility in E. splendens rhizosphere, and inoculating roots with bacteria almost doubles the shoot Cu concentration10,11. Therefore, bacterial communities within rhizosphere, especially the metal-tolerant plants such as E. splendens, become attractive and numerous researches attempt to elucidate their influencing factors.
Metals can alter bacterial abundance, community structure and diversity12,13. Smit et al. found that Cu affects the bacterial composition and reduces the microbial diversity severely by comparing the patterns of Amplified Ribosomal DNA Restriction Analysis (ARDRA) between clean and contaminated soils14. Though recovered to some extent attributing to microbial adaptation, the alteration of bacterial community composition and declining diversity are linked to metals in long-term contaminated soils15,16,17. The laboratory-based work suggested different results led by the metal-tolerant plant rhizospheric environment that E. splendens has higher capability than the non-Cu-accumulator Trifolium repens to maintain rhizospheric bacterial activities and composition18. Nevertheless, the laboratory investigation involves the conditions differing considerably from real situations in the field19. Besides, though several Cu-tolerate bacteria have been isolated from E. splendens rhizosphere, such as Actinobacteria, Firmicutes and Proteobacteria, the relationship between Cu and rhizospheric microbes still remains unclear18,20. To date, few studies give a comprehensive view regarding the influence of Cu on the composition, diversity and gene abundance of the rhizospheric bacterial community associated with E. splendens growing in situ at Cu mine sites.
Other environmental factors in soils, such as total organic carbon (TOC), total nitrogen (TN) and pH, also affect indigenous bacterial community profiles. For example, pH explains the varying diversity and richness of soil bacterial communities in two ecosystems across North and South America (r2 = 0.70 and r2 = 0.58)21; carbon resources and nitrogen depositions alter bacterial communities and form diversity patterns22,23,24. However, limited information about the roles of these individual environmental factors on E. splendens rhizospheric bacterial community is available at Cu mine sites, although their impacts on bacterial community derived from a hyperaccumulating plant rhizosphere have been investigated in several studies25,26,27.
In this study, we sampled 21 soils with different Cu concentrations from three provinces in China with the aim to illustrate a comprehensive picture about E. splendens rhizospheric bacterial communities including composition, diversity and gene profiles, and to disclose which environmental factor is the most important one influencing rhizospheric bacterial community. Tag pyrosequencing of the V4 region of the 16S rRNA gene was performed to investigate bacterial community composition and provided much more detailed insights. The metagenomic content and abundance of gene families in 21 samples were predicted using PICRUSt method28. These data allow our more comprehensive understanding on not only the gene profiles in rhizospheric soils but also the relationship between them and environmental factors. According to our knowledge, this is the first time investigating the bacterial community within E. splendens rhizosphere in situ at Cu mine sites, which extends our horizons on the rhizospheric bacteria of metal-tolerant plants.
Results
Soil physicochemical characteristics
The soil samples covered a wide range of both total and extractable Cu concentrations, ranging from 728 ± 24 (Mean ± S.D.) to 5,300 ± 430 mg kg−1 and 5.19 ± 0.13 to 92.1 ± 1.03 mg kg−1, respectively (Supplementary Table S1). All soil samples were of low fertility with TOC < 8.80‰ and TN < 1.17‰ (Supplementary Table S1). The pH values varied from 3.7 to 6.8 with huge differences. No significant relationship between pH and extractable Cu concentrations (p = 0.936) was discovered.
Composition and diversity of rhizospheric bacterial communities
After removing low-quality sequences, 1,706,691 sequences were generated from 21 samples, with an average of 81,271 ± 15,318 (Mean ± S.D.) and a range from 51,403 to 106,985 sequences per sample. Based on the classifiable sequences, 47 phyla of bacteria were identified. The major lineages (>1%) of total sequences were in the following order: Proteobacteria (26.7%) > Actinobacteria (13.9%) > Chloroflexi (10.3%)> Verrucomicrobia (9.80%)> Acidobacteria (9.52%)> Bacteroidetes (6.43%)> Planctomycetes (4.28%)> Cyanobacteria (3.54%)> Firmicutes (3.53%)> Gemmatimonadetes (2.17%)> TM7 (1.60%) (Fig. 1). Moreover, 5.04% of sequences were classified as unknown phyla affiliated with bacteria. The rare phyla (abundance <1%) contained <3.09% of the total sequences.
Figure 1. Relative abundances (%) of 11 dominant bacterial phyla across all soil samples.
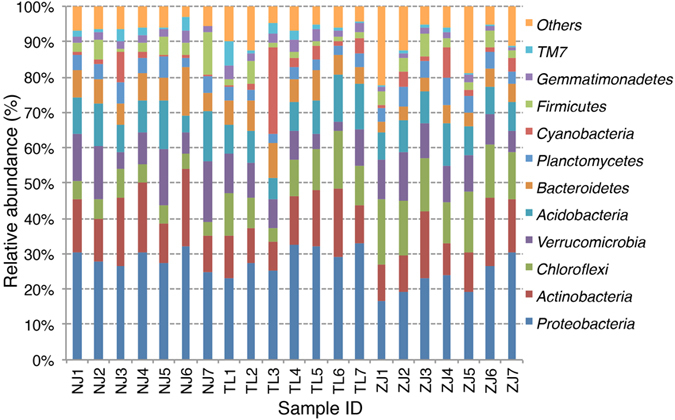
Others composed of 36 rare phyla (<1%) and unclassified bacteria.
