

Abstract
The existence of cancer stem cells has been well established in acute myeloid leukemia. Initial proof of the existence of leukemia stem cells (LSCs) was accomplished by functional studies in xenograft models making use of the key features shared with normal hematopoietic stem cells (HSCs) such as the capacity of self-renewal and the ability to initiate and sustain growth of progenitors in vivo. Significant progress has also been made in identifying the phenotype and signaling pathways specific for LSCs. Therapeutically, a multitude of drugs targeting LSCs are in different phases of preclinical and clinical development. This review focuses on recent discoveries which have advanced our understanding of LSC biology and provided rational targets for development of novel therapeutic agents. One of the major challenges is how to target the self-renewal pathways of LSCs without affecting normal HSCs significantly therefore providing an acceptable therapeutic window. Important issues pertinent to the successful design and conduct of clinical trials evaluating drugs targeting LSCs will be discussed as well.
Keywords: Leukemia stem cells, Cancer stem cells, Acute myeloid leukemia, Stem cell niche, Xenotransplantation, Plerixafor, NF-κB, C-X-C chemokine receptor type 4
Core tip: Leukemia stem cell (LSC) directed therapy targets: (1) Cell surface markers expressed on LSC: CD33, CD44, CD123, CD47, etc.; (2) Crucial pathways for maintenance of their stemness: NF-κB, PI3K/AKT/mTOR and bcl-2; and (3) Interactions between LSC in the bone marrow niche: LSC mobilization with granulocyte-colony stimulating factor and inhibition of LSC homing to the bone marrow by interrupting the C-X-C chemokine receptor type 4-CXCL12 and VCAM-VLA4 axis.
INTRODUCTION
Despite extensive research efforts in myeloid malignancies, minimal progress has been made in introducing new effective treatment strategies for acute myeloid leukemia (AML) since the introduction of the anthracycline-cytarabine combination chemotherapy regimens (known as 7 + 3) more than 40 years ago[1]. Despite achieving complete remission (CR) with intensive induction chemotherapy in about 70% of patients with AML, relapse is frequent and the rate of 5-year disease free survival is only about 30%-40%. It has been long proposed that the high rate of relapse is due to the persistence of a rare subset of malignant cells that are not effectively eliminated by current treatment regimens, the so called leukemia stem cells (LSCs)[2-4]. LSC were first identified but tumor cells with stem cell-like behavior were later found to be also present in a variety of solid tumors[5-9]. LSC remain the best studied and characterized cancer stem cell (CSC) due to the easy accessibility of tumor tissue for (i.e., blood and bone marrow) and the availability of a number of cell surface markers that allow their prospective identification and isolation by flow cytometry followed by assays to examine their function both in vitro and in vivo[10]. This review will focus on the biology of LSC, the impact they have on current leukemia diagnosis and prognosis and treatment as well as future directions of leukemia therapy based on targeting LSC[6].
CSC VS CLONAL EVOLUTION THEORY
It is now well understood that not only tumors from different patients but also cells within a single tumor are characterized by heterogeneity in terms of the morphology, cell surface markers, genetic variations and response to therapy[11]. Why there is significant variation in genetic and epigenetic abnormalities between different cells or locations within a tumor despite the clonal origin of all tumor cells, is a question that has puzzled researchers for decades. There are essentially two different explanations for this fundamental problem of cancer biology: The hierarchy or CSC model vs the stochastic or clonal evolution model[6]. In the stochastic model, all cells in a tumor have a similar biological function but are heterogeneous (e.g., expression of cell surface markers) because of clonal evolution resulting in small but entirely random/stochastic variations triggered by external and internal factors based on Darwinian principles. Importantly, all cells within the tumor have an equal sensitivity to both intrinsic (transcription factors and signaling pathways) and extrinsic (host factors, tumor microenvironment and immune response) factors[10]. In the cancer stem cell (CSC) model, a tumor follows the principles of normal, healthy tissue development with a stem cell at the top of the hierarchy, which gives rise to all other cells in the tumor. In this model only these rare population of CSCs are able to initiate tumor growth: They possess self-renewal capacity and can be isolated from the bulk non-tumorigenic population. Importantly, both models appreciate the existence of a CSC but differ in their assessment what cells within the tumor can be CSCs. In the stochastic model CSCs are created randomly and every cell has the potential to be a CSC, whereas in the CSC model only a subset of cancer cells has the potential to behave like a stem cell[11].
Whether the stochastic model or the CSC model best reflects tumorigenesis/leukemogenesis, has significant impact on how cancer/leukemia should be treated[10]. In the stochastic model, the cells within a tumor are relatively homogeneous in terms of genetic makeup and function and therapy can be uniformly directed at the bulk of tumor cells. However, per the CSC model, tumorigenic pathways might operate differently in CSCs compared with the bulk cells and therapy must specifically target the CSCs in order to be truly effective. Most of the current targeted therapies against leukemia and cancer focuses on inhibiting the molecular drivers found in all cancer cells but do not necessarily target CSCs[11].
BIOLOGY OF LSCS
CSC characteristics
The definition of a LSC is adapted from normal HSC: It is a cell that possesses the capacity to self-renew, proliferates and gives rise to leukemic blasts, which are morphologically homogeneous but biologically heterogeneous[12]. Apart from self-renewal potential, dormancy/quiescence and a protective stem cell niche are shared characteristics between HSCs and LSCs.
Self-renewal capacity: As the definition of CSCs is a functional definition, CSCs can thus only be defined experimentally by their ability to recapitulate the generation of a continuously growing tumor. Immunodeficient mice, such as the non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mouse and newer generations of xenograft models, are used to functionally define human hematopoietic stem and progenitor cells as well as LSCs[13]. Long-term repopulating cells, thought to be LSC are able to be successfully engrafted in these mice over prolonged periods as well as in secondary recipients[2,14]. Bonnet et al[15] in the John Dick laboratory isolated subpopulations of cells from primary human AML bone marrow based on their immunophenotype and xenotransplanted them into NOD/SCID mice. It demonstrated that the CD34+CD38- expressing sub-population of AML cells were capable of being serially transplanted in these immunodeficient mice[15,16]. Reflecting the emphasis on functional assessment, these cells were named as SCID leukemia-initiating cells (SL-IC) and are considered the equivalent of LSC.
Symmetrical vs asymmetrical cell division: Similar to HSCs, LSCs have the ability to undergo symmetrical self-renewing cell division, generating identical daughter stem cells that retain self-renewal capacity (expansion), or an asymmetrical self-renewing cell division, resulting in one stem cell and one more differentiated progenitor cell (maintenance)[12,17-19]. Normal stem cells are able to switch between symmetrical and asymmetrical division based on the demands of the tissue they are meant to maintain. During early embryogenesis normal stem cells undergo symmetrical cell division in order to expand the total pool of stem cells giving rise to tissues whereas in adult tissues stem cells give rise to mature cells though asymmetrical cell division[19,20]. There is increasing amount of evidence that in CSCs this delicate balance seems to be disturbed in favor of symmetric cell division[19,21,22]. For example, CSCs isolated from ERBB2-expressing breast cancer have been demonstrated to prefer symmetric cell division compared to normal breast tissue stem cells[23]. Furthermore, the adenomatous polyposis coli tumor suppressor gene (APC) has been shown to paly a major role in regulating asymmetric cell division in drosophila and its mutational loss is suspected to lead to an expansion of CSCs by symmetric cell division[22,24,25].
Stem cell quiescence and exhaustion: Normal stem cells need to be quiescent to avoid exhaustion of a stem cell pool and to minimize the risk of oncogenic events[26]. In fact, stem cell exhaustion has been described as one reason for aging and as a consequence of the attempt of the body to prevent the development of cancer[27]. Aging leads to an accumulation of DNA damage in all cells of the body, including stem cells, which in turn leads to an increased risk of developing cancer. Aging stem cells are affected by sophisticated mechanisms cells have developed to suppress the development of cancer, mainly induction of senescence and apoptosis, which are mediated through telomere shortening and activation of tumor suppressor genes p16 and p53[28-30]. The diminished ability of aging HSC to reconstitute the hematopoietic system is demonstrated by prolonged myelosuppression after cytotoxic chemotherapy in older patients as well as age of the stem cell donor being significantly associated with overall and disease-free survival after hematopoietic stem cell transplant[31,32].
However, normal stem cells are also required to continuously replenish the cells that are lost in a tissue. In order to fulfill both purposes-avoid exhaustion as well as maintaining the cellular integrity of a tissue-stem cells undergo asymmetric cell divisions, which give rise to another stem cell as well as a rapidly dividing progenitor cells. These progenitor cells proliferate quickly for a limited amount of cell divisions and regenerate all cells in a tissue[33,34].
Similarly, LSCs are quiescent, which explains the difficulties to eradicate LSCs with standard chemotherapies that preferentially target rapid proliferating cells[35-37].
Key signaling pathways relevant for retaining stemness: Similar signaling pathways involved in the control of self-renewal of HSCs are also key elements maintaining stemness in LSCs (Figure 1). Among many others, these pathways include PI3K/Akt/mTOR[38], Wnt/beta-catenin[39,40], Hedgehog[41,42], NF-kB[43,44], Notch[45] and Bcl-2[46,47]. Several drugs targeting these pathways are in different stages of preclinical and clinical development (Figure 1).
Figure 1.
