

Abstract
Background
Recombinant human endostatin (rhES) is an angiogenesis inhibitor used as a specific drug for the treatment of non-small-cell lung cancer. As mRNA concentration affects the recombinant protein expression level, any factor affecting mRNA concentration can alter the protein expression level. Response surface methodology (RSM) based on the Box-Behnken design (BBD) is a statistical tool for experimental design and for optimizing biotechnological processes.
Objectives
This investigation aimed to predict and develop the optimal culture conditions for mRNA expression of the synthetic human endostatin (hES) gene in Escherichia coli BL21 (DE3).
Materials and Methods
The hES gene was amplified, cloned, and expressed in the E. coli expression system. Three factors, including isopropyl β-D-1-thiogalactopyranoside (IPTG) concentration, post-induction time, and cell density before induction, were selected as important factors. The mRNA expression level was determined using real-time PCR. The expression levels of hES mRNA under the different growth conditions were analyzed. SDS-PAGE and western blot analyses were carried out for further confirmation of interest-gene expression.
Results
A maximum rhES mRNA level of 376.16% was obtained under the following conditions: 0.6 mM IPTG, 7 hours post-induction time, and 0.9 cell density before induction. The level of rhES mRNA was significantly correlated with post-induction time, IPTG concentration, and cell density before induction (P < 0.05). The expression of the hES gene was confirmed by western blot.
Conclusions
The obtained results indicate that RSM is an effective method for the optimization of culture conditions for hES gene expression in E. coli.
Keywords: Endostatin, Angiogenesis Inhibitors, Gene Expression, Escherichia coli
1. Background
Recombinant human endostatin (rhES) is one of the most common inhibitors of angiogenesis. It has been reported that endostatin inhibits different kinds of tumors with low toxicity and drug resistance in long-term applications, and rhES has been approved by the state food and drug administration of China as a specific drug for non-small-cell lung cancer therapy (1). The rhES protein has been expressed by eukaryotic and prokaryotic expression systems, including yeast, mammalian cells, insect cells, baculovirus, and Escherichia coli (2, 3).
The E. coli expression system is the most widely used host for recombinant protein production (4). Culture conditions, such as induction time, inducer concentration, temperature, and post-induction time, play important roles in recombinant protein expression (5). Transcription of a cloned gene is the first step in protein expression, so each factor that influences mRNA concentration leads to increased or decreased protein expression (6). Therefore, evaluating the contributing factors is necessary to improve the yield of mRNA levels of cloned genes.
The one-factor-at-a-time technique is a conventional approach to optimizing culture conditions, in which a single parameter is evaluated while all of the other parameters are fixed at a given set of conditions. However, this method possesses some limitations; for example, it is time-consuming and it ignores the interactions between the influencing parameters (7). To overcome these limitations, other methods, such as response surface methodology (RSM), can be used. RSM can determine the individual role of each parameter, and its influence on the other parameters (8). It is a mathematical and statistical tool that has been successfully used for experimental design and modeling, for evaluating the effects of several parameters at once, and for optimizing biochemical and biotechnological processes. In addition, the Box-Behnken design (BBD) is a valuable model of RSM for evaluating different variables’ interactions, with relatively few runs (9, 10).
2. Objectives
The present study aimed to obtain the highest hES mRNA level under optimal culture conditions. The hES gene was expressed in different culture conditions based on the BBD. The hES mRNA expression level was investigated with quantitative real-time PCR. The combined effect on gene expression of different culture conditions, including cell density before induction, inducer concentration, and post-induction time, was also evaluated. In addition, for further confirmation of the expression of the gene of interest, we carried out sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analyses of optimal culture conditions.
3. Materials and Methods
3.1. Reagents
Enzymes, including NcoI, XhoI, T4DNA ligase, Pfu DNA polymerase, gene ruler DNA ladder mix, and PageRuler unstained protein, were obtained from Fermentas-Thermo Fisher Scientific (Burlington, Ontario, Canada). Mini PlusTM plasmid DNA extraction kits and Viogene® gel/PCR DNA isolation system kits were purchased from Viogene (New Taipei, Taiwan). PCR reagents were purchased from Cinnagen (Tehran, Iran). Agar, tryptone, yeast extract, kanamycin, ampicillin, and isopropyl-D-1-thiogalactopyranoside (IPTG) were obtained from Sigma-Aldrich (St. Louis, MO, USA), and prestained protein ladder was obtained from Sinaclon (Tehran, Iran). All chemicals used for SDS-PAGE were purchased from Merck (Darmstadt, Germany). TRIzol® reagent was obtained from Life technologies (Carlsbad, CA, USA), and the two-step RT-PCR kit was from Vivantis technologies (Kuala Lumpur, Malaysia). SYBR Green I stain and SYBR Green PCR master mix were purchased from applied Biosystems (Foster City, CA, USA). Rabbit anti-endostatin polyclonal antibody (ab3453), goat anti-rabbit IgG H and L (HRP) (ab6721), and ECL Western blotting substrate (ab65623) were purchased from Abcam (Cambridge, MA, USA).
