

Abstract
Warfarin is the anticoagulant of choice for venous thromboembolism (VTE) treatment, although its suppression of the endogenous clot‐dissolution complex APC:PS may ultimately lead to longer time‐to‐clot dissolution profiles, resulting in increased risk of re‐thrombosis. This detrimental effect might not occur during VTE treatment using other anticoagulants, such as rivaroxaban or enoxaparin, given their different mechanisms of action within the coagulation network. A quantitative systems pharmacology model was developed describing the coagulation network to monitor clotting factor levels under warfarin, enoxaparin, and rivaroxaban treatment. The model allowed for estimation of all factor rate constants and production rates. Predictions of individual coagulation factor time courses under steady‐state warfarin, enoxaparin, and rivaroxaban treatment reflected the suppression of protein C and protein S under warfarin compared to rivaroxaban and enoxaparin. The model may be used as a tool during clinical practice to predict effects of anticoagulants on individual clotting factor time courses and optimize antithrombotic therapy.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Warfarin represents the gold standard in VTE therapy despite its suppression of the endogenous clot‐dissolution components, possibly leading to an increased risk for re‐thrombosis.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We developed a QSP model of the human coagulation network to predict the effects of commonly used anticoagulants on the individual clotting factors to evaluate if warfarin should still be the treatment of choice for VTE.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This work shows that long‐term VKA treatment induced a 50% decrease in PC and PS levels as compared to rivaroxaban or enoxaparin, potentially leading to longer clot dissolution times and increased re‐thrombotic risk. Furthermore, it determined the time necessary for recovery of PC and PS after treatment switch from warfarin to another anticoagulant.
HOW THIS MIGHT CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS
☑ The QSP model can be used as a tool to simulate different clinical situations to predict the effects of routinely used or newly developed anticoagulants on the clotting factor levels and help optimize VTE treatment.
Venous thromboembolism (VTE) represents the third most common cardiovascular disease, with an annual incidence of about 100 people per 100,000 in the United States,1, 2 and is associated with substantial morbidity and mortality.3, 4 In managing patients with VTE, minimizing the time‐to‐clot dissolution is hypothesized to be crucial for reducing the risk of repeated VTEs. Thus, there is a critical and growing need to understand the pathophysiology of re‐thrombosis and the anticoagulatory effects of major anticoagulants, like warfarin, rivaroxaban, and enoxaparin.
The initial standard‐of‐care therapy of VTE comprises either low‐molecular‐weight heparin, such as enoxaparin, unfractionated heparin, or fondaparinux, followed by a combination of low‐molecular‐weight heparin/unfractionated heparin/fondaparinux and warfarin for at least 5 days until the international normalized ratio is larger than two for two consecutive days. Thereafter, patients remain only on vitamin K antagonist (VKA) treatment, such as warfarin or phenprocoumon.5, 6
Warfarin constitutes the most commonly prescribed anticoagulant for adults in North America.7 Through inhibition of the vitamin K epoxide reductase in the vitamin K (VK) cycle, the VKAs inhibit the reduction of VK 2,3‐epoxide to VK and subsequently slow down the production of the coagulation factors FII, FVII, FIX, and FX, as well as of the endogenous clot resolution components protein C (PC) and protein S (PS),8, 9, 10, 11, 12 which are necessary for the formation of the APC:PS complex responsible for the activation of clot‐dissolution. PS not only has an anticoagulation activity through the PC pathway, but it also directly stimulates the tissue factor pathway inhibitor.13, 14 Therefore, a decrease in PS levels causes an additional distortion of the balance between procoagulant and anticoagulant factors, leading to a higher risk of thrombosis.13, 15, 16
Enoxaparin is approved by the US Food and Drug Administration for the use in preoperative and postoperative thrombosis prophylaxis and primary VTE prophylaxis in nonsurgery patients. Its main mechanism of action is an inhibitory effect through binding to antithrombin, an endogenous inhibitor within the coagulation cascade, and potentiating the action of antithrombin on thrombin (FII) and FXa inhibition. Thus, the final stage within the coagulation cascade, the lysis of fibrinogen into fibrin catalyzed by thrombin, cannot proceed normally, which results in an increased anticoagulation effect.17, 18 With regard to the endogenous clot‐dissolution components, enoxaparin shows no suppressive effect on either PC or PS, which has been seen during clinical practice, in which patients showed about 50% higher levels in both factors under enoxaparin treatment as compared to VKA treatment after switching to enoxaparin therapy (Table 1 a).
Table 1.