When grouped at a 97% similarity level, 34,214 OTUs were observed across all the samples with a range from 5,792 to 9,144 OTUs per sample. The α-diversity indices (Chao1, Shannon and Simpson) and the observed species were calculated and listed in Table 1. The Chao1, Shannon index and observed species ranged from 8,561 to 12,175, 8.79 to 10.71, and 5,792 to 9,144, respectively. Owing to the high depth of Illumina sequencing, the Simpson index had an average value of 0.9926, nearly 1. To estimate the sequencing coverage, rarefaction curves were plotted (Supplementary Figure S1) based on observed OTUs. The β-diversity (weighted Unifrac distance), representing the bacterial community similarity among samples, was shown in the supplementary (Supplementary Figure S2).
Table 1. The α-diversity indices across all samples.
| Chao1 | Shannon | Simpson | Observed species | |
|---|---|---|---|---|
| NJ1 | 12175 | 10.52 | 1.00 | 8606 |
| NJ2 | 11875 | 10.57 | 0.99 | 9144 |
| NJ3 | 10912 | 9.99 | 0.99 | 7429 |
| NJ4 | 12018 | 10.20 | 0.99 | 8988 |
| NJ5 | 11352 | 10.72 | 1.00 | 9103 |
| NJ6 | 10270 | 9.26 | 0.99 | 6673 |
| NJ7 | 10860 | 9.92 | 0.99 | 8554 |
| TL1 | 9575 | 9.90 | 0.99 | 7072 |
| TL2 | 9922 | 9.86 | 0.99 | 7027 |
| TL3 | 9198 | 8.79 | 0.97 | 7282 |
| TL4 | 11103 | 9.82 | 1.00 | 7285 |
| TL5 | 11389 | 10.00 | 1.00 | 8186 |
| TL6 | 9283 | 9.59 | 0.99 | 6264 |
| TL7 | 9407 | 10.02 | 0.99 | 6404 |
| ZJ1 | 9254 | 9.41 | 0.99 | 6560 |
| ZJ2 | 10801 | 10.16 | 1.00 | 7497 |
| ZJ3 | 11204 | 10.28 | 1.00 | 8107 |
| ZJ4 | 12209 | 10.45 | 1.00 | 8966 |
| ZJ5 | 9149 | 9.96 | 0.99 | 6697 |
| ZJ6 | 11307 | 10.57 | 1.00 | 8093 |
| ZJ7 | 8562 | 9.96 | 0.99 | 5792 |
Relationship between environmental factors and bacterial community composition and diversity
Five of the eleven major phyla (>1%) were linearly correlated with Cu or other individual environmental factors (p < 0.05). Proteobacteria, the most abundant phylum across all soil samples, showed significantly positive correlations with total Cu (Fig. 2a, r2 = 0.205, p = 0.039), extractable Cu (Fig. 2b, r2 = 0.214, p = 0.035) and pH (Fig. 2c, r2 = 0.205, p = 0.039). The relative abundance of Proteobacteria increased with the Cu concentration and pH value. Chloroflexi had a positive relationship with TOC (Fig. 2d, r2 = 0.299, p = 0.010) and TN (Fig. 2e, r2 = 0.283, p = 0.013), indicating its sensitivity to nutrition. Firmicutes was negatively correlated with TOC/TN (Fig. 2f, r2 = 0.192, p = 0.047), and Verrucomicrobia showed a negative correlation with TN (Fig. 2g, r = 0.269, p = 0.016), TOC (Fig. 2h, r2 = 0.332, p = 0.006) and TOC/TN (Fig. 2i, r2 = 0.197, p = 0.044), respectively. Besides, pH influenced Gemmatimonadetes significantly (Fig. 2j, r2 = 0.286, p = 0.012). No significant impact was identified when analyzing the relationships between the OTUs in these five phyla and environmental factors based on canonical correspondence analysis (CCA).
Figure 2.

Relationship between five major lineages phyla abundance and environmental factors (p < 0.05): (a) Proteobacteria against total Cu, (b) Proteobacteria against extractable Cu, (c) Proteobacteria against pH, (d) Chloroflexi against TOC, (e) Chloroflexi against TN, (f) Firmicutes against TOC/TN, (g) Verrucomicrobia against TN, (h) Verrucomicrobia against TOC, (i) Verrucomicrobia against TOC/TN, and (j) Gemmatimonadetes against pH.
All the tested environmental factors clearly affected the bacterial community composition and multivariate regression tree (MRT) was used to reveal the most influential factor. The results suggested that pH was the most sensitive predictor of the relative abundance (Fig. 3), consistent with the conclusion from our Pearson’s correlation analysis that two phyla, Proteobacteria (the most abundance phylum) and Gemmatimonadetes, were strongly affected by pH. The ratio of TOC/TN, relating to Verrucomicrobia and Firmicutes, was identified as the second major environmental factor and showed a powerful influence on bacterial community composition. The average phyla abundances of each split in MRT nodes were presented in Table 2 and their variance explanation was exhibited in Fig. 3. Parallelly, the results of CCA showed that pH (p = 0.004) and TOC/TN (p = 0.040) explained the bacterial community most (Fig. 4).