3.2. Bacterial Strains, Plasmids, Cell Lines, and Media
The E. coli strains DH5α and BL21 (DE3) (Pasteur Institute, Iran) were used as cloning and expression hosts, respectively. The expression vector pET26b (+) was obtained from Pasteur institute (Iran). The clone JET™ PCR cloning kit was from Fermentas-Thermo Fisher Scientific (Waltham, MA, USA), and primers were from ShineGene (Shanghai, China). In addition, the synthetic construct of interest was prepared by ShineGene (Shanghai, China) as a cloned sequence in the pUC57 plasmid.
3.3. Construction of Synthetic Gene and Amplification
The hES coding sequence from the human collagen XVIIIa gene (GenBank accession no. AF184060.1) was taken for codon optimization. Gene optimization and synthesis was done by ShineGene bio-technologies company (Shanghai, China). This construct was sub-cloned in pUC57, including these sequences: NdeI restriction site, alkaline phosphate signal peptide coding sequence (GenBank accession No. M 13763.1), hES coding sequence, and XhoI restriction site.
The hES gene was amplified using pUC57-phoA sp-hES as a template. PCR was performed using the Eppendorf thermal cycler with a set of primers in a total volume of 40 μL. The reaction mixture contained 4 μL of 10 × PCR buffer, 4 μL of MgCl2 (20 mM), 0.7 μL for each dNTP (2.5 mM), 1.5 μL plasmid DNA, 0.6 μL of each primer, and 1 μL of Pfu DNA polymerase (5 units/μL). The program for PCR was one cycle at 95°C for 5 minutes, 30 cycles with denaturation at 95°C for 35 seconds, annealing at 60°C for 40 seconds, then 1 minute at 72°C, and the final extension at 72°C for 7 minutes. The amplification products were electrophoresed on the 1% agarose gel and visualized after SYBR green I staining. The sequences of primers were hES F, TCATCACCACCATGGCGCACTCTCACCG and hES R: GATCCGATAATTTGGCTCGAGTCA. The unique NcoI and XhoI sites (underlined bases) were added to the amplified hES synthetic gene using these primers. In addition, to avoid frame-shifting during the translation of the hES transcript, two nucleotides (CG, bolded bases) were added after the NcoI site.
3.4. Constructing the hES Expression Vector
The NcoI-hES-XhoI amplified fragments were purified with a Viogene® Gel/PCR DNA Isolation kit and inserted into the pJET1.2/blunt cloning vector (between nucleotides 371 and 372). The E. coli DH5α competent cells were transformed by the calcium chloride method (11) with the recombinant plasmid (pJET-hES) for plasmid amplification. For confirmation of successful transformation, a single colony of transformants was grown in 3 mL of LB medium containing ampicillin (100 µg/mL), followed by colony PCR. For further confirmation, clones containing recombinant vectors were analyzed by the restriction enzyme digestion and sequencing method, using the abovementioned primers.
Next, the pJET-hES and the pET26b (+) vector were double-digested with the NcoI and the XhoI restriction enzymes. The hES gene was then inserted just after a pelB leader sequence in the expression vector pET26b (+) with a T7 promoter to secrete the recombinant hES into the E. coli periplasm. The recombinant vectors were transformed into the competent E. coli DH5α by the calcium chloride method (11). Transformants were selected on LB agar containing kanamycin (50 µg/mL), and the clones containing the hES gene were confirmed by colony PCR. Subsequently, in order to re-confirm the clone, a single colony of E. coli cells carrying the recombinant plasmid was grown in 3 mL LB medium containing kanamycin. After extraction of plasmid, restriction enzyme digestion and sequencing were done. Following the confirmation by sequencing, the constructs were transformed into E. coli BL21 (DE3) competent cells, and were selected on LB agar as described above. The bacterial stocks were kept at -70°C in 25% (v/v) glycerol for long-term usage.
3.5. Media and Cultivation Conditions
LB agar containing kanamycin (50 µg/mL) was used to grow E. coli BL21/pET26-hES. The pH was adjusted to 7.4 ± 0.2 prior to autoclaving for 15 minutes at 121°C. For all experiments, the pre-inocula were cultured overnight at 200 rpm and 37°C using LB medium. To evaluate whether the hES gene was expressed successfully, for all experiments, a transformed single colony was inoculated in 5 mL of LB medium supplemented with kanamycin (50 µg/mL), and incubated overnight at 37°C in a shaking (200 rpm) incubator. Then, 2 ml of starter culture was transferred into 500 mL Erlenmeyer flasks with 50 mL of the same broth containing kanamycin (50 µg/mL), and incubated in shaker incubator (37°C, 200 rpm). Cell density at induction time, IPTG concentration, and post-induction (harvesting) time for each experiment were defined according to the BBD, as described below. All experiments were conducted in duplicate.