Clinical observations from therapeutic drug monitoring of PS, PC, and plasminogen for selected patients on (a) steady‐state VKA (warfarin and phenprocoumon) compared to rivaroxaban, and (b) steady‐state VKA compared to enoxaparin therapy
| a | |||
|---|---|---|---|
| Patient # | Laboratory values | Warfarin treatment | Enoxaparin treatment |
| 1 | Protein S, % | 43 | 128 |
| Protein C, % | 59 | 100 | |
| Plasminogen, % | 98 | 119 | |
| 2 | Protein S, % | 59 | 117 |
| Protein C, % | 41 | 112 | |
| Plasminogen, % | 113 | 148 | |
| 3 | Protein S, % | 52 | 102 |
| Protein C, % | 54 | 86 | |
| Plasminogen, % | 127 | 109 | |
| 4 | Protein S, % | 52 | 111 |
| Protein C, % | 63 | 100 | |
| Plasminogen, % | 120 | 124 | |
| 5 | Protein S, % | 66 | 139 |
| Protein C, % | 65 | 96 | |
| Plasminogen, % | 110 | 106 |
| b | |||
|---|---|---|---|
| Patient # | Laboratory values | Warfarin treatment | Rivaroxaban treatment |
| 1 | Protein S, % | 61 | 120 |
| Protein C, % | 59 | 112 | |
| Plasminogen, % | 96 | 101 | |
| 2 | Protein S, % | 49 | 149 |
| Protein C, % | 61 | 137 | |
| Plasminogen, % | 114 | 120 | |
| 3 | Protein S, % | 47 | 115 |
| Protein C, % | 58 | 105 | |
| Plasminogen, % | 97 | 109 | |
| 4 | Protein S, % | 62 | 103 |
| Protein C, % | 58 | 107 | |
| Plasminogen, % | – | – | |
| 5 | Protein S, % | 54 | 85 |
| Protein C, % | 58 | 81 | |
| Plasminogen, % | – | – |
The effect of the direct oral anticoagulant rivaroxaban is independent of VK and antithrombin, and therefore differs from the aforementioned anticoagulants. It solely and directly inhibits FXa, thereby diminishing the thrombin‐mediated activation of coagulation and platelets. Due to this mechanism of action, rivaroxaban shows no influence on PC and PS,19, 20 which was also observed during clinical practice: patients had about 50% higher levels of both PC and PS after being switched to rivaroxaban as compared to prior VKA treatment (Table 1 b). Additionally, rivaroxaban is considered safe and effective compared to warfarin with low rates of thromboembolic and bleeding complications.21 Furthermore, a meta‐analysis of six randomized clinical trials has shown that direct FXa inhibitors are more effective for the prevention of VTE after total knee replacement as compared with enoxaparin, decreasing the risk of deep vein thrombosis events by 40%, without increasing a major bleeding risk.22
Considering all this, current clinical guidelines for VTE may not offer the most effective treatment paradigm for patients who may then be at higher risk for conversion to symptomatic VTE. The purpose of this analysis was to develop a quantitative systems pharmacology (QSP) model of the human coagulation network to assess the benefits and risks of commonly used anticoagulants in a critical disease.
QSP is an emerging discipline focusing on the identification and validation of drug targets and understanding existing therapeutics as well as discovering new ones. It allows probing differences in the molecular mechanisms of diseases and drug actions in individual patients, and is thought to be the “personalized medicine of the future.”23, 24 The developed QSP model aims to help provide molecular level insights into the coagulation network, which will contribute to understanding the effects of different treatments on the individual coagulation factor levels. Additionally, the presented QSP model might be utilized to simulate time‐to‐clot dissolution profiles and ultimately facilitate in the optimization of current guidelines for VTE treatment.
MATERIALS AND METHODS
Patient data
The data consist of routine clinical data from 479 white adults collected during therapeutic drug monitoring (TDM) at the University Hospital of Schleswig‐Holstein, Germany. All patients are monitored on steady‐state warfarin or phenprocoumon, enoxaparin, and rivaroxaban treatment for prophylactic VTE treatment. In Germany, most adult patients receive phenprocoumon when VKA treatment is indicated; however, if patients were at heightened risk of bleeding, warfarin was preferred because of its shorter half‐life. The data used during model development contained individual clotting factor level concentrations of 112 patients on warfarin (n = 7) or phenprocoumon (n = 105) treatment (concentrations available for plasminogen, PC, PS), 13 patients on enoxaparin treatment (concentrations available for plasminogen, PC, PS), and 25 patients on rivaroxaban treatment (concentrations available for FII, FV, FVII, FIX, FX, FXI, FXI, FXII, FXIII, PC, and PS). Furthermore, observed factor level concentrations of PC and PS were available for 5 patients who were switched from warfarin to rivaroxaban treatment, as well as 15 subjects who were switched from enoxaparin to warfarin and/or back to enoxaparin. All patient characteristics are presented in Table 2 a and b. For the patients treated with phenprocoumon, the respective dosing amount was converted into the corresponding warfarin dosing (3 mg phenprocoumon equals to 5 mg warfarin) to facilitate the use in the model. This procedure is valid due to the identical mechanism of action of these two drugs and their comparable pharmacokinetics.25 As all patients were under steady‐state conditions, the difference between the two drugs regarding their half‐life can be disregarded. Henceforth, all patients treated with either warfarin or phenprocoumon in our data are referred to as being under warfarin treatment. The present study was performed in accordance with the ethical standards laid down in the updated relevant version of the Declaration of Helsinki, and informed signed consent was obtained from each study participant or the patient's parent or guardian.
Table 2.
Patient demographics: (a) model development dataset, (b) treatment switch dataset, (c) external model evaluation dataset
| a | |||
|---|---|---|---|
| Values [range] | |||
| Parameter | VKA | Rivaroxaban | Enoxaparin |
| No. of patients | 112 (61 ♂, 51 ♀) (warfarin: n = 7; phenprocoumon: n = 105) | 25 (15 ♂, 10 ♀) | 13 (2 ♂, 11 ♀) |
| Age, y | 39.7 [17–75] | 38.6 [17–66] | 36.3 [17–71] |
| Height, cm | 173.1 [152–198] | 176.1 [156–194] | 172.1 [164–185] |
| Weight, kg | 81.3 [48–170] | 86.6 [52–136] | 79.8 [52–97.5] |
| Dose, mg | 5 | 20 | 60 |
| b | ||
|---|---|---|
| Values [range] | ||
| Parameter |
Switch (VKA → rivaroxaban) |
Switch (enoxaparin → VKA → enoxaparin) |
| No. of patients | 5 (5 ♂) | 15 (4 ♂, 11 ♀) (enoxaparin → VKA: n = 6, VKA → enoxaparin: n = 12) |
| Age, y | 37.7 [19–57] | 29.8 [17–71] |
| Height, cm | 175 [165–182] | 169.1 [156–185] |
| Weight, kg | 81.8 [79–87] | 74.6 [49–97.5] |
| Dose, mg | 5 → 20 | 60 → 5 → 60 |
| c | |||
|---|---|---|---|
| Values [range] | |||
| Parameter | VKA | Rivaroxaban | Enoxaparin |
| No. of patients | 93 (49 ♂, 44 ♀) (warfarin: n = 19; phenprocoumon: n = 74) | 198 (101 ♂, 97 ♀) | 18 (7 ♂, 11 ♀) |
| Age, y | 39.7 [16–78] | 49.3 [17–88] | 32.1 [18–67] |
| Height, cm | 174.4 [150–201] | 173.1 [155–178] | 174.4 [155–198] |
| Weight, kg | 82.3 [45–170] | 85.8 [44–152] | 85.5 [52–118] |
| Dose, mg | 2.5 (n = 67), 7.5 (n = 26) | 15 (n = 31), 20 (n = 162), 30 (n = 5) | 40 (n = 2), 80 (n = 7), 120 (n = 9) |
VKA, vitamin K antagonist.
