

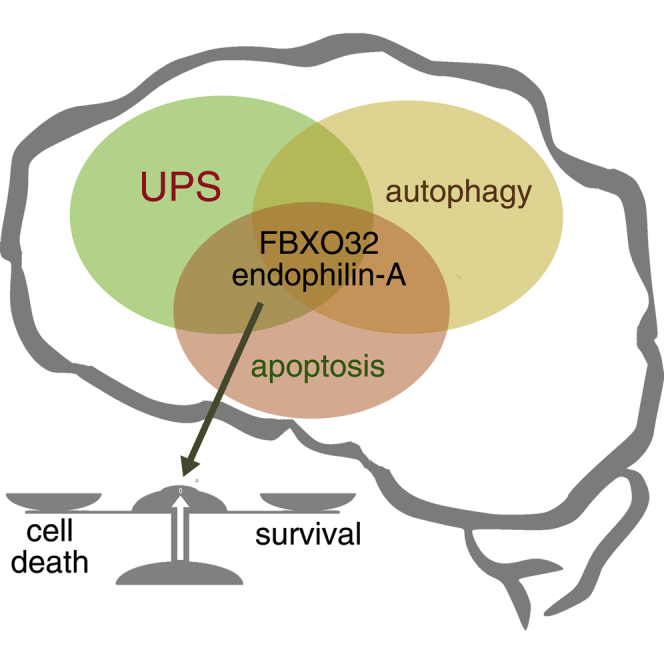
Summary
Endophilin-A, a well-characterized endocytic adaptor essential for synaptic vesicle recycling, has recently been linked to neurodegeneration. We report here that endophilin-A deficiency results in impaired movement, age-dependent ataxia, and neurodegeneration in mice. Transcriptional analysis of endophilin-A mutant mice, complemented by proteomics, highlighted ataxia- and protein-homeostasis-related genes and revealed upregulation of the E3-ubiquitin ligase FBXO32/atrogin-1 and its transcription factor FOXO3A. FBXO32 overexpression triggers apoptosis in cultured cells and neurons but, remarkably, coexpression of endophilin-A rescues it. FBXO32 interacts with all three endophilin-A proteins. Similarly to endophilin-A, FBXO32 tubulates membranes and localizes on clathrin-coated structures. Additionally, FBXO32 and endophilin-A are necessary for autophagosome formation, and both colocalize transiently with autophagosomes. Our results point to a role for endophilin-A proteins in autophagy and protein degradation, processes that are impaired in their absence, potentially contributing to neurodegeneration and ataxia.
Keywords: endophilin, FBXO32, endocytosis, autophagy, ubiquitin-proteasome system, neurodegeneration, ataxia, Parkinson’s disease, next-generation sequencing, protein homeostasis
Graphical Abstract
Highlights
-
•
Endophilin-A is needed for autophagosome formation in mammalian neurons and brain
-
•
Absence of endophilin-A upregulates the E3-ubiquitin ligase FBXO32
-
•
FBXO32-endophilin-A interaction maintains neuronal health and protein homeostasis
-
•
Endophilin-A KO mice show age-dependent ataxia, motor impairments, and neurodegeneration
Regulation of protein homeostasis and autophagy has become a promising line of research in the neurodegeneration field. Murdoch et al. now find that endophilin-A, a key factor in clathrin-mediated endocytosis, regulates protein homeostasis through the Foxo3a-Fbxo32 network.
Introduction
The nervous system relies on conserved endocytic and synaptic vesicle (SV) recycling mechanisms to sustain high activity. Defective endocytosis and SV recycling have long been implicated in neurodegeneration and neurodegenerative diseases, yet the molecular mechanisms remain unclear (Esposito et al., 2012, Heutink and Verhage, 2012, Saheki and De Camilli, 2012, Schreij et al., 2016).
Endophilin-A (henceforth endophilin) endocytic adaptors are key players in membrane dynamics at the neuronal synapse (Ringstad et al., 1999, Verstreken et al., 2002, Milosevic et al., 2011, Saheki and De Camilli, 2012). Endophilin-1 and 3 are brain-specific, whereas endophilin-2 is ubiquitous (Ringstad et al., 1997, Ringstad et al., 1999). All three contain a highly conserved BAR domain and have a central role in both clathrin-mediated endocytosis (Farsad et al., 2001, Ringstad et al., 1997, Ringstad et al., 1999, Ringstad et al., 2001, Verstreken et al., 2002, Milosevic et al., 2011) and clathrin-independent endocytosis (Boucrot et al., 2015). In clathrin-mediated endocytosis, endophilin recruits synaptojanin-1 to the necks of coated vesicles prior to scission by dynamin (Milosevic et al., 2011). Furthermore, availability of endophilin is rate limiting for vesicle uncoating and SV recycling, and neurons without endophilin show reduced neurotransmission (Milosevic et al., 2011).
Total absence of endophilin is perinatally lethal, whereas mice lacking endophilin-1 and 2 (1,2 double knockout [DKO]) show ataxia, neurodegeneration, and early lethality (Milosevic et al., 2011). Why this happens is an intriguing question. It cannot be easily explained by defective neurotransmission, because even mice with paralysis and abolished neurotransmission do not develop neurodegeneration (Heutink and Verhage, 2012). Thus, it is unlikely that neurodegeneration in endophilin 1,2 DKO mice is solely the result of defective SV recycling.
Mice without endophilin also show increased protein ubiquitination (Cao et al., 2014), and neurodegeneration is frequently associated with impaired proteolytic systems, most notably the ubiquitin-proteasome system (UPS) and autophagy (Nakamura and Lipton, 2009, Nixon, 2013, Menzies et al., 2015). Endophilin has been linked to the UPS through direct interaction with the Parkinson’s disease (PD)-related E3-ubiquitin ligase parkin (Trempe et al., 2009) and as a key component of the CBL-CIN85-endophilin complex known to downregulate receptors (Soubeyran et al., 2002, Petrelli et al., 2002).
Additional links between endophilin and neurodegeneration have recently emerged. The most commonly disrupted gene in familial PD (Cookson, 2010), LRRK2, encodes a protein that phosphorylates endophilin (Matta et al., 2012). LRRK2 appears in Lewy bodies and plays a role in apoptosis and autophagy (Iaccarino et al., 2007, Tong et al., 2012). In addition, a number of case studies described synaptojanin-1 and auxilin mutations in early-onset parkinsonism (Edvardson et al., 2012, Quadri et al., 2013, Krebs et al., 2013). Auxilin is recruited to the clathrin-coated vesicles directly after the action of synaptojanin-1, which is brought to the neck of the clathrin-coated pit by endophilin (Milosevic et al., 2011). Interestingly, endophilin is upregulated in the brains of PD patients (Shi et al., 2009). In addition, endophilin-1 and 3 have been shown to interact with ataxin-2, mutations in which lead to spinocerebellar ataxia type 2 (Ralser et al., 2005, Nonis et al., 2008).
Despite direct parkin-endophilin interaction and a striking upregulation of parkin in mice lacking all three endophilin genes (endophilin triple knockout [TKO]; Cao et al., 2014), elevated parkin could not explain the endophilin-deficient phenotype. We subjected endophilin mutants to RNA sequencing and found upregulation of another E3-ubiquitin ligase, F-box-only protein 32 (Fbxo32, also called atrogin-1/MAFbx). FBXO32 has previously been studied in muscles, where its induction occurs prior to the onset of rapid atrophy (Sandri et al., 2004). While studying how the FBXO32-endophilin-A interaction balances protein degradation and neuronal health in the brain, we discovered a direct role of endophilin-A in autophagosome formation.
Results
Endophilin-A Deficiency Results in Age-Dependent Movement Impairments in Mice
Mice lacking all three endophilin genes die perinatally, whereas endophilin 1,2 DKO animals show ataxia, neurodegenerative changes, and lethality around 3 weeks (Movie S1; Milosevic et al., 2011). Whereas single knockout (KO) and heterozygous (1HT-2HT; 1HT-2HT-3HT) mice appear phenotypically normal, endophilin 1KO-2HT, 1KO-2WT-3KO, and particularly 1KO-2HT-3KO mice exhibit shorter lifespans (∼19, ∼21, and ∼18 months, respectively; 1HT-2HT mice lived over 27 months; Figure S1A). Observing 1KO-2HT and 1KO-2HT-3KO mice revealed progressive impairments in motor coordination and increasingly obvious ataxia as these animals age.
To systematically measure the progression of ataxia, we used a composite phenotype scoring system that incorporates hindlimb clasping, ledge walking, kyphosis, and gait (Guyenet et al., 2010). When applied to four endophilin lines (1KO-2WT, 1KO-2HT, 1KO-2WT-3KO, and 1KO-2HT-3KO) and wild-type (WT) controls, it revealed phenotypic differences between endophilin mutants and within the same animals over the course of aging (Figure 1A); i.e., the onset and progression of ataxia correlate with age and severity of endophilin deficiency.
Figure 1.

Endophilin Deficiency Causes Age-Dependent Motor Impairments in Mice
(A) Endophilin mutant mice exhibit progressive ataxia in which onset depends on severity of endophilin deficiency. Mice were assessed on a 0–3 scale for ledge test, clasping, gait, and kyphosis from 3 to 18 months. Eight to twelve mice/genotype are shown. Average composite score for each indicated genotype at defined age was calculated; mean ± SEM (two-way ANOVA; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; ns > 0.05).