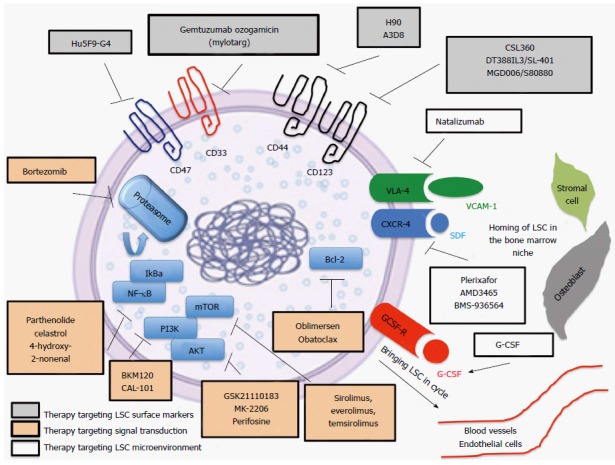
Leukemia stem cells biology and selected therapeutic strategies/agents targeting leukemia stem cell. Leukemia stem cells (LSC) directed therapy targets cell surface markers expressed on LSC (grey boxes), crucial pathways for maintenance of stemness (orange boxes) and interactions between LSC and the bone marrow niche (white boxes). Important LSC surface markers are CD33, CD44, CD123, CD47. Essential pathways are NF-κB, PI3K/AKT/mTOR and bcl-2. LSC mobilization is accomplished with G-CSF and LSC homing to the bone marrow is regulated by the CXCR4–CXCL12 and VCAM-VLA4 axis. VCAM-1: Vascular cell adhesion protein-1; VLA-4: Very late antigen-4; CXCR4: C-X-C chemokine receptor type 4; SDF: Stromal cell-derived factor 1; PI3K: Phosphatidylinositol-4,5-bisphosphate 3-kinase; AKT: Protein kinase B; mTOR: Mechanistic target of rapamycin; bcl-2: B-cell lymphoma 2; G-CSF: Granulocyte-colony stimulating factor.
Stem cell niche: The bone marrow niche is quintessential for normal HSC to maintain their quiescence but at the same time enable HSC to generate cells in the blood stream to meet the organism’s needs[48]. The stem cell niche is formed by a complex network of different cells including vascular endothelial cells, perivascular mesenchymal cells, megakaryocytes, osteoblastic lineage cells, macrophages and nerve cells[49-53]. Dysregulation of the bone marrow niche plays an important role in preventing the detection of LSC by the immune system and protecting LSC from the effects of chemotherapy[48,54]. Similar to normal HSCs, LSCs are retained in the marrow niche by interactions between CXCR4, on stem cells, and CXCL12 (SDF-1), on osteoblasts and mesenchymal cells in the bone marrow niche[55,56]. Chemokine interactions through CXCL12 can lead to up-regulation of vascular cell adhesion molecule-1 (VCAM-1) and very late antigen-4 (VLA-4) expression, which further strengthen LSC retention in the marrow niche[57,58] (Figure 1). The significance of the interaction between LSCs and the protective bone marrow niche is exemplified by the fact that elevated levels of CXCR4 and VLA-4 have been associated with poor response to chemotherapy and decreased survival[59-61]. Several therapeutic approaches attempt to break the dormancy of LSCs by induction of stem cell cycling with granulocyte-colony stimulating factor (G-CSF) and inhibition of the CXCR4-SDF-1 axis involved in LSC retention in the protective bone marrow niche[62,63] (Figure 1).
Identification of LSCs by surface markers
Recent studies have shown that LSCs may reside not only in CD34+CD38-, but also in CD34+CD38+ and CD34- CD38+ compartments demonstrating the lack of a definitive phenotype for LSCs[64-66]. Several studies have shown that the CD34+CD38+ fraction has repopulating ability when immunosuppression is applied[18,67,68]. It was demonstrated that by treating mice with immunosuppressive antibodies, the CD34+CD38+ fraction of AML samples is able to initiate leukemia in immunodeficient mice[64]. Furthermore, by transplanting sorted fractions of primary NPM-mutated AML into immunodeficient mice, it was shown that approximately one-half of cases had LICs exclusively within the CD34- fraction, whereas the CD34+ fraction contained normal multilineage hematopoietic repopulating cells[66]. Most of the remaining cases had LICs in both CD34+ and CD34- fractions and when samples were sorted based on CD34 and CD38 expression, multiple fractions initiated leukemia in primary and secondary recipients (Table 1).
Table 1.
Markers of leukemia stem cells
| Cell surface markers | Patient samples used | Mouse model used | Ref. |
| CD34+CD38- | FAB M1, M4, M5 | NOD/SCID | [15,16] |
| CD34+CD38+ | CN-AML, MLL-ENL | NOD/SCID + IVIG or anti-CD122 | [18,64,67,68] |
| CD34-CD38+ | AML with NPM1 mutation | NOD/SCID β-2 microglobulin NOD/SCID IL2 receptor γ−/− + IVIG | [66] |
| CD34+CD123+ | FAB M1, M2, M4 | NOD/SCID | [69] |
| CD34+CD38-CD96+ | CK-AML, CBFB-MYH11, PML-RARA, AML1-ETO, FAB M4 | Rag2-/- IL2RG-/- | [70] |
| CD34+CLL1+ | AMLs with FLT3-ITD | NOD/SCID | [71] |
| TIM3+ | FAB M1, M2, M4 | NOD/Rag1-/- IL2RG-/- | [72] |
| CD34+CD38- CD33+CD13+ | CN-AML, CBF-AML, MLL-ENL | NOD/SCID | [73] |
FAB: French-American-British classification system; CN: Cytogenetically normal; CK: Cytogenetically complex; MLL-ENL: Mixed-lineage leukemia-eleven nineteen leukemia; NPM1: Nucleophosmin 1; CBFB-MYH11: Core binding factor beta unit-Myosin heavy chain 11; PML-RARA: Promyelocytic leukemia-retinoic acid receptor alpha; AML1-ETO: Acute myeloid leukemia 1 protein- eight twenty one; FLT3-ITD: Fms-like tyrosine kinase 3 Internal tandem duplication; NOD/SCID: Non-obese diabetic/severe combined immunodeficiency; Rag: Recombination activating gene; IL2RG: Interleukin 2 receptor subunit gamma.
Heterogeneity within the LSC population
Over the last years several groups have found a wide variety of other markers that appear to be expressed higher in LSCs than normal HSCs[14].
These include CD123, CD96, CLL-1, TIM3, CD33, CD13, CD44, CD47 and others[69-75] (Table 1). In essence, these studies suggest that leukemogenic activity is not restricted to the CD34+CD38- fraction and there is heterogeneity among patients in leukemogenic cell phenotype. Over the last years, there has been significant advancement in the understanding of the complexity and heterogeneity of human LSC. Several important observations have been made along the way of discovery.
LSC heterogeneity within a patient: First, there is heterogeneity of the stem cell population within the same patient as not all LSC have the same self-renewal capacity[10,76]. Use of lentiviral gene marking to track the behavior of individual leukemia initiating cells following serial transplantation has revealed heterogeneity in their ability to repopulate secondary and tertiary recipients and this enabled researchers to classify long term (LT-LSC) and short term (ST-LSC) LSCs[76,77]. LT-LSCs are defined by a long-termed persistence in xenotransplantion models given an extensive self-renewal capacity while ST-LSCs have a reduced self-renewal capacity and only a transient repopulation capability in xenotransplantation models.
LSC heterogeneity based on the specific xenotransplantation model used: The LSC phenotype depends on what mouse model is used for functional assessment of stem cell properties of human AML cells[14] (Table 1). The bone marrow niche in mice differ from that of humans in terms of architecture, stromal cells, cytokines, growth factors, adhesion molecules and most importantly the immune cell composition, which potentially impairs growth of human HSC or LSC in the mouse bone marrow[78]. Normal HSCs and LSC are therefore more likely to be detected in more highly immunodeficient mice. As different xenotransplantation mouse models display different levels of immunodeficiency they are associated with different levels of engraftment of normal human HSCs and LSCs[6,14]. In nude mice T cells are absent whereas in severe combined immunodeficiency mice (SCID mice) both B and T cells are inactivated. NOD/SCID mice, which harbor defects in T, B, and macrophage activity, support higher levels of human engraftment[14]. NOD/SCID gamma (NSG) mice have almost no murine immune system left as a complete null mutation in the gene encoding the interleukin 2-receptor gamma chain blocks NK cell differentiation[79]. Similarly, NK cells can be depleted by treating NOD/SCID mice with anti-CD122 antibodies[80]. In creating a supportive bone marrow niche for engraftment of human AML cells not only a suppression of the hosts immune system is essential but also a recreation of the cytokine environment supporting stem cell self-renewal and quiescence[14]. This has led to the development of mice models that express human cytokines like human SCF, GM-SCF, IL3 and TPO[13,81].
LSC heterogeneity between patients: It has become increasingly evident that the LSC phenotype varies between patients based on the specific subtype of leukemia that they suffer from (Table 1). As mentioned above, the majority of AML cells express CD34, however in AML cells carrying a mutation in NPM1 the CD34+ percentage is very low and LSC activity is exclusively restricted to the CD34- population[66]. Furthermore, specific subtypes of AML (in particular less aggressive subtypes) are significantly more difficult to be engrafted as they may have low progenitor cell frequency or are particularly sensitive to a specific cytokine or cell type missing in the particular xenotransplantation model[14]. For example, AML samples with a t(8;21) translocation were shown to be difficult to be engrafted and found to be dependent on signaling though the TPO/mpl pathway[82,83]. Subsequently, human TPO knock-in mice were shown to have improved engraftment for t(8;21) AML samples[84].
