

Abstract
Bismuth(III) acetate is a safe, inexpensive, and selective facilitator of sequential protodeboronations, which when used in conjunction with Ir-catalyzed borylations allows access to a diversity of borylated indoles. The versatility of combining Ir-catalyzed borylations with Bi(III)-catalyzed protodeboronation is demonstrated by selectively converting 6-fluoroindole into products with Bpin groups at the 4-, 5-, 7-, 2,7-, 4,7-, 3,5-, and 2,4,7-positions and the late-stage functionalization of sumatriptan.
Graphical Abstract

Arylboronates are versatile synthetic building blocks.1,2 Iridium catalyzed C–H activation/borylation reactions are a powerful way of making such compounds as they can obviate the need for prior functionalization (e.g., halogenation), pyrophoric reagents, cryogenic conditions, etc.3 While the regioselectivity of aromatic C–H borylations is mainly driven by steric effects, C–H acidity is a secondary driver.4 For example, Ir-catalyzed borylation of unprotected indoles first installs a Bpin group at C2 and then upon further reaction at C7.5
Recently, Movassaghi and co-workers6 showed that the 2,7-diborylation of tryptophans, tryptamines, and 3-alkylindoles could be followed by in situ palladium-catalyzed C2-protodeboronation to selectively afford the C7 products (Scheme 1). While this tactic may not seem atom economical, from a strategic perspective such a borylation/protodeboronation sequence enables a streamlined approach to 7-borylated indoles that are otherwise difficult to access without additional steps and/or prefunctionalization7 We too had observed selective deborylations of a number of diborylated heterocycles, including several 2,7-diborylated indoles (Scheme 1).8 Our protodeboronations were Ir-catalyzed and for some systems could be exposing a crude Ir-catalyzed borylation mixture to protic material. Perhaps most usefully, we noted that for diborylated indoles, azaindoles, thiophenes, and benzthiophenes the first Bpin group to be installed during the Ir-catalyzed borylation was also the first Bpin to be removed in the Ir-catalyzed protodeboronation.
Scheme 1.

Prior Art
During a total synthesis project, we prepared 7-borylated 2 as shown in Scheme 1. The next step in the synthesis called for BiCl3-promoted removal of the Boc group9 (Scheme 2). Close examination of this deprotection revealed that 3 formed along with trace amounts of byproduct 4 where the C7-BPin was missing. This small amount of deborylated byproduct led us to consider whether bismuth salts could facilitate selective protodeboronation in a way similar to the previously described Ir- and Pd-catalyzed protodeboronations. Such a method would be quite attractive because bismuth salts are earth abundant, harmless, and orders of magnitude less expensive than the corresponding precious metal salts.10
Scheme 2.

Discovery of Bicatalyzed Protodeboronations
After screening BiCl3, Bi(OTf)3 and numerous other metal salts and other additives,11 Bi(OAc)3 emerged as the catalyst of choice. Subjecting purified 1 to 20 mol % Bi(OAc)3 in MeOH (127 equiv) and THF at 80 °C (sealed tube) for 7 h afforded 7-borylated 2 in 90% yield (Scheme 3).
Scheme 3.

Bi(OAc)3 Catalyzed Protodeboronation of 1
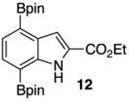
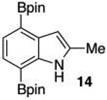
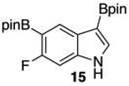
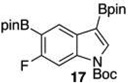
Given this favorable result, a series of indoles were subjected to multiple borylations (Table 1). Several of these Ir-catalyzed borylations are worthy of comment. Following C7 borylation, the next site for C–H borylation proved to be C4. In this way, 2,4,7-triborylated indoles 7 and 10 (entries 2 and 4)12,13 and 4,7-diborylated 12 and 14 (entries 5 and 6) were generated. We previously showed that placing a Boc14 or Bpin15 on the indole nitrogen directs borylation to the C3-position. Herein we report for the first time that, provided the C6 position is blocked, borylation of in situ N-borylated (entry 7) or N-Boc protected (entry 8) indoles occurs at the C3 and then the C5-positions, affording 15 and 17 respectively.16–18
Table 1.