(B) Rotarod performance plotted as latency to fall. Motor coordination was assessed at 1–18 months. After a training session (not shown), fall latency from the accelerating rod was measured for the same genotypes as in (A). Mean ± SEM; min. eight mice/genotype; Student’s t test; ∗∗∗p < 0.001.
(C) Highest speed that endophilin mutants were able to run on DigiGait at 3 (left) and 15 (right) months. Colors represent percentage of mice able to run at indicated speed.
(D) Percentage of time both hindlimbs touch the ground simultaneously while running at 20 cm/s, quantitated for endophilin 1KO-2WT-3KO and 1KO-2HT-3WT mice in comparison to WT. Mean ± SEM, Student’s t test, ∗p<0.05, ∗∗∗p<0.001.
(E) TUNEL assay on brain sections of mice with indicated genotypes. No apoptotic signal was observed in WTs; cell death was spotted very rarely in endophilin 1KO-2WT brains and more frequently in endophilin 1,2 DKO sections (indicated by arrows). The scale bar represents 50 μm. (Right) Quantification in motor cortex (up) and hippocampus (down) is shown. Mean ± SEM; four mice/genotype.
(F) GFAP immunofluorescence in endophilin 1,2 DKO brain sections showed more gliosis compared to controls. The scale bar represents 20 μm. (Right) Quantification is shown. Mean ± SEM; four mice/genotype.
(G) Immunostaining for cleaved caspase 3 on brain slides of the indicated genotypes. Arrows indicate cells with positive caspase 3 signal. The scale bar represents 50 μm. (Right) Quantification is shown. Mean ± SEM; five mice/genotype.
Next, we systematically evaluated locomotion by accelerating rotarod (ARR) performance test and gait analysis. Whereas the performance of 1KO-2WT mice on the ARR was comparable to WT, fall latency in 1KO-2HT-3KO mice was already significantly lower at a very young age (Figure 1B). Intermediate phenotypes were observed in 1KO-2HT and 1KO-2WT-3KO mice, where fall latency compared to WT was decreased significantly at 3 and 15 months, respectively.
Gait analysis revealed that ability to run on a motorized treadmill and walking pattern also depended on the number of missing alleles and age (Figures 1C, 1D, and S1B; see Movie S2). 1KO-2HT-3KO gait was impaired at 3 months, whereas the littermate 1KO-2WT-3KO mice showed impaired gait at 15 months (Figure 1D). Overall, onset and progression of movement impairments correspond to age and the extent of endophilin deficiency.
Neurodegenerative Changes in the Brain of Endophilin-A Mutants
To evaluate whether neurodegenerative changes could cause these motor impairments, we inspected neuronal cell death and gliosis in our model. Whereas overall neuronal cell morphology was unaltered in these mice (Figure S1D), TUNEL assay revealed slightly increased cell death in endophilin 1KO-2WT and more prominently in 1,2 DKO motor cortex and hippocampus at postnatal day (p)18 (Figure 1E). Increased cell death was also found in 18-month 1KO-2HT-3KO motor cortex and hippocampus (Figure S1C). Glial fibrillary acidic protein immunostaining revealed prominent upregulation of gliosis in 1,2 DKO motor cortex (Figure 1F). Because endophilin TKO mice die a few hours after birth, we probed for early markers of apoptosis in endophilin TKO brains and found an increase in caspase 3 activity (Figure 1G). In summary, newborn endophilin TKOs, juvenile 1,2 DKO, and aged 1KO-2HT-3KO mice display signs of mild to moderate neurodegeneration.
Endophilin-A Deficiency Changes the Expression of Ataxia-, PD -, and Protein-Homeostasis-Related Genes
We were interested in the molecular mechanisms underlying neurodegeneration in our endophilin-deficient models, so we employed RNA sequencing (experimental strategy in Figure S2A). We first sequenced hippocampal RNA from TKO mice that show altered SV recycling and controls (n = 8–9 mice/condition). The data are of high technical quality (∼27M aligned reads) and samples clustered well according to genotype (Table S1; Figures S2B and S2C). 182 differentially expressed genes (DEGs) between endophilin TKO and littermate controls and 964 DEGs between TKO and WT were detected (adjusted p value < 0.01; log2 fold change > 0.4) (Tables S2 and S3, respectively). Given the pronounced movement defects of endophilin mutants, we first looked for movement-disorder-related genes or pathways. Interestingly, TKO versus WT DEGs showed significant enrichment of genes involved in ataxia and PD (Figures 2A and 2B; Table S4).
Figure 2.

Transcriptional Analysis of Endophilin TKO and 1,2 DKO Hippocampi
(A–D) Heatmap showing the expression of ataxia- (A: TKO; C: 1,2 DKO) and Parkinson’s-disease-related genes (B: TKO; D: 1,2 DKO).
(E) Pathway analysis of the transcriptional signatures of TKO hippocampi using the IPA software. The ten most significantly enriched pathways based on Fisher exact test p values (x axis) are presented.
(F) The RNA sequencing results verified by qRT-PCR for named genes in TKO samples.
See also Figure S2 and Tables S1, S2, S3, S4, S5, S6, S7, and S8.
Because endophilin 1,2 DKO mice present first signs of neurodegeneration by the end of the 2nd postnatal week, hippocampi from DKO, littermate 1KO-2WT, and WT mice were extracted during the 3rd week and subjected to RNA sequencing and transcriptome analysis. The data are again of high technical quality (∼28M reads; Table S1; samples clustered well according to genotype; Figures S2D and S2E). 1,479 DEGs between 1,2 DKO and littermate controls, and 1,749 DEGs between DKO and WT were detected (adjusted p value < 0.01; log2 fold change > 0.4; Tables S5 and S6, respectively). Both 1,2 DKO versus littermate and DKO versus WT lists were significantly enriched for genes involved in ataxia and PD (Table S4; Figures 2C and 2D). Ataxia-related genes include ATM interactor (Atmin), E2f1, and Rad51; PD-related genes include Park2 (encoding parkin), Lrrk2, the tyrosine hydroxylase gene (Th), and Parl. These findings further support endophilin’s association with neurodegeneration, ataxia, and PD (Arranz et al., 2015, Edvardson et al., 2012, Krebs et al., 2013, Matta et al., 2012, Milosevic et al., 2011, Quadri et al., 2013, Ralser et al., 2005, Shi et al., 2009, Trempe et al., 2009).
We next analyzed TKO versus both littermate and WT controls (genes significantly changed in the same direction in TKO against both littermates and WTs; Table S7; Figure S2F) by Ingenuity Pathway Analysis (IPA) software to identify altered pathways and networks. The resulting most-represented pathways were related to compromised synaptic transmission, protein homeostasis, phagosome maturation, mitochondrial function, dopamine metabolism, and apoptosis (Figure 2E). Altered protein homeostasis has been strongly linked to neurodegeneration (e.g., Deger et al., 2015) and might be relevant to the ataxic and neurodegenerative phenotypes observed in endophilin-deficient mice.
The differential expression of several genes was validated by quantitative real-time PCR in endophilin TKO hippocampi versus WT, and a strong correlation was observed (R2 = 0.83; Figure 2F). We also examined regulators of the transcriptional profile in endophilin TKO hippocampi and found that, among those statistically enriched, Fbxo32 presents the most-robust change in gene expression (1.9-fold increase; Table S8). This was interesting because FBXO32 is an E3-ubiquitin ligase linked to autophagy in skeletal and cardiac muscle (Sandri et al., 2004, Zaglia et al., 2014). Its expression is known to be regulated by the transcription factors FOXO1 and FOXO3A (Sandri et al., 2004, Webb and Brunet, 2014): whereas Foxo1 was unchanged, Foxo3a was significantly upregulated in TKO hippocampi (Table S8).
Absence of Endophilin Upregulates the E3-Ubiquitin Ligase Fbxo32 and Its Transcription Factor Foxo3a
Given the observed changes in the Foxo3a-Fbxo32 network and that FBXO32 is known to promote cell death and is required for muscle atrophy (Sandri et al., 2004, Wu et al., 2011, Xie et al., 2009), we decided to follow up on this network at the RNA and protein level. As in the RNA-sequencing data, Foxo3a and Fbxo32 were upregulated in TKO hippocampi when measured by qPCR (Figure 3A) whereas Foxo1 was unchanged (Figures S2G and S2H). A striking increase in FBXO32 protein levels in TKO whole-brain extracts relative to littermate controls and to WT mice was detected (Figure 3B; Figure S2I displays elevated FBXO32 in the hippocampus). Levels of FOXO3A[Ser253] phosphorylation (which represses FOXO3A binding to DNA) were also decreased (Figure 3B), suggesting an increase in the FOXO3A transcriptional activity, and the distribution of FOXO3A[Ser253]-P is altered in TKO hippocampus in comparison to controls (Figure 3C).
Figure 3.

Upregulation of the E3 Ubiquitin Ligase FBXO32 and Its Transcription Factor FOXO3A in Endophilin Mutant Mice
(A) Transcript levels of Fbxo32 measured by qRT-PCR for indicated genotypes. ∗p < 0.05; Student’s t test; mean ± SEM.
(B) Upregulation of FBXO32 and FOXO3A in endophilin TKO whole-brain extracts. (Right) Quantification is shown (min. eight samples/genotype). ∗p < 0.05; ∗∗∗p < 0.005; Student’s t test; mean ± SEM.
(C) FOXO3A-[S253]P immunofluorescence reveals its altered distribution (shown by arrows) in brain sections of endophilin TKO hippocampus. Four mice/genotype are shown. The scale bar represents 10 μm.
