

Abstract
Background
A wide class of human diseases and neurodegenerative disorders, such as Alzheimer’s disease, is due to the failure of a specific peptide or protein to keep its native functional conformational state and to undergo a conformational change into a misfolded state, triggering the formation of fibrillar cross-β sheet amyloid aggregates. During the fibrillization, several coexisting species are formed, giving rise to a highly heterogeneous mixture. Despite its fundamental role in biological function and malfunction, the mechanism of protein self-assembly and the fundamental origins of the connection between aggregation, cellular toxicity and the biochemistry of neurodegeneration remains challenging to elucidate in molecular detail. In particular, the nature of the specific state of proteins that is most prone to cause cytotoxicity is not established.
Methods:
In the present review, we present the latest advances obtained by Atomic Force Microscopy (AFM) based techniques to unravel the biophysical properties of amyloid aggregates at the nanoscale. Unraveling amyloid single species biophysical properties still represents a formidable experimental challenge, mainly because of their nanoscale dimensions and heterogeneous nature. Bulk techniques, such as circular dichroism or infrared spectroscopy, are not able to characterize the heterogeneity and inner properties of amyloid aggregates at the single species level, preventing a profound investigation of the correlation between the biophysical properties and toxicity of the individual species.
Conclusion:
The information delivered by AFM based techniques could be central to study the aggregation pathway of proteins and to design molecules that could interfere with amyloid aggregation delaying the onset of misfolding diseases.
Keywords: Misfolding diseases, Alzheimer’s disease, amyloid, protein aggregation, single molecule biophysics, afm, nanomechanical properties, infrared nanospectroscopy
INTRODUCTION
Aging of the world population has amplified the visibility of several human diseases and neurodegenerative disorders such as Parkinson’s, Alzheimer’s diseases and several forms of Ataxia [1-3]. The onset of these and of more than fifty further related pathologies is associated at the molecular level with the proliferation of insoluble fibrillar protein aggregates, termed amyloids [4], formed from the misfolding of normally soluble cellular proteins. Some of these conditions, such as Alzheimer’s (AD) and Parkinson’s (PD) diseases, are predominantly sporadic, although hereditary forms are well documented. Other conditions, such as the lysozyme and fibrinogen amyloidosis, arise from specific mutations and are hereditary. In addition to sporadic (85%) and hereditary (10%) forms, spongiform encephalopathies can also be transmissible (5%) in humans as well as in other mammals [4].
Strong evidence links the propensity of proteins and peptides to misfolding and aggregation to the pathological biology implicated in the onset of misfolding diseases [5, 6]. During their aggregation, proteins, initially in their monomeric and native forms, undergo internal structural rearrangement and misfold into conformations that are susceptible to form fibrils. From a structural point of view, amyloid fibrils are unbranched, several micrometer long and with a diameter of the order of 10 nm. Despite the obvious differences in amino acid sequences and native structure of the aggregating proteins and peptides, three criteria define a protein aggregate as an amyloid fibril: green birefringence upon staining with Congo Red, the fibrillar morphology and a universal cross β-sheet quaternary structural organization, in which β-strands are oriented perpendicular to the fibril axis. In addition, amyloids are very stable, highly resistant to proteolytic degradation and exhibit remarkable mechanical properties with Young’s moduli in the range of several GPa [5-8]. It has been supposed that the amyloid state could be adopted by many if not by all polypeptide sequences and therefore it represents an alternative to the native state of proteins. Thus, it is hypothesized that the cross-β sheet conformation is likely to be the putative cause of the general unique properties of amyloids [9, 10].
Given the high number of diseases associated with amyloid fibrils, the conversion of normally soluble proteins and peptides into intractable amyloid deposits has emerged in recent years as a subject of fundamental importance in science disciplines ranging from physics and chemistry to biology and medicine, with the aim of reaching a thorough understanding of the mechanisms by which protein aggregation occurs and in some cases induces pathogenic behavior [5]. However, the molecular origin and mechanistic link between amyloid formation and disease aetiology remain unclear. Moreover, no disease modifying therapies are available for these disorders and the conformational state of aggregates that are most prone to cause cytotoxicity is still not clear. This lack of comprehension is mainly due to the formidable experimental challenge that is associated with unraveling amyloid properties and the mechanism of their formation. Indeed, amyloid aggregate species possess nanoscale dimensions, exhibit heterogeneous structural polymorphism and are naturally transient.
In vitro biophysical studies have been fundamental to shed light into the molecular processes underlying protein aggregation and consequently misfolding diseases. However, these studies have been based widely on the application of bulk techniques, such as Thioflavin T (ThT), infrared spectroscopy (IR) and Circular Dichroism (CD) [11-13]. A significant factor, limiting the general applicability of these techniques, is their capability to provide only average information on the heterogeneous ensemble of species that are present in an aggregating amyloid solution, thus precluding a profound investigation of the correlation between the biophysical properties of the individual aggregate species and their toxicity. To assist this challenging endeavor, the development of new methodologies capable to capture shapes, size, chemical and structural properties of the species formed during amyloid fibrillization is of primary importance. In particular, Atomic Force Microscopy (AFM) has emerged in the last decades as one of the most powerful and versatile single molecule techniques, thanks to the possibility of acquiring with sub-nanometer resolution 3-D morphology maps of biological specimens on a surface [8, 14]. Initially, conventional AFM imaging has enabled to gain notable knowledge on the mechanisms of amyloid formation and polymorphism. Then, atomic force spectroscopy and more recently, new AFM-based methodologies, such as peak force quantitative nanomechanical mapping (PF-QNM), have been implemented not only to unravel the morphology of biological samples, but also their mechanical properties at the nanoscale [15-17]. These techniques have been employed to resolve the complex and heterogeneous energy landscape of protein aggregation, thus providing direct information on the aggregation pathway and on the link between morphology and mechanical properties of amyloid species that are forming during the fibrillization process. However, imaging, or in the best case mapping of a single property as stiffness and Young’s modulus, is not sufficient when studying inhomogeneous and complex materials, such as aggregating proteins. A real breakthrough has been reached with the development of Infrared (IR) Nanospectroscopy (nanoIR). This technique, exploiting simultaneously AFM and IR spectroscopy, can characterize at the nanoscale the conformational rearrangements of proteins during their aggregation. In particular, infrared nanospectroscopy has been applied to investigate at the nanoscale the misfolding process and the structure of the heterogeneous amyloid species present during the aggregation process at the individual aggregate nanoscale level [12]. This information can shed light on the structural bases of protein fibrillization and misfolding, allowing a deeper comprehension of the protein aggregation field. Indeed, the investigation of the misfolding of monomers and oligomers into fibrils and their mechanical and structural properties is central to understand amyloid stability, toxicity and mechanism of their clearance from the body. The comprehension of these fundamental processes will allow the design of pharmacological approaches to contrast the onset and progression of amyloid diseases. For this reason, all together single molecule-based AFM methodologies represent a future fruitful avenue to unravel the molecular origin underlying the onset of misfolding diseases.
Misfolding diseases
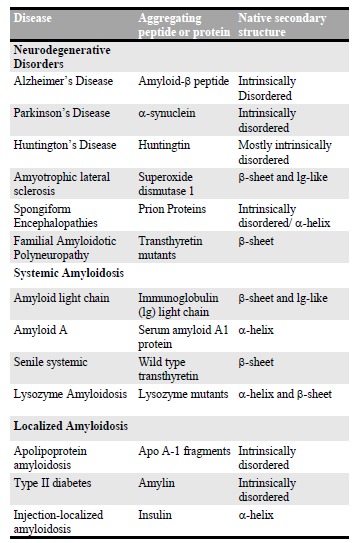
A broad range of human diseases and neurodegenerative disorders is due to the failure of a specific peptide or protein to keep its native functional conformational state. This group of debilitating pathologies has received increasing visibility because of their link with the ageing of the world population in the last century. There are currently approximately 50 illnesses associated with protein aggregation and formation of amyloid structures and they are generally indicated as protein misfolding diseases. In Table 1 [4], a selected list of amyloid misfolding diseases is shown. These diseases can be divided in three classes: neurodegenerative diseases, including PD, AD, the several forms of ataxias and the spongiform encephalopathies [18]; non-neuropathic systemic amyloidosis, such as lysozyme amyloidosis [19]; non-neuropathic localized amyloidosis, such as diabetes of type II, which is an endemic disease
Table 1.
List of Misfolding Diseases. Adapted from Ref. [4].
|
affecting approximately 6% of human beings [20-22]. The major part of these diseases is predominantly sporadic (85%), such as PD and AD, while others as lysozyme amyloidosis and Huntington’s disease arise from genetic mutations and are hereditary (10%). In addition, spongiform encephalopathies (5%) are transmissible from animals to humans and are highly infectious [23]. Despite the widespread association of amyloid formation with disease states, the mechanistic role of amyloids in cell death and disease onset has remained challenging to identify. In particular, the nature of the specific state of proteins that is most prone to cause cytotoxicity is not well established. It is clear, however, that aggregated rather than monomeric proteins are primarily implicated in the toxicity that is observed in protein misfolding diseases [7]. Initially, the prevalence of fibrillar protein aggregates in the brains of patients suffering from neurodegenerative disorders led many authors to hypothesize that these structures themselves cause cell death. Indeed, in the case of systemic amyloidosis, the major cause of the disease onset is simply the presence of large quantities, in some cases even kilograms, of amyloid deposits in vital organs, including liver, spleen and kidney [5]. Instead, in the case of neurodegenerative disorders, several successive studies revealed only a weak correlation between the quantities of accumulated amyloid fibrils and the disease symptoms. In these cases, recent evidence has increasingly suggested that prefibrillar aggregates, such as oligomers and protofibrils, might be the toxic components, rather than the final mature fibrillar products [7, 24-26]. The oligomeric toxicity could arise from their misfolded nature and at least by partial acquisition of the cross-β sheet structure. In fact, the structural rearrangements during the misfolding and/or aggregation could expose hydrophobic chemical groups, otherwise buried by the protein folding in the case of globular proteins or dispersed in the case of intrinsically disordered proteins (IDPs), which normally are not accessible to the cellular environment. The non-native structure of misfolded oligomers could, finally, interferes with several cellular components and processes. Such events could lead to the malfunctioning of the cellular machinery and of its crucial aspects as membranes integrity and trafficking, oxidative stress, sequestration of essential proteins, ions balance causing finally the cell’s death [4, 5, 27].