Figure 3. Multivariate regression tree (MRT) analysis of the relation between relative abundance of 11 dominant phyla and environmental factors.
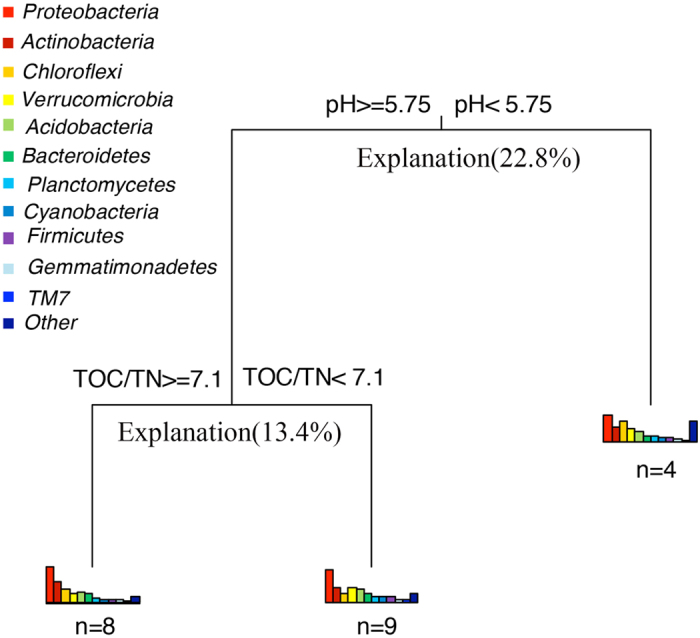
Bars plotted under each cluster represent the relative abundance of each phylum. The distribution patterns of relative abundance represent the dynamics of community composition among each split. The numbers under the bars are the number of samples in each group.
Table 2. The average phyla abundance of each node in MRT.
| Split condition | Split 1 |
Split 2 |
||
|---|---|---|---|---|
| pH> = 5.75 | pH < 5.75 | TOC/TN> = 7.1 | TOC/TN < 7.1 | |
| Proteobacteria | 0.28 | 0.21 | 0.29 | 0.27 |
| Actinobacteria | 0.14 | 0.12 | 0.17 | 0.12 |
| Chloroflexi | 0.09 | 0.16 | 0.11 | 0.07 |
| Verrucomicrobia | 0.10 | 0.10 | 0.07 | 0.12 |
| Acidobacteria | 0.10 | 0.08 | 0.09 | 0.10 |
| Bacteroidetes | 0.07 | 0.04 | 0.07 | 0.07 |
| Planctomycetes | 0.04 | 0.05 | 0.04 | 0.04 |
| Cyanobacteria | 0.04 | 0.03 | 0.03 | 0.04 |
| Firmicutes | 0.04 | 0.03 | 0.03 | 0.05 |
| Gemmatimonadetes | 0.02 | 0.01 | 0.03 | 0.02 |
| TM7 | 0.02 | 0.01 | 0.02 | 0.02 |
| Other | 0.06 | 0.16 | 0.05 | 0.07 |
Figure 4. Canonical correspondence analysis (CCA) of bacterial community composition and environmental factors.
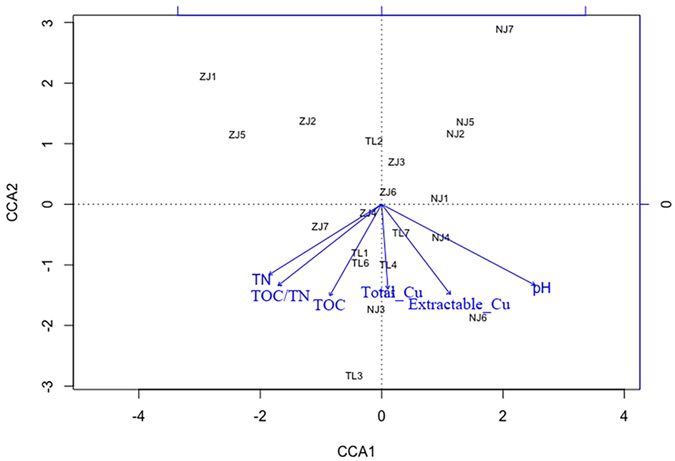
TOC/TN and pH are significantly related to bacterial community composition.
The relationship between bacterial α-diversity and environmental factors was interpreted in Supplementary Table S2. No obvious correlation was detected except for the observed species negatively correlated with TOC/TN. The Mantel test by assessing the correlation between UniFrac distance and environmental factors distance matrix, which was used in this study to estimate the phylogenetic distance affected by environmental factors, indicated that pH value was the most important factor in bacterial evolution (r2 = 0.246, p = 0.005). Meanwhile, no other environmental factor was related to the bacterial evolutionary distance.