3.6. Experimental Design Using RSM
RSM based on the BBD was employed to optimize culture conditions for the expression of the hES gene in E. coli BL21 (DE3) in a shake flask. The independent variables, including inducer concentration (X1), post-induction time (X2), and cell density before induction (X3), were tested at three different levels, as shown in Table 2. The practical level of investigation is described in Table 3. All experiments were done in triplicate, and the average rhES mRNA expression levels were considered as the experimental data. These data were evaluated statistically by regression analysis using the following second-order polynomial:
Table 2. Independent Variables and Coded Values of Variables Used in the BBD.
| Independent Variable | Symbol | Unit | Code Level | ||
|---|---|---|---|---|---|
| -1 | 0 | +1 | |||
| IPTG concentration | X1 | mM | 0.2 | 0.6 | 1 |
| Post-induction time | X2 | h | 4 | 7 | 10 |
| Cell density (OD600) | X3 | - | 0.8 | 1.1 | 1.4 |
Table 3. BBD With Measured and Predicted Response of hES mRNA Expression Levela.
| Run | X1 | X2 | X3 | Actual, %b | Predicted, % |
|---|---|---|---|---|---|
| 1 | 0.2 | 4 | 1.1 | 71.52 | 92.09 |
| 2 | 1 | 4 | 1.1 | 204.32 | 200.04 |
| 3 | 0.2 | 10 | 1.1 | 207.52 | 211.80 |
| 4 | 1 | 10 | 1.1 | 208.80 | 188.23 |
| 5 | 0.2 | 7 | 0.8 | 272.96 | 261.13 |
| 6 | 1 | 7 | 0.8 | 295.84 | 308.86 |
| 7 | 0.2 | 7 | 1.4 | 92.64 | 79.62 |
| 8 | 1 | 7 | 1.4 | 104.48 | 116.27 |
| 9 | 0.6 | 4 | 0.8 | 289.92 | 281.19 |
| 10 | 0.6 | 10 | 0.8 | 306.24 | 313.78 |
| 11 | 0.6 | 4 | 1.4 | 80.32 | 72.77 |
| 12 | 0.6 | 10 | 1.4 | 139.36 | 148.1 |
| 13 | 0.6 | 7 | 1.1 | 376.16 | 332.64 |
| 14 | 0.6 | 7 | 1.1 | 312.80 | 332.64 |
| 15 | 0.6 | 7 | 1.1 | 308.96 | 332.64 |
aX1, IPTG concentration; X2, post-induction time; X3, cell density before induction (OD600).
bMean of duplicate observations.
| Equation 1. |
Where Y is the predicted response (rhES mRNA expression level); X1, X2, and X3 are independent variables (inducer concentration, post-induction time, and cell density, respectively); β0 is a constant coefficient; β1, β2, and β3 are linear coefficients; β12, β13, and β33 are the interaction coefficients; and β11, β22, and β33 are the quadratic coefficients. For one-way analysis of variance (ANOVA) and RSM, MinitabTM version 17.1 software (Minitab Inc., PA, USA) was used. Also, in order to understand the optimal culture conditions and the interactions of different variables, a three-dimensional (3D) response surface plot and contour and correlation plots were generated.
3.7. RNA Extraction and Reverse Transcription
A volume of 107 cells was sampled from the flasks under the different growth conditions, and pelleted by centrifugation. Then, 1 mL of TRIzol® Reagent was added to the resulting cells and the extraction process was performed according to the manufacturer’s instructions. The concentration and quantity of RNA were evaluated based on OD260/280 ratio measurements and agarose gel (1%) electrophoresis, respectively. In order to synthesize cDNA from total RNA, reverse transcription of RNA was performed in a final volume of 20 μL containing Vivantis RT enzyme mix I (0.5 µL), random hexamer primers (0.5 µL from 100 uM), buffer RT enzyme (2 µL), and 1 μg of total RNA. Subsequently, the samples were incubated at 65°C for 10 minutes and 42°C for 30 minutes. Reverse transcriptase was inactivated by heating at 85°C for 5 minutes, followed by cooling at 4°C for 5 minutes.
3.8. Quantitative Real-Time PCR Assay
The level of hES mRNA expression was determined with the quantitative real-time PCR technique using power SYBR-green PCR master mix, by means of the step-one real-time PCR system (applied biosystems, Foster city, CA, USA). For real-time PCR, hES primers were designed based on optimized hES sequences, and 16s rRNA were used. These primers were designed using Primer3 (version 0.4.0). The forward and reverse primer sequences of hES and 16s RNA are shown in Table 1. The 16s rRNA mRNA was calculated as the internal standard control gene by specific primers.
Table 1. Primers Used in This Study.
| Primers | Sequence | Product Size |
|---|---|---|
| hES | 592 bp | |
| Forward | 5′TCATCACCACCATGGCGCACTCTCACCG3′ | |
| Reverse | 5′GATCCGATAATTTGGCTCGAGTCA3′ | |
| hES a | 146 bp | |
| Forward | 5′GCTGTCTGGTGGTATGCGTG3′ | |
| Reverse | 5′CACGGTCAGCACGACGAAC3′ | |
| 16s rRNA a | 98 bp | |
| Forward | 5′ATTGACGTTACCCGCAGAAG3′ | |
| Reverse | 5′CTTTACGCCCAGTAATTCCGAT3′ |
aPrimers used for real-time PCR
A 146-bp amplicons were generated for hES, and 98-bp amplicons were generated for 16s rRNA, in 20-μL reaction mixtures containing 5 pmol of each of the forward and reverse primers for 16s rRNA or for hES; 10 μL of Power SYBR green PCR master mix and 2 μL of the cDNA was used. The program for the real-time PCR reaction included an initial denaturation step at 95°C for 5 minutes and 40 cycles of denaturation (95°C for 15 seconds), then annealing (60°C for 60 seconds).