Protein Misfolding and Amyloid Formation
The native state of a protein was initially associated with a compact globular conformation possessing a rigid and highly ordered 3-D structure. It was demonstrated that the structure of globular proteins is encoded in their amino acids sequences and that these proteins spontaneously fold following a diffusional research of a conformational free energy minimum, which corresponds to the native state [28, 29]. Later on, it was found that this assumption is not accurate, only a part of proteins possesses a globular conformation and one-third to one-half of the human proteome has been reported to contain proteins with more than 40 consecutive disordered residues. These proteins are termed intrinsically disordered proteins (IDPs) and they lack a well-defined 3-D structure under physiological conditions. They can sample a wide range of conformations that range from completely extended to ones that are more compact. Due to the lack of a precise structure, IDPs are characterized by a high flexibility that allows them to interact with multiple partners and hence to exert multiple biological functions [30]. Moreover, in order to be functional, in most of the cases specific regions within IDPs undergo an induced folding that is triggered by either binding to their biological partner(s) or due to environmental changes. Such interactions with biological partner(s) are often accompanied by the formation of complexes that possess a more ordered structure than the original components and lower free energy minima than the native state [31-36]. This implies that proteins can acquire other biologically relevant conformational states different from their native one. Globular proteins can also adopt intermediate conformations simply because of thermodynamical fluctuations, which correspond to local minima in their energy landscape [37, 38]. In addition, many proteins possess both structured and unstructured and highly dynamic fragments (termini, loops, etc.) [39, 40]. It is also supposed that in some cases, partially unfolded states could retain a biological function, such as cellular trafficking and translocations through mitochondrial and nuclear membranes [41-43]. The partially folded/unfolded states of a protein, independently of its globular or naturally unfolded conformation, are vulnerable to misfolding and to aggregation into amyloid structures [4, 5, 27, 29, 44, 45]. This condition is promoted by conditions that destabilize the native fold of the protein, such as high temperature, high pressure, low pH, organic solvents, natural or post-translational mutations [45-47]. During their aggregation, proteins initially in their native monomeric forms undergo internal structural rearrangement, and misfold into conformations that are susceptible to form fibrils [48]. Besides, several coexisting aggregate species are formed, giving rise to a highly heterogeneous reaction mixture. The fibrillation process typically takes the form of a nucleation-dependent polymerization reaction [5, 49, 50]. This model supposes that the formation of oligomeric structures is necessary to nucleate the first protofibrillar structures, ultimately leading to the formation of the mature amyloid fibrils. This process is typically described by a sigmoidal reaction time course, commonly measured by ThT fluorescence and light scattering assays (Fig. 1) [51, 52].
Fig. (1).

Schematic nucleation dependent process of amyloid fibrils formation.
According to the classical nucleation process, a primary nucleation step is necessary for the formation of aggregates, but in the case of amyloid fibrils several secondary steps can also be involved [53]. In these secondary processes, the formation of the nuclei is catalyzed either by the fragmentation of the already formed fibrils or through a surface-catalyzed secondary nucleation mechanism, whereby the existing fibrils catalyze the nucleation of further
nuclei at their surface (Fig. 2) [5]. The availability of integrated laws describing protein aggregation allows to extrapolate the microscopic parameters determining amyloid fibrillization and to individuate if primary or secondary nucleation processes dominate [53-57]. The conventional “first-misfolding-then-aggregation” paradigm is the generally accepted process of amyloid formation, but several observations have shown that the misfolding process could take place after a first step in which native monomers aggregate, i.e. “first-aggregation-then-misfolding” [58-61]. These native oligomers undergo a structural misfolding to form the early cross-β sheet aggregate whereas the final amyloid fibrillar structures only form in a second step. Despite the initial difference, both pathways are conceptually similar: a misfolded state, monomeric or oligomeric, is necessary to nucleate the formation of the universal amyloidogenic cross-β sheet structure. Notwithstanding the great success of the kinetic descriptions of the mechanistic details of amyloid formation at the microscopic level [54, 56, 62-65], the full elucidation of the process requires the identification of all the conformational and oligomeric states adopted by the protein and of its possible aggregates. Indeed, structural polymorphism can be encountered at all aggregation levels, and it originates as a consequence of the glassy, frustrated energy landscape that underlies misfolding and aggregation [66, 67]. Furthermore, oligomeric and protofibrillar structures can form on- and off-pathway during the formation of mature fibrils [68-70]. In Fig. (3), a schematic depiction is shown describing the different conformational states that arepopulated by a protein and that lead to the formation of
Fig. (2).

Possible processes of amyloid nucleation. A) Primary nucleation when monomers interact to form an oligomeric nucleus. B) Fragmentation whereby a nucleus breaks down into two nuclei. C) Surface-catalyzed secondary nucleation whereby fibrils surface catalyze the formation of more nuclei.
Fig. (3).

Schematic representation of distinct protein conformational states that can lead to the formation of amyloid structures. From [5].
oligomeric and fibrillar structures. These latter structures can be either biologi-cally functional or disease-related. All these conformational states are regulated in the cell environment by molecular chaperones, degradatory and quality control systems [4, 5, 44, 45, 71-73].
The case of Alzheimer’s disease
Alzheimer’s disease (AD) is one of over 50 related amyloid disorders that are characterized by the misfolding of soluble proteins and their subsequent conversion into amyloid fibrils [4, 74, 75]. The number of people affected by AD is continuously increasing as the global population ages. It is estimated that 44 million people currently suffer from AD and that this number will grow to more than 135 million by 2050 [76]. AD is characterized by increasing memory loss, behavioral changes, paranoia, loss of social awareness and impaired language function, ultimately resulting in the death [77]. For this reason, the pathology represents a major public health concern and it has been identified as a research priority. The onset and progression of AD are associated with the abnormal accumulation of protein deposits in the ageing brain and over the past 25 years, it has become clear that the proteins forming the deposits are central to the disease process. Indeed, senile plaques and neurofibrillary tangles that are formed by insoluble extracellular deposits and intracellular inclusions, respectively, were found in post mortem analysis of the brain of AD patients [78, 79]. Two proteins or protein fragments thereof have been identified as the main components of these deposits. A protein fragment called the Amyloid-β (Aβ) peptide, has been in particular identified as the major plaque component and the protein Tau as the major tangle component. The Aβ peptide has a variable length and molecular weight of about 4 kDa and it has an intrinsically disordered structure in solution [78, 79]. The biological role of the Aβ peptides it is still not clear, though it has been suggested that they could be part of the mechanism controlling the synaptic excitatory activity [80, 81]. Aβ proteins are produced in the brain by a sequential proteolytic cleavage of the type I transmembrane amyloid precursor protein (APP) by β- and γ-secretases. As a function of the site of APP cleavage by the secretase, proteins of different chain lengths are generated [79]. Tau is a microtubule-binding protein that can also aggregate into filaments, which are amyloid in nature and possess cross-β structure [82]. Tau possesses a microtubule-binding domain that is positively charged and that allows its interaction with the negatively charged surfaces of microtubules. Besides, Tau is rich in several repeat regions of roughly 30 residues. The presence of hexapeptide motifs in two of the repeat regions is considered crucial for the aggregation process and for the core of the filaments. The aggregation of full-length Tau in vitro is in general slow and therefore aggregation studies have rather focused either on constructs formed from the aggregation-prone repeat domains or in the presence of polyanion cofactors, such as heparin and RNA, that compensate for the positive charges. The amyloid cascade hypothesis suggests that the aggregation and deposition of Aβ triggers neuronal dysfunction and cellular death in the brain of AD patients [83]. In the original hypothesis, the neuronal dysfunction and death was thought to be a toxic effect of the total fibrillar load. However, several studies have showed that the number and size of plaques in post mortem AD brains do not correlate with the severity of the pathology and that amyloid plaques were found in the cortex of cognitively normal elder people [84, 85]. As the knowledge of pathological effects of AD has increased, research has been focused on the specific alterations in Aβ processing, such as the cleavage of amyloid precursor protein (APP) into Aβ peptides, the most abundant of which are the Aβ1-40 (90%) and Aβ1-42 (10%) in humans. Despite its minor abundance in human plasma and cerebrospinal fluid, the Aβ1-42 peptide has higher aggregation propensity and toxicity than Aβ1-40 [86]. The ratio of these two isoforms is influenced by the pattern of cleavage from APP by β and γ secretases and it is more critical than the total amount of Aβ produced [87, 88]. In vitro and in vivo studies have shown that Aβ oligomers reduce glutamatergic synaptic transmission strength and plasticity, posing them as the putative toxic species [89-91]. Indeed, since the protein is normally expressed in the organism and many different events could cause fibrils formation and disease, similarly as PD, it is now generally accepted that the prefibrillar intermediates could be the triggering factor of the disease [18, 92]. Fibrillar aggregate formation could be even a protective mechanism by the organism to sequester intermediate aggregates, which could be toxic because of their misfolded structure. Thus, specific oligomeric species are likely to be the primary toxic agents [91, 93-99]. From a mechanistic point of view, the successful application of chemical kinetics to the aggregation of the Aβ peptide has unraveled the key microscopic step that is responsible for the generation of the toxic species. Indeed, it has been shown that the dominant mechanism for catalyzing the formation of toxic Aβ42 species is surface-catalyzed secondary nucleation. In other words, once a small but critical concentration of Aβ42 aggregates has been generated through primary nucleation of monomers, surface-catalyzed secondary nucleation becomes the dominant process where the surface of the existing fibrils serve as catalytic sites for the generation of toxic oligomeric species [54, 57]. Furthermore, the role of intrinsic and extrinsic factors on the aggregation process of Aβ42 has been partly unveiled and a great effort has been focused on drug development against Aβ42 aggregation, which has proven to be very difficult [100, 101]. In agreement, no compound has cleared clinical trials and entered clinical use [102, 103]. One of the main reasons that have led to this gnawing clinical failure is the incomplete knowledge of the molecular mechanisms underlying the process by which small molecules interfere with the aggregation pathway. Indeed, inhibiting Aβ aggregation per se, without an accurate knowledge of the underlying microscopic processes, could have unexpected consequences on the toxicity, as it could not only decrease it, but also leave it unaffected, or even increase it [104]. Therefore, promising effective therapeutic strategies must be aimed at targeting precise microscopic steps during the aggregation process.
Biophysical Investigation of Amyloid Structure and Formation
Biophysics is an extremely interdisciplinary science that uses methods and theories developed in the fields of physics, mathematics, and chemistry with the main aim of studying biological systems. Nowadays, biophysical approaches scan all biological scales from atoms and molecules to cells, organisms, and environments. Experimental biophysics is based on several methodologies, which can be divided in two categories: bulk and single molecule techniques. The first category includes techniques, such as Circular Dichroism (CD), Thioflavin T (ThT) fluorescence, Dynamic Light scattering (DLS), Infrared Spectroscopy (IR). These techniques are able to determine only average properties of biomolecules and living systems. On the other hand, single molecule techniques enable the accurate measurements of single molecule properties. Among all the possible single molecules techniques, AFM has become one of the most powerful available to investigate the structure of biomolecules, but also for their manipulation, at the single molecule scale.
Spectroscopic Bulk techniques: Measuring Average Properties
Circular Dichroism
Circular Dichroism (CD) is based on the differential absorption of left‐ and right-handed circularly polarized light [105]. In the case of chiral biomolecules, the CD absorption of light in the far-UV (wavelength between 180-240 nm) derives from the peptide bond (amide bond) electronic transitions: π → π* and n → π* around 190 nm and 220 nm, respectively [106]. These transitions are highly sensitive to the molecule conformation and to its secondary structure. As we can observe in Fig. (4), when polarized light is absorbed by a protein, its electronic structure gives rise to characteristics bands in the far-UV. The dichroic signature corresponding to: β-sheet has ππ* positive maximum near 195 nm and nπ* negative maximum near 217 nm; random coil state a strong ππ* negative maximum near 200 nm; and the α-helix structure has a strong maximum near 190 nm and a double negative maxima at approximately 208 nm and 222 nm [11]. The capacity of CD to give a representative structural signature makes it a powerful tool in modern biochemistry, especially for detecting conformation changes during the fibrillization of amyloidogenic proteins as a function of time, temperature or different solvents [40, 59, 107, 108]. However, CD provides an average of the properties of all the species present in the heterogeneous aggregating solution. In other words, it is not possible to assign the CD signal of a particular species out of a mixture of co-existing species, such as fibrils and oligomers, as all these species will contribute to the signal with the contribution being directly related to their abundance. Furthermore, a CD signal reflects simultaneously protein content in secondary structures and solubility. Hence, two protein samples with same secondary structure content but with different solubility lead to different CD spectra. Therefore, CD spectroscopy is only suitable for qualitative estimation of fibril formation since amyloids possess typical β-sheet signatures but is not able to unravel subtle differences in the mechanism of fibril formation at the molecular level. Finally, CD has poor structural resolution, sensitivity to conformational changes and it is not able to measure concentrated samples [109].