Quantitative systems pharmacology model
A QSP model of the human coagulation network was developed in Matlab (MathWorks, version 2014a), using the coagulation network model developed by Wajima et al.26 as a starting point.27 Each compartment represents an active or inactive clotting factor with specific input and output functions. Changes in factor concentrations were described by a series of ordinary differential equations (ODEs). The general form of the equations used for the enzyme‐catalyzed stimulation was based on a turnover model and mass‐balance principles via Michaelis‐Menten kinetics. The rate of transformation of a substance y by another factor C is described by Eq. 1:
| (1) |
with V max being the maximum possible rate at which a reaction can occur and Km being the concentration of the catalyzing factor for which half of the V max rate is achieved.
Complex formations, such as formation of the Xa:Va complex from FXa and FVa, were represented by a stoichiometric equation in which the components were assumed to combine in a 1:1 ratio and the complex formation would result in removal of the participating factors from their respective compartments. Separate dosing compartments for the drugs used in the model (warfarin, rivaroxaban, and enoxaparin) were added to the model according to their specific mechanism of action.
The warfarin and enoxaparin effects were added into the model as in the study by Wajima et al.26 For the rivaroxaban effect, the set of ODEs was extended by equations describing dosing (Eq. 2) and concentration compartments (Eq. 3) for rivaroxaban, as well as the direct inhibitory effects of rivaroxaban on unbound FXa (Eq. 4) and the Xa:Va‐complex (Eq. 5):
| (2) |
| (3) |
| (4) |
| (5) |
with ri indicating the transformation rate, as described in Eq. (1), and dj describing the degradation rate of the respective coagulation factor, Imax being the maximum inhibitory effect of the drug, IC50 the concentration necessary to achieve 50% of the maximum possible inhibitory effect, and C the substance concentration.
All initial parameter values in the model were adopted from Wajima et al.26 Given that our patient observations consisted of steady‐state treatment data, standard physiological conditions of all initial factor concentrations were used to simulate steady‐state scenarios for all patients. Information for the additional pathways and pharmacokinetic parameters for rivaroxaban were adopted from literature.28, 29
Parameter estimation
All available data, as described in Table 2 a (rivaroxaban: 20 mg; VKA: 5 mg; and enoxaparin: 60 mg), were used for model development. All factor rate constants (Vmax and Km) and production rates (pi) were estimated in Matlab using the function “fmincon.” The remaining parameters (initial factor concentrations, degradation constants, and pharmacokinetic parameters) were fixed to values obtained from literature.26, 28 The difference between the observed and predicted data was minimized using the least square method. Multiple initial estimates were used (exploiting the Matlab function “multistart”) to ensure the identification of a global minimum. A lower boundary of zero was set for each parameter. Estimation of interindividual variability (IIV) in Matlab was not possible due to the sparseness of the available data and the large number of parameters that needed to be estimated.
Model simulations/predictions
In order to calculate the concentration of each clotting factor over time, the ODEs were numerically solved using a stiff ODE solver in Matlab, “ode15s.” The system was initialized under steady‐state nonclotting conditions, with the assumption that all activated factors have an initial concentration of zero. To predict long‐term anticoagulant therapy, routine dosing was incorporated for warfarin, enoxaparin, and rivaroxaban, with dose amount and interval set to the dosing regimens used in the parameter estimation. To assess the importance of IIV in model parameters, Monte Carlo (MC) simulations were performed in Matlab by adding a 20% variability on the estimated production rates (FII, FV, FVII, FIX, FX, FXI, FXII, FXIII, PC, and PS), thereby accounting for IIV. For each treatment option, 500 subjects were simulated using the estimated parameter values and the median as well as the 5th and 95th percentile of the concentrations of the clotting factors calculated; residual unexplained variability was not added. Additional MC simulations were performed using MatVPC,30 a new computational tool that provides simulation and evaluation of QSP models. Similar to the simulations performed for generating visual predictive checks, MatVPC30 was used to simulate 500 observation‐specific datasets to automatically plot the 90% confidence intervals of the 5th, 50th, and 95th percentiles of the predictions.
Comparison of observations and predictions
The observed factor levels were superimposed onto the model predictions of the clotting factor time courses. The simulated dosing regimens for rivaroxaban included once‐daily oral dosing of 20 mg over 1.5 years. For warfarin, an oral dose of 5 mg per day over 2 years was predicted. The predictions for enoxaparin were performed for a once daily s.c. dosing of 60 mg for 14 days. To describe the therapy switch from warfarin to rivaroxaban in silico, predictions started out with a once‐daily oral dose of 5 mg warfarin for 52 days, followed by a 20 mg dose of rivaroxaban (oral, once‐daily) for 20 days. For the treatment switch from enoxaparin to warfarin and back to enoxaparin, the predictions were initiated with a 10 day enoxaparin dosing (60 mg s.c., once‐daily), followed by a once‐daily oral dose of 5 mg warfarin for 72 days. Subsequently, another dosing regimen of 60 mg enoxaparin was given for 21 days. A commonly used variability of 20% was added to the estimated production rates to account for IIV. The predictions were overlaid with the respective observations.
Sobol sensitivity analysis
Sobol sensitivity analysis, a global sensitivity analysis, was performed to identify the parameters that have the greatest influence on the activation of clot‐dissolution under warfarin, enoxaparin, and rivaroxaban treatment. The procedure was performed as described previously.31 In brief, 96 parameters (all rate constants and production rates) were varied simultaneously over a range of ±20% of their estimated value, generating a total number of 1,940,000 parameter sets that were used to simulate the model output. Based on the simulation results, total‐order sensitivity indices were calculated and used to evaluate the overall contribution of each parameter on the desired output. Sobol indices with values greater than 0.05 are generally considered to be statistically relevant.