(D) Silencing TKO neurons with Foxo3a siRNA (left: efficiency of knockdown by western blotting) decreases transcript levels of Fbxo32 measured by qRT-PCR. n = 3. ∗∗p < 0.01; Student’s t test; mean ± SD.
(E) Upregulation of FBXO32 in endophilin 1,2 DKO brains, at p0 (whole brain) and p14–p21 mice (cortex). (Right) Quantification is shown (eight samples/genotype). ∗p < 0.05; ∗∗p < 0.01; Student’s t test; mean ± SEM.
(F) Upregulation of FBXO32 in endophilin 1KO-2HT-3KO cortical extracts in 18-month mice. (Right) Quantification is shown (three samples/genotype). ∗p < 0.05; Student’s t test; mean ± SD.
See also Figures S2 and S3.
To determine whether FOXO3A activation is directly responsible for the upregulation of FBXO32, Foxo3a was silenced in TKO primary hippocampal neurons by small interfering RNA (siRNA) knockdown, using two siRNA constructs. Near-complete ablation of FOXO3A affects neuronal survival, so we used a milder condition (45% reduction) to probe for FBXO32 levels (Figure 3D). The elevated FBXO32 levels in TKO neurons were lowered upon Foxo3a knockdown, implying that FOXO3A is a sufficient regulator of FBXO32 in this system (Figure 3D). FBXO32 was also strongly upregulated in 1,2 DKO brain samples, both at p0 (whole brain) and 3 weeks (cortex; Figure 3E), as well as in the cortex of 1KO-2HT-3KO mice at 18 months (Figure 3F). Thus, the Foxo3a-Fbxo32 network originally described in muscle atrophy is operative in the brain of endophilin mutant mice.
Proteasome Saturation in the Absence of Endophilin-A
Given the reported role of FBXO32 in the regulation of protein homeostasis via the UPS, we investigated whether the ubiquitin system is also affected in endophilin mutants. The PD-related E3-ubiquitin ligase parkin monoubiquitinates endophilin, and TKO brains show increased levels of parkin and ubiquitinated proteins (Trempe et al., 2009, Cao et al., 2014). However, 1,2 DKO mice in which the parkin gene Park2 was also knocked out displayed a similar neurological phenotype to 1,2 DKO mice (Cao et al., 2013), suggesting that other ubiquitin ligases might be involved. Given that FBXO32 is also an E3-ubiquitin ligase, we hypothesized that its robust upregulation in endophilin-deficient brain might contribute to the loss of protein homeostasis in endophilin knockouts. First, we tested whether FBXO32 can (mono)ubiquitinate endophilin-1 and 2, but this was not the case (Figures S3A and S3B). We observed that 1,2 DKO brains also display increased levels of ubiquitinated proteins (Figure 4A), similar to TKO brains (Cao et al., 2013). To distinguish whether this was due to proteasome-related or localization-/signaling-related ubiquitination, we measured K48-(proteasome-related) and K63-(signaling-related)-linkage-specific polyubiquitination (Herhaus and Dikic, 2015). Whereas K48-linked ubiquitination was prominently increased in 1,2 DKO cortical extracts (Figure 4B), there was no significant change in the levels of K63-linked ubiquitination (Figure 4C), suggesting that the accumulation of ubiquitinated proteins is associated with defective processing by the proteasome. A similar result was observed in TKO brains (Figures S3C and S3D).
Figure 4.

Proteasomal Saturation in Endophilin Mutant Mice
(A–C) Western blots for polyubiquitin (A), K48 linked (B; proteasome targeting), and K63 linked (C; proteasome unrelated) in 1,2 DKO cortex. (Right) Quantification is shown (min. six samples/genotype). ∗p < 0.05; ∗∗p < 0.01; Student’s t test; mean ± SEM.
(D and E) TKO MEFS (D) or neurons (E) transfected with ubiquitin-G76V-GFP (modified GFP targeted for proteasomal degradation). The scale bar represents 10 μm. (Right) Quantification of GFP fluorescence is shown. In MEFs, proteasome inhibitor MG132 was used as control. ∗p < 0.05; Student’s t test; mean ± SEM.
(F) Proteasomal activity (chymotrypsin-like) in brain extracts of TKOs relative to littermate controls. p > 0.05; Student’s t test; mean ± SEM.
(G) Activity of caspases 3 and 7 reveals that apoptosis is increased in TKO MEFs transfected with ubiquitin-G76V-GFP. ∗∗∗p < 0.001; Student’s t test; mean ± SD.
See also Figure S3.
Given the apparent inability of the proteasome to cope with the level of ubiquitinated proteins in endophilin knockouts, we probed the proteasomal system using a ubiquitinated substrate that is targeted to the proteasome and degraded (ubiquitin-G76V-GFP; Dantuma et al., 2000), as well as by measuring the enzyme activity of the proteasome itself. We observed an accumulation of GFP-fluorescence originating from the substrate in TKO primary hippocampal neurons as well as fibroblasts (mouse embryonic fibroblasts [MEFs]) in comparison to respective controls (Figures 4D and 4E), denoting proteasome saturation in the endophilin KO cells. Interestingly, we noticed that the increase in ubiquitinated proteins elicited by the expression of ubiquitin-G76V-GFP results in significantly more apoptosis in the TKO MEFs than in controls (Figure 4G). Altogether, these results suggest a causal link between proteasome saturation in endophilin mutants, higher levels of ubiquitinated proteins, and increased cell death susceptibility.
To determine whether the proteasome saturation was due to decreased activity of the proteasome, we measured the proteasome enzyme activity. While there was a trend toward a decrease in proteasomal activity, we observed no significant difference in the proteasome activity per se in the brain extracts (Figure 4F). Thus, the proteasome system in TKO brains seems functional but unable to keep up with the high demand.
FBXO32 Interacts with Three Members of the Endophilin-A Family
In light of the upregulation of FBXO32 in the absence of endophilin, we sought to understand the connection between these proteins. First, we tested whether FBXO32 interacts with endophilin family members by immunoprecipitation and pull-downs. An interaction between FBXO32 and all three endophilins in vivo was revealed by specific enrichment of FBXO32 in anti-GFP immunoprecipitates from HeLa expressing FBXO32-mCherry and endophilin 1, 2, or 3-EGFP (Figures 5A–5C). Similar results were obtained by glutathione S-transferase (GST) pull-downs from mouse brain lysate using the GST-FBXO32 as bait (Figure S3E). We demonstrated that FBXO32 is able to pull-down purified endophilin-1 protein (Figure 5D), suggesting a direct interaction. Furthermore, the interaction is mediated through endophilin’s SH3 domain, as revealed by another set of pull-downs from mouse brain lysate using the GST-SH3 domains of endophilin-1 and 3 as baits (an enrichment of FBXO32 with stronger binding to endophilin-1 and 3 SH3s was observed; Figure S3F).
Figure 5.

FBXO32 and Endophilin-A Interact and Colocalize Transiently on Organelles and Tubular Structures
(A–C) Endophilin-1 (A), 2 (B), and 3 (C) interact with FBXO32. Anti-GFP immunoprecipitation from HeLa cell lysate probed with anti-RFP antibody is shown (three experiments).
(D) FBXO32-GFP-coupled beads pull-down purified endophilin-1.
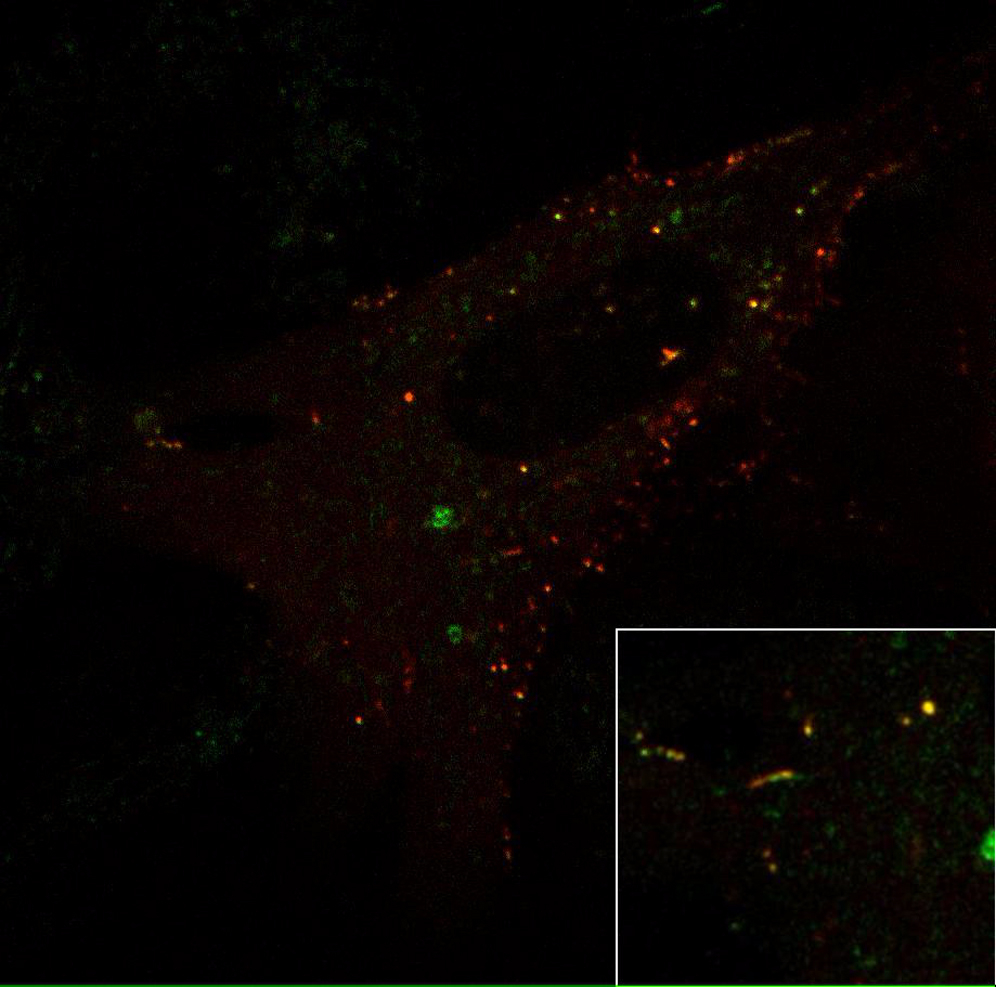
(E) MEF overexpressing FBXO32-EGFP (left) and endophilin A2-RFP (middle); magnified in the inset. Note that FBXO32 and endophilin-2 colocalize on organellar and tubular structures. The scale bar represents 7 μm.