Ir-Catalyzed Borylation of Indoles
| entry | Starting indole | product | time (h) | yield (%)a |
|---|---|---|---|---|
| 1b | 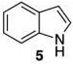 |
 |
48 | 77 |
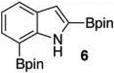
| 2c | 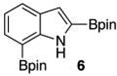 |
 |
12 | 96 |
| 3b | 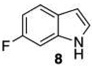 |
 |
24 | 82 |
| 4c | 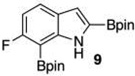 |
 |
12 | 92 |
| 5b | 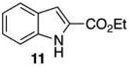 |
 |
12 | 84 |
| 6b | 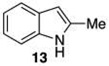 |
 |
24 | 71 |
| 7d,e | 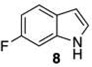 |
 |
5 | 90 |
| 8e | 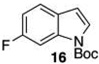 |
 |
3 | 80 |
Isolated yields.
Borylations ran with 2.0 equiv B2pin2, 2.8 mol % HBpin, 0.5 mol % [Ir(OMe)COD]2, 1 mol % dtbpy, at 80 °C.
Borylations ran as described above, but with 1.0 equiv B2pin2.
Substrate stirred in neat HBpin (4 equiv) at rt for 1 h before being subjected to the borylation conditions.
Borylation ran with 2.0 equiv B2pin2, 3 mol % [Ir(OMe)COD]2, 6 mol % dtbpy, at 80 °C.
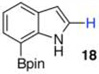
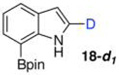
With the borylated indoles in hand, we explored their Bi(OAc)3 mediated protodeboronations (Table 2). Examining first 2,7-diborylated indole (6), we found that heating this compound with 20 mol % Bi(OAc)3 and 125 equiv of ACS grade MeOH in THF afforded the 7-borylated indole (17) in 82% yield after 17 h (entry 1). Curiously, when we looked to deuterate 6, the reaction was complete (83% isolated yield, 87% deuterium incorporation19,20) after stirring with 60 equiv of 99.8% CD3OD for 12 h at room temperature (entry 2). A closer look into these differences revealed that, as we had observed with some of the Ir-catalyzed deboronations,8 the grade of MeOH could significantly impact the reaction rate. For example, protodeboronation of 6 was complete in less than 3 h when anhydrous MeOH that came in sealed bottles was employed. Notably, reactions with either grade of methanol were reproducible. To highlight the method’s relative robustness and economy, we chose to continue our study with the lower grade methanol.21
Table 2.
Bi(OAc)3 Catalyzed Protodeboronations
| entry | starting indole | product | conditions and yielda |
|---|---|---|---|
| 1 | 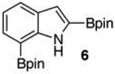 |
 |
20 mol % Bi(OAc)3, 125 equiv  , ,THF, 80 °C, 17 h, 82% |
| 2 | 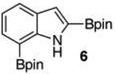 |
 |
20 mol % Bi(OAc)3, 40 equiv  , ,THF, 80 °C, 2.5 h, 83% |
| 3 | 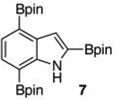 |
 |
20 mol % Bi(OAc)3, 125 equiv ,THF, 80 °C, 17 h, 75% |
| 4 | 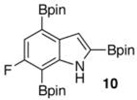 |
 |
20 mol % Bi(OAc)3, 250 equiv ,THF, 80 °C, 15 h, 80% |
| 5 | 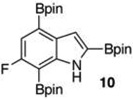 |
 |
20 mol % Bi(OAc)3, 60 equiv ,THF, 80 °C, 5 h, 67% |
| 6 | 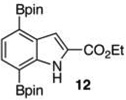 |
 |
40 mol % Bi(OAc)3, 375 equiv ,THF, 80 °C, 16 h, (54:9:41 22: 11:12)b Ref 8 Ir-catalysis (33:67 22:11)b Modifiedd Ir-catalysisc 54% 22, 13% 11 |
| 7 | 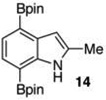 |
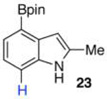 |
40 mol % Bi(OAc)3, 500 equiv ,THF, 80 °C, 16 h, (52:5:43 23:13:14)b Ref 8 Ir-catalysisc 74% (91:9 23:14)b |
| 8 | 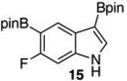 |
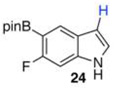 |
20 mol % Bi(OAc)3, 50 equiv ,THF, 80 °C, 3 h, 88% Ref 8 Ir-catalysisc 66% (87:13 24:8)b |
| 9 | 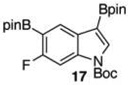 |
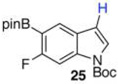 |
40 mol % Bi(OAc)3, 500 equiv ,THF, 80 °C, 16 h, (no reaction)b Ref 8 Ir-catalysisc 47% (60:40 25:16)b |
Isolated yields.