To verify the specificity of the fragments, melting curves were determined for each reaction from 60 - 95°C, with data collection at each 0.3°C increment. The 16s rRNA gene, expressed at a constant rate under our conditions, was used as reference gene to normalize the data. After normalization to the reference gene 16s rRNA, the expression level of hES for each different growth condition was analyzed with the 2-ΔΔCT method. Each DNA sample was divided so that hES and 16s rRNA could be amplified in parallel, and the reactions were done in duplicate with equal amounts of starting material.
3.9. Protein Expression Analysis
The expressed rhES of optimal culture conditions was checked using SDS-PAGE under reduced conditions (11). In addition, western blot analysis was applied for further identification of produced rhES using rabbit anti-endostatin polyclonal antibody and goat anti-rabbit IgG H and L (HRP). Proteins were visualized using incubation with ECL Western Blotting Substrate.
4. Results
4.1. Construction and Amplification of the Synthetic hES Gene
The wild-type hES gene has 31 rare codons, including 3CGG, 3AGG for arginine, 8CCC for proline, 4 TCC, 2 TCA, 1 AGT, 4TCG for serine, and 6 GGG for glycine. These codons were replaced with preferred E. coli codons in synthetic hES. The codon adaptation index (CAI) used to estimate the adaptation of codon-optimized hES to E. coli was 0.878. The optimized gene was 75.8% identical to the wild-type hES gene. Subsequently, the hES gene was amplified using specific primers and pUC57-phoA sp-hES as a template, showing a 592-bp band on 1% agarose gel (Figure 1A). This fragment contained the hES coding sequence (555 bp) and the NcoI and XhoI restriction sites.
Figure 1. Amplification of hES Coding Sequence and Construction of Expression Vector.

A, Amplification of hES coding sequence using Pfu DNA polymerase. Lane 1: gene ruler DNA ladder mix (thermo scientific); lane 2: negative control; lane 3: amplified ORF of hES (592 bp). B, Construction of the pET26b-hES vector.
4.2. Construction of the pET26b-rhES Expression Vector
The plasmid pET26b (+) was used to construct expression vectors. The protein-coding region of the hES gene was inserted between the NcoI and XhoI sites of the pET26 (+). The recombinant pET26b (+) - hES contained a T7 promoter, a Lac operon, an N-terminal pelB signal sequence, multiple cloning sites, the rhES encoding gene, and the T7 terminator. Following the transformation of E. coli BL21 (DE3), the positive clones were selected on LB agar containing kanamycin (50 µg/mL). A sequencing analysis of the recombinant pET26b (+) - hES plasmid with the abovementioned primers confirmed that there were no amplification errors or frame shifts in the sequence of the cloned hES gene. The construction of the expression vector is presented in Figure 1B.
4.3. Evaluation of Culture Conditions for the mRNA Expression Level Using RSM
RSM was applied to evaluate optimal culture conditions for the mRNA expression level of the synthetic hES gene in E. coli BL21 (DE3). BBD, an efficient design tool for fitting second-order models under RSM, was used to investigate the effect of three variables, including cell density before induction, IPTG concentration, and post-induction time, which influence the mRNA expression level of hES.
The actual and predicted results for hES mRNA expression levels are summarized in Table 3. The hES mRNA expression levels varied, in the range of 71.52% - 376.16%. The ANOVA for the adjusted model revealed that hES expression was significantly affected by the linear and quadratic coefficients and their interactions, except for the interaction factors of X1X3 and X2X3 (Table 4). The effects of the inducer concentration (coefficient = 21.09, P = 0.001) and post-induction time were positive (coefficient = 26.98, P < 0.001), whereas the cell density before induction was negative (coefficient = -93.52, P < 0.001), and the same results were observed with X1X1, X2X2, X3X3, and X1X2. To estimate the optimal region of hES mRNA expression, second-order models were fitted to the hES mRNA observed results (Table 3). The model is described by Equation 2, where the variables are specified in their original units as follows:
Table 4. ANOVA for Response Surface Quadratic Model for hES mRNA Expression Levela.
| Source | D.F. | S.S. | M.S. | F-Value | Coef | SE Coef | T-Value | P-Value |
|---|---|---|---|---|---|---|---|---|
| Model | 9 | 270569 | 30063 | 65.07 | - | - | - | < 0.001 |
| Constant | - | - | - | 332.64 | 8.77 | 37.91 | < 0.001 | |
| X 1 | 1 | 7120 | 7120 | 15.41 | 21.09 | 5.37 | 3.93 | 0.001 |
| X 2 | 1 | 11646 | 11646 | 25.21 | 26.98 | 5.37 | 5.02 | < 0.001 |
| X 3 | 1 | 139947 | 139947 | 302.92 | -93.52 | 5.37 | -17.4 | < 0.001 |
| X 1 X 1 | 1 | 54672 | 54672 | 118.34 | -86.04 | 7.91 | -10.88 | < 0.001 |
| X 2 X 2 | 1 | 39955 | 39955 | 86.48 | -73.56 | 7.91 | -9.30 | < 0.001 |
| X 3 X 3 | 1 | 22441 | 22441 | 48.58 | -55.13 | 7.91 | -6.97 | < 0.001 |
| X 1 X 2 | 1 | 8649 | 8649 | 18.72 | -32.88 | 7.6 | -4.33 | < 0.001 |
| X 1 X 3 | 1 | 61 | 61 | 0.13 | -2.77 | 7.6 | -0.36 | 0.719 |
| X 2 X 3 | 1 | 913 | 913 | 1.98 | 10.68 | 7.6 | 1.41 | 0.175 |
| Lack of fit | 3 | 3536 | 1179 | 3.51 | - | - | - | 0.038 |
| Pure error | 17 | 5704 | 336 | - | - | - | - | - |
| Total | 29 | 279809 | - | - | - | - | - | - |
aX1, IPTG concentration; X2, post-induction time; X3, cell density in OD600; D.F., degrees of freedom; S.S, sum of squares; M.S., mean of squares; Coef., coefficient; SE.Coef., standard error coefficient; F-value, Fisher’s test; P-value, probability distribution value. The correlation coefficient (R2) was 0.967 and the adjusted R2 was 0.9521.