Cell of origin of LSCs
It is important to distinguish the concept of the cell of origin from the CSC[10]. The CSC has stem cell like properties and is capable of initiating and sustaining tumor growth, whereas the cell of origin refers to the normal cell in which the initial transforming event occurs. Importantly, cancer and LSCs do not have to arise from a normal stem cell, in fact, it is not entirely clear what the cell of origin for most LSCs is[11,12]. One hypothesis is that LSCs are only able to arise from normal HSCs but not from committed progenitor cells[10,15]. This theory is supported by the observation that LSCs and HSCs share many characteristics like self-renewal capacity controlled by genes like Bmi1 and PTEN and quiescence[35,85,86]. On the contrary, transformation might occur in a variety of cell types in the hematopoietic hierarchy, including HSCs and committed progenitors[10,87]. Experimental evidence in mice shows that LSCs may arise either through neoplastic changes initiated in normal self-renewing HSCs or downstream progenitors cells[10,11,88]. Some oncogenes including MOZ-TIF, MLL-AF9 and MLL-ENL can induce LSCs regardless of what target cell population they are expressed in[88-90]. Other oncogenes like BCR-ABL, FLT3-ITD, Hoxa9 and Meis1 were found to be oncogenic when expressed in HSCs but not when expressed in progenitor cells[39,89,91]. However, experimental data in murine studies might be confounded by non-physiologic levels of expression from exogenous promoters, such as transgenes or retroviral vectors[11]. This was demonstrated by the recent finding that in an MLL-AF9 knock-in model of the same construct shown to initiate disease in both HSCs and progenitor cells by retroviral expression only initiated leukemia from HSCs when expressed from the endogenous MLL promoter[92]. In vivo clonality studies in humans suggest variations in the cells of origin and is was demonstrated that in patients with t(8;21) AML primitive CD34+CD90-CD38- HSC like cells from leukemic bone marrow give rise to normally differentiating progenitors, whereas more mature CD34+CD90-CD38+ multi-potent progenitor like cells form exclusively leukemic blast colonies[93-95]. These observations suggest that the truth about the cell of origin might be reflected by a combination of both theories depicted above: Although the initial genetic mutation might happen in HSCs subsequent events occur in the committed progenitor pool, giving rise to LSCs[11].
IMPACT OF LSC ON CURRENT TREATMENT AND PROGNOSIS
Impact on prognosis
The LSC burden of AML patient is suggested to be a strong biomarker for clinical outcome in AML[96-100]. The ability of cells from AML patients to engraft NOD/SCID mice and the LSC frequency (simplistically characterized as CD34+CD38- frequency) are associated with worse clinical outcomes[99-101]. AML patients with greater than 3.5% of CD34+CD38- AML cells show a median relapse free survival of 5.6 mo vs 16 mo in those with a lower percentage of CD34+CD38- cells[96]. Furthermore, poor clinical outcome seems to correlate with the degree to which the LSCs matched normal HSC gene expression[98].
It is noted that it is controversial whether the simplistically phenotypically defined LSC frequency (characterized as CD34+CD38-) in AML is prognostic and correlates with xenograft potential[14]. Also, as described above, LSCs can be found outside of the CD34+CD38- cell fraction. An improved characterization of subpopulations of LSCs is expected to be associated with improved prediction of prognosis.
Impact on current therapies
It is thought that LSCs have a significant role in the relapse of leukemia as induction chemotherapy targets the bulk of blast cells but not LSC[102]. Minimal residual disease (MRD) is an important determinant for relapse and poor outcomes in AML and it is likely that the MRD cell population contains LSCs[103-105]. Thus, in order to improve outcomes in AML, MRD needs to be reduced to prevent disease relapse. LSCs seem to be only minimally affected by traditional chemotherapy[35,106]. Several reasons for chemotherapy resistance have been proposed, which are related to the key features of LSCs discussed above. LSCs are quiescent in the G0 phase of the cell cycle but chemotherapy is only effective in killing rapidly cycling cells[36,37]. LSCs are supported by a stem cell niche in the bone marrow protecting them from the effect of classical chemotherapy[65]. Furthermore, LSCs express high levels of ATP transporters, which are involved in extrusion of chemotherapeutic drugs from LSCs[107-109].
To improve survival in AML, traditional chemotherapy targeting the blast population needs to be combined with therapy specifically targeting LSCs to maintain prolonged remission.
FUTURE DIRECTIONS FOR THERAPY
Despite the recent increased interest in LSCs, experimental studies have not been translated into improved survival outcomes for cancer patients. However, several new agents targeting LSC specific surface molecules and pathways as well as the LSC microenvironment remains under different stages of preclinical and clinical development (Table 2 and Figure 1). To rationally design clinical trials testing drugs for efficacy against LSCs, it is important to appreciate the fundamental differences between drug design targeting blast cells and LSCs[102]. Principles and challenges faced by targeting LSCs will be discussed first followed by an overview of various new therapeutic options targeting LSCs.
Table 2.
Emerging therapy targeting leukemia stem cells
| Drug | Mechanism | Selected clinical trials | Phase | Ref. |
| Therapy targeting cell surface markers | ||||
| GO | Anti-CD33 monoclonal antibody conjugated with calicheamicin, a potent antitumor anthracycline antibiotic | NCT00882102 NCT01869803 NCT00968071 NCT01409161 NCT00766116 NCT02724163 NCT00658814 NCT02473146 NCT00895934 NCT00006265 NCT00860639 NCT00927498 NCT00085709 NCT00195000 NCT00893399 NCT00017589 | Phase I-III | [124,126, 130,132,133] |
| Hu5F9-G4 | Anti-CD47 monoclonal antibody | NCT02678338 | Phase I | [74,141] |
| CSL360 | Anti-CD123 monoclonal antibody | NCT00401739 | Phase I | [69,134] |
| DT388IL3/ SL-401 | Anti-CD123 recombinant immunotoxin created by the fusion of diphtheria toxin with a ligand targeting the IL-3 receptor | NCT02113982 NCT00397579 | Phase I-II | [69,134,136] |
| MGD006/ S80880 | Anti-CD3 and CD123 DART | NCT02152956 | Phase I | [137] |
| H90 | Anti-CD44 monoclonal antibody | N/A | N/A | [75,139] |
| A3D8 | anti-CD44 monoclonal antibody | N/A | N/A | [139] |
| Therapy targeting LSC-specific molecular pathways | ||||
| Bortezomib | Proteasome inhibitor inhibits the degradation of the IkBa creating an anti-NF-κB effect | NCT00789256 NCT00382954 NCT01127009 NCT00666588 NCT00703300 NCT01534260 NCT00383474 | Phase I-III | [36,143-147, 175-177] |
| Parthenolide | Inhibitor of NF-κB | N/A | N/A | [149] |
| Celastrol | Inhibitor of Hsp90 and by extension NF-κB | N/A | N/A | [150] |
| 4-hydroxy- 2-nonenal | Product of lipid peroxidation, inhibiting the proteasome and NF-κB function | N/A | N/A | [151,152] |
| BKM120 CAL-101 | PI3K inhibitors | NCT01396499 NCT01833169 NCT00710528 | Phase I-II | [38,153,154] |
| GSK21110183 MK-2206 Perifosine | AKT inhibitors | NCT00881946 NCT01253447 NCT01231919 NCT00301938 | Phase I-II | [38,155-157] |
| Sirolimus, everolimus, temsirolimus | mTOR inhibitors | NCT01184898 NCT01611116 NCT01074086 NCT01074086 NCT01154439 NCT00775593 NCT02583893 NCT01869114 NCT01822015 | Phase I-II | [38,158] |
| Oblimersen (Genasense, G3139) | bcl-2 antisense oligodeoxynucleotide | NCT00085124 NCT00039117 NCT00017589 | Phase I-III | [46,159,160] |
| Obatoclax Mesylate (GX15-070MS) | Small molecule bcl-2 inhibitor | NCT00438178 NCT00684918 NCT00684918 | Phase I-II | [161-163] |
| Therapy targeting the LSC microenvironment | ||||
| G-CSF | Mobilization of LSC from the protective bone marrow niche - > increased susceptibility to traditional chemotherapy | NCT00820976 NCT00602225 NCT00199147 NCT01723657 NCT01101880 NCT00943943 NCT00906945 | Phase I-III | [165-168] |
| Plerixafor (AMD3100) | CXCR4 inhibitor Decreased homing to the bone marrow | NCT00943943 NCT00906945 NCT01236144 NCT00512252 NCT01319864 NCT01352650 NCT02416908 | Phase I-II | [61,135,178] |
| AMD3465 | CXCR4 inhibitor Decreased homing to the bone marrow | N/A | N/A | [61,135,169,179] |
| BMS-936564 | Anti-CXCR4 antibody Decreased homing to the bone marrow | NCT01120457 | Phase I | [172] |
| Natalizumab | Anti-VLA4 antibody Decreased homing to the bone marrow | N/A | N/A | [174] |
GO: Gemtuzumab ozogamicin; DART: Dual-affinity retargeting molecule; N/A: Not available; LSC: Leukemia stem cell; IkBa: Inhibitor of kappa B alpha; CXCR4: C-X-C chemokine receptor type 4; mTOR: Mechanistic target of rapamycin; G-CSF: Granulocyte-colony stimulating factor.