Fig. (4).

Typical CD spectra of proteins. Signal indicating α-helical (red), random coil (green) and β-sheet (light blue) structures.
Thioflavin T Fluorescence
The fluorescent benzothiazole Thioflavin T has become among the most widely used dyes for selectively staining and identifying amyloid fibrils both in vivo and in vitro [110]. Indeed, under appropriate conditions, it selectively stains amyloid structures undergoing characteristic spectral alterations [111]. ThT fluorescence originates from the dye binding to cross-β sheet rich structures, which causes a dramatic enhancement of its fluorescence emission. When
associated with amyloid fibrils, the dye displays a dramatic shift of its excitation maximum, from 385 nm to 450 nm, and of its emission maximum, from 445 nm to 480 nm [112]. This method notably enables to investigate the kinetics of amyloid fibrils formation. In addition, many histological and bio-imaging studies mainly rely on measuring ThT fluorescence [52, 57, 110]. A paramount progress has been made in this area thanks to the development of ThT-based chemical kinetics tools that, unlike CD, allow deciphering at the microscopic level the mechanistic details of the aggregation of amyloid-forming proteins. Indeed, although the general pathways for aggregation and the resulting fibrils appear remarkably similar for different amyloid disorders, major differences are present at the level of the microscopic steps leading to the fibrils. Kinetic measurements reflect a nucleation-growth process leading to the common fibril cross-β structure. The nucleation-growth mechanism is supported by a wealth of data, including: i) the kinetics of aggregation showing a sigmoidal profile characteristic of nucleation-dependent polymerization, and ii) the observation that the addition of sufficient quantities of pre-formed fibrils (i.e. seeds) can abolish the lag phase [54]. Thanks to the substantial advances made in the application of chemical kinetics to the protein misfolding problem, it becomes now possible to connect macroscopic kinetics measurements of a protein to the specific microscopic steps in the mechanism of aggregation and accordingly to interpret the macroscopic measurements in terms of the rates of the individual microscopic processes underlying amyloid formation [57, 113, 114]. In the case of AD, as described above, the application of chemical kinetics has tremendously improved our understanding of the molecular mechanisms of the onset and progression of the disease [57, 100]. In addition, chemical kinetics has proven very powerful in drug discovery. Indeed, it allowed a thorough understanding of the mechanism of action of drug-like molecules on Aβ aggregation by providing details at the microscopic level. This level of control was otherwise lacking since it is impossible by means of bulk techniques to decipher how a small molecule interferes with the aggregation process at a single microscopic step scale. Interestingly, very recently, an FDA approved anti-cancer molecule, bexarotene, has been found to inhibit specifically the nucleation processes in Aβ42 aggregation with no effect observed on the elongation of the fibrils. These findings were further confirmed in neuroblastoma cells and in C. elegans models of Aβ-mediated neurotoxicity [115]. Despite its widespread use, the structural basis for binding specificity and for the changes to the photophysical properties of the ThT dye remain poorly understood [116]. In addition, ThT is not perfectly amyloid specific and it may not undergo a spectroscopic change upon binding to precursor monomers or small oligomers, even if with a high β-sheet content [117]. Moreover, some amyloid fibers such as the polyglutamine fibrils that are associated with Huntingtin disease do not affect ThT fluorescence, raising the prospect of false negative results and the necessity of couple studies based on this technique with other methodologies. Finally, although ThT-based chemical kinetics has been a major advance in the field, being capable of providing a detailed understanding of the molecular mechanisms underlying the generation of toxic species during amyloid formation, detailed structural analysis of the transient and unstable species still elusive so far.
Mass Spectrometry
Mass spectrometry (MS) in general, and more precisely ion mobility coupled to electrospray ionization mass spectrometry (ESI-IM-MS), has emerged as a powerful method for the characterization of transient and heterogeneous biomolecular complexes [118-120]. ESI-IM-MS allows the simultaneous determination of size, relative population and shape of heterogeneous mixture of covalent and/or non-covalent multimers and conformers [121]. In particular, ion mobility (IM) allows the separation of species according to their collisional cross-section, providing an additional dimension where multimers and/or conformers of identical mass to charge ratio (m/z) are effectively separated. This collisional cross-section is informative of overall shape and molecular size, delivering insights into the structure of the analyzed molecules, which can be rationalized through molecular dynamics simulations [122-124]. Furthermore, ESI-IM-MS is ideal for studying highly dynamic and transient molecular systems such as Aβ aggregation, since samples are analysed in the extremely short millisecond time frame. Importantly, ESI-IM-MS studies have been directed to the characterization of Aβ oligomers formed at the early stages of aggregation. Note however that, some species are not detected mainly due to their highly hydrophobic and heterogeneous nature, which ultimately preclude their ionization. In addition, in some cases cross-linking techniques are required prior to mass analysis for the stabilization of the transient oligomeric species [125]. Notwithstanding its huge potential, MS possesses some major drawbacks limiting the investigation of the structure and formation of amyloid aggregates. Amongst the limitations, the analyte must be able to be ionized and the technique is destructive. Then, to obtain structural information such as secondary structure or atomic level information, time-consuming and often challenging MD simulations are necessary. These simulations become more challenging as the molecules become larger. In particular, when dealing with large self-assembled protein aggregates, the complexity brought by the range of molecular shapes and dimensions that arise poses serious limitations to the applicability of such algorithms. The main limitation is the inability of MD to deal with particular type of intra- and intermolecular interactions because of the extremely demanding computational costs [126].
Infrared Spectroscopy
Infrared spectroscopy is a general established tool for the determination of secondary structure of proteins [127, 128]. Moreover, the technique has been extensively used to observe the conformational transition from monomers to cross β-sheet amyloid structures during amyloid formation and to investigate the aggregates structural properties in solution [51, 129, 130]. IR spectroscopy is based on the molecular vibrations produced, under light exposition, by stretching, deformational motions, bending and rotations of chemical bonds. The amide I, amide II and amide III modes are the most commonly used to study the structural properties of polypeptides. The amide I mode arises mainly from backbone C=O stretching vibrations (>80%) with frequencies at 1700-1600 cm-1. The exact band position is determined by the backbone conformation and thus by the secondary structure of the protein [127, 131]. The amide II band reflects a combination of backbone N-H bending and C-N stretching and has frequencies within 1580-1510 cm-1. The amide III band at 1350-1200 cm-1 reflects a combination of different modes such as C-N stretching, N-H bending, C-C stretching and C=O bending. We could associate the position of these two bands to the secondary structure of the protein, but the analysis is far from trivial because of the various contributions. The amide I band is thus the most frequently used to infer the secondary structure of peptides. From several studies, a consensus has emerged for the assignment of the secondary structure IR peaks in this spectral region [132, 133]. As shown in , there are several components concurring to the shape and position of the Amide I band: intermolecular antiparallel β-sheet are located within 1695-1680 cm-1; intramolecular β-turn within 1680-1665 cm-1; α-helical between 1660-1650 cm-1; random coil between 1645-1635 cm-1; intramolecular low density native β-sheet within 1635-1620 cm-1; intermolecular high density amyloid β-sheets within 1625-1610 cm-1. In particular, several studies showed that amyloidogenic β-sheet are located at lower energy in the IR absorption spectrum than native β-sheet, reflecting likely the presence of stronger intermolecular hydrogen bonds in the cross-β structure than the intramolecular ones in the native globular protein [108, 129, 133, 134]. Respect to CD or Raman spectroscopies, in IR spectroscopy the β-sheet contribution has the highest absorption coefficient. Therefore, IR spectroscopy is a particularly suitable technique to analyze β-sheet-rich amyloid aggregates [135]. However, the technique shares with all these bulk techniques the major limitation of being capable to measure only average properties of a population.
Single molecule investigation by Atomic Force Microscopy techniques
In order to address the challenge of understanding amyloids aggregation, single molecule measurements possess increased robustness than bulk measurements measuring heterogeneous populations. AFM has emerged in the last decades as one of the most powerful and versatile single molecule techniques because of the possibility to acquire 3-dimensional (3D) morphology maps of specimens on a surface [136]. This capability has been widely used in the field of protein aggregation and amyloid fibrils formation and a huge amount of notable results has been obtained so far. Indeed, a simple AFM map provides extremely valuable information on the structure of amyloid fibrils at the nanometer scale, such as height, width, periodicity, flexibility and packing of single protofilaments inside mature fibrils [8, 14]. Moreover, the recent application of new AFM-based methodologies, such as quantitative nanomechanical mapping and infrared nanospectroscopy, represents a fruitful avenue to unravel the process of monomer misfolding and to elucidate the molecular mechanisms of amyloid polymorphism and formation. The combination of these AFM-based methodologies enables to correlate at the nanoscale the morphological, mechanical and structural properties of amyloids at the individual aggregate scale [12, 61, 137-139].
Atomic Force Microscopy: Working Principle
A microscope is an instrument designed to produce magnified images of objects too small to be seen with naked eyes. The first idea that could come in mind in association with the word “microscope” is that the instrument manipulates light to obtain a magnified image of the object. Indeed, for a long time after the invention of the early microscope, which happened centuries ago, microscopy was based on a light source that emits photons that are used as probes directed onto or emitted from the specimen. Nowadays, there are different types of microscopes: optical, electron and scanning probe microscopy. Optical and Electron microscopy are based on the scheme “source-specimen-detector-analyzer”, where particles are probes sent to the sample, then registered by a detector and subsequently analyzed bringing information on the sample morphology. Optical microscopy uses photons as probes and it can reach a resolution r as small as approximately the half of the wavelength of the light interacting with the sample (~200 nm), as stated by Abbe’s principle r = λ/2NA where NA is the numerical aperture of the microscope [140]. Electron microscopy, invented in the early 20th century, uses an accelerated electron beam to magnify and observe the specimen and, thanks to the much shorter wavelength of fast electrons, has a very significantly higher resolution than optical microscopy. A state-of-the-art electron microscope is capable of acquiring images with sub-angstrom resolution. The main limitation of this technique is that sample has to be conductive. Non-conductive samples, as biomolecules, must be coated with a conductive material, which could alter the sample or mask its properties. In scanning probe microscopy (SPM), a sharp tip interacts with the sample and scans its surface in a raster way. In other words, the probe interacts with the sample moving along sequential parallel lines. Every line is divided in pixels, which can record several types of signal as currents, voltages and forces. The first scanning probe technique was the scanning tunneling microscopy (STM), whose invention was awarded with the Nobel Prize [141]. This system is based on the quantum tunnel effect, which causes the passage of a current between two conductive objects placed close each other. First, a metallic tip is brought close to a conductive surface, causing an electronic current flow depending exponentially on the distance between the two elements. Successively, the tip scans the surface. As depicted in Fig. (5), by means of a feedback system controlling piezoelectric elements that displace the sample with respect to the tip, the system can keep the tip-sample distance or the electronic current constant [142]. The signal from the feedback mechanism allows retrieving the topography of the sample with a resolution along the z direction of approximately 0.01 nm and along the XY directions with sub-nanometer resolution. This was a conceptual revolution in the field of microscopy, but unfortunately, the limitation of the investigation to only conducting or semiconducting samples remained. For this reason, STM is not suitable for biological samples. In order to overcome the difficulties in investigating dielectric samples, in the 1986 Binnig et al. invented AFM. They combined the principles at the base of the STM and of the so-called stylus profilometer. The probe of the AFM is a cantilever with a microfabricated sharp tip at its end, which is the sensing device. AFM probes are normally made of silicon or silicon nitride. The cantilevers have usually rectangular or triangular dimensions, typical length within 2-200 μm and they can be considered as springs with elastic constant k in the range of 0.01-100 N/m. Tips at the end of the lever typically have an apical radius between 1 and 50 nm. Similarly to STM, by means of a piezo-actuators
Fig. (5).