(F) Time-lapse images from (E) showing dynamic tubular structure with FBXO32 and endophilin-2. Note FBXO32’s association with membrane tubules independently of endophilin-2. Inset size is 4 μm.
(G and H) Primary hippocampal neurons expressing FBXO32-EGFP and endophilin-1-mRFP (G) or endophilin-2-mRFP (H), transfected by electroporation. (Up) Neuronal processes are shown (scale bar 3 μm). (Bottom) Neuronal cell bodies are shown (scale bar 7 μm).
(I) FBXO32-EGFP colocalization with LC3-mRFP in starved MEF. A representative cell shows the overlay between FBXO32-EGFP and LC3-mRFP (imaged by near-TIRF microscopy). Panels on the right show dynamic FBXO32-labeled tubule. Movie S4 shows trafficking of FBXO32 and LC3 in the same cell. The scale bar represents 3 μm.
(J) HeLa cell expressing LC3-EGFP (left) and endophilin-A2 protein (middle), starved for 2 or 3 hr. The scale bar represents 3 μm.
(K) Endophilin-2-EGFP transiently colocalized with ATG5-mCherry (early autophagosomal marker) in starved HeLa cell. Magnified is shown in the inset. The scale bar represents 5 μm.
(L and M) Quantification of colocalization of endophilins with FBXO32 (L) and LC3 (M). Random colocalization has been subtracted. Mean ± SD.
(N) Kymograph of image series (200 s) showing a transient colocalization of endophilin-3 (green) and LC3 (red) in a HeLa cell imaged by spinning-disk microscopy.
We examined the intracellular localization of FBXO32 and its overlap with endophilins in cultured clonal cells (HeLa and MEFs were used to control for cell line variability) and primary hippocampal neurons. It is noteworthy that expressing FBXO32 (either FBXO32-EGFP or FBXO32-mCherry) induced cell death: ∼50% of Fbxo32-transfected HeLa and ∼40% of Fbxo32-transfected MEFs survived 30 hr post-transfection; less than 15% of Fbxo32-transfected primary neurons were detected on day in vitro (DIV) 14 (detailed below). FBXO32 distribution showed substantial cell-to-cell variability: although mainly cytosolic, FBXO32 was enriched at dynamic organelle-like structures of various sizes and often accumulated in the nucleus (Movie S3). Native FBXO32 distribution was similar to that of expressed FBXO32: immunostaining against FBXO32 revealed nuclear as well as cytosolic protein distribution in HeLa cells (Figure S4A). Surprisingly, both in HeLa cells and MEFs, FBXO32-labeled tubular structures were observed (examples in Figures 5F, 5I, and S4C; see also Movie S5). Given that FBXO32 interacts with endophilins, and the propensity of endophilins to tubulate membranes, we examined the colocalization of FBXO32 with endophilin 1, 2, and 3. The distribution of endophilins is mainly cytosolic: sparse puncta were attributed to endophilin’s role in endocytosis (Milosevic et al., 2011, Perera et al., 2006, Boucrot et al., 2015, Renard et al., 2015).
When endophilin-1 and FBXO32 were coexpressed at similar levels, FBXO32 protein distribution changed: FBXO32 became predominantly cytoplasmic and excluded from the nucleus (Figures S4B and S6E; also see Figures 7B and 7D). Interestingly, we also observed that cell death due to FBXO32 overexpression was abolished upon endophilin coexpression, which we detail further below. FBXO32 and endophilin-1 were also found to colocalize sparsely at dynamic organelle-like structures (Figures 5L and S4B). These were found throughout the cytoplasm and not only in the vicinity of the plasma membrane (as would be expected for endophilin-1 when being recruited to endocytic structures).
Figure 7.

FBXO32 Upregulation Causes Apoptosis in Non-neuronal and Neuronal Cells that Can Be Rescued by Endophilin-1 and 2
(A and B) MEFs cotransfected with FBXO32-EGFP and mRFP (A) or EGFP and endophilin-1-mRFP (B) for 30 hr. Note that FBXO32-expressing cells look healthy if coexpressing endophilin-1. The scale bar represents 20 μm.
(C) Quantification of EGFP-positive cells in (A) and (B) normalized to DAPI. Three experiments are shown, with at least 48 cells/condition. ∗∗∗p < 0.001; ∗p < 0.05. Student’s t test; mean ± SEM.
(D) Quantification of EGFP signal in the nucleus in (A) and (B) normalized to total transfected cells. ∗p < 0.05; Student’s t test.
(E) Activity of caspases 3 and 7 reveals that apoptosis is increased upon Fbxo32 expression but abolished when cells coexpress endophilin-1 (three experiments). ∗∗∗p < 0.001; Student’s t test; mean ± SEM.
(F and G) Primary cortical neurons cotransfected with FBXO32-EGFP and endophilin-1-mRFP (F) or endophilin-2-mRFP (G). The scale bar represents 20 μm.
(H) Quantification of survival of FBXO32-transfected neurons in primary culture (as shown in F and G; three independent experiments). ∗∗p < 0.01; ∗∗∗p < 0.001; Student’s t test, mean ± SD.
See also Figure S6.
When endophilin-2 and FBXO32 were coexpressed, they colocalized transiently at both organelle-like structures and tubuli (Figures 5E, 5L, and S4C; Movie S5). We further observed that endophilin-2 colocalized with FBXO32 on dynamic tubulated structures, although endophilin-2 was not needed for the recruitment of FBXO32 to tubuli (Figure 5F at 24 s; Movie S5). Similarly to endophilins, FBXO32 also directly binds and tubulates membranes in vitro (B. Kroppen, M. Meinecke, and I.M., unpublished data). Lastly, FBXO32 and endophilin-3 colocalized partially on organelle-like structures (Figures 5L and S4D).
We examined the overlap between FBXO32 and endophilin-1 or endophilin-2 in primary hippocampal cultures. FBXO32-EGFP and endophilin-1-mRFP or endophilin-2-mRFP signal overlapped partially both in the cell bodies and in neuronal processes (Figures 5G and 5H; again FBXO32 colocalized to a greater extent with endophilin-2 than endophilin-1).
In sum, the data show that FBXO32 interacts directly with endophilin-A proteins (possibly through their SH3 domain), both proteins are involved in membrane tubulation, and both transiently colocalized on dynamic organelle-like structures of various sizes.
FBXO32 and Endophilins-A Associate Transiently with Autophagosomes
In addition to being an E3-ubiquitin ligase, FBXO32 is known to regulate autophagy in muscle, raising the question of whether the FBXO32-labeled organelle-like structures were autophagosomes. We coexpressed FBXO32-EGFP together with LC3-mRFP and observed a prominent colocalization of the two probes in starved cells (Figures 5I and 5M; Movie S4). A similar result was observed when FBXO32 was immunostained (Figure S4A), irrespective of type of cell (HeLa cells were also used) or fluorescent labeling (Figure S4E). Thus, FBXO32 is detected at autophagosomes.
Next, when endophilin-2 was coexpressed with the autophagosomal marker LC3, sparse but significant colocalization was observed in starved HeLa cells, suggesting that endophilin-2, like FBXO32 albeit to a lesser extent, transiently associates with autophagosomes (Figures 5J and 5M). Similar observations were made for endophilin-1 and endophilin-3, which were also occasionally seen on LC3-labeled autophagosomes (Figures 5M, S4F, and S4G; please note that the values represent colocalization above coincidental colocalization, which is prominent in this system given that endophilins are cytosolic proteins). For example, a kymograph from endophilin-3- and LC3-expressing HeLa cell shows that endophilin-3 colocalizes with an LC3-labeled autophagosome transiently (Figure 5N; arrows indicate colocalization). Importantly, FBXO32, LC3, and endophilin-2 could be simultaneously detected at the same dynamic organelle (Movie S6). Notably, endophilin-2 showed much stronger overlap with ATG5 (an early autophagosomal marker) than with LC3 (Figure 5K; 43% colocalization; see Movie S7), implying a role for endophilin-A in autophagosome formation.