General principles and challenges faced by targeting LSCs
Limiting side effects: As LSCs and HSCs have many similar properties (see above), therapeutic approaches targeting LSCs also have the potential of causing severe side effects by eliminating healthy HSCs. To develop novel therapies with limited side effects, unique properties of LSCs have to be identified[102,110]. While expression of several surface markers is similar between normal HSCs and LSCs (CD34, CD38, CD71 and HLA-DR), other surface antigens are only displayed on LSCs (CD33, CD90, CD117 and CD123)[110]. Apart from a similar immunophenotype, HSCs and LSCs share many pathways important for maintaining features of “stemness” like quiescence and self-renewal capacity[111]. Pathways, which are up-regulated in LSCs compared to normal HSCs, are the ideal target for therapeutic approaches directed towards LSCs. For example, the active form of NF-κB and bcl-2, which are associated with anti-apoptotic activity in cancer cells, are overexpressed in LSCs compared to normal HSC and drugs targeting both NF-κB and bcl-2 are in clinical development[36,46,112].
Using biomarkers for LSC eradication: To assess the efficacy of investigational therapies targeting LSCs, precise diagnostic methods are needed to assess the quantity of LSCs present in leukemia patients. Unfortunately, current characterization of LSC phenotype is not precise enough to permit real-time tracking of LSCs in vivo[113]. As discussed above, current strategies for purification do not yield functionally homogeneous population: The frequency of LSCs within the CD34+CD38- fraction in AML ranges from 1 in 104 to 1 in 5 × 106 cells and several other populations contain LSCs as well[15]. Functional assessment of LSC frequency with xenotransplantation models offers a more robust method to evaluate eradication of LSCs but might not be feasible in large clinical trials[102]. Similarly, methods for detecting MRD might guide decisions by detecting patients who do require additional therapy to prevent relapse. However, detecting MRD does not distinguish persistent LSCs, which may cause relapse, from residual blasts and normal HSCs that do not have tumor-initiating activity. Distinguishing residual LSCs from residual blasts might be accomplished by gene expression analysis showing reactivation of self-renewal genes in LSCs but not in blast cells[88,114]. In preclinical development, the recently published Connectivity Map could be investigated for agents that attenuate a stem cell gene signature or induce a differentiated state[115,116].
Timing of LSC targeted therapy: Therapy targeting LSCs is effective in eradicating a small amount of leukemia initiating cells but not the bulk of blasts cells in the blood and bone marrow[102]. By combining drugs eradicating LSCs with standard chemotherapy targeting the bulk of the disease, both the aggressive proliferating process as well as the root of the leukemia can be targeted[117]. An example serves the successful combination of the anti-CD33 immunoconjugate antibody gemtuzumab ozogamicin (GO) with standard chemotherapy[118]. This is associated with challenges in a meaningful design of clinical trials in terms of the correct timing of these therapies. LSC targeting therapy can either be given after reduction of the bulk population with standard chemotherapy as remission therapy or concomitant with chemotherapy as an induction regimen[102]. Upfront combination would allow assessing for additive and/or synergistic properties between drugs and would allow targeting of LSCs early on in the disease process, which might improve outcomes[102]. On the other hand, LSC targeted therapy might be particular valuable as post-consolidation therapy as no current post-consolidation intervention has led to improved OS for patients with AML[102,119,120]. LSC targeting therapies have the potential to fill the gap as they eradicate the cells responsible for relapses of AML.
Assessing clinical endpoints: Classical response criteria like CR and hematologic improvement might not be the best parameters to assess the efficacy of therapeutic approaches targeting LSCs as these drugs do not eradicate the bulk of blast cells but rather eliminate the rare population of LSCs[102]. Progression-free survival (PFS), event free survival and overall survival (OS) may be a more relevant endpoint for assessing the effectiveness of LSC elimination than tumor response as they better account for whether the root of the leukemia has been eliminated[113]. Importantly, while LSC frequency was found to be prognostic for survival, response rates did not correlate with LSC burden[96]. Subsequently, drugs targeting LSCs may show little activity if tested in traditional phase I/II trials as a proper assessment of endpoints relevant for LSCs, like PFS and OS, is generally only feasible in a phase III trial with a larger numbers of patients and long-term follow-up[113,121].
One example for the importance of assessing relevant endpoints for LSC targeting therapy, is inconsistency of clinical trials evaluating the efficacy of GO[102]. Single agent studies of GO showed overall response rates only approaching 30% at best and GO was voluntarily withdrawn from the United States market in 2010 after a study showed no improvements in outcomes when used in combination therapy as well as increased fatal toxicity[122-124]. In contrast, other large clinical trials showed improvement in outcomes more relevant for therapies targeting LSC- event-free survival, disease-free survival and OS- despite no differences in disease response rates[125-128].
Targeting LSC surface molecules
Anti-CD33 antibodies: CD33 is found on LSCs although it is not a consistent feature of all LSCs studied[73,118,129]. As discussed above, there have been conflicting reports surrounding the efficacy and safety of GO and currently GO is not available on the market in the United States or Europe[130]. Apart from the different endpoints studied, there are additional explanations for the discrepancies observed: First, the dose of daunorubicin as the combination partner of GO did vary between trials, although it is known that treatment with daunorubicin-based schedules of 90 mg/m2 for 3 d is more effective than similar schedules with daunorubicin at 45 mg/m2[131]. In the SWOG trial, which questioned the efficacy of GO, single bolus combined with daunorubicin at 45 mg/m2 was studied against a control group with daunorubicin at 60 mg/m2[124]. However, the best effect of GO was seen when higher dose of GO (3 d at 3 mg/m2 for 2 cycles) was added to a daunorubicin regimen of 60 mg/m2 in both comparator groups[126]. Furthermore, GO seems to be quite active in acute promyelocytic leukemia (APL) as APL cells express high levels of CD33[132,133]. These results have prompted calls to reconsider the approval status of GO[130].
Anti-IL-3 receptor (CD123) antibodies: The interleukin-3 receptor alpha chain (IL-3Rα or CD123) is strongly expressed in CD34+/CD38- LSCs and can be targeted with monoclonal antibodies[69,134]. The blockage of CD123 has pleiotropic anti-leukemic effects including inhibition of LSC homing to the bone marrow, activation of innate immunity and inhibition of intracellular signaling events[135]. Several different agents targeting CD123 are currently evaluated in clinical trials: CD123 targeting antibodies can either be naked antibodies or be conjugated to toxins (e.g., diphteriod toxin) or chemotherapeutic agents (chemo-immune conjugates) or be the backbone of a bi-specific T cell engager (BITE, e.g., CD3-CD123)[134,136,137] (Table 2).
Anti-CD44 antibodies: CD44 regulates interaction between LSCs and the bone marrow niche by controlling cell-cell adhesion and cell-matrix interaction through binding to hyaluronic acid, osteopontin, collagens and others[138].
Inhibition of CD44 with monoclonal antibodies was shown to reduce the numbers of LSCs in NOD/SCID mice and to increase the survival of the primary recipient mice as well prevent engraftment into the secondary receipt mice[75,139] (Table 2).
Anti-CD47 antibodies: CD47 is overexpressed on LSCs and high expression of CD47 is associated with worse outcomes[74]. By interaction with the extracellular region of signal-regulatory protein alpha (SIRPα) on phagocytic cells, LSCs deliver a “do not eat me” message to these phagocytic cells[140]. Antibodies blocking the interaction between CD47 and SIRPα promote LSC phagocytosis and are in development (Table 2)[74,141].
Targeting LSC-specific molecular pathways
NF-κB signaling pathway: Bortezomib is able to suppress the NF-κB signaling pathway by inhibiting the destruction of IκB, a cellular inhibitory protein of NFκB, by the ubiquitin-proteasome pathway[142]. Several clinical trials are examining the efficacy of Bortezomib targeting AML LSCs (Table 2): Two clinical trials combining Bortezomib with Cytarabine and Anthracyclines resulted in CR rates of 61% and 65%[143,144], whereas other trials that co-administrated Bortezomib with other drugs did not show encouraging CR rates[145-147]. Several other inhibitors of NF-κB signaling are in different phases of development (Table 2)[148-152].
PI3K/AKT/mTOR pathway: The PI3K/AKT/mTOR pathway is of utmost importance in regulating cellular growth, survival, and metabolism and is frequently dysregulated in cancers and AML[38]. A multitude of PI3K inhibitors[153,154], AKT inhibitors[155-157] and mTOR inhibitors[158] is currently investigated for their efficacy targeting LSCs in clinical trials (Table 2).
Bcl-2 pathway: LSCs, similar to other tumor cells, are able to avoid apoptosis due to overexpression of bcl-2[46]. Currently, bcl-2 inhibition is investigated in clinical trials in form of the bcl-2 antisense oligodeoxynucleotide oblimersen[159,160] and the small molecule inhibitor of bcl-2 obatoclax[161-163] (Table 2).
Targeting the LSC microenvironment
Approaches targeting the interactions of LSCs with the bone marrow niche focus on breaking the dormancy of LSCs in the bone marrow in order to make them sensitive to traditional chemotherapy[62,164].