External model evaluation
An external dataset, as described in Table 2 c, was leveraged for external model evaluation after development of the QSP model was concluded. External model evaluation was performed by simulating clotting factor concentrations for dosing regimens and patients other than the ones used during model development. The performed simulations included oral once‐daily dosing regimens of 15 mg and 30 mg for rivaroxaban, once‐daily s.c. dosing of 40 mg, 80 mg, and 120 mg for enoxaparin, as well as 2.5 mg and 7.5 mg (oral, once/day) for warfarin. The simulations were carried out using MatVPC,30 as described above, and were overlaid with the observations for model evaluation. Furthermore, the coagulation factor rate constants and production rates were re‐estimated using the 20 mg rivaroxaban data of the evaluation dataset, as described above, and the observed patient data were superimposed onto the model simulations of the coagulation factor time courses using the new estimates. MC simulations for these dosing regimens were performed using MatVPC.30
RESULTS
Model development
A QSP model of the human coagulation network was developed using the network model by Wajima et al.26 as a starting point. The final QSP model consists of 56 compartments, including the extrinsic and intrinsic coagulation pathways, the VK cycle, as well as components necessary to simulate drug therapy profiles of steady‐state VKA (warfarin/phenprocoumon), enoxaparin, and rivaroxaban treatment for different dose levels based on the available TDM data, as described in Table 2 a. The balance equations for each component were described by a series of ODEs. A scheme of the full QSP model is shown in Figure 2.
Figure 2.
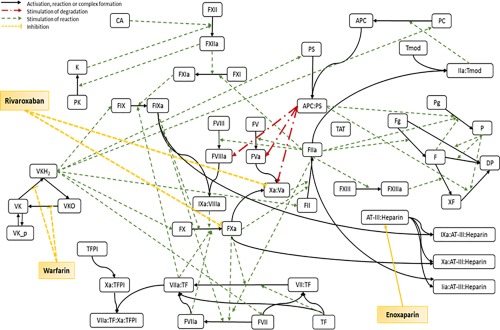
Scheme of the human coagulation network model. Solid black lines represent activation processes, complex formation, reduction, or oxidation. Broken red lines represent stimulation of degradation. Broken green lines represent stimulation of reaction. Broken yellow lines represent inhibition of reaction. APC, activated protein C; CA, activator of contact system; D, degradation product; F, fibrin; Fg, fibrinogen; FII, prothrombin; FIIa, thrombin; K, kallikrein; P, plasmin; PC, protein C; PS, protein S; Pg, plasminogen; TAT, thrombin‐antithrombin‐complex; TF, tissue factor; TFPI, tissue factor pathway inhibitor; Tmod, thrombomodulin; VK, vitamin K; VKH2, vitamin K hydroquinone; VKO, vitamin K epoxide; XF, cross‐linked fibrin.
Parameter estimation
The rate constants based on Michaelis‐Menten kinetics (Vmax and Km) and production rates of the coagulation factors were estimated based on the available patient clotting factor concentrations under steady‐state VKA, enoxaparin, and rivaroxaban treatment by global optimization. The final parameter estimates are shown in Supplementary Table S1. The estimated parameter values were used for all following model simulations.
Prediction of steady‐state effects of rivaroxaban on the individual clotting factor levels
The developed QSP model allowed for predictions of steady‐state effects of rivaroxaban administration on the individual clotting factor concentrations. The model predicted that rivaroxaban treatment has no effect on the inactivated coagulation factor levels FII, FV, FVII, FIX, FX, FXI, FXII, FXIII, PC, and PS, which concurs with the observed data obtained from the TDM patients (Figure 3). IIV was accounted for by adding a variability of 20% to the estimated production rates of FII, FV, FVII, FIX, FX, FXI, FXII, FXIII, PC, and PS, which captured the variability in the observed factor levels well. The respective MC simulations based on the final estimates using MatVPC30 depicting additional confidence intervals for the 5th and 95th percentiles can be found in Supplementary Figure S1.
Figure 3.
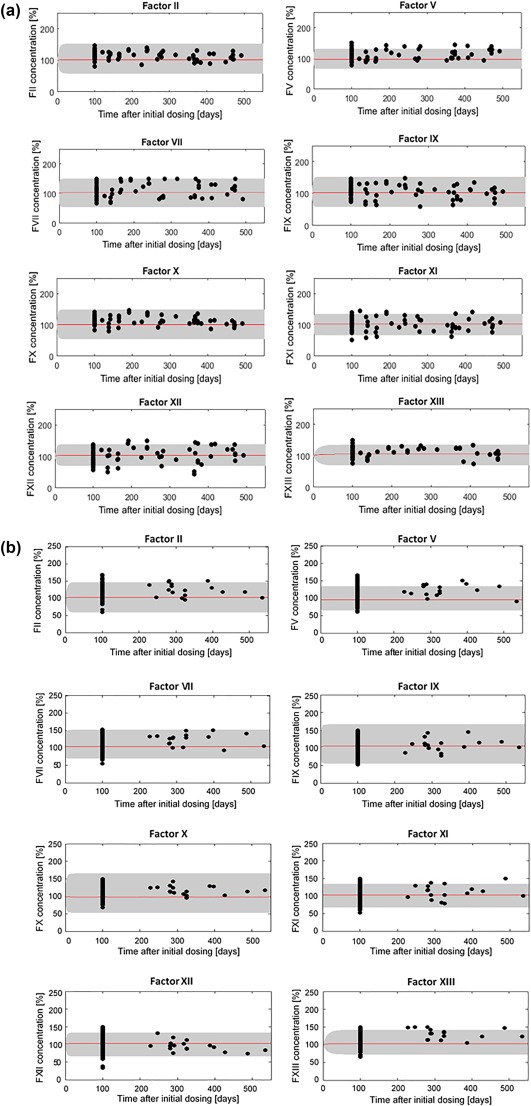
Monte Carlo simulations using Matlab of respective individual clotting factor level time courses based on the developed model under steady‐state rivaroxaban treatment (20 mg once‐daily; red line: median, lower, and upper bounds of grey shaded area: 5th and 95th percentiles, respectively). Simulations are overlaid with the observed individual patient coagulation factor level concentrations (a) developmental dataset, (b) evaluation dataset (observations: black dots). The gray‐shaded area stems from the 20% variability that was added to the estimated production rates of FII, FV, FVII, FIX, FX, FXI, FXII, FXIII, protein C (PC), and protein S (PS) to account for interindividual variability. Factor concentrations are expressed as percent of the initial concentration obtained from literature assumed to represent physiological values.