Metagenome prediction and the relationship between environmental factors and functional gene profiles
PICRUSt approach was applied to infer the metagenomic content of the samples, and to evaluate the functional potential of the bacterial community’s metagenome from its 16S profile. Although this method is limited by the number of available genomes, it has been shown to replicate metagenomes to a high degree of accuracy28. The Nearest Sequenced Taxon Index (NSTI) quantified the availability of nearby genome representatives for each sample, and its value was 0.17 ± 0.02 (Mean ± S.D.) in Langille’s study. Our work showed a similar NSTI value (0.17 ± 0.01, Supplementary Table S3), indicating that enough and closely related reference genomes were available for the dataset. Based on the predicted metagenomes, 41 of level 2 KEGG Orthology (KO) groups were found and the gene families belonging to amino acid metabolism, carbohydrate metabolism and membrane transport were identified as the major gene families (Fig. 5). To elucidate the relationship between environmental factors and gene families, the Pearson’s correlation was performed. Total Cu was negatively related with carbohydrate (p = 0.042) and lipid (p = 0.012) metabolism gene abundance. With the decreasing pH value, the gene abundances of “biosynthesis of secondary metabolites” (p = 0.010), “signaling molecules and interaction” (p = 0.001) and “transcription” (p = 0.019) increased significantly.
Figure 5. Gene profiles of bacterial community in E. splendens rhizosphere predicted using PICRUSt.

Discussion
In this study, we presented the composition of bacterial community in E. splendens rhizosphere at Cu mine sites. The community composition (Fig. 1) provided much deeper information in situ comparing with previous studies. Although the laboratory research gave us some insight in bacterial composition in E. splendens rhizosphere18, no more than 19 bacterial species were identified due to the limited resolution of denatured gradient gel electrophoresis (DGGE). Here, with the help of pyrosequencing, 47 phyla were found and the abundance of 11 phyla was larger than 1%. The results indicated that the bacterial communities in E. splendens rhizosphere are quite diversified and more complicated than we detected before18.
To understand the relationship between environmental factors and bacterial community in E. splendens rhizosphere, the Pearson’s correlation analysis illustrated the increasing abundance of Proteobacteria with the Cu concentration, showing the capability of the bacteria affiliated to this phylum to tolerate Cu contamination in rhizosphere. This tolerance may be attributed to an array of metal transport systems in Proteobacteria, as reported in a deep-sea genome analysis29. In addition, several isolated integrons and plasmids are found to be broadly disseminated among Proteobacteria30,31, which allows the resistance genes transfer from one bacterium to another. Such horizontal gene transfer mechanisms might explain the increasing abundance of Proteobacteria with Cu pressure.
Surprisingly, the present study found weak influence of Cu on rhizospheric bacterial community, different from previous studies. Here, MRT and CCA illustrated limited effects of Cu on the change of bacterial community composition (Figs 3 and 4). There was also no relationship discovered between Cu concentration and bacterial diversity, including α- and β-diversity. Additionally, the rarefaction curve (Supplementary Figure S1) also revealed the high diversity level, which suggested that Cu does not have significant impacts on rhizospheric bacterial community diversity. While, previous researches reported that metals can influence the bacterial community composition greatly32. High concentrations of metals decrease the Chao133 and Shannon index34, and alter the Simpson index35. Comparing to these studies, the unique feature of our samples, E. splendens rhizosphere, may explain the difference. We speculated that E. splendens is responsible for mitigating the effects of Cu on the rhizospheric bacterial community composition and diversity in heavily Cu-contaminated soils. Firstly, E. splendens decreases the concentration of extractable Cu in rhizospheric soils due to the direct uptake of roots36. Secondly, E. splendens may reduce the free Cu2+ activity via bonding Cu with the higher dissolved organic carbon (DOC) in the rhizosphere37, consequently reducing Cu toxicity. In addition to our study, it is also revealed by other studies that plants exhibit the ability to harbor bacterial composition and diversity in metals contaminated soils25. Sedum alfredii, a Zn/Cd-hyperaccumulator native to China, possesses more bacteria, actinomycetes and fungi in its rhizosphere compared with bulk soils25. Besides, it is noted that the low pH does not improve the extractable Cu concentration in our study, which may also be explained by the rhizospheric effects of E. splendens. Beyond that, horizontal gene transfer and local adaptation may also explain the little influence of Cu on the rhizospheric bacterial community composition and diversity in this study. The change of bacterial community and loss of bacterial diversity might be compensated by the horizontal gene transfer in metal resistance38, especially when long-term metal-contamination provides bacteria more time and spatial opportunities for local adaptation to the metal stress39. Proteobacteria, Actinobacteria and Chloroflexi, with strong resistance to metals in different studies40,41, were the three most abundant phyla (>10%) in this study, further supporting our hypothesis. Additionally, bacteria can release exopolymers into soils, which are strongly bound to the metals and decrease their toxicity, especially Cu42. All the above reasons result in the tiny change of bacterial diversity in E. splendens rhizosphere and the weak influence on bacterial community composition of heavy Cu contamination. However, Cu still maintains its toxicity on bacteria with active carbohydrate and lipid metabolism by lipid peroxidation and inhibiting the carbohydrate metabolism enzymes43, which is presented in the weak part of bacteria under Cu stress.
Soil pH, which is observed to be a key factor for the construction of soil bacterial communities, affects the phyla abundance (Fig. 2c,j), bacterial community profiles, diversity and metagenomic content in E. splendens rhizosphere. This is a foreseeable result and other studies hold the same opinion44,45,46. Soil pH can directly change the physiological status of indigenous bacteria, alter their ecological niches, and reduce the abundance of individual species that are difficult to survive in soil with unsuitable pH44. Besides, pH also indirectly influences bacterial community by regulating soil nutrient bioavailability, affecting the TOC/TN ratio of soil organic matters, altering nitrification efficiency, changing plant primary productivity, and impacting the mobility and sorption of metals47,48,49. These features explain the reason why pH is such powerful in affecting bacterial community in E. splendens rhizosphere.