LSC mobilization: LSC mobilization from the marrow niche can be achieved by nonspecific stimulators like G-CSF, Interferon-α and Arsenic trioxide[62]. Using the NOD/SCID/IL2rgamma (null) mouse model, Saito et al[165] showed that quiescent human AML LSCs, at first resistant to cytarabine, start proliferating and become susceptible to cytarabine once exposed to G-CSF. Combining chemotherapy with G-CSF leads to significantly increased survival of secondary recipients after transplantation of leukemia cells compared with chemotherapy alone. Furthermore, they showed that treatment with G-CSF before cytarabine did not increase apoptosis of normal HSCs making this approach a particular attractive option for targeting LSCs but at the same time avoiding side effects from depletion of HSCs. The data from clinic trials using G-CSF priming in combination with chemotherapy are conflicting. Löwenberg et al[166] randomized 640 newly diagnosed AML patients to receive cytarabine plus idarubicin with G-CSF (321 patients) or without G-CSF (319 patients) for the first cycle of induction of chemotherapy. Patients in CR after induction chemotherapy plus G-CSF had a higher rate of disease-free survival than patients who did not receive G-CSF (42% vs 33% at four years, P = 0.02), owing to a reduced probability of relapse (relative risk, 0.77; P = 0.04). Other studies did not show a benefit of adding G-CSF to traditional chemotherapy regimens[167,168]. These different responses to G-CSF might be explained by differences in the group of patients included in these trials[63]. In the trial by Löwenberg et al[166] patients with standard-risk AML benefited from G-CSF therapy whereas G-CSF did not improve the outcome in the subgroup with an unfavorable prognosis. In the trials without improvement with G-CSF, patients had a more unfavorable prognosis based on age, cytogenetic abnormalities or response to previous treatment. Several clinical trials are ongoing to investigate the efficacy of G-CSF in combination of chemotherapy in different risk groups of AML (Table 2).
Inhibition of homing: LSC dormancy can be targeted by specifically interrupting the CXCR4-CXCL12 and VCAM-VLA4 axis as well as inhibiting CD44 and CD123 on LSCs to prevent homing of LSCs to the bone marrow.
CXCR4-CXCL12 axis: SDF-1 was shown to promote survival of AML cells, whereas addition of neutralizing CXCR4 antibodies, SDF-1 antibodies, or AMD3100 significantly decreased their survival[169]. Furthermore, pretreatment of primary human AML cells with neutralizing CXCR4 antibodies blocked their homing into the BM and spleen of transplanted NOD/SCID/B2mnull mice[169]. Additionally, CXCR4 inhibition with AMD3465 was shown to increase the sensitivity of FLT3-mutated leukemic cells to the apoptogenic effects of the FLT3 inhibitor sorafenib[170]. Recently a phase 1/2 study examined the efficacy of the CXCR4 inhibitor plerixafor in combination with mitoxantrone, etoposide, and cytarabine in 52 patients with relapsed or refractory AML[171]. Overall CR was found to be 46% and correlative studies demonstrated a 2-fold mobilization in leukemic blasts into the peripheral circulation without evidence of symptomatic hyperleukocytosis or delayed count recovery. BMS-936564, a fully human IgG4 monoclonal antibody against CXCR4, exhibits antitumor activity in cytarabine-resistant mouse xenograft models of AML and is currently tested in a phase I clinic trial (Table 2)[172].
VCAM-VLA4 axis: Integrin alpha4beta1 (VLA4) mediates adhesion of LSCs to stromal cells and extracellular matrix in the marrow niche and can be blocked by the monoclonal antibody Natalizumab[59,69,173]. AML cells were shown to de-adhere from a layer of immobilized human VCAM1 expressing human stromal cells when exposed to Natalizumab and NSG mice transplanted with human AML cells survived significantly longer when they received intraperitoneal Natalizumab injections[174].
CONCLUSION
AML remains one of the most difficult malignancies to treat. Despite significant advancements in the understanding of disease biology, this has not been translated yet into new treatment modalities improving outcomes. The relapse of AML is frequent and is responsible for the inability to cure AML. LSCs are understood to be the root of relapse and their presence has been found to be prognostic for the disease course. Unfortunately, LSCs are not easy to target as they are quiescent, able to self-renew and well protected by a supportive bone marrow niche. Furthermore, their inconsistent phenotype and similarity to normal HSCs hamper specific drug development. Nevertheless, a multitude of potential targets have been identified and are currently tested in different phases of clinical and preclinical development. Successful eradication of LSCs will require combination of different strategies including targeting LSC specific surface molecules and pathways as well as interactions of LSCs with the microenvironment. Furthermore, clinical trials have to be designed in a way that they are able to detect a specific effect of LSCs, which is easy to miss in a traditional trial design. Overall, targeting LSCs has the promise to not only effectively reduce disease burden but to eradicate the root of leukemia itself.
Footnotes
Conflict-of-interest statement: Amer M Zeidan receives Honoraria from Ariad, Pfizer, Incyte and Celgene. Maximilian Stahl and Tae Kon Kim have no conflict of interests to declare for this article.
Manuscript source: Invited manuscript
Specialty type: Cell and tissue engineering
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: May 23, 2016
First decision: July 6, 2016
Article in press: August 29, 2016
P- Reviewer: Kita K, Li ZJ, Ramírez M, Shao R S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Roboz GJ. Novel approaches to the treatment of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2011;2011:43–50. doi: 10.1182/asheducation-2011.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Felipe Rico J, Hassane DC, Guzman ML. Acute myelogenous leukemia stem cells: from Bench to Bedside. Cancer Lett. 2013;338:4–9. doi: 10.1016/j.canlet.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crews LA, Jamieson CH. Selective elimination of leukemia stem cells: hitting a moving target. Cancer Lett. 2013;338:15–22. doi: 10.1016/j.canlet.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Lutz C, Hoang VT, Buss E, Ho AD. Identifying leukemia stem cells--is it feasible and does it matter? Cancer Lett. 2013;338:10–14. doi: 10.1016/j.canlet.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18:460–466. doi: 10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 8.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 10.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–4807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- 12.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 13.Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, Mulloy JC. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24:1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood. 2015;125:2630–2640. doi: 10.1182/blood-2014-11-570218. [DOI] [PubMed] [Google Scholar]
- 15.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 16.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 17.Till JE, Mcculloch EA, Siminovitch L. A stochastic model of stem cell proliferation, based on the growth of spleen colony-forming cells. Proc Natl Acad Sci USA. 1964;51:29–36. doi: 10.1073/pnas.51.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenzie JL, Gan OI, Doedens M, Wang JC, Dick JE. Individual stem cells with highly variable proliferation and self-renewal properties comprise the human hematopoietic stem cell compartment. Nat Immunol. 2006;7:1225–1233. doi: 10.1038/ni1393. [DOI] [PubMed] [Google Scholar]
- 19.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 20.Huttner WB, Kosodo Y. Symmetric versus asymmetric cell division during neurogenesis in the developing vertebrate central nervous system. Curr Opin Cell Biol. 2005;17:648–657. doi: 10.1016/j.ceb.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 21.Boman BM, Wicha MS, Fields JZ, Runquist OA. Symmetric division of cancer stem cells--a key mechanism in tumor growth that should be targeted in future therapeutic approaches. Clin Pharmacol Ther. 2007;81:893–898. doi: 10.1038/sj.clpt.6100202. [DOI] [PubMed] [Google Scholar]
- 22.Powell AE, Shung CY, Saylor KW, Müllendorff KA, Weiss JB, Wong MH. Lessons from development: A role for asymmetric stem cell division in cancer. Stem Cell Res. 2010;4:3–9. doi: 10.1016/j.scr.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 24.Yamashita YM, Mahowald AP, Perlin JR, Fuller MT. Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science. 2007;315:518–521. doi: 10.1126/science.1134910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita YM, Jones DL, Fuller MT. Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science. 2003;301:1547–1550. doi: 10.1126/science.1087795. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. 2014;141:4656–4666. doi: 10.1242/dev.106575. [DOI] [PubMed] [Google Scholar]
- 27.Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 28.Allsopp RC, Morin GB, DePinho R, Harley CB, Weissman IL. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood. 2003;102:517–520. doi: 10.1182/blood-2002-07-2334. [DOI] [PubMed] [Google Scholar]
- 29.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 31.Lenhoff S, Hjorth M, Westin J, Brinch L, Bäckström B, Carlson K, Christiansen I, Dahl IM, Gimsing P, Hammerström J, et al. Impact of age on survival after intensive therapy for multiple myeloma: a population-based study by the Nordic Myeloma Study Group. Br J Haematol. 2006;133:389–396. doi: 10.1111/j.1365-2141.2006.06042.x. [DOI] [PubMed] [Google Scholar]
- 32.Kollman C, Howe CW, Anasetti C, Antin JH, Davies SM, Filipovich AH, Hegland J, Kamani N, Kernan NA, King R, et al. Donor characteristics as risk factors in recipients after transplantation of bone marrow from unrelated donors: the effect of donor age. Blood. 2001;98:2043–2051. doi: 10.1182/blood.V98.7.2043. [DOI] [PubMed] [Google Scholar]