Endophilin-A and FBXO32 Are Needed for Autophagosome Formation in Neurons and in Mammalian Brain
Given that both FBXO32 and endophilins-A were occasionally found on LC3-positive autophagosomes, we examined whether autophagy was altered in brains of TKO and 1,2 DKO mice. Notably, both LC3B-II (lower band is the active form), a classical marker for autophagosomes, and ATG5, a marker for early autophagosomes, were prominently decreased in TKO brains (Figure 6A) and in aged endophilin 1KO-2HT-3KO mice (Figures 6C and 6D). Interestingly, the ATG5 protein level in endophilin 1KO-2HT-3KO mice decreases progressively between 3 and 18 months of age (Figure 6D). A reduction in LC3B-II and ATG5 was also observed in 1,2 DKO cortex (Figure 6B), suggesting that endophilin-3 cannot effectively compensate for the loss of endophilin-1 and 2 in autophagy.
Figure 6.

Number of Autophagosomes Is Decreased in Endophilin TKO and 1,2 DKO Mutant Brains and Neurons
(A and B) Decrease in autophagosomal markers LC3B-II, ATG5, and CDK5 in TKO brain (A; p0) or 1,2 DKO cortical extracts (B; p14–p21). (Right) Quantification is shown (min. six samples/genotype). ∗p < 0.05; ∗∗∗p < 0.001; Student’s t test; mean ± SEM.
(C) Decrease in the autophagosomal marker LC3B-II in the cortex of aged endophilin 1KO-2HT-3KO mice. (Right) Quantification is shown (three cortices/genotype). ∗∗p < 0.01; Student’s t test; mean ± SEM.
(D) Decrease in the early autophagosomal marker ATG5 in the cortex of 3-, 9-, and 18-month-old endophilin 1KO-2HT-3KO mice. (Right) Quantification is shown (min. three samples/genotype). ∗p < 0.05; Student’s t test; mean ± SEM.
(E) Endophilin TKO and control hippocampal neurons transfected with LC3-EGFP and treated with 250 nM Torin-1 in the EBSS buffer for 4 hr were co-stained for synaptobrevin-2. Note that less LC3 signal can be detected at TKO synapses when compared to WTs (circles indicate synapses). Also note weaker synaptobrevin-2 staining in TKO samples is shown, in agreement with Milosevic et al. (2011). The scale bar represents 2 μm.
(F) Quantification of LC3 puncta in (C). ∗p < 0.05; t test; mean ± SEM.
(G) Endophilin-B1 in TKO brains. (Right) Quantification is shown (eight samples/genotype); mean ± SEM.
(H) Autophagosomal markers ATG9A and ATG16L are not changed in endophilin TKO brains (p0). (Right) Quantification is shown (min. four samples/genotype). ∗p < 0.05; ∗∗∗p < 0.001; Student’s t test; mean ± SEM.
See also Figure S5.
We prepared primary neuronal cultures from endophilin TKO and WT hippocampi and labeled autophagosomes either by mildly expressing LC3-EGFP (Figures 6E and S5C) or by immunostaining for LC3 (Figure S5E). We detected an overall decrease in the number of LC3-labeled autophagosomes when autophagy was simultaneously induced by starvation and mTOR inhibitor Torin-1 (Figures 6E, 6F, S5C, and S5D). Interestingly, the effect was more obvious at the synapses, as apparent from colocalization of LC3 and synaptobrevin-2, a presynaptic marker (Figure 6E; insets in Figure S5C). Altogether, the biochemical and imaging data suggest that autophagy depends on endophilin in vivo and in vitro and imply a role for endophilin in the formation and/or maturation of autophagosomes.
Notably, endophilin-B has a known role in autophagosome formation, and its CDK5-mediated phosphorylation is required for induced autophagy in models of PD (Wong et al., 2011). Endophilin-A proteins are closely related to endophilin-B: they share an identical domain structure with a high homology, and both protein subfamilies are enriched at the synapses and able to tubulate membranes (Farsad et al., 2001, Ringstad et al., 2001). Thus, we tested the levels of endophilin-B, CDK5, and several autophagy initiators. No change in the protein level of endophilin-B in endophilin-A TKO brains was detected (Figure 6G), whereas the levels of CDK5 were decreased (Figure 6A). However, in 1,2 DKO brains, the CDK5 level was unchanged (Figure 6B). Protein levels of UVRAG, BECLIN, ATG3, and HDAC6 were not altered, either in endophilin TKO brains or endophilin 1,2 DKO cortex (Figures S5A and S5B). These results imply that endophilin-B cannot compensate for autophagosome formation in the absence of endophilin-A and that autophagy is not altered due to repression of signaling pathways, because the key autophagy initiation regulators are not affected by absence of endophilin-A.
The key autophagy factors ATG9A and ATG16L have been shown to undergo clathrin-mediated endocytosis (Ravikumar et al., 2010a, Puri et al., 2014). In the light of endophilins’ well-established role in endocytosis, it is possible that the observed phenotype is an indirect effect of defective endocytosis in endophilin mutants. However, we found that both ATG9A and ATG16L were not significantly changed in our mutants (Figures 6H and S5F), indicating that availability of these two factors does not influence the findings we describe here.
To further characterize the role of FBXO32 in autophagy, we employed the Drosophila melanogaster neuromuscular junction (NMJ) to see whether FBXO32 is relevant for autophagy in neuronal cells in vivo. When Fbxo32 is silenced in Drosophila in vivo, there was a very obvious decrease in the number of autophagosomes detected at the NMJ (Figures S6A–S6C; quantification in Figure S6D). Thus, FBXO32 seems necessary for autophagosome formation at the fruit fly NMJ.
Given that, in the endophilin mutant brains there is more FBXO32 and yet still less autophagosome formation, the interaction between endophilin and FBXO32 may play a key role in this process.
Fbxo32 Upregulation Causes Apoptosis in Cells and Neurons that Can Be Rescued by Endophilin-A
To determine whether upregulation of Fbxo32 is an important step in the brain pathology of endophilin mutants, we explored the consequences of Fbxo32 upregulation in clonal (HeLa and MEF) and primary (neurons) cell lines. It has been reported that FBXO32 elevation induces apoptosis in cardiomyocytes and cancer cells (Wu et al., 2011, Xie et al., 2009). We observed that expression of FBXO32 causes substantial cell death in HeLa and MEFs, as determined by the number of remaining EGFP-positive cells and by the activity of caspases 3 and 7 (Figures 7A–7C, 7E, and S6E). The remaining cells looked unhealthy: they were smaller and often had vacuolar structures. The detrimental effects of FBXO32 were independent of its fluorescent tag and cell-transfection method (electroporation/Lipofectamine/Fugene).
Strikingly, when endophilin-1 was coexpressed with FBXO32 in MEFs, FBXO32-induced cell death was rescued, as demonstrated by the number of EGFP-positive cells (normalized [norm.] to total DAPI count) 30 hr after transfection and healthy cell appearance (Figure 7B; also in HeLa cells: Figure S6E), the quantification of cell survival (Figure 7C), and the activity of caspases 3 and 7 (Figure 7E). Importantly, the levels of FBXO32 expression were elevated when FBXO32 and endophilin-1 were coexpressed, implying that the rescue of FBXO32-induced apoptosis is not due to a reduction in FBXO32 levels (Figures 7A and 7B). However, coexpression with endophilin-1 seemed to relocate FBXO32-EGFP out of the nucleus (Figures 7D and S6E) in agreement with the data presented in Figure 5. Thus, FBXO32-induced apoptosis is associated with aberrant intracellular FBXO32 localization, and both the localization and the apoptosis susceptibility can be rescued by simultaneous upregulation of endophilin-1. These data show that FBXO32 and endophilin-1 interact not only at the protein but also at the genetic level.
In neurons, the effect was even more striking. When either WT primary hippocampal or cortical neurons were transfected with FBXO32-EGFP, less than 15% of all transfected neurons were detected at DIV14 (Figures 7F–7H). The surviving FBXO32-expressing neurons displayed a lower number of processes and appeared unhealthy (Figure S6F). TKO hippocampal and cortical neurons in culture were comparable to WT and littermate endophilin 1KO-2WT-3KO cells and did not show increased cell death under standard culturing conditions. However, when TKO cortical neurons were transfected with Fbxo32, less than 2% of transfected neurons were detected at DIV14 (in comparison to EGFP-expressing TKO neurons), with the surviving cells presenting a reduction in the number of processes and vacuoles in cell bodies. Lastly, coexpression of endophilin-1 or endophilin-2 was able to rescue FBXO32-induced apoptosis in cortical neurons: when endophilin-1-mRFP or endophilin-2-mRFP were coexpressed with FBXO32-EGFP, either in WT and endophilin TKO cortical neurons, the majority of neurons thrived (over 80% and 67% of all Fbxo32-transfected neurons, respectively; Figures 7F–7H). Imbalance in FBXO32/endophilin-A levels is likely a key mechanistic step in the brain pathology of endophilin mutants, and FBXO32-endophilin interplay seems important for maintaining neuronal health and overall protein homeostasis in the brain.
Discussion
To date, the known roles of endophilin-A have been limited to endocytosis and SV recycling. Here, we show that endophilin also affects the UPS and autophagy in the brain. The robust induction of the Foxo3a/Fbxo32 network (known to mediate muscle atrophy) in the brain of mice with endophilin deficiency promotes apoptosis and likely mediates brain pathology. Similarly to muscle, the Foxo/Fbxo32 network seems relevant for regulating protein degradation in the brain and for maintaining neuronal health. If FBXO32 is expressed in cell lines and primary neurons, it causes apoptosis that can be rescued by simultaneous coexpression of endophilin. Thus, balancing the FBXO32-endophilin interaction seems essential for sustaining neuronal viability.