| Equation 2. |
Where Y is the response variable (hES mRNA level), and X1, X2, and X3 are the independent variables (inducer concentration, post-induction time, and cell density before induction, respectively). The fit of the model was evaluated by the determination coefficient (R2). The R2 value was 0.967, indicating that 96.7% of the total variability in the response could be well-described by the new model. The adjusted determination coefficient (adj. R2) was 0.9521, and the F-value was 65.07 (D.F = 9) (Table 4). The scatterplot of the predicted versus actual rhES mRNA expression level is shown in Figure 2. The actual value is the result obtained for a specific treatment, and the predicted value is obtained from the independent variables in the Box-Behnken model. The model predicted a maximum level of 374.54% hES mRNA, with an inducer concentration (IPTG) of 0.64 mM, a post-induction time of 7.27 hours, and cell density before induction (OD600) of 0.85. Therefore, the ANOVA results indicated the satisfactory adjustment of the quadratic models to the actual data.
Figure 2. Diagnostic Plots for Estimating the Adequacy of the Regression Model.

A, Correlation between predicted and actual values for hES mRNA expression; B; the studentized and normal percentage probability plot of hES mRNA expression.
The interactive effects of variables on hES expression were plotted in the form of contour and 3D response surfaces, as illustrated in Figure 3A - C. Response surface contour plots and 3D graphs demonstrated accurate information about the relationship between the actual levels and the response of each independent variable. The interactions of IPTG and post-induction time with hES mRNA gene expression are shown in Figure 3A. The optimum condition for hES expression was found to be at an IPTG concentration of 0.6 mM and a post-induction time of 7 hours. Moreover, the combined impact of IPTG concentration and cell density on hES expression is shown in Figure 3B; the combined impact of post-induction time and cell density on hES mRNA gene expression is depicted in Figure 3C. These plots show that an IPTG concentration of < 0.6 mM and a post-induction time of 7 hours were favorable for hES mRNA expression, while increased OD600 resulted in decreased hES mRNA expression.
Figure 3. Response Surface Plots (left) and Contour Plots (right) of rhES mRNA Revealed the Relationship Between the Response and Actual Levels of Each Independent Variable.

A, The effect of inducer concentration (IPTG) and post-induction time on rhES mRNA; B, the effect of inducer concentration (IPTG) and cell density before induction time on rhES mRNA; C, the effect of post-induction time and cell density before induction time on rhES mRNA.
The expressed rhES presented approximately 20 kDa on SDS-PAGE (Figure 4A), which was compatible with the theoretical molecular mass, and it was found to be immune-reactive when evaluated by western blot using rabbit anti-endostatin polyclonal antibody (Figure 4B). Finally, the results of SDS-PAGE and western blot confirmed the successful expression of hES mRNA in E. coli BL21 (DE3).
Figure 4. SDS-PAGE and Western Blot Analysis of Expressed hES Gene.