- 33.Watt FM, Jensen KB. Epidermal stem cell diversity and quiescence. EMBO Mol Med. 2009;1:260–267. doi: 10.1002/emmm.200900033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. 2011;2011:pii: 396076. doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terpstra W, Ploemacher RE, Prins A, van Lom K, Pouwels K, Wognum AW, Wagemaker G, Löwenberg B, Wielenga JJ. Fluorouracil selectively spares acute myeloid leukemia cells with long-term growth abilities in immunodeficient mice and in culture. Blood. 1996;88:1944–1950. [PubMed] [Google Scholar]
- 36.Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, Luger SM, Jordan CT. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–2307. doi: 10.1182/blood.V98.8.2301. [DOI] [PubMed] [Google Scholar]
- 37.Guan Y, Gerhard B, Hogge DE. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML) Blood. 2003;101:3142–3149. doi: 10.1182/blood-2002-10-3062. [DOI] [PubMed] [Google Scholar]
- 38.Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol Cell Ther. 2015;3:2. doi: 10.1186/s40591-015-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lento W, Congdon K, Voermans C, Kritzik M, Reya T. Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb Perspect Biol. 2013;5:pii: a008011. doi: 10.1101/cshperspect.a008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mar BG, Amakye D, Aifantis I, Buonamici S. The controversial role of the Hedgehog pathway in normal and malignant hematopoiesis. Leukemia. 2011;25:1665–1673. doi: 10.1038/leu.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irvine DA, Copland M. Targeting hedgehog in hematologic malignancy. Blood. 2012;119:2196–2204. doi: 10.1182/blood-2011-10-383752. [DOI] [PubMed] [Google Scholar]
- 43.Zhou J, Ching YQ, Chng WJ. Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: from molecular pathogenesis to therapeutic target. Oncotarget. 2015;6:5490–5500. doi: 10.18632/oncotarget.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, Kobayashi H, Saito T, Iwakura Y, Kurokawa M. Positive feedback between NF-κB and TNF-α promotes leukemia-initiating cell capacity. J Clin Invest. 2014;124:528–542. doi: 10.1172/JCI68101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu N, Zhang J, Ji C. The emerging roles of Notch signaling in leukemia and stem cells. Biomark Res. 2013;1:23. doi: 10.1186/2050-7771-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Domen J, Weissman IL. Hematopoietic stem cells need two signals to prevent apoptosis; BCL-2 can provide one of these, Kitl/c-Kit signaling the other. J Exp Med. 2000;192:1707–1718. doi: 10.1084/jem.192.12.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schepers K, Campbell TB, Passegué E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell. 2015;16:254–267. doi: 10.1016/j.stem.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, Seandel M, Shido K, White IA, Kobayashi M, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 52.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20:1315–1320. doi: 10.1038/nm.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood. 2009;114:1150–1157. doi: 10.1182/blood-2009-01-202606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 56.Ara T, Tokoyoda K, Sugiyama T, Egawa T, Kawabata K, Nagasawa T. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity. 2003;19:257–267. doi: 10.1016/S1074-7613(03)00201-2. [DOI] [PubMed] [Google Scholar]
- 57.Juneja HS, Schmalsteig FC, Lee S, Chen J. Vascular cell adhesion molecule-1 and VLA-4 are obligatory adhesion proteins in the heterotypic adherence between human leukemia/lymphoma cells and marrow stromal cells. Exp Hematol. 1993;21:444–450. [PubMed] [Google Scholar]
- 58.Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, Shido K, Jin DK, Dias S, Zhang F, Hartman TE, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004;10:64–71. doi: 10.1038/nm973. [DOI] [PubMed] [Google Scholar]
- 59.Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, Akiyama T, Kuroda H, Kawano Y, Kobune M, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158–1165. doi: 10.1038/nm909. [DOI] [PubMed] [Google Scholar]
- 60.Rombouts EJ, Pavic B, Löwenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104:550–557. doi: 10.1182/blood-2004-02-0566. [DOI] [PubMed] [Google Scholar]
- 61.Spoo AC, Lübbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007;109:786–791. doi: 10.1182/blood-2006-05-024844. [DOI] [PubMed] [Google Scholar]
- 62.Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 2010;4:443–450. doi: 10.1016/j.molonc.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mikkola HK, Radu CG, Witte ON. Targeting leukemia stem cells. Nat Biotechnol. 2010;28:237–238. doi: 10.1038/nbt0310-237. [DOI] [PubMed] [Google Scholar]
- 64.Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 65.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 66.Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood. 2010;115:1976–1984. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Civin CI, Almeida-Porada G, Lee MJ, Olweus J, Terstappen LW, Zanjani ED. Sustained, retransplantable, multilineage engraftment of highly purified adult human bone marrow stem cells in vivo. Blood. 1996;88:4102–4109. [PubMed] [Google Scholar]
- 68.Hogan CJ, Shpall EJ, Keller G. Differential long-term and multilineage engraftment potential from subfractions of human CD34+ cord blood cells transplanted into NOD/SCID mice. Proc Natl Acad Sci USA. 2002;99:413–418. doi: 10.1073/pnas.012336799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, Meyerrose T, Rossi R, Grimes B, Rizzieri DA, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777–1784. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 70.Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, Krensky AM, Weissman IL. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci USA. 2007;104:11008–11013. doi: 10.1073/pnas.0704271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, Stigter-van Walsum M, Zweegman S, Ossenkoppele GJ, Jan Schuurhuis G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659–2666. doi: 10.1182/blood-2007-03-083048. [DOI] [PubMed] [Google Scholar]
- 72.Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, Takenaka K, Teshima T, Tanaka T, Inagaki Y, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708–717. doi: 10.1016/j.stem.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 73.Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, Luongo JL, Danet-Desnoyers GA, Bonnet D. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106:4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, van Rooijen N, Weissman IL. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 76.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 77.Misaghian N, Ligresti G, Steelman LS, Bertrand FE, Bäsecke J, Libra M, Nicoletti F, Stivala F, Milella M, Tafuri A, et al. Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia. 2009;23:25–42. doi: 10.1038/leu.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Manz MG. Human-hemato-lymphoid-system mice: opportunities and challenges. Immunity. 2007;26:537–541. doi: 10.1016/j.immuni.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 79.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 80.McDermott SP, Eppert K, Lechman ER, Doedens M, Dick JE. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood. 2010;116:193–200. doi: 10.1182/blood-2010-02-271841. [DOI] [PubMed] [Google Scholar]
- 81.Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, Saito Y, Marches F, Halene S, Palucka AK, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. 2014;32:364–372. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chou FS, Griesinger A, Wunderlich M, Lin S, Link KA, Shrestha M, Goyama S, Mizukawa B, Shen S, Marcucci G, et al. The thrombopoietin/MPL/Bcl-xL pathway is essential for survival and self-renewal in human preleukemia induced by AML1-ETO. Blood. 2012;120:709–719. doi: 10.1182/blood-2012-01-403212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pulikkan JA, Madera D, Xue L, Bradley P, Landrette SF, Kuo YH, Abbas S, Zhu LJ, Valk P, Castilla LH. Thrombopoietin/MPL participates in initiating and maintaining RUNX1-ETO acute myeloid leukemia via PI3K/AKT signaling. Blood. 2012;120:868–879. doi: 10.1182/blood-2012-03-414649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chin DW, Watanabe-Okochi N, Wang CQ, Tergaonkar V, Osato M. Mouse models for core binding factor leukemia. Leukemia. 2015;29:1970–1980. doi: 10.1038/leu.2015.181. [DOI] [PubMed] [Google Scholar]
- 85.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 86.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 87.Griffin JD, Löwenberg B. Clonogenic cells in acute myeloblastic leukemia. Blood. 1986;68:1185–1195. [PubMed] [Google Scholar]
- 88.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 89.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 90.Cozzio A, Passegué E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–3035. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kvinlaug BT, Chan WI, Bullinger L, Ramaswami M, Sears C, Foster D, Lazic SE, Okabe R, Benner A, Lee BH, et al. Common and overlapping oncogenic pathways contribute to the evolution of acute myeloid leukemias. Cancer Res. 2011;71:4117–4129. doi: 10.1158/0008-5472.CAN-11-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen W, Kumar AR, Hudson WA, Li Q, Wu B, Staggs RA, Lund EA, Sam TN, Kersey JH. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell. 2008;13:432–440. doi: 10.1016/j.ccr.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fialkow PJ, Singer JW, Raskind WH, Adamson JW, Jacobson RJ, Bernstein ID, Dow LW, Najfeld V, Veith R. Clonal development, stem-cell differentiation, and clinical remissions in acute nonlymphocytic leukemia. N Engl J Med. 1987;317:468–473. doi: 10.1056/NEJM198708203170802. [DOI] [PubMed] [Google Scholar]