We also report that partial endophilin absence in mice results in neurodegeneration and in perturbations of a significant number of ataxia- and PD-related genes. These observations corroborate a number of recent findings that indirectly link endophilin to neurodegeneration and PD (Arranz et al., 2015, Edvardson et al., 2012, Krebs et al., 2013, Matta et al., 2012, Milosevic et al., 2011, Quadri et al., 2013, Shi et al., 2009, Trempe et al., 2009). Endophilin models may also be of relevance for neurodegeneration research because they may provide insights into signaling pathways affected prior to overt manifestation of neurodegeneration.
FBXO32 was thus far studied mainly in skeletal muscle, where this E3-ubiquitin ligase was found to mediate rapid atrophy as a result of FOXO3A transcriptional activation. Interestingly, FOXO3A activation is associated with cellular stress responses and neurodegeneration (Webb and Brunet, 2014). FOXO3A stimulates autophagy in neurons (Xu et al., 2011), and FBXO32 is necessary for autophagy; activation of the FoxO-Fbxo32 axis should thus lead to increased autophagy. Nevertheless, this is not the case when endophilin is lacking: the number of autophagosomes is sharply decreased. Also, endophilin colocalizes transiently with mature and even more impressively with early autophagosomes, suggesting that endophilin-A has a previously unreported role in the formation and/or maturation of autophagosomes. This is supported by the recent identification of endophilin-A as one of the regulators of autophagosome formation (Strohecker et al., 2015). Interestingly, mice without endophilin also display the same pattern of perinatal lethality observed in mice lacking proteins necessary for autophagosome formation (Kuma et al., 2004).
Given endophilin’s involvement in endocytic processes, the decrease in the number of autophagosomes might also result from compromised endocytosis. Specifically, it has been shown that blockage of endocytosis upstream of clathrin coat assembly results in decreased autophagosome formation (Ravikumar et al., 2010a, Ravikumar et al., 2010b). However, endophilin deficiency results in the accumulation of clathrin-coated vesicles, and the levels of clathrin are not altered (Milosevic et al., 2011). Thus, clathrin is available to interact with ATG16L1, and RNA and protein levels of ATG16L1 are not altered in this model. In addition, the RNA and protein levels of ATG9, a key autophagic protein known to traffic via the plasma membrane and endosomal compartments (Mari et al., 2010, Puri et al., 2014) are not altered in endophilin mutant brains. Altogether, whereas we cannot entirely exclude endophilin’s role in endocytosis also affecting autophagy, we consider other mechanism(s) besides endocytic defects to be underlying the decrease in autophagosomes in endophilin mutants.
Decreased autophagosome availability and, consequently, lower autophagic flux are known to affect protein homeostasis (Korolchuk et al., 2009). We observed an accumulation of K48-linked ubiquitinated proteins in the cytoplasm, suggesting saturation of the UPS (due to overloading rather than deficiencies in the proteasome). There is possibly a double hit on this pathway: decreased autophagy and increased availability of ubiquitin ligases (FBXO32 and parkin). Both FBXO32 and parkin are involved in selective autophagy of ubiquitinated proteins (Zaglia et al., 2014), and both function under the regulation of FOXOs (Webb and Brunet, 2014). Upregulation of FBXO32 and parkin may thus be a compensatory response to defective autophagy, an attempt to shunt more of the burden to the UPS, or possibly both.
Interestingly, the lower abundance of autophagosomes in the absence of endophilin-A cannot be (fully) compensated for by a related protein with the same domain organization, endophilin-B, an established mediator of autophagosome formation (Takahashi et al., 2011). Endophilin-A primarily operates in the endocytic processes at the plasma membrane, whereas endophilin-B has prominent roles in apoptosis, mitochondrial membrane dynamics, and autophagosome formation (Kjaerulff et al., 2011). Furthermore, CDK5-mediated phosphorylation of endophilin-B is required for inducing autophagy in models of PD (Wong et al., 2011). More research is needed to understand endophilin-A and endophilin-B links to autophagy, neurodegeneration, and PD.
We propose a model in which endophilin’s absence, likely due to its membrane-curvature-inducing/stabilizing properties, leads to reduced availability of membranes for autophagosome formation and decreased autophagic activity. Interestingly, FBXO32 and endophilin interact at both the gene and protein level and both are required for the autophagy process. Like endophilin, FBXO32 can tubulate membranes and is recruited to membrane tubules in vivo. Thus, endophilin and FBXO32 membrane-tubulating activity may contribute to the formation/maturation of autophagosomes. This model is in concordance with the reported role of endophilin-A as necessary and sufficient for macroautophagy at presynaptic terminals of fruit flies (Soukup et al., 2016).
In summary, our study addresses the origin of neurodegeneration in a key mammalian endocytic model and reveals that the FOXO/FBXO32/endophilin network may play a role in maintaining neuronal health by balancing autophagy and the UPS in the brain. The results are consistent with the following plausible scenario: reduced endophilin availability results in impaired capacity to form autophagosomes, resulting in decreased autophagy, accumulation of ubiquitinated proteins in the cytoplasm, proteasome saturation, and further accumulation of unwanted proteins over time that eventually affect neuronal function and health, leading to neurodegeneration. Accordingly, we show that imbalanced autophagy also develops in aged endophilin heterozygote mice that display age-dependent progressive ataxia. Our data reveal a role in autophagy for a key endocytic adaptor previously thought to be strictly dedicated to endocytosis and invite further research addressing the physiological and pathological implications of these findings.
Experimental Procedures
Reagents and Gene-Targeting Strategies
Unless otherwise stated, reagents were purchased from Sigma-Aldrich. Supplemental Experimental Procedures detail gene-targeting and cloning strategies and list the antibodies and plasmids used.
Behavior Experiments
Animal experiments were conducted according to the European Guidelines for animal welfare (2010/63/EU) with approval by the Lower Saxony Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES), registration number 14/1701.
For rotarod experiments, 6–20 males/group were tested on a setup by TSE Systems (3375-5). A training phase consisted of four trainings over 2 days (intertrial interval [ITI] was 2–5 hr; 180 s at 5 rpm). The testing phase consisted of four tests over 2 days (ITI 2–5 hr). Mice were placed on the rod, which then accelerated from 5 to 40 rpm in 180 s. Fall latency was measured. Student’s t test was applied.
A simple composite phenotype scoring system for evaluating mouse models of ataxia was applied as described (Guyenet et al., 2010). Eight to twelve mice were used per condition, and scoring was performed by two researchers independently. Data were analyzed using two-way ANOVA.
Ventral plane videography for gait analysis was performed using instrumentation (DigiGait) and software from Mouse Specifics, according to manufacturer’s instructions. For each group, 6–20 males were used. Student’s t test was applied.
Fluorescence Imaging
TUNEL assay was performed on cryosections of the brains of perfused mice to test for apoptotic and necrotic cells. Mice were anesthetized with chloral hydrate (40 mg/mL) before cardiac perfusion with 4% paraformaldehyde (PFA). Harvested brains were post-fixed in PFA. After slow dehydration with sucrose, brains were embedded in OCT (Tissue Tek) and frozen in liquid N2. Sagittal brain sections were cut with a cryostat (10 μm thick). Sections were post-fixed and permeabilized with 0.1% Triton X-100. Positive controls were incubated with DNase I (833 U/mL). In Situ Cell Death Detection Kit (Fluorescein) was used according to manufacturer’s instructions. Slides were counterstained with DAPI, embedded with Mowiol 4-88, and imaged with a LSM 710 confocal setup (Zeiss). Immunofluorescence of frozen brain sections and cultured neurons (DIV 14–20) was carried out as described (Ringstad et al., 2001, Milosevic et al., 2011). Live MEFs, HeLa cells, and neurons were imaged either using a Zeiss AxioObserver Z1 total internal reflection fluorescence (TIRF) microscope with an Evolve charge-coupled device (CCD) camera (Photometrics) and 100× Plan Apo Zeiss objective or Perkin Elmer Ultraview spinning-disk confocal setup that consists of Nikon Ti-E Eclipse inverted microscope equipped with Perfect Focus, 60× CFI Plan Apo VC Nikon objective, and 14-bit electron-multiplied CCD camera (Hamamatsu C9100). Autophagy induction and colocalization analysis was performed as detailed in the Supplemental Experimental Procedures and below.
RNA Sequencing
RNA sequencing was performed as described in Halder et al. (2016). The pathway analysis was performed as described in Raimundo et al. (2012). These methods are detailed in Supplemental Experimental Procedures.
Biochemical Procedures
Whole-brain or cortex samples for western blotting were prepared by homogenization of brain tissue (whole brain for p0 animals; one cortex for p14–p21 animals and aged animals) as detailed in Supplemental Experimental Procedures. For Trap-GFP immunoprecipitation, HeLa cells were transfected with endophilin (1, 2, or 3)-EGFP and mCherry/FBXO32-mCherry using Lipofectamine 3000 (Invitrogen). We used Chromotek Trap-GFP agarose beads (Chromotek) and followed manufacturer’s protocol. Alternatively, HeLa cells were transfected with FBXO32-EGFP and coupled to Chromotek Trap-GFP agarose beads. Purified endophilin-1 was added, and all fractions (input, non-bound, and bound) were collected. Samples were subjected to SDS-PAGE electrophoresis and immunoblotted against indicated proteins.