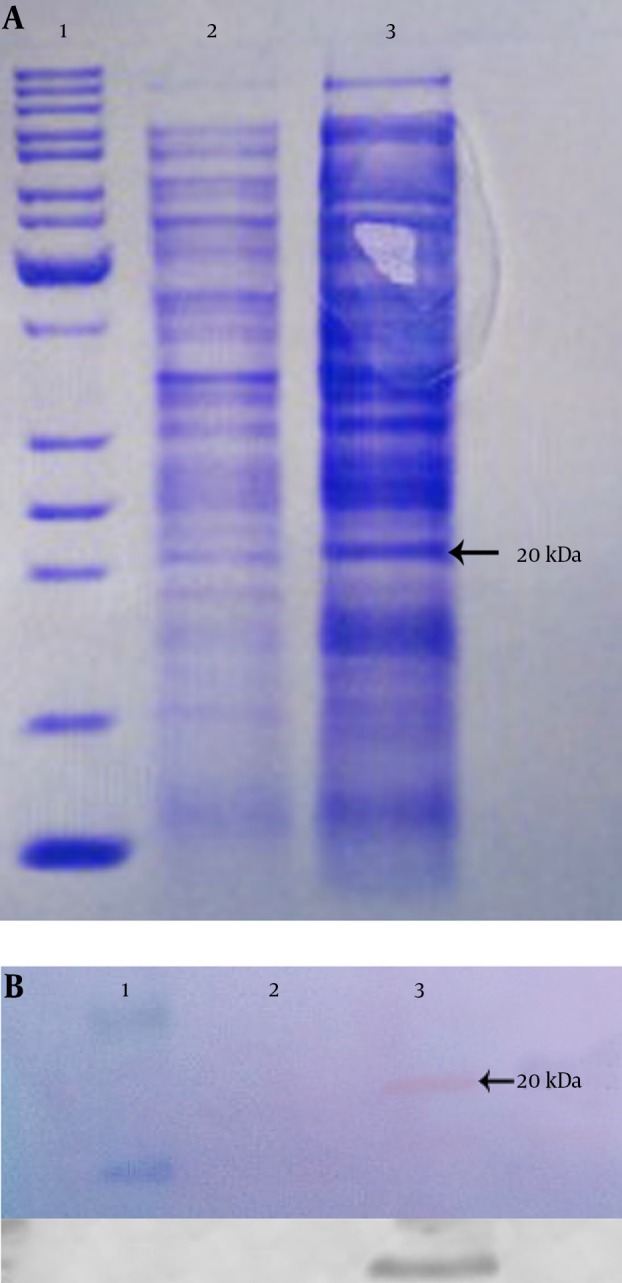
A, SDS-PAGE (12%); B, Western blot. Lane 1, protein marker; lane 2, total protein before induction; lane 3, total protein after induction in recombinant pET26b.
5. Discussion
Recombinant human endostatin protein is an attractive candidate as an anti-cancer agent with high efficacy, few side effects, and no reported drug resistance (12). Transcription of a cloned gene is the first step in gene expression that leads to protein production (6). To obtain high yields of a recombinant protein, the protein-coding gene is usually expressed at the highest possible level (13), so improving the involved factors in gene expression can be useful. A few studies have employed qRT-PCR to study the effects of the involved factors in gene expression in eukaryotic cells (14, 15). For the first time, we utilized this technique to evaluate effective factors in the expression of synthetic hES gene, which is involved in the synthesis of rhES protein. In the present study, hES gene expression, which was cloned in pET26b plasmid, was assessed under the experimental conditions using the BBD. The interest-gene expression was also confirmed by western blot. This is the first attempt to consider the influence of cell density before induction, inducer concentration, and post-induction time alone, as well as each factor’s interactions with the others, with regard to increasing hES mRNA expression levels.
Several hosts, such as E. coli, yeast, mammalian cells, and baculovirus, have been used to produce rhES (2, 3). Expressing heterologous proteins in the E. coli system has some advantages over other systems, including a high expression level, rapid growth, and low cost (16, 17). However, despite the many advantages of the E. coli expression system, high-level expression is not easily achieved. Several common factors attenuate protein expression by E. coli, including codon bias, gene product toxicity, insolubility, mRNA secondary structure, and mRNA instability (18-20). In contrast, several reports have proposed the use of synthetic genes to increase expression efficiency (21). Considering the factors involved in heterologous protein expression, the optimized synthetic hES gene was applied to amplify the hES gene, followed by cloning and expression in E. coli BL21 (DE3) in the current study.
Researchers have established that culture conditions significantly affect the recombinant protein expression level (22, 23) that results from mRNA expression. The altered protein expression level could be related to the expression level of the protein-coding gene (13). In order to optimize the culture conditions for hES gene expression, three factors were selected. The conventional method is time-consuming, analyzes only one variable at one time, and is unable to analyze the interactions between the different factors (8). Therefore, the BBD was applied instead.
The BBD helps in the analysis of many variables using the lowest number of examinations, testing the statistical significance of the variable and validating the interactions between the variables (8, 21). Therefore, this method was applied to achieve the optimal conditions for the expected responses and to reduce the number of required examinations, and was used to predict the hES mRNA expression level. In this experiment, the BBD results revealed that the level of hES mRNA expression was significantly correlated with post-induction time, IPTG concentration, and cell density before induction (P < 0.05). The BBD results are given by Equation 2.
It has been proven that a large F-value indicates that most of the variation can be described by a regression equation, and a P < 0.05 demonstrates that the model is considered to be statistically significant (8). Our ANOVA results confirmed that the model was adequate for representing the experimental data in the current study. The fit of the model can be confirmed by the correlation coefficient (R2) along with an acceptable agreement with the adjusted correlation coefficient R2adj (24). The values of R2 and R2adj were close to 1, which clearly indicates that the obtained experimental responses adequately fit with the model (Figure 2B). These findings indicated a strong correlation between the experimental and predicted values (Figure 2A) (8).
In the present study, the hES mRNA expression level was markedly improved at the optimum point of culture conditions given by the BBD model, and any change in the values of the optimized factors resulted in decreased hES mRNA expression. As previously reported, any change in the optimum culture conditions results in an altered growth rate of E. coli, which in turn affects the gene expression levels (22, 24). The aforementioned factors in the present investigation are completely explained as follows:
IPTG was found to be an effective factor in hES expression. Its optimum concentration was 0.64 mM, and any change in this concentration caused decreased hES mRNA levels. It has been reported that the concentration of IPTG for the induction of gene expression varies from 0.005 mM to 5 mM (7). In addition, low IPTG concentrations result in ineffective induction, while high IPTG concentrations result in a significantly reduced growth rate of E. coli (25, 26). Therefore, it is suggested that the determination of the optimum IPTG concentration is important in gene expression.