Schematic depiction of Scanning Probe Techniques. From Ref. [143].
system, the probe is kept at a distance in the order of the nanometer from the surface and it scans the surface in a raster way. In the easiest configuration, during the scanning, interaction forces between the tip and the sample produce a bending of the cantilever, which can be directly measured. There are several methods to measure the cantilever deformation. However, the most commonly used is the optical lever method, where a laser beam is focused on the back of the lever and the reflected beam is detected by a four-quadrant photodiode (Fig. 5). This configuration enables to measure vertical and lateral (torsion) deformations of the lever. Finally, the feedback electronics drives the piezoactuator in order to compensate the deviation of the lever deflection from the chosen setpoint and to keep a constant deformation of the cantilever. The bending Δx of the cantilever is proportional to the interaction force, as described by the Hook’s law: F = k·Δx, where k is the elastic constant of the lever [136]. In a typical force microscope, cantilever deflection ranges from 0.1 nm to micrometers. This enable to routinely measure forces ranging from 10-13 to 10-4 N [143]. The forces relevant to AFM are ultimately of electromagnetic origin, which are due to the interaction between tip and sample atoms. However, different intermolecular, surface and macroscopic effects give rise to interactions with distinctive distance dependencies. In the absence of external electric and magnetic fields, the dominant forces are van der Waals interactions, short-range repulsive interactions, capillary and adhesive forces [136, 143]. If the tip is in strong contact with the sample, elastic forces can occur. All the described forces can be divided roughly in two categories: repulsive and attractive. The former dominate at a close tip-sample distance, while the latter are long-range interactions. The sum of these forces is generally approximated by means of a Lennard-Jones potential, which combines short-range repulsive and long-range attractive forces, as represented in Fig. (6).
Fig. (6).

AFM tip-sample potential energy representation and related AFM scanning modes.
The sum of these contributions typically leads to the non-monotonic force vs. distance dependence schematized in Fig. (7). According to the regime of interaction, there are two main operational modes of AFM: static, termed also contact mode, and dynamic mode. In contact mode, the tip is brought at close contact with the sample and scanned across the surface. In this modality, the tip-sample interaction is mainly due to strong repulsive forces (~1-10 nN). As described above, the imaging of the sample is generally obtained by maintaining a constant deflection (force) of the lever during the scanning. This is possible through the extension or retraction of the piezoactuator along the z direction by the feedback
Fig. (7).

AFM Schematic illustration of the convolution of the shape of the AFM tip with the shape of the feature or particle being scanned. (a) Blunt tip, (b) sharp tip.
electronics loop, which adjusts the tip-sample distance in order to have a constant deflection of z adjustment of the piezo is used to display the sample heightΔthe lever. The for each pixel of the XY plane, so retrieving a 3D map of the morphology of the sample (Fig. 5). Static mode was historically the first operational mode of AFM and it allows straightforwardly to obtain morphology images with atomic resolution. However, huge tip-sample lateral forces are present during scanning and they could introduce damage or artifacts into the specimen. For this reason, the dynamic mode was successively introduced. In this configuration, the tip is oscillated, at a frequency equal or close to the cantilever resonance, over the surface of the sample and typically at a distance from few to tens of nanometers. At this distance, the tip-sample interaction is mainly due to the much weaker attractive forces, such as van der Waals and electrostatic interactions, which are typically below 1 nN. However, according to the absolute value of amplitude oscillations and tip-sample separation, also the other forces described above, such as repulsive, frictional and elastic deformation, can have important contribution. As shown in Fig. 1, two main dynamic regimes can be considered: the tapping mode, in which the equilibrium separation of the tip and sample is smaller than the amplitude of the cantilever oscillation, in which case the tip is periodically brought in contact with the sample; the non-contact mode, where tip-sample mechanical contact is avoided, deformation and friction effects are eliminated, and the tip is mainly submitted to attractive forces [144]. Finally, the instrument resolution deserves a detailed discussion. AFM generates three-dimensional images of the sample surface. Although not always independent, two different resolutions should be distinguished: lateral and vertical. Vertical resolution is limited by both noise from the detection system and thermal fluctuations of the cantilever. In general, the thermal noise of the cantilever is the largest source of noise in AFM [144, 145]. Damping systems have become so effective that mechanical vibrations represent a negligible perturbation of the cantilever oscillation. For a cantilever with a force constant of 40 N/m, the thermal fluctuations of the cantilever are below 0.01 nm. Therefore, vertical resolution of AFM along the z direction is easily below sub-nanometer scale. A notable effect, especially in air conditions, is that AFM measurements of biomolecules report heights that are always smaller than their nominal values. Smaller heights are a consequence of different processes such as dehydration, sample deformation due to repulsive tip-sample forces and differential tip-surface-sample interaction. In the case of the lateral resolution, three main factors should be considered: the instrumental resolution, defined by the ratio between size and number of pixels of the image, the precision of the piezo along the XY direction, and the radius of the scanning tip. The instrumental resolution is generally sufficient, since it is possible to use an elevate number of pixels to scan and the piezoelectric system possesses sub-nanometer precision, while the geometrical shape of the tip is mainly determining the lateral resolution. If the apical radius of the tip is of the same order or bigger of the dimensions of the object under investigation, an effect of lateral broadening will be always visible in the AFM image (convolution effect). In Fig. (7), we can observe a depiction of the convolution effect in the case of scanning by tips with broad or sharp radius of curvature. Only if the tip has comparable dimensions with the object under investigation, the broadening effect will be minimum [146].
Conventional AFM Imaging Unravel Amyloid Formation and Polymorphism
AFM morphology 3-D maps furnish a sub-nanometer resolution view of the structural and morphological characteristics of biomolecules aggregates [147]. This enables to study the morphological conformation of the heterogeneous and polymorphic species present during the process of amyloid aggregation, such as monomeric proteins, oligomers, protofibrillar structures and the final mature amyloid fibrils (Fig. 8) [8, 14, 148, 149]. It has been shown that AFM, though at the limit of its resolution (objects with smaller height than < 1 nm), is capable to visualize the species present during the lag-phase of the aggregation. During this initial time frame, the species that are mainly present in solution are monomeric and early oligomeric forms of the aggregating proteins [70, 150-152]. AFM empowers the possibility to characterize statistically the morphological properties of these early oligomeric aggregates, such as their height and diameter, and to distinguish and weight the morphological different populations, such as dimeric, trimeric aggregates. This has been demonstrated to be fundamental to compare the oligomerization process of mutated forms of a protein, such as in the case of α-synuclein and the exon1 of the huntingtin protein (Fig. 8A) [150, 153, 154]. Together with the statistical assessment of the cross-sectional dimensions (height and length) of the forming fibrillar species (Fig. 8B), these studied has allowed to study the pathway of fibrillar formation and/or to compare quantitatively the kinetics of aggregation of different fibrillating proteins [153, 155]. For instance, it was possible to assess the effect of mutations, causing the early onset of Parkinson’s disease, on the aggregation propensity of α-synuclein or to study the effect of post-translational modifications on the propensity of aggregation of the huntingtin protein [155, 156]. Amyloid fibrils appear at the AFM as unbranched elongated structures just few nanometers in diameter, but with length up to several micrometers. As demonstrated by X-ray diffraction, the amyloid characteristic fingerprint is the universal cross-β sheet quaternary structure [48, 157]. The repeating subunit of such structure consists of β-strands, forming continuous hydrogen-bonded β-sheets, which are oriented perpendicularly to the fibril axis [158]. The β-sheet ribbons are associated via side chain interactions that serve to stabilize the structure, which is closely packed and highly ordered [159]. The possibility to analyze the morphology at several time points, during the process of amyloid aggregation, enables to shed light on the pathway of aggregation. Several AFM investigations confirmed the observation that mature amyloid fibrils are formed by the hierarchical self-assembly [160] of cross-β protofilaments, composed by a pair of β-sheets, twisting together through specific side chains interactions (Fig. 9) [161]. Although the basic structural arrangement of the cross-β structure is conserved for different fibrils, there are different ways how protofilaments can pack into the fibril 3D structure and accordingly causing a range of structurally different amyloid fibrils [162]. Morphologically, fibrils can diverge in the nature and number of component protofilaments, in their arrangement inside the fibrils and in several structural properties, such as the cross-sectional thickness, helical periodicity and chirality [149, 163-166]. This heterogeneity is termed polymorphism and it implies that, for the same peptide, fibril formation can lead to many different patterns of inter- or intra-residue interactions [66, 149, 162]. Furthermore, AFM imaging enables investigating the persistence length of amyloid fibrillar structures (Fig. 10) [149, 167]. The persistence length lp is the length above which thermal fluctuations can bend a rod-like polymer and it is therefore a measure of its elastic properties [168, 169]. This quantity can be determined directly from AFM images, by means of the bond correlation function (1) for semi-flexible polymers in a two-dimensional conformation:
| (1) |
As shown in Fig. (10), θ is the angle between the tangent vectors to the chain at two points separated by a contour distance s. The factor 2 is used to rescale the exponential decay in order to consider the two-dimensional nature of protein fibrils absorbed on a surface [149]. These fluctuations depend on the fundamental physical properties of the polymer, such as its bending rigidity B, which is defined as the product of the Young’s elastic modulus E and the cross-sectional area moment of inertia I of the polymer: B=EI [169]. Then, the measured lp can be used to calculate the elastic modulus E of a semi-flexible polymer if I is known. Indeed, E and lp are directly linked by the relationship (2), where T is the temperature and kb the Boltzmann constant.
Fig. (10).

Persistence length of a flexible amyloid fibril. A) Schematic depiction of a semi-flexible polymer and the parameter used to calculate its persistence length. B) Example of flexible (top, α-lactalbumin) and rigid (bottom, TTR 105-115) amyloid fibrils. Adapted from ref [170].
| (2) |
Therefore, the study of the statistical mechanics properties of polymers by means of the measurement of the persistence lp has been a valuable route towards understanding the nanoscale mechanics of polymers and other linear structures enabling to measure their intrinsic stiffness [148, 149, 170]. However, a limiting step in the measurements of the intrinsic Young’s modulus of amyloid fibrillar aggregates on a surface is the correct evaluation of the cross-sectional moment of inertia I [148]. Recently, it was presented a general approach based on theory of elasticity and an innovative calculation of the polymorphic fibrillar aggregates cross-sectional moment of inertia I in order to evaluate correctly the nanomechanical properties of amyloids. This method enables to calculate bending rigidities B and matching the measured experimental values of Young’s modulus of amyloid fibrils [149, 165]. However, fibril imaging by AFM requires deposition on a surface and drying, which can potentially lead to artifacts in the evaluation of the persistence length and bending rigidity.