Effect of steady‐state rivaroxaban administration on PC and PS concentrations
The model was used to predict the effects of long‐term rivaroxaban administration on the levels of PC and PS. The predictions of steady‐state rivaroxaban treatment as well as the observations of the respective patients showed that neither PC nor PS levels changed compared to physiological concentrations of these factors obtained from literature (Figure 4 a).
Figure 4.
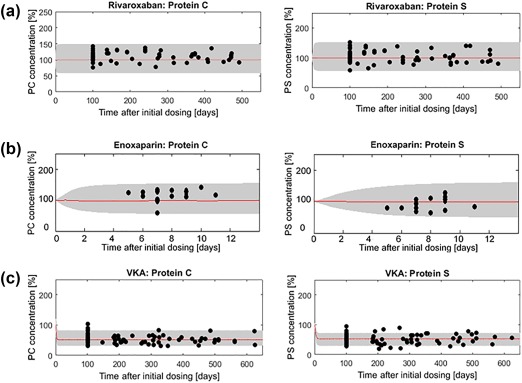
Monte Carlo simulations of protein C (PC) and protein S (PS) under steady‐state (a) rivaroxaban (20 mg), (b) enoxaparin (60 mg), and (c) vitamin K antagonist (VKA; warfarin/phenprocoumon; 5 mg treatment; red line indicates the median, lower, and upper bounds of the gray‐shaded area: 5th and 95th percentiles, respectively). Simulations are overlaid with the observed individual patient coagulation factor level concentrations (black dots). The gray‐shaded area stems from the 20% variability that was added to the estimated production rates of FII, FV, FVII, FIX, FX, FXI, FXII, FXIII, PC, and PS to account for interindividual variability. Factor concentrations are expressed as percent of the initial concentration obtained from literature assumed to represent physiological values.
Effect of steady‐state administration of enoxaparin on PC and PS concentrations
Similar to the predictions of steady‐state rivaroxaban treatment, the model was utilized to predict the effects of steady‐state enoxaparin administration on PC and PS. Both the predictions and the observations of the respective patients demonstrate that the concentrations of PC and PS remain at levels seen under physiological conditions during enoxaparin administration (Figure 4 b). The respective MC simulations utilizing MatVPC30 are depicted in Supplementary Figure S3.
Prediction of PC and PS concentrations under steady‐state warfarin treatment
The model was used in a similar fashion as for the prediction of steady‐state rivaroxaban and enoxaparin treatment to predict the effects of VKA (warfarin/phenprocoumon) on the endogenous clot‐dissolution components. In contrast to rivaroxaban and enoxaparin, the levels of PC and PS were decreased by about 50% under VKA treatment compared to physiological concentrations known from literature, which fits well with the patient observations (Figure 4 c). Additionally, when directly compared to the factor levels observed during rivaroxaban or enoxaparin administration, PC and PS concentrations were about 50% lower during steady‐state VKA treatment. For the respective MC simulations using MatVPC30 depicting additional confidence intervals for the 5th and 95th percentiles, see Supplementary Figure S4.
Effect of rivaroxaban administration following warfarin treatment on PC and PS levels
A treatment switch from warfarin to rivaroxaban was simulated based on the final predicted estimates to understand the time course of the concentration recovery of PS and PC. Warfarin treatment was terminated after 52 days and immediately followed by once‐daily rivaroxaban administration. The developed QSP model predicted that a therapy switch leads to a 50% increase of PC and PS concentrations within 9 and 11 days, respectively, after the start of rivaroxaban treatment (Figure 5 a), which concurs with the observed data of patients who were switched from steady‐state warfarin to rivaroxaban treatment.
Figure 5.
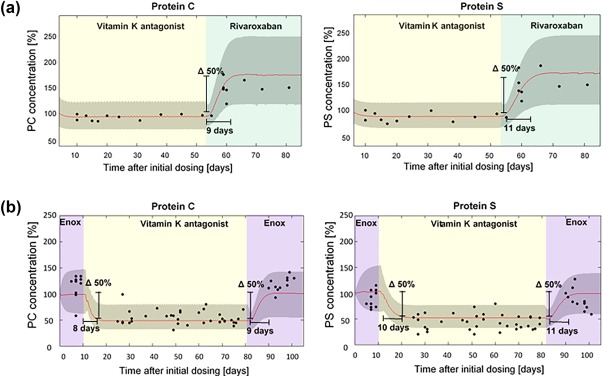
Monte Carlo simulations of protein C (PC) and protein S (PS) under therapy switch from (a) steady‐state vitamin K antagonist (VKA; warfarin/phenprocoumon; 5 mg p.o. once‐daily) to steady‐state rivaroxaban (20 mg p.o. once‐daily) treatment, and (b) steady‐state enoxaparin (60 mg s.c., once‐daily) to steady‐state VKA (5 mg p.o, once‐daily) to steady‐state enoxaparin (60 mg s.c., once‐daily; red line: median, lower, and upper bounds of the gray‐shaded area: 5th and 95th percentiles, respectively). Simulations are overlaid with the observed individual patient coagulation factor level concentrations (black dots). The gray‐shaded area stems from the 20% variability added to the estimated production rates of FII, FV, FVII, FIX, FX, FXI, FXII, FXIII, PC, and PS in order to account for interindividual variability. Factor concentrations are expressed as percent of the initial concentration obtained from literature assumed to represent physiological values.