Methods
Soil sample collection
Twenty-one E. splendens rhizospheric soil samples with different Cu concentrations were collected from three Chinese Cu mines located in Tongling city in Anhui Province (TL) (30°54′1″N, 117°49′15″E), Zhuji city in Zhejiang Province (ZJ) (29°43′23″N, 119°59′9″E), and Nanjing city in Jiangsu Province (NJ) (32°4′24″N, 119°5′33″E) in November, 2013 (Supplementary Figure S3). The Cu-accumulation ability of E. splendens in these three provinces had been previously reported50,51,52. There were seven samples in each province. We collected rhizospheric soils adhering to the roots by vigorous shaking and combined duplicates from four adjacent plants as one sample. The ice packs were then used for the 2-day transport of samples from field to laboratory. After removing small gravels and roots, soil samples (about 50~100 g per sample) were sieved through a 2-mm mesh, blended and kept in a refrigerator. Soil samples used for the analysis of physicochemical properties were kept at 4 °C, and the remaining soils for DNA extraction were stored at −20 °C.
Physicochemical analysis
Total organic carbon (TOC) and total nitrogen (TN) were determined following Hedges’ method53 with some modifications. Briefly, the 1.0 g of soil sample was freeze-dried, followed by blending and treating twice with 30 mL of organic-free 1 M HCl in a 50 mL centrifuge tube. Next, the soil was washed with ultrapure water to a final pH value of 6–7 and freeze-dried again. TOC and TN were then analyzed with an elemental analyzer (vario EL cube, Elementar Analysensysteme GmbH). Soil total Cu, composed of all types of Cu including the fractions hardly absorbed by bacteria or plants, was determined using the flame-atomic absorption spectrometer (AAS; novAA 400, Analytik Jena AG) after homogenization and strong acid digestion (4:1 concentrated HNO3 and HClO4, v/v) for 32 h. The CaCl2 extractable Cu, representing the bioavailable Cu, was determined by flame-AAS after shaking 2.5 g of soil with 12.5 mL of a 0.01 M CaCl2 solution for 2 h. The standard sample (GBW07410) was measured to estimate the recovery efficiency and the recovery was 94.5% for Cu. Soil pH was measured in a suspension with 1:5 (w/v) soil/0.01 M CaCl2 solution using a pH meter3.
DNA extraction
Soil DNA was extracted from 0.5 g of the homogeneous soil using a PowerSoil DNA Isolation Kit (MO BIO Laboratories) with a final elution in 50 μL deionized water according to the manufacturer’s instruction. Triple times extraction was performed per sample. Then, the DNA solutions were combined and the concentration/quality was measured using a NanoDrop 2000 Spectrophotometer (NanoDrop Technologies). The DNA sample was stored at −20 °C for further analysis.
Amplification and bar-coded pyrosequencing of bacterial 16S rRNA genes
The V4 hypervariable region (~300 bp) was amplified to provide sufficient resolution for accurately classifying the taxonomy of bacterial sequences. Polymerase chain reaction (PCR) was performed using the universal primer (515F 5′-GTGCCAGCMGCCGCGGTAA-3′ and 806R 5′-GGACTACHVGGGTWTCTAAT-3′) for nearly all-bacterial taxa54. A unique 12-bp barcode was incorporated into the 806R primer to distinguish the amplified products. The link code and pad sequence are located between the barcode and primer. The sample IDs, primers, and their congruent relationships are shown in Supplementary Table S4. PCR mixtures (50 μL) contained the following components: 50–100 ng (1 μL) of template DNA, 25 μL of rTaq premix buffer (TaKaRa), 100 nM of each primer (1 μL), and 22 μL ultrapure water. The amplification in duplicates followed the procedure: 94 °C for 5 min; 28 cycles at 94 °C for 30 s, 55 °C for 30 s, 72 °C for 90 s; and a final extension at 72 °C for 5 min. Duplicate PCR products for each sample were pooled and purified using the MicroElute Cycle-Pure Kit (Omega Bio-Tek) following the manufacturer’s instruction. Then, the purified PCR products for pyrosequencing were quantified as previously and combined with approximately equimolar amounts. Before sequencing, the DNA length was detected by agarose gel electrophoresis, and the DNA concentration and quality were confirmed using a Qubit fluorometer (Thermo Fisher Scientific). Sequencing was then performed at Macrogen (Seoul, South Korea) using the Miseq PE200 sequencer.
Processing of pyrosequencing data
The raw reads have been deposited in the National Center for Biotechnology Information (NCBI, BioProject ID: PRJNA306969, Accession number: SAMN04370756~SAMN04370776). Raw data were processed and analyzed according to Mothur55 and Quantitative Insights Into Microbial Ecology (QIIME)56. Briefly, low-quality reads with a quality score <25 or a length <250 bp were removed. Sequences were assembled based on overlap and assigned to samples according to their unique barcodes. Then, the singletons were removed which reduce the error rate with a small reduction in sensitivity. Operational taxonomic units (OTUs) with 97% similarity were picked out using Uclust57. Next, the representative sequence set was picked and aligned, and the chimeric sequences identified by the UCHIME algorithm58 were then discarded. The taxonomic classification of phylotypes was assigned to the Greengenes 13.5 database using “assign_taxonomy.py” in QIIME with default sets. The relative abundance of each taxon within each community was determined by comparing the number of sequences with the total sequences obtained from each sample.