- 94.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8; 21 chromosomal translocation. Proc Natl Acad Sci USA. 2000;97:7521–7526. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1:635–645. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, van der Pol MA, Waisfisz Q, Ossenkoppele GJ, Schuurhuis GJ. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11:6520–6527. doi: 10.1158/1078-0432.CCR-05-0468. [DOI] [PubMed] [Google Scholar]
- 97.Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304:2706–2715. doi: 10.1001/jama.2010.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 99.Pearce DJ, Taussig D, Zibara K, Smith LL, Ridler CM, Preudhomme C, Young BD, Rohatiner AZ, Lister TA, Bonnet D. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood. 2006;107:1166–1173. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Terwijn M, Zeijlemaker W, Kelder A, Rutten AP, Snel AN, Scholten WJ, Pabst T, Verhoef G, Löwenberg B, Zweegman S, et al. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS One. 2014;9:e107587. doi: 10.1371/journal.pone.0107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vargaftig J, Taussig DC, Griessinger E, Anjos-Afonso F, Lister TA, Cavenagh J, Oakervee H, Gribben J, Bonnet D. Frequency of leukemic initiating cells does not depend on the xenotransplantation model used. Leukemia. 2012;26:858–860. doi: 10.1038/leu.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pollyea DA, Gutman JA, Gore L, Smith CA, Jordan CT. Targeting acute myeloid leukemia stem cells: a review and principles for the development of clinical trials. Haematologica. 2014;99:1277–1284. doi: 10.3324/haematol.2013.085209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Feller N, van der Pol MA, van Stijn A, Weijers GW, Westra AH, Evertse BW, Ossenkoppele GJ, Schuurhuis GJ. MRD parameters using immunophenotypic detection methods are highly reliable in predicting survival in acute myeloid leukaemia. Leukemia. 2004;18:1380–1390. doi: 10.1038/sj.leu.2403405. [DOI] [PubMed] [Google Scholar]
- 104.Venditti A, Buccisano F, Del Poeta G, Maurillo L, Tamburini A, Cox C, Battaglia A, Catalano G, Del Moro B, Cudillo L, et al. Level of minimal residual disease after consolidation therapy predicts outcome in acute myeloid leukemia. Blood. 2000;96:3948–3952. [PubMed] [Google Scholar]
- 105.van Rhenen A, Moshaver B, Kelder A, Feller N, Nieuwint AW, Zweegman S, Ossenkoppele GJ, Schuurhuis GJ. Aberrant marker expression patterns on the CD34+CD38- stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia. 2007;21:1700–1707. doi: 10.1038/sj.leu.2404754. [DOI] [PubMed] [Google Scholar]
- 106.Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, Gastaut JA, Olive D. Human acute myeloid leukemia CD34+/CD38- progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000;60:4403–4411. [PubMed] [Google Scholar]
- 107.de Grouw EP, Raaijmakers MH, Boezeman JB, van der Reijden BA, van de Locht LT, de Witte TJ, Jansen JH, Raymakers RA. Preferential expression of a high number of ATP binding cassette transporters in both normal and leukemic CD34+CD38- cells. Leukemia. 2006;20:750–754. doi: 10.1038/sj.leu.2404131. [DOI] [PubMed] [Google Scholar]
- 108.Bunting KD. ABC transporters as phenotypic markers and functional regulators of stem cells. Stem Cells. 2002;20:11–20. doi: 10.1002/stem.200011. [DOI] [PubMed] [Google Scholar]
- 109.de Jonge-Peeters SD, Kuipers F, de Vries EG, Vellenga E. ABC transporter expression in hematopoietic stem cells and the role in AML drug resistance. Crit Rev Oncol Hematol. 2007;62:214–226. doi: 10.1016/j.critrevonc.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 110.Jordan CT. Unique molecular and cellular features of acute myelogenous leukemia stem cells. Leukemia. 2002;16:559–562. doi: 10.1038/sj.leu.2402446. [DOI] [PubMed] [Google Scholar]
- 111.Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–740. doi: 10.1038/aps.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guzman ML, Swiderski CF, Howard DS, Grimes BA, Rossi RM, Szilvassy SJ, Jordan CT. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci USA. 2002;99:16220–16225. doi: 10.1073/pnas.252462599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang JC. Evaluating therapeutic efficacy against cancer stem cells: new challenges posed by a new paradigm. Cell Stem Cell. 2007;1:497–501. doi: 10.1016/j.stem.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 114.Krivtsov AV, Feng Z, Armstrong SA. Transformation from committed progenitor to leukemia stem cells. Ann N Y Acad Sci. 2009;1176:144–149. doi: 10.1111/j.1749-6632.2009.04966.x. [DOI] [PubMed] [Google Scholar]
- 115.Michnick SW. The connectivity map. Nat Chem Biol. 2006;2:663–664. doi: 10.1038/nchembio1206-663. [DOI] [PubMed] [Google Scholar]
- 116.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 117.Huff CA, Matsui WH, Smith BD, Jones RJ. Strategies to eliminate cancer stem cells: clinical implications. Eur J Cancer. 2006;42:1293–1297. doi: 10.1016/j.ejca.2006.01.045. [DOI] [PubMed] [Google Scholar]
- 118.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119:6198–6208. doi: 10.1182/blood-2011-11-325050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cassileth PA, Lynch E, Hines JD, Oken MM, Mazza JJ, Bennett JM, McGlave PB, Edelstein M, Harrington DP, O’Connell MJ. Varying intensity of postremission therapy in acute myeloid leukemia. Blood. 1992;79:1924–1930. [PubMed] [Google Scholar]
- 120.Hewlett J, Kopecky KJ, Head D, Eyre HJ, Elias L, Kingsbury L, Balcerzak SP, Dabich L, Hynes H, Bickers JN. A prospective evaluation of the roles of allogeneic marrow transplantation and low-dose monthly maintenance chemotherapy in the treatment of adult acute myelogenous leukemia (AML): a Southwest Oncology Group study. Leukemia. 1995;9:562–569. [PubMed] [Google Scholar]
- 121.Jones RJ, Matsui WH, Smith BD. Cancer stem cells: are we missing the target? J Natl Cancer Inst. 2004;96:583–585. doi: 10.1093/jnci/djh095. [DOI] [PubMed] [Google Scholar]
- 122.Sievers EL, Larson RA, Stadtmauer EA, Estey E, Löwenberg B, Dombret H, Karanes C, Theobald M, Bennett JM, Sherman ML, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19:3244–3254. doi: 10.1200/JCO.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- 123.Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, Roy S, Sridhara R, Rahman A, Williams G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7:1490–1496. [PubMed] [Google Scholar]
- 124.Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, Larson RA, Erba HP, Stiff PJ, Stuart RK, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121:4854–4860. doi: 10.1182/blood-2013-01-466706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Burnett AK, Hills RK, Milligan D, Kjeldsen L, Kell J, Russell NH, Yin JA, Hunter A, Goldstone AH, Wheatley K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29:369–377. doi: 10.1200/JCO.2010.31.4310. [DOI] [PubMed] [Google Scholar]
- 126.Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie JN, Legrand O, Thomas X, Turlure P, Reman O, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379:1508–1516. doi: 10.1016/S0140-6736(12)60485-1. [DOI] [PubMed] [Google Scholar]
- 127.Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, Estey EH, Dombret H, Chevret S, Ifrah N, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15:986–996. doi: 10.1016/S1470-2045(14)70281-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Burnett AK, Russell NH, Hills RK, Kell J, Freeman S, Kjeldsen L, Hunter AE, Yin J, Craddock CF, Dufva IH, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol. 2012;30:3924–3931. doi: 10.1200/JCO.2012.42.2964. [DOI] [PubMed] [Google Scholar]
- 129.Bernstein ID. Monoclonal antibodies to the myeloid stem cells: therapeutic implications of CMA-676, a humanized anti-CD33 antibody calicheamicin conjugate. Leukemia. 2000;14:474–475. doi: 10.1038/sj.leu.2401663. [DOI] [PubMed] [Google Scholar]
- 130.Rowe JM, Löwenberg B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood. 2013;121:4838–4841. doi: 10.1182/blood-2013-03-490482. [DOI] [PubMed] [Google Scholar]
- 131.Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, Racevskis J, Dewald GW, Ketterling RP, Bennett JM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361:1249–1259. doi: 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lo-Coco F, Cimino G, Breccia M, Noguera NI, Diverio D, Finolezzi E, Pogliani EM, Di Bona E, Micalizzi C, Kropp M, et al. Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood. 2004;104:1995–1999. doi: 10.1182/blood-2004-04-1550. [DOI] [PubMed] [Google Scholar]
- 133.Breccia M, Cimino G, Diverio D, Gentilini F, Mandelli F, Lo Coco F. Sustained molecular remission after low dose gemtuzumab-ozogamicin in elderly patients with advanced acute promyelocytic leukemia. Haematologica. 2007;92:1273–1274. doi: 10.3324/haematol.11329. [DOI] [PubMed] [Google Scholar]
- 134.Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, Guthridge MA, Thomas D, Barry EF, Boyd A, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 135.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29:591–599. doi: 10.1200/JCO.2010.31.0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Frankel A, Liu JS, Rizzieri D, Hogge D. Phase I clinical study of diphtheria toxin-interleukin 3 fusion protein in patients with acute myeloid leukemia and myelodysplasia. Leuk Lymphoma. 2008;49:543–553. doi: 10.1080/10428190701799035. [DOI] [PubMed] [Google Scholar]
- 137.Al-Hussaini M, Rettig MP, Ritchey JK, Karpova D, Uy GL, Eissenberg LG, Gao F, Eades WC, Bonvini E, Chichili GR, et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood. 2016;127:122–131. doi: 10.1182/blood-2014-05-575704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhou J, Chng WJ. Identification and targeting leukemia stem cells: The path to the cure for acute myeloid leukemia. World J Stem Cells. 2014;6:473–484. doi: 10.4252/wjsc.v6.i4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gadhoum Z, Delaunay J, Maquarre E, Durand L, Lancereaux V, Qi J, Robert-Lezenes J, Chomienne C, Smadja-Joffe F. The effect of anti-CD44 monoclonal antibodies on differentiation and proliferation of human acute myeloid leukemia cells. Leuk Lymphoma. 2004;45:1501–1510. doi: 10.1080/1042819042000206687. [DOI] [PubMed] [Google Scholar]