Post-induction time was another effective factor with a positive influence in this study. The hES mRNA expression level was increased with a longer post-induction time, but post-induction times beyond the optimum point caused decreased mRNA levels. It has been established that the effects of post-induction time depend on case-specific expression (4). Therefore, optimizing the post-induction time is strongly suggested.
With regard to cell density before induction, a lower cell density caused a lower level of hES mRNA in this study. Induction is usually performed at the early or mid-log phase. However, some reports recommend induction in the late-log phase or even in the stationary phase (27). However, Manderson et al. reported that the effects of cell density before induction are case-specific (28). Therefore, it is recommended that cell density should be optimized in each case.
Based on these findings, we conclude that optimizing the three important factors of IPTG concentration, post-induction time, and cell density before induction can improve hES mRNA yield in the E. coli expression system.
Acknowledgments
The authors are thankful to their colleagues at the tuberculosis and lung disease research center of Tabriz University of Medical Sciences. The authors are also thankful to their coworkers at the drug applied research center of Tabriz University of Medical Sciences. We thank Dr. Sarvin Sanyi for assistance in editing this manuscript.
Footnotes
Authors’ Contribution:This work was a part of the PhD thesis of Abbas Mohajeri, who wrote the paper; Nosratollah Zarghami was the thesis supervisor and edited the manuscript, directed the research, and coordinated the study; Abbas Mohajeri performed the plasmid design and molecular work, carried out the analysis, and wrote the draft manuscript; Yones Pilehvar-Soltanahmadi participated in practical activities; Jalal Abdolalizadeh and Pouran Karimi participated as technical assistance advisors. All of the authors read and approved the final manuscript.
Financial Disclosure:The authors declare no financial disclosure.
Funding/Support:This research was financially supported by a grant (Project Number 5/76/520) from the tuberculosis and lung disease research center, Tabriz University of Medical Sciences. Tabriz, Iran.
References
- 1.Ma Y, Jin XB, Chu FJ, Bao DM, Zhu JY. Expression of liver-targeting peptide modified recombinant human endostatin and preliminary study of its biological activities. Appl Microbiol Biotechnol. 2014;98(18):7923–33. doi: 10.1007/s00253-014-5818-0. [DOI] [PubMed] [Google Scholar]
- 2.Xu YF, Zhu LP, Hu B, Rong ZG, Zhu HW, Xu JM, et al. A quantitative method for measuring the antitumor potency of recombinant human endostatin in vivo. Eur J Pharmacol. 2007;564(1-3):1–6. doi: 10.1016/j.ejphar.2007.01.086. [DOI] [PubMed] [Google Scholar]
- 3.Du C, Yi X, Zhang Y. Expression and purification of soluble recombinant Human Endostatin in Escherichia coli. Biotechnol Bioprocess Eng. 2010;15(2):229–35. doi: 10.1007/s12257-009-0100-5. [DOI] [Google Scholar]
- 4.Muntari B, Amid A, Mel M, Jami MS, Salleh HM. Recombinant bromelain production in Escherichia coli: process optimization in shake flask culture by response surface methodology. AMB Express. 2012;2(1):12. doi: 10.1186/2191-0855-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papaneophytou CP, Kontopidis GA. Optimization of TNF-alpha overexpression in Escherichia coli using response surface methodology: Purification of the protein and oligomerization studies. Protein Expr Purif. 2012;86(1):35–44. doi: 10.1016/j.pep.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Dressaire C, Picard F, Redon E, Loubiere P, Queinnec I, Girbal L, et al. Role of mRNA stability during bacterial adaptation. PLoS One. 2013;8(3):e34091. doi: 10.1371/journal.pone.0059059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akbari V, Sadeghi HMM, Jafarian-Dehkordi A, Chou CP, Abedi D. Optimization of a single-chain antibody fragment overexpression in Escherichia coli using response surface methodology. Res pharma sci. 2015;10(1):75–83. [PMC free article] [PubMed] [Google Scholar]
- 8.Maharjan S, Singh B, Bok J, Kim J, Jiang T, Cho C, et al. Exploring codon optimization and response surface methodology to express biologically active transmembrane RANKL in E. coli. PloS one. 2014;9(5):e34091. doi: 10.1371/journal.pone.0096259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Lima CJB, Coelho LF, Contiero J. The use of response surface methodology in optimization of lactic acid production: focus on medium supplementation, temperature and pH control. Food Technol Biotechnol. 2010;48(2):175–81. [Google Scholar]