Effect of warfarin treatment following enoxaparin on PC and PS levels
Therapy switch from enoxaparin to warfarin was predicted similarly to the switch from warfarin to rivaroxaban. After an initial phase of enoxaparin administration for 10 days, the patients were started on warfarin‐only treatment for 72 days. Subsequently, warfarin dosing was stopped and the patients were continued on enoxaparin for 21 days. The presented QSP model predicted a 50% decrease in PC and PS levels occurring within 8 and 10 days, respectively, after the start of warfarin treatment. When patients were switched back to enoxaparin, the QSP model predicted a period of 9 days for PC and 11 days for PS to recover by 50% to the levels assumed to be equivalent to physiological concentrations26, 32 (Figure 5 b).
Sobol sensitivity analysis
Global sensitivity analysis determined the production rate of VK to be the most influential parameter with regard to the activation of clot‐dissolution under VKA, enoxaparin, or rivaroxaban treatment (Figure 1). This result is in concordance with the physiology of the coagulation network: by inhibition of vitamin K epoxide reductase, the VKAs decrease VK concentrations necessary for the production of PC and PS, leading to less available concentrations of these proteins to form the APC:PS complex. This result does explain the decrease in PC and PS levels under steady‐state VKA treatment, which could not be observed under rivaroxaban or enoxaparin treatment.
Figure 1.
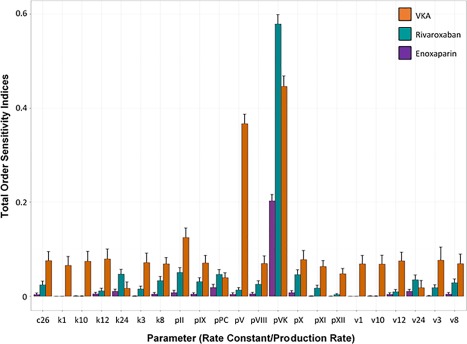
Total‐order sensitivity indices (bars) with their respective confidence intervals (error bars) of the Sobol sensitivity analysis in regard to activation of the clot‐dissolution complex APC:PS for rivaroxaban (green), enoxaparin (purple), and vitamin K antagonist (VKA; warfarin/phenprocoumon) treatment (orange). Ci, rate constant for complex formation; ki, Km; vi, Vmax; pj, production rate.
External model evaluation
An external model evaluation dataset, described in Table 2 c, was leveraged for external model evaluation. Parameter re‐estimation for the 20 mg rivaroxaban data of the evaluation dataset led to almost identical values for the factor rate constants and production rates as estimated for the development rivaroxaban dataset (Supplementary Table S1). MC simulations utilizing Matlab (Figure 3 b) and MatVPC30 (Supplementary Figure S3) using these estimates show that the QSP model captured the observed data well.
For the remaining dose regimens (rivaroxaban: 15 mg, 30 mg; enoxaparin: 40 mg, 80 mg, 120 mg; and warfarin: 2.5 mg, 7.5 mg), MC simulations were performed using MatVPC and overlaid with the respective patient data. The model was able to simulate the time courses of the clotting factor concentrations under VKA (warfarin/phenprocoumon), enoxaparin, and rivaroxaban treatment adequately well for all data that were not used during parameter estimation (Supplementary Figures S2, S4, and S6).
DISCUSSION
The developed QSP model of the human coagulation network includes the extrinsic and intrinsic pathways as well as subsystems for the VK cycle and the antithrombin‐III: heparin complex. The current model allows for the simulation of several individual clotting factor concentrations under VKA, enoxaparin, and rivaroxaban treatment. In contrast to other recently published systems pharmacology models of the coagulation network,33, 34 this work presents the first attempt to estimate parameters of the coagulation network using individual clotting factor concentrations from TDM data.
The model was successfully applied to adequately predict the time courses of individual clotting factor levels under steady‐state treatment of the extensively used anticoagulants warfarin/phenprocoumon, enoxaparin, and rivaroxaban. With regard to the endogenous clot‐dissolution components, PC and PS, the model was able to accurately describe the significant suppression of both factors under steady‐state VKA treatment, which did not occur during rivaroxaban or enoxaparin administration. Concordantly, predictions of the factor level time courses of PC and PS during therapy switch revealed that after switching from warfarin to rivaroxaban, the factor levels increased by 50% compared to their levels during warfarin administration, within 9 and 11 days, respectively, after the start of rivaroxaban treatment. Similarly, prediction of a treatment switch from enoxaparin to warfarin led to a 50% decrease in both PC and PS levels under warfarin administration compared with the levels during enoxaparin within 8 and 10 days, respectively, after the start of warfarin administration. Switching from warfarin back to enoxaparin was accompanied by a concentration increase of 50% for PC within 9 days and PS within 11 days after enoxaparin treatment was initiated. Overall, we did not detect any differences in the behavior of enoxaparin and rivaroxaban regarding their influence on the clot‐dissolution components.
These results, along with the fact that low levels of the endogenous clot‐dissolution components are thought to be connected to a higher probability of developing a blood clot,35, 36 and the hypothesis that due to its fibrinolytic activity37, 38 low levels of PC may cause slower clot‐dissolution, indicate that rivaroxaban and enoxaparin might be considered as valid alternatives to VKA for VTE treatment. However, further clinical studies are necessary to confirm these findings. Although all data used for this model stemmed from the same laboratory, the QSP model can be used for data collected from various laboratories, as coagulation factor levels are generally normalized against an internal reference sample to make values received from different sources comparable. Sobol sensitivity analysis demonstrated VK to be the most influential parameter for the QSP model with respect to the activation of the clot‐dissolution complex APC:PS. This finding is in agreement with the physiology of the coagulation system and provides an explanation why only VKA, as direct interferer of the VK cycle, but neither rivaroxaban nor enoxaparin displays the suppressive effect on the components of the endogenous clot dissolution cascade PC and PS.