Cell Culture
siRNA-based knockdown of Foxo3a was performed as in Milosevic et al. (2011). Primary hippocampal and cortical cultures were prepared as previously described (Ferguson et al., 2007). Primary neurons expressed constructs, following Amaxa (Lonza)-based transfection, under the control of the chicken-β-actin promoter to allow for long-term expression. A minimum of 15 images from three to five experiments were analyzed for each genotype/condition. Given that the majority of respective fluorescent signals were cytosolic, colocalization was evaluated semi-automatically using object-based overlap analysis and ImageJ JACoP plugin (Bolte and Cordelières, 2006). In short, the image was first segmented into objects and background (i.e., bright fluorescent objects are segmented from the image), and then their spatial relationship (overlap) was measured. The same analysis was performed with the mirror image, and random colocalization was subtracted in each case. Complementary manual colocalization analysis was also performed, and it yielded similar results. Here, circles were superimposed on bright fluorescent spots in the FBXO32 (or LC3 or ATG5) channel and transferred to identical image locations in the endophilin channel. If the fluorescence intensity maximum in the endophilin channel was located in the same circle and the morphology of the signal resembled that of the FBXO32 signal, the circle was rated as positive (colocalized); if not, it was rated as negative (not colocalized). To correct for accidental colocalization, circles were also transferred to a mirror image of the endophilin channel (detailed in Supplemental Experimental Procedures). Fluorescent puncta were quantified as in Hayashi et al. (2008). Data are presented as puncta per 100 μm2, normalized to controls. Student’s t test was used unless otherwise stated.
Proteasome Activity Assay
We used WT and endophilin TKO MEFs, immortalized with E6/E7 viral proteins, grown on supplemented DMEM at 5% CO2 at 37°C. The cells were about 70%–80% confluent when harvested. Cells were pelleted and resuspended with n-dodecylmaltoside 1.5% in PBS with Complete Protease Inhibitor Cocktail (Roche) and PhosSTOP phosphatase inhibitor cocktail (Roche) and lysed at 4°C in a rotator for 30 min. The lysate was then centrifuged to pellet debris and placed in 96-well glass-bottomed plates for use in the assay. The fluorimetric proteasome assay (Proteasome 20S Activity Assay Kit) was performed according to manufacturer’s instructions using a Synergy H1 plate reader (Biotek) at 37°C.
Caspase Activity Assay
Caspase 3/7 apoptosis assay was performed with the ApoTox-Glo Triplex Assay (Promega) kit according to a modified version of the manufacturer’s protocol for 96-well plates, detailed in Supplemental Experimental Procedures.
Author Contributions
Conceptualization, N.R. and I.M.; Methodology, J.D.M., N.R., and I.M.; Resources, P.V., S.B., A.F., N.R., and I.M.; Investigation and Analysis, J.D.M., C.M.R., S.G., A.S.A., S.F.S., S.F., R.V., V.C., P.V., S.B., N.R., and I.M. (J.D.M.: Figures 2E [with N.R.], 3A, 3B, 3E, and 6A–6C; C.M.R.: Figures 1 [with I.M.], 3C, 3F, 4A–4C, 6D, 6G, and 6H); Supervision, P.V., S.B., N.R., and I.M.; Writing – Original Draft, J.D.M., N.R., and I.M.; Writing – Review & Editing, C.M.R., J.D.M., N.R., and I.M.
Acknowledgments
We thank M. König, E. Rizou, J. Liebig, and S. Burkhardt for superb technical assistance; A. Cepeda for help with protein expression and purification; B. Flix for cloning; and J. Kroll for video processing. This work was supported in part by grants from Schram-Stiftung T287/25457 and Deutsche Forschungsgemeinschaft (DFG) (Emmy Noether Young Investigator Award MI 1702/1 and SFB889/2/A08) to I.M., ERC Starting Grant 337327 to N.R., an Alexander von Humboldt postdoctoral fellowship to J.D.M., and a SySy fellowship to S.G.
Published: October 6, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, eight tables, and seven movies and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.09.058.
Contributor Information
Nuno Raimundo, Email: nuno.raimundo@med.uni-goettingen.de.
Ira Milosevic, Email: imilose@gwdg.de.
Accession Numbers
The accession number for the NGS data reported in this paper is GEO: GSE85702.
Supplemental Information
Table S1. RNA Sequencing of Endophilin TKO and Endophilin 1,2 DKO Samples, Related to Figure 2
Table S2. Comparison of Endophilin TKO and Littermate 1KO-2WT-3KO Samples, Related to Figure 2
Table S3. Comparison of Endophilin TKO and WT Samples, Related to Figure 2
Table S4. Crossing of TKO and 1,2 DKO Differentially Expressed Genes with Gene Lists of Ataxia and Parkinson’s Disease, Related to Figure 2
Table S5. Comparison of Endophilin 1,2 DKO and 1KO-2WT Samples, Related to Figure 2
Table S6. Comparison of Endophilin 1,2 DKO and WT Samples, Related to Figure 2
Table S7. List of DEGs when Endophilin TKO Samples Are Compared against All Controls Pooled Together, Related to Figure 2
Table S8. Upstream Regulators of TKO Transcriptome, Related to Figure 2
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Imaged by spinning-disc confocal microscopy. Note that FBXO32 (green) and endophilin (red) colocalized on trafficking organelles and tubular structures. Inset shows detail with dynamic tubule labelled with both FBXO32-EGFP and endophilin-2-mRFP. Time-lapse images were captured every 4s, video runs at 10 frames/s.
{kind=link}
{kind=link}
{kind=link}
References
- Arranz A.M., Delbroek L., Van Kolen K., Guimarães M.R., Mandemakers W., Daneels G., Matta S., Calafate S., Shaban H., Baatsen P. LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J. Cell Sci. 2015;128:541–552. doi: 10.1242/jcs.158196. [DOI] [PubMed] [Google Scholar]
- Bolte S., Cordelières F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Boucrot E., Ferreira A.P., Almeida-Souza L., Debard S., Vallis Y., Howard G., Bertot L., Sauvonnet N., McMahon H.T. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature. 2015;517:460–465. doi: 10.1038/nature14067. [DOI] [PubMed] [Google Scholar]
- Cao, M., Milosevic, I., Giovedi, S., and De Camilli, P. (2013). Genetic interaction between endophilin and Parkin. In Society for Neuroscience (SfN) Annual Meeting 2013, p. 423.12.
- Cao M., Milosevic I., Giovedi S., De Camilli P. Upregulation of Parkin in endophilin mutant mice. J. Neurosci. 2014;34:16544–16549. doi: 10.1523/JNEUROSCI.1710-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma N.P., Lindsten K., Glas R., Jellne M., Masucci M.G. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- Deger J.M., Gerson J.E., Kayed R. The interrelationship of proteasome impairment and oligomeric intermediates in neurodegeneration. Aging Cell. 2015;14:715–724. doi: 10.1111/acel.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S., Cinnamon Y., Ta-Shma A., Shaag A., Yim Y.I., Zenvirt S., Jalas C., Lesage S., Brice A., Taraboulos A. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE. 2012;7:e36458. doi: 10.1371/journal.pone.0036458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G., Ana Clara F., Verstreken P. Synaptic vesicle trafficking and Parkinson’s disease. Dev. Neurobiol. 2012;72:134–144. doi: 10.1002/dneu.20916. [DOI] [PubMed] [Google Scholar]
- Farsad K., Ringstad N., Takei K., Floyd S.R., Rose K., De Camilli P. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J. Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S.M., Brasnjo G., Hayashi M., Wölfel M., Collesi C., Giovedi S., Raimondi A., Gong L.W., Ariel P., Paradise S. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- Guyenet S.J., Furrer S.A., Damian V.M., Baughan T.D., La Spada A.R., Garden G.A. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp. 2010;(39):1787. doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder R., Hennion M., Vidal R.O., Shomroni O., Rahman R.U., Rajput A., Centeno T.P., van Bebber F., Capece V., Garcia Vizcaino J.C. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci. 2016;19:102–110. doi: 10.1038/nn.4194. [DOI] [PubMed] [Google Scholar]
- Hayashi M., Raimondi A., O’Toole E., Paradise S., Collesi C., Cremona O., Ferguson S.M., De Camilli P. Cell- and stimulus-dependent heterogeneity of synaptic vesicle endocytic recycling mechanisms revealed by studies of dynamin 1-null neurons. Proc. Natl. Acad. Sci. USA. 2008;105:2175–2180. doi: 10.1073/pnas.0712171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herhaus L., Dikic I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015;16:1071–1083. doi: 10.15252/embr.201540891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heutink P., Verhage M. Neurodegeneration: new road leads back to the synapse. Neuron. 2012;75:935–938. doi: 10.1016/j.neuron.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Iaccarino C., Crosio C., Vitale C., Sanna G., Carrì M.T., Barone P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum. Mol. Genet. 2007;16:1319–1326. doi: 10.1093/hmg/ddm080. [DOI] [PubMed] [Google Scholar]
- Kjaerulff O., Brodin L., Jung A. The structure and function of endophilin proteins. Cell Biochem. Biophys. 2011;60:137–154. doi: 10.1007/s12013-010-9137-5. [DOI] [PubMed] [Google Scholar]