- 10.Morowvat MH, Babaeipour V, Rajabi Memari H, Vahidi H. Optimization of Fermentation Conditions for Recombinant Human Interferon Beta Production by Escherichia coli Using the Response Surface Methodology. Jundishapur J Microbiol. 2015;8(4):e34091. doi: 10.5812/jjm.8(4)2015.16236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yari K, Afzali S, Mozafari H, Mansouri K, Mostafaie A. Molecular cloning, expression and purification of recombinant soluble mouse endostatin as an anti-angiogenic protein in Escherichia coli. Mol Biol Rep. 2013;40(2):1027–33. doi: 10.1007/s11033-012-2144-4. [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Zheng L, Shi H, Chen X, Wan Y, Zhang H, et al. Suppression of peritoneal tumorigenesis by placenta-derived mesenchymal stem cells expressing endostatin on colorectal cancer. Int J Med Sci. 2014;11(9):870–9. doi: 10.7150/ijms.8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlegel S, Rujas E, Ytterberg AJ, Zubarev RA, Luirink J, de Gier JW. Optimizing heterologous protein production in the periplasm of E. coli by regulating gene expression levels. Microb Cell Fact. 2013;12:24. doi: 10.1186/1475-2859-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Ni Z, Duan Z, Wang G, Li F. Altered expression of hypoxia-inducible factor-1alpha (HIF-1alpha) and its regulatory genes in gastric cancer tissues. PLoS One. 2014;9(6):e34091. doi: 10.1371/journal.pone.0099835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruhrmund DW, Arfsten AE, Dolim K, Qin X, Buckman BO, Shea BS, et al. Impact Of LPA And LPA1 Receptor Antagonists On Gene And Protein Expression In Human Primary Cells. Am J Respir Crit Care Med. 2016;189:2014. [Google Scholar]
- 16.Jaliani HZ, Farajnia S, Safdari Y, Mohammadi SA, Barzegar A, Talebi S. Optimized condition for enhanced soluble-expression of recombinant mutant anabaena variabilis phenylalanine ammonia lyase. Adv Pharm Bull. 2014;4(3):261–6. doi: 10.5681/apb.2014.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi S, Paek SY. The orientation-dependent expression of angiostatin-endostatin hybrid proteins and their characterization for the synergistic effects of antiangiogenesis. Biotechnol Bioprocess Eng. 2010;20(10):1430–5. doi: 10.4014/jmb.1004.04040. [DOI] [PubMed] [Google Scholar]
- 18.Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22(7):346–53. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Hernandez V, Maldonado L, Rivero E, de la Rosa A, Acevedo L, Rodríguez A. Optimization of human interferon gamma production in Escherichia coli by response surface methodology. Biotechnology and Bioprocess Engineering. 2008;13:7–13. doi: 10.1007/s12257-007-0126-5. [DOI] [Google Scholar]
- 20.Wu X, Jornvall H, Berndt KD, Oppermann U. Codon optimization reveals critical factors for high level expression of two rare codon genes in Escherichia coli: RNA stability and secondary structure but not tRNA abundance. Biochem Biophys Res Commun. 2004;313(1):89–96. doi: 10.1016/j.bbrc.2003.11.091. [DOI] [PubMed] [Google Scholar]
- 21.Maldonado LM, Hernandez VE, Rivero EM, Barba de la Rosa AP, Flores JL, Acevedo LG, et al. Optimization of culture conditions for a synthetic gene expression in Escherichia coli using response surface methodology: the case of human interferon beta. Biomol Eng. 2007;24(2):217–22. doi: 10.1016/j.bioeng.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Cao W, Li H, Zhang J, Li D, Acheampong DO, Chen Z, et al. Periplasmic expression optimization of VEGFR2 D3 adopting response surface methodology: antiangiogenic activity study. Protein Expr Purif. 2013;90(2):55–66. doi: 10.1016/j.pep.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Li B, Wang L, Su L, Chen S, Li Z, Chen J, et al. Glycine and Triton X-100 enhanced secretion of recombinant α-CGTase mediated by OmpA signal peptide in Escherichia coli. Biotechnol Bioprocess Eng. 2012;17(6):1128–34. [Google Scholar]
- 24.Kahyaoglu T. Optimization of the pistachio nut roasting process using response surface methodology and gene expression programming. Lebensm Wiss Technol. 2008;41(1):26–33. [Google Scholar]
- 25.Papaneophytou CP, Kontopidis G. Statistical approaches to maximize recombinant protein expression in Escherichia coli: a general review. Protein Expr Purif. 2014;94:22–32. doi: 10.1016/j.pep.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 26.Larentis AL, Nicolau JF, Esteves Gdos S, Vareschini DT, de Almeida FV, dos Reis MG, et al. Evaluation of pre-induction temperature, cell growth at induction and IPTG concentration on the expression of a leptospiral protein in E. coli using shaking flasks and microbioreactor. BMC Res Notes. 2014;7:671. doi: 10.1186/1756-0500-7-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranjbari J, Babaeipour V, Vahidi H, Moghimi H, Mofid MR, Namvaran MM, et al. Enhanced Production of Insulin-like Growth Factor I Protein in Escherichia coli by Optimization of Five Key Factors. Iran J Pharm Res. 2015;14(3):907–17. [PMC free article] [PubMed] [Google Scholar]
- 28.Manderson D, Dempster R, Chisti Y. Production of an active recombinant Aspin antigen in Escherichia coli for identifying animals resistant to nematode infection. Enzyme Microb Technol. 2006;38(5):591–8. doi: 10.1016/j.enzmictec.2005.03.029. [DOI] [Google Scholar]