The presented QSP model may be used as a tool to simulate different clinical situations to predict the effects of various anticoagulants on the individual clotting factors and, hence, help optimize antithrombotic therapy. The model can be adjusted to incorporate additional anticoagulants or compounds within the early stage of drug development to predict biomarker responses like prothrombin time, activated partial thromboplastin time, D‐dimer protein, PC, PS, or thrombin generation.
In conclusion, the model developed in this study is the first description of the in vivo coagulation network in humans with parameters estimated using TDM data from VKA, rivaroxaban, and enoxaparin therapy for VTE prophylaxis. The model accurately described the effects of the anticoagulants warfarin/phenprocoumon, enoxaparin, and rivaroxaban on the individual clotting factor levels. In addition, the model predicted the suppressive effect of VKA on the concentrations of PC and PS adequately well, which play a central role in the regulation of clot dissolution. The PC and PS concentrations after treatment switch from VKA to rivaroxaban or from enoxaparin to VKA and back were well‐described by the developed QSP model. The model estimated that a period of 9 (PC) and 11 (PS) days after switching to either rivaroxaban or enoxaparin treatment is needed in order for PC and PS levels to recover by 50% toward the physiological values of these endogenous clot‐dissolution components compared with levels under warfarin treatment. The model may be used as a tool during clinical practice to predict the effects of anticoagulant therapy on the individual clotting factor time courses and, therefore, help optimize antithrombotic therapy. Furthermore, the QSP model might be utilized in the future to serve as basis during drug development and regulatory guidance development as well as decision‐making on VTE treatment.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Author Contributions
S.H. wrote the manuscript. M.N.T. and U.N.‐G. designed the research. M.N.T., S.H., and K.B. performed the research. M.N.T., S.H., and U.N.‐G. analyzed the data. M.N.T., S.H., K.B., and L.J.L. contributed new reagents/analytical tools.
Conflict of Interest
The authors declared no conflict of interest.
References
- 1. Heit, J.A. Epidemiology of venous thromboembolism. Nat. Rev. Cardiol. 12, 464–474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldhaber, S.Z. Venous thromboembolism: epidemiology and magnitude of the problem. Best Pract. Res. Clin. Haematol. 25, 235–242 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Beckman, M.G. , Hooper, W.C. , Critchley, S.E. & Ortel, T.L. Venous thromboembolism: a public health concern. Am. J. Prev. Med. 38(4 suppl.), S495–S501 (2010). [DOI] [PubMed] [Google Scholar]
- 4. Naess, I.A. , Christiansen, S.C. , Romundstad, P. , Cannegieter, S.C. , Rosendaal, F.R. & Hammerstrøm, J . Incidence and mortality of venous thrombosis: a population‐based study. J. Thromb. Haemost. 5, 692–699 (2007). [DOI] [PubMed] [Google Scholar]
- 5. Wells, P.S. , Forgie, M.A. & Rodger, M.A. Treatment of venous thromboembolism. JAMA 311, 717–728 (2014). [DOI] [PubMed] [Google Scholar]
- 6. McRae, S.J. & Ginsberg, J.S. Initial treatment of venous thromboembolism. Circulation 110(24 suppl. 1), IV33 (2004). [DOI] [PubMed] [Google Scholar]
- 7. Garcia, D.A. & Spyropoulos, A.C. Update in the treatment of venous thromboembolism. Semin. Respir. Crit. Care Med. 29, 40–46 (2008). [DOI] [PubMed] [Google Scholar]
- 8. Esmon, C.T. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J. Biol. Chem. 264, 4743–4746 (1989). [PubMed] [Google Scholar]
- 9. O'Brien, A.E. , Tate, G.M. & Shiach, C. Evaluation of protein C and protein S levels during oral anticoagulant therapy. Clin. Lab. Haematol. 20, 245–252 (1998). [DOI] [PubMed] [Google Scholar]
- 10. Thompson, A.R. Factor IX antigen by radioimmunoassay. Abnormal factor IX protein in patients on warfarin therapy and with hemophilia B. J. Clin. Invest. 59, 900–910 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weiss, P. , Soff, G.A. , Halkin, H. & Seligsohn, U. Decline of proteins C and S and factors II, VII, IX and X during the initiation of warfarin therapy. Thromb. Res. 45, 783–790 (1987). [DOI] [PubMed] [Google Scholar]
- 12. Zivelin, A. , Rao, L.V. & Rapaport, S.I. Mechanism of the anticoagulant effect of warfarin as evaluated in rabbits by selective depression of individual: procoagulant vitamin K‐dependent clotting factors. J. Clin. Invest. 92, 2131–2140 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hackeng, T.M. , Seré, K.M. , Tans, G. & Rosing, J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc. Natl. Acad. Sci. USA 103, 3106–3111 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosing, J. , Maurissen, L.F. , Tchaikovski, S.N. , Tans, G. & Hackeng, T.M. Protein S is a cofactor for tissue factor pathway inhibitor. Thromb. Res. 122 suppl. 1, S60–S63 (2008). [DOI] [PubMed] [Google Scholar]
- 15. Haran, M.Z. , Lichman, I. , Berebbi, A. , Weinmann, E. & Rosenberg, N. Unbalanced protein S deficiency due to warfarin treatment as a possible cause for thrombosis. Br. J. Haematol. 139, 310–311 (2007). [DOI] [PubMed] [Google Scholar]