Finally, the presented approaches based on AFM imaging have enabled to gain notable insights into the molecular processes underlying the misfolding, early aggregation and fibrillization process of Aβ42, Aβ40 and Aβ peptide fragments (Fig. 11) [171-174]. Initially, AFM demonstrated to be capable to identify first time pre-fibrillar species as protofibrillar aggregates and oligomeric species,
which were suggested to be toxic [175]. Furthermore, a recent study was able to confirm the mechanistic insights obtained by ThT chemical kinetics analysis and to prove independently with high-resolution that secondary nucleation events are the mechanism dominating the kinetics of Aβ42 fibrillization. In the same study, the kinetics of Aβ42 aggregation were reconstructed in the presence and absence of monomeric and protofibrillar seeds, which play fundamental role in determining the structural properties and heterogeneity of the fibrils formed. For the first time, the secondary nucleation events could be directly visualized at the fibrils surface and the polymorphism of the aggregated fibrillar structures was successfully studied [53]. Finally, several studies have shown that AFM is able to investigate the effect of antibodies and small molecules on inhibiting the formation of Aβ42 fibrillar aggregates and to differentiate the morphology of the aggregated species in each different condition [176, 177].
Force Spectroscopy Measurements and Peak Force Quantitative Nanomechanical Imaging
A cantilever is an extremely precise and sensitive force sensor with piconewton resolution [136, 178]. Consequently, it can be used to measure mechanical properties of biological samples. The basic principle of this measurement relies on performing force-distance curves, in a trace-retrace manner, due to the vertical motion of the lever on the sample under investigation. A single force-distance curve is the plot of the tip-sample force versus the piezoelectric z displacement, as depicted in Fig. (12) [136]. The process can be illustrated as made of six fundamental regions [136]. The 1-2 line represents the zero-extension line, when tip is approaching out of contact. Line 2-3 corresponds to the jump-in-contact, also called snap-in, which happens when gradient of attractive forces is higher than cantilever spring constant. The line 3-4 corresponds to the deflection of the cantilever in contact with the sample; in this region the lever passes from being bent downward, through the zero deflection, to being bent upward. This region is purely linear for perfectly stiff samples, while is not linear in the case of elastic or plastic deformation. The line 4-5 indicates the beginning of the retracting and it should be ideally superimposed and with same slope of the corresponding approaching line 3-4. However, eventual distance displacement and different slope of the two curves related to the plastic indentation of the sample can take place. Line 5-6 represents the moment of the loss of tip-sample contact, termed snap-out, and it is generally linked to adhesive forces. Finally, the line 6-7 represents the retracting out of contact. It is worth to underline that the instrument does not measure directly forces and distances, but it measures directly the displacement of the piezo and the deflection of the laser by means of a photodiode. Therefore, in order to convert the read voltage by the photodiode into a distance, the photodiode sensitivity needs to be calibrated and this calibration is performed by relating the vertical movement of piezo relatively to the cantilever deflection on a stiff substrate. Successively, in order to convert the measured deflection into a force, the elastic constant of the lever is measured. A common method to perform this measurement is the thermal tuning [179].
Fig. (12).
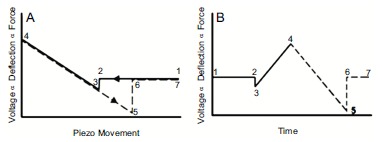
Ideal force-displacement curve depiction. Ideal (A) force-distance and (B) force-time curves representation. Adapted from Ref. [179].
Once the force-distance curve is obtained by means of its analysis, the mechanical properties of the sample can be retrieved. Several methods have been implemented to study mechanical properties of biomolecules as amyloids. Nanoindentation, quantitative nanomechanical mapping and force spectroscopy pulling are part of this category (Fig. 13) [8, 168].
In force spectroscopy measurements, the tip is able to pick up and manipulate the macromolecule on the surface. In this configuration, upon retraction of the tip once a binding is created, it has been shown that amyloid molecules can be stretched or unzipped [180-182]. The resultant force curves and the stretching behaviour can be analyzed by theoretical models, as the worm like chain (WLC) [183, 184]. This methodology has been applied to characterize the nanomechanical response of functional adhesive amyloid fibrils exploited by the algae P. Linearis [185]. Fitting the elastic response present in the force curves, it has been found that these functional amyloids possess a persistence length of 0.34 ± 0.18 nm, similar to the one of proteins such as titin (0.4 nm) and tenascin (0.42 nm) [186]. Instead, it was measured that mature amyloid fibrils formed by the glucagon peptide have an average persistence length of 0.70 ± 0.15 nm. This value is twice as bigger than the value for functional amyloids, suggesting that they possess more mature amyloidogenic nature. Furthermore, force spectroscopy studies of different mature amyloid fibrils have been presented [180, 181]. These studies tried to quantify the interaction forces within β-sheet filaments inside a fibril of different fragments of Aβ42, finding substantially smaller forces necessary to unzip subunit sheets of Aβ42 mature fibrils in comparison with Aβ40 ones. They well highlighted the importance of these works for understanding the details of amyloid formation and pointed out how single molecule manipulation methods allow to study the mechanics and structural dynamics of amyloid formation. However, the weak point of these zipping experiments is that the measurements were based on nonspecific binding between the tip and the fibrils. Therefore, the variability between individual measurements likely results from random and multiple attachment sites of the protein to the tip [187].
Nanoindentation is one of the most common methodologies to retrieve the Young’s modulus and hardness of a sample. Indeed, if the sample is soft relatively to the tip stiffness, during the approach the tip will indent the sample (Fig. 14A). The subtraction of the force-distance curve obtained from an undeformable stiff substrate allows retrieving the indentation curve (Fig. 14B). The fit of this curve, by means of a mechanical model [136], enables to extract the intrinsic Young’s modulus of the object. Nanoindentation measurements, in conjunction with mechanical modeling such as Hertz model of elasticity, have been able to measure the Young’s modulus of amyloid fibrils [168, 188]. This methodology was able to measure the elastic moduli of glucagon fibrils, which was ranging from 0.72 ± 0.80 GPa to 1.26 ± 0.62 GPa under small compressive forces. Similarly, this approach has measured a Young’s modulus of 1.3 ± 0.4 GPa for α-synuclein fibrils and of 5-50 MPa for insulin amyloid fibrils [188]. Notwithstanding the usefulness of the information provided by nanoindentation, the values of stiffness obtained are systematically smaller that the generically accepted and measured ones by other methods [168].
A further possible modality to perform nanoindentation studies is the force-volume mode, in which a map of force-distance curves is collected in a raster way over a selected area of the sample. However, a single force curve requires several tens of milliseconds to be completed, so a typical force-volume image is characterized by long completion times (from several minutes to several hours) and low spatial resolution (32×32 or 64×64 pixel images are common). An improvement of the force-volume mapping is the Quantitative Imaging (QI), which allows acquiring maps of mechanical properties of a sample with a resolution up to 512 x 512 pixels contemporarily with morphology maps [61, 190, 191]. Finally, PF-QNM, a similar and relatively new technique, is able to map mechanical properties with higher speed and spatial resolution. The methodology is based on a new tapping mode in which every tapping of the probe on the surface produces a force-distance curve. This technique uses the peak force value of the force-distance curve as imaging feedback, diversely than AM-AFM that uses the amplitude as feedback parameter. This mode can be used with standard tips and cantilevers, the drive frequency is notably below the resonance one and the typical amplitude oscillation is approximately 100 nm. In Fig. (15A), it is represented the tip trajectory as a function of time over a tapping cycle.
Fig. (15).
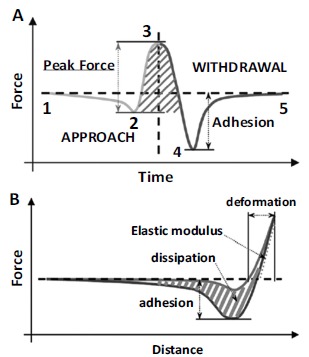
Principle of operation of PF-QNM. (A) Principles of peak force tapping. (B) Force-distance curve and indicative representation of the calculated mechanical properties. From Ref. [191].
Initially (1), the tip is far from the surface and it is brought in contact with the sample (2). Successively, the tip indents the sample until the maximum peak force in reached (3). Then, it is pulled off until the adhesion force is maximum (4) and it finally recovers its original position (5). Using the maximum peak force for the feedback calculation, a force-distance curve is generated at every pixel. Since the z position of the modulation and the deflection of the cantilever are controlled as a function of time, it is possible to eliminate the time variable, from Fig. (15A), and a force-distance plot can be generated, as in Fig. (15B). From those curves, the usually measured quantities by force spectroscopy can be probed: adhesion, dissipation and elastic modulus. This last parameter can be extrapolated by fitting the retraction curve by the Derjaguin-Mueller-Toporov (DMT) model [136]. Since a force curve is obtained for each pixel, the resolution of channel is identical to the
topography image. This technique has been applied successfully to measure the intrinsic elastic moduli of different classes of mature amyloid fibrils including β-lactoglobulin, α-synuclein, Aβ42, bovine serum albumin, insulin, lysozyme, ovalbumin, and Tau protein in GPa range [188, 191-193], which are consistent with the values of the Young’s moduli obtained by statistical analysis of AFM images [170, 192]. Furthermore, this technique is capable to measure the intrinsic Young’s modulus of fibrils independently on their polymorphic state and their cross-sectional properties [191]. In one of its latest application, PF-QNM allowed to investigate at the single molecule level the nanomechanical properties of different products of amyloids aggregation process. In particular, it was able to measure the evolution of Young’s modulus during fibrillation of α-synuclein and Aβ42. The elastic modulus of the forming amyloidogenic species evolved monotonically during the process of aggregation as a function of the maturity of the fibrillation process. In fact, oligomers, protofibrils and mature fibrils showed an increasing value of the intrinsic stiffness in the order of GPa (Fig. 16). It was mentioned above that the hydrogen bonds between β-sheets plays a key role in the mechanical properties of amyloid fibrils and the
presence of β-sheets is a characteristic feature of amyloid fibrils. It has also been reported that during the aggregation process, oligomers or eventually present protofibrils contain partial β-sheet conformations [70]. This suggests that the content of β-sheets in the assembled structures during fibrillization process is an important parameter affecting the mechanical properties of these structures.
Infrared Nanospectroscopy Structural Characterization
The invention of AFM was crucial to the development of nanoscience, due to the possibility to acquire the morphology of samples at unprecedented resolution. However, the study of macromolecules at nanoscale remains challenging, especially when truly quantitative information is required. The main reason is that imaging, or in the best case mapping of a single property, such as Young’s modulus, is not sufficient when studying inhomogeneous and complex materials, such as aggregating amyloidogenic proteins.
A real breakthrough was reached with the development of Infrared nanospectroscopy (nanoIR). The technique exploits the combination of AFM and IR (AFM-IR) and it can be efficiently used to characterize at the nanoscale the structural properties of biologically relevant material and to study specific processes such as amyloid aggregation (Fig. 17). As described above, infrared spectroscopy (IR) is a key method for studying conformational properties of proteins and their structural conversion during amyloid fibrillation. While, as we extensively described, AFM-based methods can provide useful information on the morphology and mechanical properties of the species formed along the aggregation pathway at the nanoscale, these conventional techniques do not allow characterizing separately the chemical and structural properties of individual amyloid aggregates, nor the correlation with their nanomechanical properties. Their combination however, in infrared nanospectroscopy, enables a direct measurement and correlation of the morphological, chemical and structural properties and the discrimination of objects in the same AFM map by identifying their different secondary structure and chemical signature with nanoscale resolution [15, 129, 195]. This ability is one of the main advantages of AFM-IR compared to conventional bulk techniques and in particular to IR, which have spatial resolution limited by the smallest achievable IR spot size (~20 μm) [196].