To compare the bacterial diversity at the same level, a subset of 50,000 sequences per sample was randomly selected to normalize the sequences. The community α-diversity was determined using Chao1, Shannon and Simpson indices. The rarefaction curves were plotted based on the observed OTUs. The β-diversity characterized by UniFrac in this study, measuring the phylogenetic distance between samples, was used to determine whether the communities differ significantly. Here, weighted UniFrac matrices were calculated using QIIME scripts.
Predictive functional profiling of bacterial communities using 16S rRNA gene
The prediction analysis was performed using PICRUSt software package. Briefly, the closed-reference OTU was picked against the Greengenes database (13.5). To reflect the accuracy abundance of gene, the OTU number was normalized by “normalize_by_copy_number.py” script. Then the metagenomes were predicted against KEGG database and the accuracy of metagenome predictions were estimated by the Nearest Sequenced Taxon Index (NSTI)28. KEGG has been organized into 4 levels of hierarchies. The level one is the most general categories and the level four is the most specific in each KO terms. The analyses based on PICRUSt prediction were conducted on the level 2 after grouping the predicted KOs into a higher level of categorization.
Statistical analysis
Correlations between bacteria communities and environmental factors were determined using the taxonomy-supervised analysis (phyla abundance as variables), which is more tolerant of sequencing errors and often used to analyze the bacterial community attributing to no requirement for exhaustive computation on the alignment and clustering59. Eleven dominant phyla (>1%) of bacteria, pH, TOC, TN, TOC/TN, total Cu and extractable Cu were analyzed using Pearson’s correlations (two-tail) running on SPSS to examine the relationship between the relative abundance of dominant phyla and environmental factors. SPSS was also used for calculating the relationship between α-diversity and environmental factors. Additionally, the relationship between gene families abundance and environmental factors was executed using the same method described above to understand the influential factors of gene abundance. CCA and the permutation test were performed using “vegan” package based on R. MRT constructed using the R package “mvpart” with default sets was applied to understand the correlation between the relative abundance of the phyla and the environmental factors60. The Mantel test, a versatile statistical test that was checked by the “compare_distance_matrices.py” in QIIME package with two-tailed test and 999 times permutations, was used to describe the relationship between environmental factors and phylogenetic distance.
Additional Information
How to cite this article: Jiang, L. et al. Exploring the Influence of Environmental Factors on Bacterial Communities within the Rhizosphere of the Cu-tolerant plant, Elsholtzia splendens. Sci. Rep. 6, 36302; doi: 10.1038/srep36302 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Nos. 41322008 & 41673111).
Footnotes
Author Contributions C.L. and Z.S. designed the experiment; L.J. and M.S. performed the laboratory experiment; L.Y. and Y.S. collected the plants and soils in the field; L.J. wrote the manuscript; G.Z. and D.Z. revised the manuscript.
References
- Leng X. P. et al. Comparative transcriptome analysis of grapevine in response to copper stress. Sci Rep 5, 17749, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. R. et al. Copper induced oxidative stresses, antioxidant responses and phytoremediation potential of Moso bamboo (Phyllostachys pubescens). Sci Rep 5, 13554, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C. L., Shen Z. G. & Li X. D. Enhanced phytoextraction of Cu, Pb, Zn and Cd with EDTA and EDDS. Chemosphere 59, 1–11, (2005). [DOI] [PubMed] [Google Scholar]
- Peng H. Y., Kroneck P. M. H. & Kupper H. Toxicity and Deficiency of Copper in Elsholtzia splendens Affect Photosynthesis Biophysics, Pigments and Metal Accumulation. Environ Sci Technol 47, 6120–6128, (2013). [DOI] [PubMed] [Google Scholar]
- Yang X., Feng Y., He Z. L. & Stoffella P. J. Molecular mechanisms of heavy metal hyperaccumulation and phytoremediation. J Trace Elem Med Bio 18, 339–353, (2005). [DOI] [PubMed] [Google Scholar]
- Ke W. S., Xiong Z. T., Chen S. J. & Wang Z. H. Differences of Cu accumulation and cu-induced ATPase activity in roots of two populations of Elsholtzia haichowensis Sun. Environ Toxicol 23, 193–199, (2008). [DOI] [PubMed] [Google Scholar]
- Xia Y. et al. Cloning and characterization of a type 1 metallothionein gene from the copper-tolerant plant Elsholtzia haichowensis. Acta Physiol Plant 34, 1819–1826, (2012). [Google Scholar]
- Giller K. E., Witter E. & McGrath S. P. Toxicity of heavy metals to microorganisms and microbial processes in agricultural soils: A review. Soil Biol Biochem 30, 1389–1414, (1998). [Google Scholar]
- Verma S. C., Ladha J. K. & Tripathi A. K. Evaluation of plant growth promoting and colonization ability of endophytic diazotrophs from deep water rice. J Biotechnol 91, 127–141, (2001). [DOI] [PubMed] [Google Scholar]