- 140.Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, Weissman IL. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, Willingham S, Howard M, Prohaska S, Volkmer J, et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS One. 2015;10:e0137345. doi: 10.1371/journal.pone.0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shen M, Schmitt S, Buac D, Dou QP. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin Ther Targets. 2013;17:1091–1108. doi: 10.1517/14728222.2013.815728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Attar EC, De Angelo DJ, Supko JG, D’Amato F, Zahrieh D, Sirulnik A, Wadleigh M, Ballen KK, McAfee S, Miller KB, et al. Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clin Cancer Res. 2008;14:1446–1454. doi: 10.1158/1078-0432.CCR-07-4626. [DOI] [PubMed] [Google Scholar]
- 144.Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, Kolitz JE, Powell BL, Voorhees P, Wang ES, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol. 2013;31:923–929. doi: 10.1200/JCO.2012.45.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Blum W, Schwind S, Tarighat SS, Geyer S, Eisfeld AK, Whitman S, Walker A, Klisovic R, Byrd JC, Santhanam R, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012;119:6025–6031. doi: 10.1182/blood-2012-03-413898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lancet JE, Duong VH, Winton EF, Stuart RK, Burton M, Zhang S, Cubitt C, Blaskovich MA, Wright JJ, Sebti S, et al. A phase I clinical-pharmacodynamic study of the farnesyltransferase inhibitor tipifarnib in combination with the proteasome inhibitor bortezomib in advanced acute leukemias. Clin Cancer Res. 2011;17:1140–1146. doi: 10.1158/1078-0432.CCR-10-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Walker AR, Klisovic RB, Garzon R, Schaaf LJ, Humphries K, Devine SM, Byrd JC, Grever MR, Marcucci G, Blum W. Phase I study of azacitidine and bortezomib in adults with relapsed or refractory acute myeloid leukemia. Leuk Lymphoma. 2014;55:1304–1308. doi: 10.3109/10428194.2013.833333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, Becker MW, Bennett JM, Sullivan E, Lachowicz JL, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. 2007;110:4427–4435. doi: 10.1182/blood-2007-05-090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hassane DC, Sen S, Minhajuddin M, Rossi RM, Corbett CA, Balys M, Wei L, Crooks PA, Guzman ML, Jordan CT. Chemical genomic screening reveals synergism between parthenolide and inhibitors of the PI-3 kinase and mTOR pathways. Blood. 2010;116:5983–5990. doi: 10.1182/blood-2010-04-278044. [DOI] [PubMed] [Google Scholar]
- 150.Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood. 2007;109:2727–2735. doi: 10.1182/blood-2006-10-050807. [DOI] [PubMed] [Google Scholar]
- 151.Page S, Fischer C, Baumgartner B, Haas M, Kreusel U, Loidl G, Hayn M, Ziegler-Heitbrock HW, Neumeier D, Brand K. 4-Hydroxynonenal prevents NF-kappaB activation and tumor necrosis factor expression by inhibiting IkappaB phosphorylation and subsequent proteolysis. J Biol Chem. 1999;274:11611–11618. doi: 10.1074/jbc.274.17.11611. [DOI] [PubMed] [Google Scholar]
- 152.Siow RC, Ishii T, Mann GE. Modulation of antioxidant gene expression by 4-hydroxynonenal: atheroprotective role of the Nrf2/ARE transcription pathway. Redox Rep. 2007;12:11–15. doi: 10.1179/135100007X162167. [DOI] [PubMed] [Google Scholar]
- 153.Allegretti M, Ricciardi MR, Licchetta R, Mirabilii S, Orecchioni S, Reggiani F, Talarico G, Foà R, Bertolini F, Amadori S, et al. The pan-class I phosphatidyl-inositol-3 kinase inhibitor NVP-BKM120 demonstrates anti-leukemic activity in acute myeloid leukemia. Sci Rep. 2015;5:18137. doi: 10.1038/srep18137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lu JW, Lin YM, Lai YL, Chen CY, Hu CY, Tien HF, Ou DL, Lin LI. MK-2206 induces apoptosis of AML cells and enhances the cytotoxicity of cytarabine. Med Oncol. 2015;32:206. doi: 10.1007/s12032-015-0650-7. [DOI] [PubMed] [Google Scholar]
- 156.Konopleva MY, Walter RB, Faderl SH, Jabbour EJ, Zeng Z, Borthakur G, Huang X, Kadia TM, Ruvolo PP, Feliu JB, et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin Cancer Res. 2014;20:2226–2235. doi: 10.1158/1078-0432.CCR-13-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Krawczyk J, Keane N, Swords R, O’Dwyer M, Freeman CL, Giles FJ. Perifosine--a new option in treatment of acute myeloid leukemia? Expert Opin Investig Drugs. 2013;22:1315–1327. doi: 10.1517/13543784.2013.826648. [DOI] [PubMed] [Google Scholar]
- 158.Altman JK, Sassano A, Platanias LC. Targeting mTOR for the treatment of AML. New agents and new directions. Oncotarget. 2011;2:510–517. doi: 10.18632/oncotarget.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Moore J, Seiter K, Kolitz J, Stock W, Giles F, Kalaycio M, Zenk D, Marcucci G. A Phase II study of Bcl-2 antisense (oblimersen sodium) combined with gemtuzumab ozogamicin in older patients with acute myeloid leukemia in first relapse. Leuk Res. 2006;30:777–783. doi: 10.1016/j.leukres.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 160.Marcucci G, Moser B, Blum W, Stock W, Wetzler M, Kolitz JE, Thakuri M, Carter T, Stuart RK, Larson RA. A phase III randomized trial of intensive induction and consolidation chemotherapy { /-} oblimersen, a pro-apoptotic Bcl-2 antisense oligonucleotide in untreated acute myeloid leukemia patients >60 years old. J Clin Oncol. 2007;25(June 20 Supplement):7012. [Google Scholar]
- 161.Rahmani M, Aust MM, Attkisson E, Williams DC, Ferreira-Gonzalez A, Grant S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood. 2012;119:6089–6098. doi: 10.1182/blood-2011-09-378141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xie C, Edwards H, Caldwell JT, Wang G, Taub JW, Ge Y. Obatoclax potentiates the cytotoxic effect of cytarabine on acute myeloid leukemia cells by enhancing DNA damage. Mol Oncol. 2015;9:409–421. doi: 10.1016/j.molonc.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schimmer AD, Raza A, Carter TH, Claxton D, Erba H, DeAngelo DJ, Tallman MS, Goard C, Borthakur G. A multicenter phase I/II study of obatoclax mesylate administered as a 3- or 24-hour infusion in older patients with previously untreated acute myeloid leukemia. PLoS One. 2014;9:e108694. doi: 10.1371/journal.pone.0108694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Adams GB, Martin RP, Alley IR, Chabner KT, Cohen KS, Calvi LM, Kronenberg HM, Scadden DT. Therapeutic targeting of a stem cell niche. Nat Biotechnol. 2007;25:238–243. doi: 10.1038/nbt1281. [DOI] [PubMed] [Google Scholar]
- 165.Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28:275–280. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Löwenberg B, van Putten W, Theobald M, Gmür J, Verdonck L, Sonneveld P, Fey M, Schouten H, de Greef G, Ferrant A, et al. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N Engl J Med. 2003;349:743–752. doi: 10.1056/NEJMoa025406. [DOI] [PubMed] [Google Scholar]
- 167.Estey EH, Thall PF, Pierce S, Cortes J, Beran M, Kantarjian H, Keating MJ, Andreeff M, Freireich E. Randomized phase II study of fludarabine + cytosine arabinoside + idarubicin +/- all-trans retinoic acid +/- granulocyte colony-stimulating factor in poor prognosis newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Blood. 1999;93:2478–2484. [PubMed] [Google Scholar]
- 168.Milligan DW, Wheatley K, Littlewood T, Craig JI, Burnett AK. Fludarabine and cytosine are less effective than standard ADE chemotherapy in high-risk acute myeloid leukemia, and addition of G-CSF and ATRA are not beneficial: results of the MRC AML-HR randomized trial. Blood. 2006;107:4614–4622. doi: 10.1182/blood-2005-10-4202. [DOI] [PubMed] [Google Scholar]
- 169.Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, Shemtov N, Deutsch V, Naparstek E, Nagler A, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64:2817–2824. doi: 10.1158/0008-5472.CAN-03-3693. [DOI] [PubMed] [Google Scholar]
- 170.Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, Levis M, Rubin JB, Negrin RR, Estey EH, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–6224. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, Kulkarni S, Abboud CN, Cashen AF, Stockerl-Goldstein KE, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–3924. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C, Cao F, Niekro W, Kempe T, Henning KA, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19:357–366. doi: 10.1158/1078-0432.CCR-12-2333. [DOI] [PubMed] [Google Scholar]
- 173.Zohren F, Toutzaris D, Klärner V, Hartung HP, Kieseier B, Haas R. The monoclonal anti-VLA-4 antibody natalizumab mobilizes CD34+ hematopoietic progenitor cells in humans. Blood. 2008;111:3893–3895. doi: 10.1182/blood-2007-10-120329. [DOI] [PubMed] [Google Scholar]
- 174.Hsieh YT, Jiang E, Pham J, Kim HN, Abdel-Azim H, Khazal S, Bug G, Spohn G, Bonig H, Kim YM. VLA4 Blockade In Acute Myeloid Leukemia. Blood. 2013;122:3944. [Google Scholar]
- 175.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, Jordan CT. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–4169. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Liesveld JL, Rosell KE, Lu C, Bechelli J, Phillips G, Lancet JE, Abboud CN. Acute myelogenous leukemia--microenvironment interactions: role of endothelial cells and proteasome inhibition. Hematology. 2005;10:483–494. doi: 10.1080/10245330500233452. [DOI] [PubMed] [Google Scholar]
- 178.Mohty M, Duarte RF, Croockewit S, Hübel K, Kvalheim G, Russell N. The role of plerixafor in optimizing peripheral blood stem cell mobilization for autologous stem cell transplantation. Leukemia. 2011;25:1–6. doi: 10.1038/leu.2010.224. [DOI] [PubMed] [Google Scholar]
- 179.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]