From a technical point of view, the combination of AFM and IR spectroscopy is based on a photo-thermal induced resonance effect (PTIR) [198]. In the initially introduced configuration of the system, a pulsed laser beam passes through a ZnSe prism and undergoes total internal reflection (Fig. 17). At the same time, the AFM tip is brought in contact with the sample in the area of interest. An evanescent wave, with typical length of the order of the wavelength of incident light, interacts with the sample deposited over the ZnSe prism. If at a given wavelength the IR pulse is absorbed by the sample, the local temperature rise leads to local thermal expansion. This deformation excites mechanical resonances in the AFM cantilever, which is in contact with the sample. Total internal reflection in the ZnSe prism avoids direct excitation of the AFM cantilever via the incoming laser pulse. After excitation, the cantilever
resonates with a ringdown shape, with a time decay of the oscillations in the order of hundreds of μs. The cantilever displacement is read via the usual AFM optical detection system, with a laser beam reflected on the back of the cantilever towards a 4-quadrant photodetector. The Fast Fourier Transform (FFT) of this signal contains several peaks corresponding to the cantilever eigenfrequencies. It has been found that both the peak-to-peak maximum of the deflection vs. time ringdown and the Fourier transformed amplitude of eigenmodes are directly proportional to IR absorption [197]. The pulsed laser is tunable, commercially available instruments span the mid-IR range typically from 900 to 3600 cm-1 and can achieve a spectral resolution of 0.1 cm-1. The local absorption spectrum is obtained scanning the pulsed laser in the IR range and plotting the amplitude of the fundamental resonance (or the ringdown amplitude) as a function of the wavelength. The system allows also mapping the IR absorption of the biological sample at fixed wavelength. Summarizing, the AFM detection of the temporary expansion of the scanned region enables nanoscale resolution IR imaging and acquisition of local chemical spectra. The AFM-IR spectra generated from this technique contain the same kind of information with respect to molecular structure as conventional IR spectroscopy measurements. Moreover, as illustrated in Fig. (18) in the case of collagen fibrils, simultaneously with the acquisition of IR-absorption and topography images, the system is able to obtain qualitative stiffness maps [139]. The mapping of the mechanical properties of the sample is based on contact-resonance frequency measurements. A cantilever, in contact with a sample surface, acts like a coupled spring system, wherein the sample elasticity changes the frequency at which the cantilever resonates will change as well. Therefore, the system enables the generation of stiffness maps of the sample, where higher contact resonance frequency means higher stiffness. The stiffness map is evaluated tracking the variation of the contact resonance frequency of the cantilever [197].
Fig. (18).
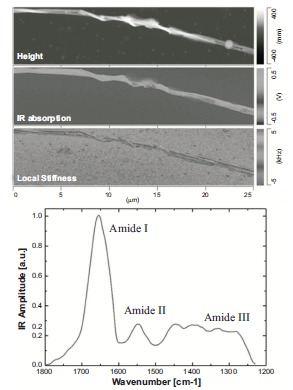
AFM-IR images and spectra of a bundle of collagen fibrils. In the figure are shown on the left the morphology, local stiffness and IR absorption maps and on the right an IR spectrum in the protein range (1800-1200 cm-1) is acquired at a specific location of the maps (indicated by a circle) [139].
The resolution of the instrument in the case of the morphology maps is determined by the usual parameters in standard AFM. While, the resolution of the stiffness, IR maps and IR spectra are however unrelated with the topographic channel. In particular, the ultimate lateral resolution and minimal thickness (i.e. height, Z direction) measurable are determined by independent factors. The minimal measurable thickness is defined by the minimal detectable photo-thermal expansion of the sample. On the other hand, the lateral resolution of an isolated object is only in principle limited by the sharpness of the AFM lever. In the case of non-isolated objects, thermal diffusion and mechanical properties of the sample can limit lateral spatial resolution. It is fundamental to specify that the absorption depends linearly on the thickness of the sample only up to 1 μm, while for higher thicknesses a consequent diminution of the absorption signal with increasing thickness is observed [199]. This was demonstrated on polymethylmethacrylate structures, finding that the absorption signal linearly increases with thickness until 1μm thickness, while for higher thicknesses a downturn and a consequent diminution of the absorption signal is observed. This qualitative behavior is generally reproduced by other kind of samples. However, also on samples thicker than 1 μm, this does not limit the capability of the instrument to chemically and structural investigate their heterogeneity. AFM-IR can be implemented by using different kinds of IR laser sources, such as optical parametric oscillators (OPO) or quantum cascade laser (QCL). The use of OPO lasers enable to measure routinely biological samples possessing an minimal thickness of approximately 50 nm with a lateral resolution as small as 20 nm [200]. Recently, the use of QCL has led to a notable improvement of the technique resolution. QCL allows tuning the repetition frequency of the pulsed laser in order to match the selected eigenfrequencies mode of the AFM cantilever. In this way, an increase of the oscillating amplitude of the cantilever is observed leading to a better signal-to-noise ratio. This can be combined to a local field enhancement obtained by using the coupling of gold-coated AFM tips and gold substrates [201]. A lateral resolution of less than 20 nm and on approx. 2 nm thick mono-layered samples has been demonstrated, opening new possibilities to the application of the AFM-IR technique to characterize the structural and chemical properties of single molecules.
The technique has been increasingly applied in the fields of biology, biochemistry and biophysics. First results regarded the localization of structures with specific IR signature inside cells. Candida albicans fungi cells were characterized in air and liquid environment [202]. This study showed the correlation between the topography and the spectral maps obtained from the absorption in the glycogen band at 1080 cm-1 and a lateral resolution for absorption maps of less than 100 nm. Similarly, it was reported the possibility of localizing viruses inside infected bacteria with a resolution of circa 200 nm [203]. Localization of viruses was performed using both the amide I mode of the virus capsid proteins (at 1650 cm-1) and the maximum of the DNA band (at 1080 cm-1). Similar studies allowed identifying polyhydroxybutyrate polymers inside Rhodobacter capsulatus cells and the influence of acetone and glucose on their expression [204, 205]. Moreover, the technique has been implemented to detect the presence of exogenous elements such as metal-carbonyl compounds in human cells and to study the variations in the primate osteonal bone composition [206, 207]. More recently, it was possible to correlate the amide I signal map to the topography of E. Coli and human HeLa cells in order to investigate the distribution of protein-rich material inside the cells [208]. The technique was also successfully employed to characterize the lipid content in human stratum corneum and human hair [209, 210]. Localization of lipids in strains of Streptomyces (soil bacteria of interest for the production of biodiesel) was performed in an analogous way [211]. The combination of topography, IR absorption and stiffness maps helped elucidating the formation of lipoprotein complexes in chloroplast lipid membranes having a central role in photosynthesis processes in plants. These measurements were obtained with a lateral resolution of less than 20 nm [139, 212, 213].
One of greatest benefits of the AFM-IR technique is the fact that chemical analysis can be performed from the micron scale to the nanometer scale. Thus, the technique requires very small quantities of biological specimens (less than picograms or sub-femtomoles are sufficient). This is a major advantage when working with biological materials whose production is expensive and time costly such as certain amyloid proteins. For this reason, reduction of sample quantity to perform routinely IR and secondary structure analysis is a problem of extreme importance. For these reasons, AFM-IR has posed itself as a powerful tool to the study of the conformational changes and the heterogeneity of aggregating amyloid solutions. In the first proof of concept demonstrated, the system was used to analyze proteins deposited as patterned micro-droplets by means of a microfluidic system. The off-line assay of individual micrometer-sized droplets of biomolecules is an efficient way to obtain high-throughput information about many biologically relevant processes. In particular, the compartmentalisation of biomolecules into monodisperse, micrometer-sized droplets allows for quantitative and high-throughput biochemical studies on amyloid formation. In this work, integrating droplet microfluidics and AFM-IR, microdroplets with microns scale diameter of monomeric and aggregate solutions of lysozyme were patterned over the ZnSe prism of a nano-IR microscope. The instrument enabled to study their chemical and structural properties in the characteristic region of proteins, where amide I, II and III bands are present (1200-1800 cm-1). Spectra from the micro-sized droplets containing monomeric and aggregated lysozyme were obtained and found to be readily distinguishable. In particular, a shift in the amide I band toward lower wavenumbers allowed to identify an α-to-β structure transition associated with amyloid formation [214]. Notably, the volume needed to investigate the structural conformation of the droplets was extremely tiny, i.e. as small as picograms or sub-femtomoles. Similarly, infrared nanospectroscopy enabled a structural characterization of monomeric and macro-molecular aggregates of α-synuclein. Monomers did not self-organize on the prism surface, while fibrils formed rigid rod-like structures with micrometer dimensions. The monomeric protein showed an amide I band indicating a dominating random coil conformation, while the fibrillar aggregates were predominantly β-sheet. Thus, the comparison of the spatially localized IR spectra showed a net variation of the amide I mode conformation from the monomeric to fibrillar state, which well correlated with a random coil to β-sheet transition [214]. A subsequent study aimed to apply AFM-IR exploiting polarized light to demonstrate the amyloidogenic nature of the Erα17p peptide of the estrogen receptor α (ERα) [137]. This receptor is involved in breast cancer and it has been found that it can quickly form hydrogels, which are receiving increasing attention due to their possible use in several medical related applications, such as drug delivery, immobilized antimicrobial surfaces and tissue engineering. To identify the highly organized cross-β structure of amyloids, this work exploited the possibility to acquire nanoscale spectra with polarized light. Spectra in the range of amide I and II bands were acquired to characterize the protein secondary structure and amide I band indicated a strong β-sheet and β-turn signatures. Moreover, in the amide band I, the work showed a strong dependence on the polarization of the IR absorption. This was a proof that Erα17p naturally aggregates in orientated, well-ordered structures. In its latest application, infrared nanospectro-scopy has been applied to characterize at the nanoscale the conformational rearrangements of proteins during their aggregation. In particular, the work demonstrates that AFM-IR can individually characterize the oligomeric and fibrillar species formed along the amyloid aggregation using as model system the Josephin domain of the ataxin-3 protein, which is implicated in spinocerebellar ataxia-3. Similarly to the above-described studies, the secondary structure rearrangements of the amyloidogenic species present during the aggregation process are reported, monitoring at the individual amyloid aggregate nanoscale an alpha-to-beta transition. These results suggested that the aggregation of Josephin proceeds from the monomer state to the formation of spheroidal intermediates with a native structure. Only successively, these intermediates evolved into misfolded aggregates and into the final fibrils. This finding was of fundamental importance because it demonstrated that proteins were still in their native conformation at the earliest state of aggregation. This result was independently confirmed by nanomechanical measurements. Representative spectra for the different maturation phases, together with a sketch of the proposed aggregation pathway and the results of Young’s modulus, are shown in Fig. (19). For the first time, AFM-based techniques were able to correlate at the single aggregated species nanoscale the biophysical properties of amyloids, in particular correlating their intrinsic stiffness with their secondary structure content. Furthermore, this work gave for the first direct evidence to ‘first-aggre-gation-and-then-misfolding’ pathway of amyloid formation [61].
Fig. (19).