- Wang F. Y., Lin M. G. & Yin R. Role of microbial inoculation and chitosan in phytoextraction of Cu, Zn, Pb and Cd by Elsholtzia splendens - a field case. Environ Pollut 147, 248–255, (2007). [DOI] [PubMed] [Google Scholar]
- Chen Y. X., Wang Y. P., Lin Q. & Luo Y. M. Effect of copper-tolerant rhizosphere bacteria on mobility of copper in soil and copper accumulation by Elsholtzia splendens. Environ Int 31, 861–866, (2005). [DOI] [PubMed] [Google Scholar]
- Sun F. L., Fan L. L. & Xie G. J. Effect of copper on the performance and bacterial communities of activated sludge using Illumina MiSeq platforms. Chemosphere 156, 212–219, (2016). [DOI] [PubMed] [Google Scholar]
- Zhang C. et al. Effects of heavy metals and soil physicochemical properties on wetland soil microbial biomass and bacterial community structure. Sci Total Environ 557, 785–790, (2016). [DOI] [PubMed] [Google Scholar]
- Smit E., Leeflang P. & Wernars K. Detection of shifts in microbial community structure and diversity in soil caused by copper contamination using amplified ribosomal DNA restriction analysis. FEMS Microbiol Ecol 23, 249–261, (1997). [Google Scholar]
- Moffett B. F. et al. Zinc contamination decreases the bacterial diversity of agricultural soil. FEMS Microbiol Ecol 43, 13–19, (2003). [DOI] [PubMed] [Google Scholar]
- Liu Y. R., Wang J. J., Zheng Y. M., Zhang L. M. & He J. Z. Patterns of Bacterial Diversity Along a Long-Term Mercury-Contaminated Gradient in the Paddy Soils. Microb Ecol 68, 575–583, (2014). [DOI] [PubMed] [Google Scholar]
- Zhou X. X. et al. Long-Term Use of Copper-Containing Fungicide Affects Microbial Properties of Citrus Grove Soils. Soil Sci Soc Am J 75, 898–906, (2011). [Google Scholar]
- Wang Y. P. et al. Assessment of microbial activity and bacterial community composition in the rhizosphere of a copper accumulator and a non-accumulator. Soil Biol Biochem 40, 1167–1177, (2008). [Google Scholar]
- He L. Y. et al. Characterization of copper-resistant bacteria and assessment of bacterial communities in rhizosphere soils of copper-tolerant plants. Appl Soil Ecol 44, 49–55, (2010). [Google Scholar]
- Sun L. N. et al. Genetic diversity and characterization of heavy metal-resistant-endophytic bacteria from two copper-tolerant plant species on copper mine wasteland. Bioresource Technol 101, 501–509, (2010). [DOI] [PubMed] [Google Scholar]
- Fierer N. & Jackson R. B. The diversity and biogeography of soil bacterial communities. P Natl Acad Sci 103, 626–631, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. Z. et al. Spatial and resource factors influencing high microbial diversity in soil. Appl Environ Microb 68, 326–334, (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn M. E. et al. Ecological effects of nitrogen deposition in the western United States. Bioscience 53, 404–420, (2003). [Google Scholar]
- Sun X. L., Zhao J., You Y. M. & Sun O. J. Soil microbial responses to forest floor litter manipulation and nitrogen addition in a mixed-wood forest of northern China. Sci Rep 6, 19536, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X. X., Zhang Y. G., Jun D. & Zhou Q. X. Zinc, Cadmium and Lead Accumulation and Characteristics of Rhizosphere Microbial Population Associated with Hyperaccumulator Sedum Alfredii Hance Under Natural Conditions. B Environ Contam Tox 82, 460–467, (2009). [DOI] [PubMed] [Google Scholar]
- Epelde L. et al. Impact of Metal Pollution and Thlaspi caerulescens Growth on Soil Microbial Communities. Appl Environ Microb 76, 7843–7853, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelde L., Becerril J. M., Blanco F., Kowalchuk G. A. & Garbisu C. Links between pseudometallophytes and rhizosphere microbial communities in a metalliferous soil. Pedobiologia 55, 219–225, (2012). [Google Scholar]
- Langille M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–821, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S. et al. Deep-sea vent epsilon-proteobacterial genomes provide insights into emergence of pathogens. P Natl Acad Sci USA 104, 12146–12150, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosewarne C. P., Pettigrove V., Stokes H. W. & Parsons Y. M. Class 1 integrons in benthic bacterial communities: abundance, association with Tn402-like transposition modules and evidence for coselection with heavy-metal resistance. FEMS Microbiol Ecol 72, 35–46, (2010). [DOI] [PubMed] [Google Scholar]
- van Elsas J. D., Gardener B. B. M., Wolters A. C. & Smit E. Isolation, characterization, and transfer of cryptic gene-mobilizing plasmids in the wheat rhizosphere. Appl Environ Microb 64, 880–889, (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan T. S., McBride M. B. & Thies J. E. Soil bacterial and archaeal community composition reflects high spatial heterogeneity of pH, bioavailable Zn, and Cu in a metalliferous peat soil. Soil Biol Biochem 66, 102–109, (2013). [Google Scholar]
- Chodak M., Golebiewski M., Morawska-Ploskonka J., Kuduk K. & Niklinska M. Diversity of microorganisms from forest soils differently polluted with heavy metals. Appl Soil Ecol 64, 7–14, (2013). [Google Scholar]