Ratio determined by 1H NMR of the crude reaction mixture.
See Supporting Information for details.
3 mol % [Ir(OMe)COD]2, 40 equiv MeOH, THF at rt.
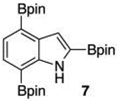
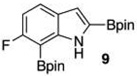
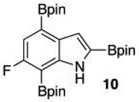
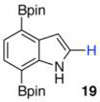
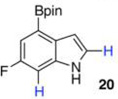
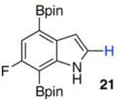
Within these parameters, 2,4,7-triborylated indole (7) was monoprotodeboronated to afford 4,7-diborylated indole (19) in 75% yield under similar conditions (entry 3). Attempts at the selective C2/C7 diprotodeboronation of 7 were disappointing. While 4-borylated indole (20) was formed as the major product, NMR analysis of the crude reaction mixture revealed a 50/45/5 mixture of 20/19/indole.22 In contrast, 2,4,7triborylated-6-fluoroindole (10) underwent clean diprotodeboronation to afford 20 in 80% yield (entry 4) when the amount of MeOH was increased. A qualitative feature of deborylating 10 is that upon completion the reaction mixture takes on a slight pink color. Monoprotodeboronation of 10 (entry 5) provided further indication that these reactions are in part substrate dependent, as relative to 7, trisboylated 10 required less time and equivalents of methanol to achieve the selective deborylation of the Bpin at C2 in similar yields.
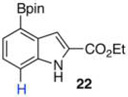
The protodeboronation of 4,7-diborylated-2-carboethoxyindole 12 (entry 6) was instructive for comparing the synthetic efficiency of Bi vs Ir-catalyzed deboronations. After 24 h at 80 °C, indole 12 and 40 mol % Bi(OAc)3 in MeOH/THF gave monoprotodeboronated 22 as the major product along with fully protodeboronated 11 and unreacted 12 in a 54/9/41 ratio per NMR analysis of the crude reaction product. Our recently published Ir-mediated conditions of 2 h at 1.5 mol % [Ir(OMe)COD]2 in 2:1 MeOH/CH2Cl2 at 60 °C8 performed worse, giving the fully protodeboronated 11 and 22 in a ratio of 67:33. However, Ir catalysis in MeOH/THF (~1:6) at rt reacted best, affording 22 in 54% isolated yield along with 13% 11. 4,7-Diborylated-2-methyindole 14 under the Bi(OAc)3 conditions also afforded a 52/5/43 mixture of monoprotodeboronated 23 to 13 to 14, respectively. Indole 14 was another substrate where Ir-mediated protodeboronation proved superior, giving a 91/9 mixture of 23 and starting material 14 with 23 being isolated in 74% yield (entry 7).
The 3,5-diborylated indoles (15 and 17) were informative substrates in their own right. 15 was exclusively monoprotodeboronated at C3 by 20 mol % Bi(OAc)3 in MeOH/THF after 3 h at 80 °C, affording 24 in 88% yield (entry 8). Deboronation of 15 under our published Ir-catalyzed protodeboronation conditions proved less selective. With Ir, the crude reaction product contained 13% of fully deboronated 6-fluoroindole (8) and 24 was isolated in 66% yield. Attempts to optimize Ir-catalyzed deboronation of 15 never met with the selectivity observed with Bi(OAc)3 unless the reaction was stopped prior to complete consumption of starting material.22
In contrast to 15, Boc-protected 17 failed to undergo any deboronation by the action of Bi(OAc)3 (entry 9). Indole 17 was susceptible to Ir-catalyzed deboronation, but again those conditions proved too harsh, giving the N-Boc protected 6-fluoroindole as the major product (21/79 25/16 in the crude reaction mixture). The ratio of 25/16 improved to 60/40 (47% isolated yield of 25) when the protodeboronation was run with 3 mol % Ir in MeOH/THF at room temperature for 10 h.
The reactivity difference between unprotected and N-Boc-indoles was probed further. 4,7-Diborylated-6-fluoroindole 21 was converted to its Boc derivative (26) and then subjected to both the Bi and Ir deboronation conditions (Scheme 4). Again, there was no reaction by Bi(OAc)3. However, under the Ir-catalyzed protodeboronation conditions, using CD3OD as the protic material, afforded the C4 deuterated product 27 in 78% yield. This result demonstrates that the general order of the first boron “on” being the first boron “off” in Ir-catalyzed deboronations can be altered subsequent to borylation by introducing nearby functionality that is sterically demanding.