- 16. Binymin, K.A. , Nasher, M. & Patel, D. Warfarin‐induced deep vein thrombosis. Int. Med. Case Rep. J. 7, 123–125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Padilla, A. , Gray, E. , Pepper, D.S. & Barrowcliffe, T.W. Inhibition of thrombin generation by heparin and low molecular weight (LMW) heparins in the absence and presence of platelet factor 4 (PF4). Br. J. Haematol. 82, 406–413 (1992). [DOI] [PubMed] [Google Scholar]
- 18. Fareed, J. , Walenga, J.M. , Hoppensteadt, D. , Huan, X. & Racanelli, A. Comparative study on the in vitro and in vivo activities of seven low‐molecular‐weight heparins. Haemostasis 18 suppl. 3, 3–15 (1988). [DOI] [PubMed] [Google Scholar]
- 19. Mani, H. , Hesse, C. , Stratmann, G. & Lindhoff‐Last, E. Ex vivo effects of low‐dose rivaroxaban on specific coagulation assays and coagulation factor activities in patients under real life conditions. Thromb. Haemost. 109, 127–136 (2013). [DOI] [PubMed] [Google Scholar]
- 20. Martinelli, I. et al Anticoagulant treatment with rivaroxaban in severe protein S deficiency. Pediatrics 132, e1435–e1439 (2013). [DOI] [PubMed] [Google Scholar]
- 21. Coleman, C.M. et al Novel oral anticoagulants for DC cardioversion procedures: utilization and clinical outcomes compared with warfarin. Pacing Clin. Electrophysiol. 38, 731–737 (2015). [DOI] [PubMed] [Google Scholar]
- 22. Ma, G. , Zhang, R. , Wu, X. , Wang, D. & Ying, K. Direct factor Xa inhibitors (rivaroxaban and apixaban) versus enoxaparin for the prevention of venous thromboembolism after total knee replacement: a meta‐analysis of 6 randomized clinical trials. Thromb. Res. 135, 816–822 (2015). [DOI] [PubMed] [Google Scholar]
- 23. Sorger, P.K. et al Quantitative and systems pharmacology in the post‐genomic era: new approaches to discovering drugs and understanding therapeutic mechanisms. QSP White Paper. <https://www.nigms.nih.gov/Training/Documents/SystemsPharmaWPSorger2011.pdf> (2011). [Google Scholar]
- 24. Trame, M.N. , Biliouris, K. , Lesko, L.J. & Mettetal, J.T. Systems pharmacology to predict drug safety in drug development. Eur. J. Pharm. Sci.; e‐pub ahead of print 2016. [DOI] [PubMed] [Google Scholar]
- 25. Ufer, M. Comparative pharmacokinetics of vitamin K antagonists warfarin, phenprocoumon and acenocoumarol. Clin. Pharmacokinet. 44, 1227–1246 (2005). [DOI] [PubMed] [Google Scholar]
- 26. Wajima, T. , Isbister, G.K. & Duffull, S.B. A comprehensive model for the humoral coagulation network in humans. Clin. Pharmacol. Ther. 86, 290–298 (2009). [DOI] [PubMed] [Google Scholar]
- 27. Hartmann, S. , Biliouris, K. , Nowak‐Göttl, U. & Trame, M.N. Development of a systems pharmacology model to predict the effects of warfarin and rivaroxaban on the human coagulation network. Clin. Pharmacol. Ther. 99 (suppl. 1), S5–S107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mueck, W. , Lensing, A.W. , Agnelli, G. , Decousus, H. , Prandoni, P. & Misselwitz, F. Rivaroxaban: population pharmacokinetic analyses in patients treated for acute deep‐vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin. Pharmacokinet. 50, 675–686 (2011). [DOI] [PubMed] [Google Scholar]
- 29. Perzborn, E. , Roehrig, S. , Straub, A. , Kubitza, D. , Mueck, W. & Laux, V. Rivaroxaban: a new oral factor Xa inhibitor. Arterioscler. Thromb. Vasc. Biol. 30, 376–381 (2010). [DOI] [PubMed] [Google Scholar]
- 30. Biliouris, K. , Lavielle, M. & Trame, M.N. MatVPC: a user‐friendly MATLAB‐based tool for the simulation and evaluation of systems pharmacology models. CPT Pharmacometrics Syst. Pharmacol. 4, 547–557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang, X.‐Y. , Trame, M.N. , Lesko, L.J. & Schmidt, S. Sobol sensitivity analysis: a tool to guide the development and evaluation of systems pharmacology models. CPT Pharmacometrics Syst. Pharmacol. 4, 69–79 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tanos, P.P. , Isbister, G.K. , Lalloo, D.G. , Kirkpatrick, C.M. & Duffull, S.B. A model for venom‐induced consumptive coagulopathy in snake bite. Toxicon 52, 769–780 (2008). [DOI] [PubMed] [Google Scholar]
- 33. Nayak, S. et al Using a systems pharmacology model of the blood coagulation network to predict the effects of various therapies on biomarkers. CPT Pharmacometrics Syst. Pharmacol. 4, 396–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou, X. , Huntjens, D. & Gilissen, R. A systems pharmacology model for predicting anticoagulant effects of factor Xa inhibitors in healthy subjects: assessment of drug pharmacokinetic and binding kinetic properties. CPT Pharmacometrics Syst. Pharmacol. 4, 650–659 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soare, A.M. & Popa, C. Deficiencies of proteins C, S and antithrombin and activated protein C resistance–their involvement in the occurrence of arterial thromboses. J. Med. Life 3, 412–415 (2010). [PMC free article] [PubMed] [Google Scholar]
- 36. Folsom, A.R. , Aleksic, N. , Wang, L. , Cushman, M. , Wu, K.K. & White, R.H. Protein C, antithrombin, and venous thromboembolism incidence: a prospective population‐based study. Arterioscler. Thromb. Vasc. Biol. 22, 1018–1022 (2002). [DOI] [PubMed] [Google Scholar]
- 37. Roemisch, J. , Diehl, K.H. , Reiner, G. & Paques, E.P. Activated protein C: antithrombotic properties and influence on fibrinolysis in an animal model. Fibrinolysis 5, 191–196 (1991). [Google Scholar]
- 38. Sakata, Y. , Loskutoff, D.J. , Gladson, C.L. , Hekman, C.M. & Griffin, J.H. Mechanism of protein C‐dependent clot lysis: role of plasminogen activator inhibitor. Blood 68, 1218–1223 (1986). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information