- Korolchuk V.I., Mansilla A., Menzies F.M., Rubinsztein D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C.E., Karkheiran S., Powell J.C., Cao M., Makarov V., Darvish H., Di Paolo G., Walker R.H., Shahidi G.A., Buxbaum J.D. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat. 2013;34:1200–1207. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yoshimori T., Ohsumi Y., Tokuhisa T., Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Mari M., Griffith J., Rieter E., Krishnappa L., Klionsky D.J., Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010;190:1005–1022. doi: 10.1083/jcb.200912089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S., Van Kolen K., da Cunha R., van den Bogaart G., Mandemakers W., Miskiewicz K., De Bock P.J., Morais V.A., Vilain S., Haddad D. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- Menzies F.M., Fleming A., Rubinsztein D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- Milosevic I., Giovedi S., Lou X., Raimondi A., Collesi C., Shen H., Paradise S., O’Toole E., Ferguson S., Cremona O., De Camilli P. Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron. 2011;72:587–601. doi: 10.1016/j.neuron.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T., Lipton S.A. Cell death: protein misfolding and neurodegenerative diseases. Apoptosis. 2009;14:455–468. doi: 10.1007/s10495-008-0301-y. [DOI] [PubMed] [Google Scholar]
- Nixon R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Nonis D., Schmidt M.H., van de Loo S., Eich F., Dikic I., Nowock J., Auburger G. Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell. Signal. 2008;20:1725–1739. doi: 10.1016/j.cellsig.2008.05.018. [DOI] [PubMed] [Google Scholar]
- Perera R.M., Zoncu R., Lucast L., De Camilli P., Toomre D. Two synaptojanin 1 isoforms are recruited to clathrin-coated pits at different stages. Proc. Natl. Acad. Sci. USA. 2006;103:19332–19337. doi: 10.1073/pnas.0609795104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli A., Gilestro G.F., Lanzardo S., Comoglio P.M., Migone N., Giordano S. The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature. 2002;416:187–190. doi: 10.1038/416187a. [DOI] [PubMed] [Google Scholar]
- Puri C., Renna M., Bento C.F., Moreau K., Rubinsztein D.C. ATG16L1 meets ATG9 in recycling endosomes: additional roles for the plasma membrane and endocytosis in autophagosome biogenesis. Autophagy. 2014;10:182–184. doi: 10.4161/auto.27174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri M., Fang M., Picillo M., Olgiati S., Breedveld G.J., Graafland J., Wu B., Xu F., Erro R., Amboni M., International Parkinsonism Genetics Network Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum. Mutat. 2013;34:1208–1215. doi: 10.1002/humu.22373. [DOI] [PubMed] [Google Scholar]
- Raimundo N., Song L., Shutt T.E., McKay S.E., Cotney J., Guan M.X., Gilliland T.C., Hohuan D., Santos-Sacchi J., Shadel G.S. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell. 2012;148:716–726. doi: 10.1016/j.cell.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralser M., Nonhoff U., Albrecht M., Lengauer T., Wanker E.E., Lehrach H., Krobitsch S. Ataxin-2 and huntingtin interact with endophilin-A complexes to function in plastin-associated pathways. Hum. Mol. Genet. 2005;14:2893–2909. doi: 10.1093/hmg/ddi321. [DOI] [PubMed] [Google Scholar]
- Ravikumar B., Moreau K., Jahreiss L., Puri C., Rubinsztein D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B., Moreau K., Rubinsztein D.C. Plasma membrane helps autophagosomes grow. Autophagy. 2010;6:1184–1186. doi: 10.4161/auto.6.8.13428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard H.-F., Garcia-Castillo M.D., Chambon V., Lamaze C., Johannes L. Shiga toxin stimulates clathrin-independent endocytosis of the VAMP2, VAMP3 and VAMP8 SNARE proteins. J Cell Sci. 2015;128:2891–2902. doi: 10.1242/jcs.171116. [DOI] [PubMed] [Google Scholar]
- Ringstad N., Nemoto Y., De Camilli P. The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc. Natl. Acad. Sci. USA. 1997;94:8569–8574. doi: 10.1073/pnas.94.16.8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N., Gad H., Löw P., Di Paolo G., Brodin L., Shupliakov O., De Camilli P. Endophilin/SH3p4 is required for the transition from early to late stages in clathrin-mediated synaptic vesicle endocytosis. Neuron. 1999;24:143–154. doi: 10.1016/s0896-6273(00)80828-4. [DOI] [PubMed] [Google Scholar]
- Ringstad N., Nemoto Y., De Camilli P. Differential expression of endophilin 1 and 2 dimers at central nervous system synapses. J. Biol. Chem. 2001;276:40424–40430. doi: 10.1074/jbc.M106338200. [DOI] [PubMed] [Google Scholar]
- Saheki Y., De Camilli P. Synaptic vesicle endocytosis. Cold Spring Harb. Perspect. Biol. 2012;4:a005645. doi: 10.1101/cshperspect.a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S.H., Goldberg A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreij A.M., Fon E.A., McPherson P.S. Endocytic membrane trafficking and neurodegenerative disease. Cell. Mol. Life Sci. 2016;73:1529–1545. doi: 10.1007/s00018-015-2105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M., Bradner J., Bammler T.K., Eaton D.L., Zhang J., Ye Z., Wilson A.M., Montine T.J., Pan C., Zhang J. Identification of glutathione S-transferase pi as a protein involved in Parkinson disease progression. Am. J. Pathol. 2009;175:54–65. doi: 10.2353/ajpath.2009.081019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubeyran P., Kowanetz K., Szymkiewicz I., Langdon W.Y., Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature. 2002;416:183–187. doi: 10.1038/416183a. [DOI] [PubMed] [Google Scholar]
- Soukup S., Kuenen S., Vanhauwaert R., Manetsberger J., Hernández-Díaz S., Swerts J., Schoovaerts N., Vilain S., Gounko N.V., Vints K. A LRRK2-dependent EndophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron. 2016;92 doi: 10.1016/j.neuron.2016.09.037. Published online October 6, 2016. [DOI] [PubMed] [Google Scholar]
- Strohecker A.M., Joshi S., Possemato R., Abraham R.T., Sabatini D.M., White E. Identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel autophagy regulator by high content shRNA screening. Oncogene. 2015;34:5662–5676. doi: 10.1038/onc.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Meyerkord C.L., Hori T., Runkle K., Fox T.E., Kester M., Loughran T.P., Wang H.G. Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy. 2011;7:61–73. doi: 10.4161/auto.7.1.14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y., Giaime E., Yamaguchi H., Ichimura T., Liu Y., Si H., Cai H., Bonventre J.V., Shen J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 2012;7:2. doi: 10.1186/1750-1326-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe J.F., Chen C.X., Grenier K., Camacho E.M., Kozlov G., McPherson P.S., Gehring K., Fon E.A. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol. Cell. 2009;36:1034–1047. doi: 10.1016/j.molcel.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Verstreken P., Kjaerulff O., Lloyd T.E., Atkinson R., Zhou Y., Meinertzhagen I.A., Bellen H.J. Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell. 2002;109:101–112. doi: 10.1016/s0092-8674(02)00688-8. [DOI] [PubMed] [Google Scholar]
- Webb A.E., Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci. 2014;39:159–169. doi: 10.1016/j.tibs.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A.S., Lee R.H., Cheung A.Y., Yeung P.K., Chung S.K., Cheung Z.H., Ip N.Y. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat. Cell Biol. 2011;13:568–579. doi: 10.1038/ncb2217. [DOI] [PubMed] [Google Scholar]
- Wu C.L., Kandarian S.C., Jackman R.W. Identification of genes that elicit disuse muscle atrophy via the transcription factors p50 and Bcl-3. PLoS ONE. 2011;6:e16171. doi: 10.1371/journal.pone.0016171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P., Guo S., Fan Y., Zhang H., Gu D., Li H. Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation. J. Biol. Chem. 2009;284:5488–5496. doi: 10.1074/jbc.M806487200. [DOI] [PubMed] [Google Scholar]
- Xu P., Das M., Reilly J., Davis R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011;25:310–322. doi: 10.1101/gad.1984311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaglia T., Milan G., Ruhs A., Franzoso M., Bertaggia E., Pianca N., Carpi A., Carullo P., Pesce P., Sacerdoti D. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Invest. 2014;124:2410–2424. doi: 10.1172/JCI66339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. RNA Sequencing of Endophilin TKO and Endophilin 1,2 DKO Samples, Related to Figure 2
Table S2. Comparison of Endophilin TKO and Littermate 1KO-2WT-3KO Samples, Related to Figure 2
Table S3. Comparison of Endophilin TKO and WT Samples, Related to Figure 2
Table S4. Crossing of TKO and 1,2 DKO Differentially Expressed Genes with Gene Lists of Ataxia and Parkinson’s Disease, Related to Figure 2
Table S5. Comparison of Endophilin 1,2 DKO and 1KO-2WT Samples, Related to Figure 2
Table S6. Comparison of Endophilin 1,2 DKO and WT Samples, Related to Figure 2
Table S7. List of DEGs when Endophilin TKO Samples Are Compared against All Controls Pooled Together, Related to Figure 2
Table S8. Upstream Regulators of TKO Transcriptome, Related to Figure 2
Imaged by spinning-disc confocal microscopy. Note that FBXO32 (green) and endophilin (red) colocalized on trafficking organelles and tubular structures. Inset shows detail with dynamic tubule labelled with both FBXO32-EGFP and endophilin-2-mRFP. Time-lapse images were captured every 4s, video runs at 10 frames/s.