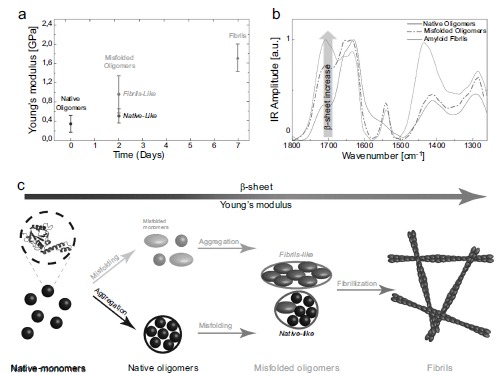
AFM-IR can reconstruct the pathway of protein misfolding and aggregation. a) Young modulus of Josephin protein aggregates as a function of the maturation time. b) Spectra of native oligomers, misfolded oligomers and fibrils showing an increase of the β-sheet content in time. c) Sketch of the two proposed pathways of Josephin aggregation. From Ref. [140].
CONCLUSION
A key reason for the longstanding and widespread interest in the biophysical and biochemical studies of amyloid formation is the strict link with several human illnesses and neurodegenerative diseases. Despite its fundamental role in biological function and malfunction, the mechanism of protein aggregation and the fundamental origins of the connection between the aggregation process and cellular toxicity have remained challenging to elucidate at the molecular level. Strong evidence intimately correlated the physical and structural properties of intermediate and final forms of amyloids to the biochemistry of neurodegeneration. However, the molecular origins of the process of neurodegeneration and its connection with amyloid formation remain unclear, which is strongly supported by the current lack of a cure to the related diseases. Therefore, unraveling the mechanisms of amyloid formation and polymorphism, as well as the mechanical and structural properties of the products of their aggregation is fundamental to understand their stability, toxicity and mechanism of clearance from the body. This is central to further design new therapeutic strategies against amyloid-related diseases. Furthermore, it has been recently found that it exists an always-bigger class of functional amyloids occurring naturally in several disease-unrelated biological processes and that many artificial peptides can form amyloid-like structures in-vitro. For this reason, in order to appreciate the full potential of amyloid fibrils also as biomaterials for future biomedical and nanotechnology applications, it is important to measure and quantify the structural and mechanical properties of amyloid fibrils and understand how they are formed.
We believe that the development of new biophysical methodologies, which are able to bridge together the investigation of morphological and ultrastructural properties of amyloid at the nanoscale, represents a fruitful avenue to address the challenge of solving the problem of protein self-assembly, to unravel monomer misfolding and to elucidate the molecular mechanisms of amyloid polymorphism and formation. Such an understanding at the molecular level of the key (non)-pathogenic processes is crucial in order to provide the basis to answer the proposed above scientific questions. Indeed, their elucidation is fundamental for establishing approaches for the rational design of pharmacological approaches and consequently the prevention of the onset of the misfolding diseases.
Finally, characterizing in a quantitative manner how small molecules can modify specific steps in the aggregation process of proteins by binding to specific toxic species is considered key for the development of drug candidates against misfolding diseases. Indeed, chemical kinetics has been tremendously effective and powerful in providing exquisite molecular insights into the specific microscopic steps that are inhibited by small molecules. However, combining chemical kinetics to AFM-based methods, and more specifically AFM-IR, is expected to provide a complete knowledge of the mechanism of action of small molecules from both, mechanistic and structural point of views. In addition, the application of these two methods concomitantly will allow the establishment of a correlation between the targeted microscopic steps (i.e. primary nucleation, secondary nucleation or elongation) and the structure of species involved in this particular step and will provide structural insights into the transiently occurring species during the aggregation pathway that have remained elusive so far. These advances will enable a quantitative control on the delay of the onset of aggregation and the slowing down of its progression.
Fig. (8).

Statistical characterization of α-synuclein amyloid aggregates cross-sectional dimensions. A) Monomeric and early oligomeric structures. B) Protofibrillar and fibrillar aggregates. Adapted from ref. [151].
Fig. (9).

Fibrils Hierarchical self-assembly and polymorphism. Hierarchical self-assembly of β-lactoglobulin. Polymorphism can be due to different number, arrangement and structure of protofilaments composing the fibril. From Ref. [150].
Fig. (11).

The mechanistic pathway of Aβ42 aggregation. a-d) AFM images describing the aggregation process. The arrows indicate the secondary nucleation events. e) The proposed model of Aβ42 fibrillogenesis. Adapted from Ref. [54].
Fig. (13).
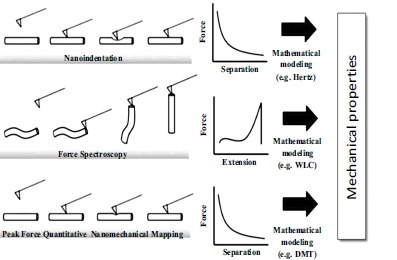
AFM-based methods to investigate the nanomechanical properties of biomolecules and amyloid aggregates. From Ref. [168].
Fig. (14).
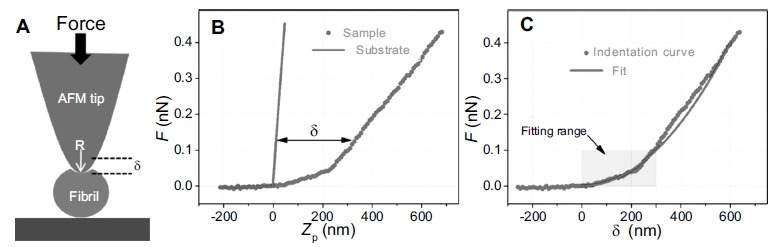
Nanoindentation measurements. (A) Schematic depiction of the indentation mechanism, (B) force-displacement curves on the sample and on an undeformable sample, where δ is the indentation depth. Adapted from Ref. [189].
Fig. (16).
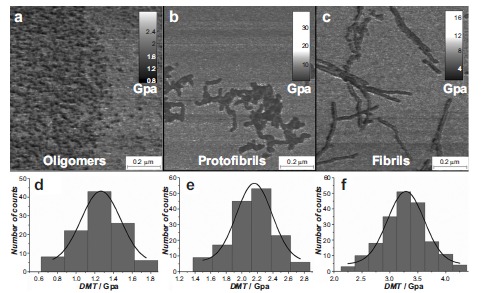
Young’s modulus evolution of Aβ42 species present during its fibrillization process. A-C) Morphology AFM maps. D-F) DMT Young’s modulus maps. G-I) Young’s modulus histograms. From Ref. [194].
Fig. (17).
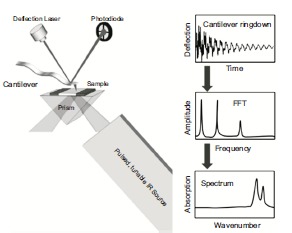
Schematic depiction of the working principle of the nanoscale infrared spectroscopy [197].
Table 2.
Amide I band secondary structure components.
| Amide I Structural assignment | Wavenumber (cm-1) |
|---|---|
| Antiparallel β-sheets (Intermolecular) | 1695-1680 |
| β-turn (Intramolecular) | 1680-1665 |
| α-helix | 1660-1650 |
| Random coil | 1645-1635 |
| Low density Native β-sheets (Intramolecular) | 1640-1625 |
| High density Amyloid β-sheets (Intermolecular) | 1625-1610 |
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Goedert M. Familial Parkinson's disease. The awakening of alpha-synuclein. Nature. 1997;388(6639):232–233. doi: 10.1038/40767. [DOI] [PubMed] [Google Scholar]
- 3.Zoghbi H.Y., Orr H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 4.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 5.Knowles T.P., Vendruscolo M., Dobson C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014;15(6):384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 6.Dobson C.M. Protein folding and misfolding. Nature. 2003;426(6968):884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 7.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2012;20(14):38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adamcik J., Mezzenga R. Study of amyloid fibrils via atomic force microscopy. Curr. Opin. Colloid Interface Sci. 2012;17(6):369–376. [Google Scholar]
- 9.Lashuel H.A., Overk C.R., Oueslati A., Masliah E. The many faces of [alpha]-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013;14(1):38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suh Y., Checler F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol. Rev. 2002;54(3):469–525. doi: 10.1124/pr.54.3.469. [DOI] [PubMed] [Google Scholar]
- 11.Whitmore L., Wallace B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers. 2008;89(5):392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 12.Ruggeri F.S., Adamcik J., Jeong J.S., Lashuel H.A., Mezzenga R., Dietler G. Influence of the β-Sheet content on the mechanical properties of aggregates during amyloid fibrillization. Angew. Chem. 2015;127(8):2492–2496. doi: 10.1002/anie.201409050. [DOI] [PubMed] [Google Scholar]
- 13.Zandomeneghi G, Krebs MR, McCammon MG, Fändrich M. FTIR reveals structural differences between native β -sheet proteins and amyloid fibrils. Prot Sci. 2004;1(13):3314–21. doi: 10.1110/ps.041024904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sokolov D.V. Atomic force microscopy for protein nanotechnology. Methods Mol. Biol. 2013;996:323–367. doi: 10.1007/978-1-62703-354-1_19. [DOI] [PubMed] [Google Scholar]
- 15.Dazzi A, Dazzi R, Prater CB, et al. AFM-IR: combining atomic force microscopy and infrared spectroscopy for nanoscale chemical characterization. 2012. [DOI] [PubMed]
- 16.Mingdong D., Mads Bruun H., Wael M., Sailong X., Daniel Erik O., Flemming B. AFM-based force spectroscopy measurements of mature amyloid fibrils of the peptide glucagon. Nanotechnology. 2008;19(38):384013. doi: 10.1088/0957-4484/19/38/384013. [DOI] [PubMed] [Google Scholar]
- 17.Zhang S., Aslan H., Besenbacher F., Dong M. Quantitative biomolecular imaging by dynamic nanomechanical mapping. Chem. Soc. Rev. 2014;43(21):7412–7429. doi: 10.1039/c4cs00176a. [DOI] [PubMed] [Google Scholar]
- 18.Lansbury P.T., Lashuel H.A. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443(7113):774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- 19.Granel B., Valleix S., Serratrice J., et al. Lysozyme amyloidosis: report of 4 cases and a review of the literature. Medicine. 2006;85(1):66–73. doi: 10.1097/01.md.0000200467.51816.6d. [DOI] [PubMed] [Google Scholar]
- 20.Jaikaran E.T., Clark A. Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta. 2001;1537(3):179–203. doi: 10.1016/s0925-4439(01)00078-3. [BBA]. [DOI] [PubMed] [Google Scholar]
- 21.Hull R.L., Westermark G.T., Westermark P., Kahn S.E. Islet amyloid: A critical entity in the pathogenesis of type 2 diabetes. J. Clin. Endocrinol. Metab. 2004;89(8):3629–3643. doi: 10.1210/jc.2004-0405. [DOI] [PubMed] [Google Scholar]
- 22.Meetoo D., McGovern P., Safadi R. An epidemiological overview of diabetes across the world. Br. J. Nurs. 2007;16(16):1002–1007. doi: 10.12968/bjon.2007.16.16.27079. [Mark Allen Publishing]. [DOI] [PubMed] [Google Scholar]
- 23.Prusiner S.B. Prions. Proc. Natl. Acad. Sci. USA. 1998;95(23):13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uversky V., Eliezer D. Biophysics of Parkinsons Disease: Structure and Aggregation of-Synuclein. Curr. Protein Pept. Sci. 2009;10(5):483–499. doi: 10.2174/138920309789351921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lashuel H.A., Lansbury P.T. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 2006;39(02):167. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]
- 26.Lashuel H.A., Hartley D., Petre B.M., Walz T., Lansbury P.T. Neurodegenerative disease - Amyloid pores from pathogenic mutations. Nature. 2002;418(6895):291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 27.Dobson C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999;24(9):329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 28.Anfinsen C.B. Principles that govern the folding of protein chains. Science. 1973;181(4096):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 29.Dobson CM. Protein folding and misfolding. 2003 doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 30.Habchi J., Tompa P., Longhi S., Uversky V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014;114(13):6561–6588. doi: 10.1021/cr400514h. [DOI] [PubMed] [Google Scholar]