Scheme 4.

Changing the Sequence of Protodeboronation
To demonstrate this chemistry in late-stage functionalization, we applied the one-pot diborylation/deboronation sequence to 5-HT receptor agonist sumatriptan (28) (Scheme 5). Thus, indole 28 was thus converted to the 2,7-diborylated product. Selective Bi(OAc)3 catalyzed deboronation of the crude product was then achieved in 85% yield by quantitative HPLC. However, the highly polar nature of 29 coupled with the hydrolytic instability of the Bpin ester made purification a challenge and the isolated yield of 29 was only 28%.
Scheme 5.

Functionalization of Sumatriptan
Questions on the mechanism of these deboronations remain. Analysis of the crude reaction material showed a mixture of boronates but no evidence of MeOBpin. The above examples do point to an interaction with the indole nitrogen as being important to achieving selectivity and gaining reactivity. Given Movassaghi and co-workers’ Pd-catalyzed C2 protodeboronation of indoles with HOAc as the proton source,6 we questioned if HOAc, either residual in the Bi(OAc)3 or in situ generated, was playing a part in our bismuth-catalyzed protodeboronations. Toward this end, we examined the reactivity of diborylated 10 with 0.6 equiv of HOAc, which would correspond to the theoretical amount of acetic acid available from 20 mol % of Bi(OAc)3 (Scheme 6). Under these conditions no protodebornation was observed. Increasing the amount of HOAc to 40 equiv had no affect as again only starting 10 was observed after 5 h at 80 °C. The next set of experiments was performed with Bi(OAc)3 that had been washed with CCl4 until the washings showed no HOAc by NMR. Somewhat surprisingly, HOAc free Bi(OAc)3 exhibited enhanced reactivity, as washed Bi(OAc)3 afforded a 3:1 mixture of 21 and 20 while the same reaction with unwashed Bi(OAc)3 gave no 20. While not quantified, it appears that adventitious HOAc lowers the relative reactivity of the unwashed Bi(OAc)3, perhaps by interfering with a putative Bi/indole nitrogen interaction.
Scheme 6.

Exploring the Potential Role of HOAc
In conclusion, bismuth(III) acetate is a safe, shelf stable, inexpensive, and operationally simple alternative to Ir and Pd for the catalytic protodeboronations of indoles. Where as the conditions for deboronations with Ir8 and Pd6 call for an inert atmosphere, Bi(III)-catalyzed deboronations can be run under air. Furthermore, while reaction times are dependent on the grade of methanol employed, solvents need not be distilled or degassed. In general, sequential deboronations with Bi(OAc)3 occur in the same order in which the Bpin groups are installed via Ir-catalyzed borylation. Relative to related methods, Bi(OAc)3 tends to offer greater selectivity in protodeboronations of di- and triborylated indoles. Thus, by tuning the C–H borylation and deboronation conditions, one can access a variety of boron substitution patterns from a single starting indole.
Supplementary Material
Acknowledgments
We thank the NIH (GM63188), NSF (GOALI-1012883), Merck, and the ACS/GCI Pharmaceutical Roundtable for financial support, BoroPharm, Inc., for supplying B2pin2, and Judy Morris (Merck) for assistance with preparative separations of Sumatriptan derivatives. A Thomas J. Pinnavaia Fellowship supported F.S. LRMS data were obtained from MSU’s Molecular Metabolism and Disease Collaborative Mass Spectrometry Core facility.
Footnotes
ASSOCIATED CONTENT
Supporting Information
- Experimental details and product characterization data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors approved the final version of this manuscript.
The authors declare the following competing financial interest(s): MRS and REM acknowledge financial interest in BoroPharm, Inc..