- Jose J., Giridhar R., Anas A., Bharathi P. A. L. & Nair S. Heavy metal pollution exerts reduction/adaptation in the diversity and enzyme expression profile of heterotrophic bacteria in Cochin estuary, India. Environ Pollut 159, 2775–2780, (2011). [DOI] [PubMed] [Google Scholar]
- Wakelin S. A., Chu G. X., Lardner R., Liang Y. C. & McLaughlin M. A single application of Cu to field soil has long-term effects on bacterial community structure, diversity, and soil processes. Pedobiologia 53, 149–158, (2010). [Google Scholar]
- Jiang L. Y., Yang X. E. & He Z. L. Growth response and phytoextraction of copper at different levels in soils by Elsholtzia splendens. Chemosphere 55, 1179–1187, (2004). [DOI] [PubMed] [Google Scholar]
- Song J., Zhao F. J., Luo Y. M., McGrath S. P. & Zhang H. Copper uptake by Elsholtzia splendens and Silene vulgaris and assessment of copper phytoavailability in contaminated soils. Environ Pollut 128, 307–315, (2004). [DOI] [PubMed] [Google Scholar]
- Epelde L., Lanzen A., Blanco F., Urich T. & Garbisu C. Adaptation of soil microbial community structure and function to chronic metal contamination at an abandoned Pb-Zn mine. FEMS Microbiol Ecol 91, (2015). [DOI] [PubMed] [Google Scholar]
- Li J. et al. Initial Copper Stress Strengthens the Resistance of Soil Microorganisms to a Subsequent Copper Stress. Microbiol Ecol 67, 931–941, (2014). [DOI] [PubMed] [Google Scholar]
- Azarbad H. et al. Microbial community composition and functions are resilient to metal pollution along two forest soil gradients. FEMS Microbiol Ecol 91, (2015). [DOI] [PubMed] [Google Scholar]
- Gremion F., Chatzinotas A. & Harms H. Comparative 16S rDNA and 16S rRNA sequence analysis indicates that Actinobacteria might be a dominant part of the metabolically active bacteria in heavy metal-contaminated bulk and rhizosphere soil. Environ Microbiol 5, 896–907, (2003). [DOI] [PubMed] [Google Scholar]
- Kunito T., Saeki K., Nagaoka K., Oyaizu H. & Matsumoto S. Characterization of copper-resistant bacterial community in rhizosphere of highly copper-contaminated soil. Eur J Soil Biol 37, 95–102, (2001). [Google Scholar]
- Dupont C. L., Grass G. & Rensing C. Copper toxicity and the origin of bacterial resistance-new insights and applications. Metallomics 3, 1109–1118, (2011). [DOI] [PubMed] [Google Scholar]
- Lauber C. L., Hamady M., Knight R. & Fierer N. Pyrosequencing-Based Assessment of Soil pH as a Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl Environ Microb 75, 5111–5120, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terahara T. et al. Molecular diversity of bacterial chitinases in arable soils and the effects of environmental factors on the chitinolytic bacterial community. Soil Biol Biochem 41, 473–480, (2009). [Google Scholar]
- Hartman W. H., Richardson C. J., Vilgalys R. & Bruland G. L. Environmental and anthropogenic controls over bacterial communities in wetland soils. P Natl Acad Sci USA 105, 17842–17847, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmitt S. J., Wright D., Goulding K. W. T. & Jones D. L. pH regulation of carbon and nitrogen dynamics in two agricultural soils. Soil Biol Biochem 38, 898–911, (2006). [Google Scholar]
- Chen X. B., Wright J. V., Conca J. L. & Peurrung L. M. Effects of pH on heavy metal sorption on mineral apatite. Environ Sci Technol 31, 624–631, (1997). [Google Scholar]
- Chuan M. C., Shu G. Y. & Liu J. C. Solubility of heavy metals in a contaminated soil: Effects of redox potential and pH. Water Air Soil Poll 90, 543–556, (1996). [Google Scholar]
- Xia Y. & Shen Z. G. Comparative studies of copper tolerance and uptake by three plant species of the genus Elsholtzia. B Environ Contam Tox 79, 53–57, (2007). [DOI] [PubMed] [Google Scholar]
- Yang M. J., Yang X. E. & Romheld V. Growth and nutrient composition of Elsholtzia splendens Nakai under copper toxicity. J Plant Nutr 25, 1359–1375, (2002). [Google Scholar]
- Tang S. R., Wilke B. M. & Huang C. Y. The uptake of copper by plants dominantly growing on copper mining spoils along the Yangtze River, the People’s Republic of China. Plant Soil 209, 225–232, (1999). [Google Scholar]
- Hedges J. I. & Stern J. H. Carbon and Nitrogen Determinations of Carbonate-Containing Solids. Limnol Oceanogr 29, 657–663, (1984). [Google Scholar]
- Bates S. T. et al. Examining the global distribution of dominant archaeal populations in soil. ISME J 5, 908–917, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P. D. et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl Environ Microb 75, 7537–7541, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, (2010). [DOI] [PubMed] [Google Scholar]
- Edgar R. C., Haas B. J., Clemente J. C., Quince C. & Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sul W. J. et al. Bacterial community comparisons by taxonomy-supervised analysis independent of sequence alignment and clustering. P Natl Acad Sci USA 108, 14637–14642, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De’Ath G. Multivariate regression trees: a new technique for modeling species-environment relationships. Ecology 83, 1105–1117, (2002). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.