REFERENCES
- 1.Zhichkin PE, Krasutsky SG, Beer CM, Rennells WM, Lee SH, Xiong JM. Synthesis. 2011;2011:1604–1608. [Google Scholar]
- 2.Reck F, Zhou F, Eyermann CJ, Kern G, Carcanague D, Ioannidis G, Illingworth R, Poon G, Gravestock MB. J. Med. Chem. 2007;50:4868–4881. doi: 10.1021/jm070428+. [DOI] [PubMed] [Google Scholar]
- 3.(a) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem. Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (b) Preshlock SM, Ghaffari B, Maligres PE, Krska SW, Maleczka RE, Jr, Smith MR., III J. Am. Chem. Soc. 2013;135:7572–7582. doi: 10.1021/ja400295v. [DOI] [PubMed] [Google Scholar]
- 4.(a) Vanchura BA, II, Preshlock SM, Roosen PC, Kallepalli VA, Staples RJ, Maleczka RE, Jr, Singleton DA, Smith MR., III Chem. Commun. 2010;46:7724–7726. doi: 10.1039/c0cc02041a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tajuddin H, Harrisson P, Bitterlich B, Collings JC, Sim N, Batsanov AS, Cheung MS, Kawamorita S, Maxwell AC, Shukla L, Morris J, Lin Z, Marder TB, Steel PG. Chem. Sci. 2012;3:3505–3514. [Google Scholar]
- 5.For representative examples, see: Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N. Tetrahedron Lett. 2002;43:5649–5651. Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew. Chem., Int. Ed. 2002;41:3056–3058. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. Ishiyama T, Takagi J, Nobuta Y, Miyaura N. Org. Synth. 2005;82:126–133. Paul S, Chotana GA, Holmes D, Reichle RC, Maleczka RE, Jr, Smith MR., III J. Am. Chem. Soc. 2006;128:15552–15553. doi: 10.1021/ja0631652. Meyer F-M, Liras S, Guzman-Perez A, Perreault C, Bian J, James K. Org. Lett. 2010;12:3870–3873. doi: 10.1021/ol1015674. Homer JA, Sperry J. Tetrahedron Lett. 2014;55:5798–5800.
- 6.Loach RP, Fenton OS, Amaike K, Siegel DS, Ozkal E, Movassaghi M. J. Org. Chem. 2014;79:11254–11263. doi: 10.1021/jo502062z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For a recent selective synthesis of a monoborylated indazole by selective deborylation of a diborylated indazole using KOH, see: Sadler SA, Hones AC, Roberts B, Blakemore D, Marder TB, Steel PG. J. Org. Chem. 2015;80:5308–5314. doi: 10.1021/acs.joc.5b00452.
- 8.Kallepalli VA, Gore KA, Shi F, Sanchez L, Chotana GA, Miller SL, Maleczka RE, Jr, Smith MR., III J. Org. Chem. 2015;80:8341–8353. doi: 10.1021/acs.joc.5b01588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Procedure adapted from Navath, R S, Pabbisetty KB, Hu L. Tetrahedron Lett. 2006;47:389–393.
- 10.Mohan R. Nat. Chem. 2010;2:336. doi: 10.1038/nchem.609. [DOI] [PubMed] [Google Scholar]
- 11.Details of the screening studies will be presented elsewhere.
- 12.Indole and 6-fluoroindole can be triborylated directly, but the overall yields and combined catalyst loads are better if 6 and 9 are isolated and then converted to 7 and 10.22
- 13.For a report of other Ir-catalyzed trisborylations, see: Eastabrook AS, Sperry J. Aust. J. Chem. 2015;68:1810–1814.
- 14.Kallepalli VA, Shi F, Paul S, Onyeozili EN, Maleczka RE, Jr, Smith MR., III J. Org. Chem. 2009;74:9199–9201. doi: 10.1021/jo901822b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Preshlock SM, Plattner DL, Maligres PE, Krska SW, Maleczka RE, Jr, Smith MR., III Angew. Chem., Int. Ed. 2013;52:12915–12919. doi: 10.1002/anie.201306511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For the 3,5-diborylation of 7-azaindole, see ref 8.
- 17.Ir-catalyzed borylation of 3-borylated-N-Boc-indole afforded an ~1:1 mixture of 3,5- and 3,6-bisborylated-N-Boc-indole
- 18.For a selective Ir-catalyzed C–H borylation of a fully protected tryptophan and N-TIPS protected indoles, see: Feng Y, Holte D, Zoller J, Umemiya S, Simke LR, Baran PS. J. Am. Chem. Soc. 2015;137:10160–10163. doi: 10.1021/jacs.5b07154.
- 19.10% Deuterium incorporation was initially observed at C3. Washing with H2O reprotonated this carbon
- 20.The percent deuterium incorporation was determined by integration of the 1H NMR spectrum
- 21.We suspect the protodeboronations are slowed by materials leaching from the plastic bottle and/or common MeOH impurities such as formaldehyde, DMAc, and dimethyl acetals of simple alkanones and/or alkanals: Guella G, Ascenzi D, Franceschi P, Tosi P. Rapid Commun. Mass Spectrom. 2007;21:3337–3344. doi: 10.1002/rcm.3222.
- 22.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.