- 31.Dunker A.K., Cortese M.S., Romero P., Iakoucheva L.M., Uversky V.N. Flexible nets - The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272(20):5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 32.Uversky N.V. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002;11(4):739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uversky V.N., Gillespie J.R., Fink A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41(3):415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 34.Uversky V.N., Li J., Fink A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001;276(14):10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 35.Lara H.A. Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions. J. Clin. Invest. 2002;110(9):1221–1232. doi: 10.1172/JCI16781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turoverov K.K., Kuznetsova I.M., Uversky V.N. The protein kingdom extended: Ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog. Biophys. Mol. Biol. 2010;102(2-3):73–84. doi: 10.1016/j.pbiomolbio.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Onuchic J.N., Luthey-Schulten Z., Wolynes P.G. Theory of protein folding: The energy landscape perspective. Annu. Rev. Phys. Chem. 1997;48(1):545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 38.Baldwin A.J., Knowles T.P., Tartaglia G.G., et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011;133(36):14160–14163. doi: 10.1021/ja2017703. [DOI] [PubMed] [Google Scholar]
- 39.Fändrich M., Forge V., Buder K., Kittler M., Dobson C.M., Diekmann S. Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proc. Natl. Acad. Sci. USA. 2003;100(26):15463–15468. doi: 10.1073/pnas.0303758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santambrogio C., Frana A.M., Natalello A., et al. The role of the central flexible region on the aggregation and conformational properties of human ataxin-3. FEBS J. 2012;279(3):451–463. doi: 10.1111/j.1742-4658.2011.08438.x. [DOI] [PubMed] [Google Scholar]
- 41.Neupert W., Herrmann J.M. Translocation of Proteins into Mitochondria. Annu. Rev. Biochem. 2007;76(1):723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 42.Goers J., Manning-Bog A.B., McCormack A.L., et al. Nuclear localization of α-synuclein and its interaction with histones. Biochemistry. 2003;42(28):8465–8471. doi: 10.1021/bi0341152. [DOI] [PubMed] [Google Scholar]
- 43.Chua C.E., Chua C.E., Tang B.L. Rabs, SNAREs and α-synuclein — Membrane trafficking defects in synucleinopathies. Brain Res. Brain Res. Rev. 2011;67(1-2):268–281. doi: 10.1016/j.brainresrev.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Stefani M., Dobson C.M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003;81(11):678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 45.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003;4(1):49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 46.Jahn T.R., Radford S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 2008;469(1):100–117. doi: 10.1016/j.abb.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.White D.A., Buell A.K., Knowles T.P., Welland M.E., Dobson C.M. Protein Aggregation in Crowded Environments. J. Am. Chem. Soc. 2010;132(14):5170–5175. doi: 10.1021/ja909997e. [DOI] [PubMed] [Google Scholar]
- 48.Sunde M., Serpell L.C., Bartlam M., Fraser P.E., Pepys M.B., Blake C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997;273(3):729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 49.Gillam J.E., MacPhee C.E. Modelling amyloid fibril formation kinetics: mechanisms of nucleation and growth. J. Phys. Condens. Matter. 2013;25(37):373101. doi: 10.1088/0953-8984/25/37/373101. [DOI] [PubMed] [Google Scholar]
- 50.Nayak A., Sorci M., Krueger S., Belfort G. A universal pathway for amyloid nucleus and precursor formation for insulin. Prot Struct Funct Bioinformatics. 2009;74(3):556–565. doi: 10.1002/prot.22169. [DOI] [PubMed] [Google Scholar]
- 51.Militello V., Casarino C., Emanuele A., Giostra A., Pullara F., Leone M. Aggregation kinetics of bovine serum albumin studied by FTIR spectroscopy and light scattering. Biophys. Chem. 2004;107(2):175–187. doi: 10.1016/j.bpc.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 52.Levine H. Thioflavine-T interaction with synthetic alzheimers-disease beta-amyloid peptides - detection of amyloid aggregation in solution. Protein Sci. 1993;2(3):404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeong J.S., Ansaloni A., Mezzenga R., Lashuel H.A., Dietler G. Novel mechanistic insight into the molecular basis of amyloid polymorphism and secondary nucleation during amyloid formation. J. Mol. Biol. 2013;425(10):1765–1781. doi: 10.1016/j.jmb.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 54.Cohen S.I., Cohen S.I., Vendruscolo M., Dobson C.M., Knowles T.P., Knowles T.P. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012;421(2-3):160–171. doi: 10.1016/j.jmb.2012.02.031. [DOI] [PubMed] [Google Scholar]
- 55.Michaels T.C., Knowles T.P. Mean-field master equation formalism for biofilament growth. Am. J. Phys. 2014;82(5):476–483. [Google Scholar]
- 56.Knowles T.P., Waudby C.A., Devlin G.L., et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326(5959):1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 57.Cohen S.I., Linse S., Luheshi L.M., et al. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA. 2013;110(24):9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chayen N.E. Methods for separating nucleation and growth in protein crystallisation. Prog. Biophys. Mol. Biol. 2005;88(3):329–337. doi: 10.1016/j.pbiomolbio.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 59.Bouchard M., Zurdo J., Nettleton E.J., Dobson C.M., Robinson C.V. Formation of insulin amyloid fibrils followed by FTIR simultaneously with CD and electron microscopy. Protein Sci. 2000;9(10):1960–1967. doi: 10.1110/ps.9.10.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chow M.K., Ellisdon A.M., Cabrita L.D., Bottomley S.P. Polyglutamine expansion in ataxin-3 does not affect protein stability: Implications for misfolding and disease. J. Biol. Chem. 2004;279(46):47643–47651. doi: 10.1074/jbc.M405799200. [DOI] [PubMed] [Google Scholar]
- 61.Ruggeri F.S., Longo G., Faggiano S., Lipiec E., Pastore A., Dietler G. Infrared nanospectroscopy characterization of oligomeric and fibrillar aggregates during amyloid formation. Nat. Commun. 2015;•••:6. doi: 10.1038/ncomms8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buell A.K., Dhulesia A., White D.A., Knowles T.P., Dobson C.M., Welland M.E. Detailed Analysis of the Energy Barriers for Amyloid Fibril Growth. Angew. Chem. Int. Ed. 2012;51(21):5247–5251. doi: 10.1002/anie.201108040. [DOI] [PubMed] [Google Scholar]
- 63.Knowles T.P., Shu W., Devlin G.L., et al. Kinetics and thermodynamics of amyloid formation from direct measurements of fluctuations in fibril mass. Proc. Natl. Acad. Sci. USA. 2007;104(24):10016–10021. doi: 10.1073/pnas.0610659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.White DA. Biosensor-based label-free assays of amyloid growth. FEBS Lett. 2009;583(16):2587–2592. doi: 10.1016/j.febslet.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 65.Buell A.K., Dobson C.M., Knowles T.P. The physical chemistry of the amyloid phenomenon: thermodynamics and kinetics of filamentous protein aggregation. Essays Biochem. 2014;56:11–39. doi: 10.1042/bse0560011. [DOI] [PubMed] [Google Scholar]
- 66.Pedersen J.S., Andersen C.B., Otzen D.E. Amyloid structure - one but not the same: the many levels of fibrillar polymorphism. FEBS J. 2010;277(22):4591–4601. doi: 10.1111/j.1742-4658.2010.07888.x. [DOI] [PubMed] [Google Scholar]
- 67.Vendruscolo M., Dobson C.M. Structural biology: Protein self-assembly intermediates. Nat. Chem. Biol. 2013;9(4):216–217. doi: 10.1038/nchembio.1210. [DOI] [PubMed] [Google Scholar]
- 68.Gales L., Cortes L., Almeida C., et al. Towards a structural understanding of the fibrillization pathway in machado-Joseph's Disease: Trapping early oligomers of non-expanded ataxin-3. J. Mol. Biol. 2005;353(3):642–654. doi: 10.1016/j.jmb.2005.08.061. [DOI] [PubMed] [Google Scholar]
- 69.Hinault M. Stable-synuclein oligomers strongly inhibit chaperone activity of the hsp70 system by weak interactions with J-domain co-chaperones. J. Biol. Chem. 2010;285(49):38173–38182. doi: 10.1074/jbc.M110.127753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong D., Han S., Fink A.L., Uversky V.N. Characterization of the non-fibrillar α-synuclein oligomers. Protein Pept. Lett. 2011;18(3):230–240. doi: 10.2174/092986611794578332. [DOI] [PubMed] [Google Scholar]
- 71.McLean P., Kawamata H., Shariff S. TorsinA and heat shock proteins act as molecular chaperones: suppression of α‐synuclein aggregation. J. Neurochem. 2002;83(4):846–854. doi: 10.1046/j.1471-4159.2002.01190.x. [DOI] [PubMed] [Google Scholar]
- 72.Muchowski P.J. Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron. 2002;35(1):9–12. doi: 10.1016/s0896-6273(02)00761-4. [DOI] [PubMed] [Google Scholar]
- 73.Sherman M.Y., Goldberg A.L. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29(1):15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 74.Haass C. x, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 75.Eisenberg D., Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148(6):1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alzheimer's A. 2012 Alzheimer's disease facts and figures. Alzheimers Dement. 2012;8(2):131–168. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 77.Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 78.Glenner G.G., Wong C.W. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 79.Masters C.L., Simms G., Weinman N.A., Multhaup G., McDonald B.L., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 1985;82(12):4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Busciglio J., Gabuzda D.H., Matsudaira P., Yankner B.A. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. USA. 1993;90(5):2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Palop J.J., Mucke L. Amyloid-[beta]-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat. Neurosci. 2010;13(7):812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barghorn S., Davies P., Mandelkow E. Tau paired helical filaments from Alzheimer's disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry. 2004;43(6):1694–1703. doi: 10.1021/bi0357006. [DOI] [PubMed] [Google Scholar]
- 83.Hardy J., Higgins G. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 84.Lemere C.A., Blusztajn J.K., Yamaguchi H., Wisniewski T., Saido T.C., Selkoe D.J. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis. 1996;3(1):16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 85.Terry R.D., Masliah E., Salmon D.P., et al. Physical basis of cognitive alterations in alzheimers-disease - synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 86.Hasegawa K., Yamaguchi I., Omata S., Gejyo F., Naiki H. Interaction between A beta(1-42) and A beta(1-40) in Alzheimer's beta-amyloid fibril formation in vitro. Biochemistry. 1999;38(47):15514–15521. doi: 10.1021/bi991161m. [DOI] [PubMed] [Google Scholar]
- 87.Iwatsubo T., Odaka A., Suzuki N., Mizusawa H., Nukina N., Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron. 1994;13(1):45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 88.Jan A., Gokce O., Luthi-Carter R., Lashuel H.A. The ratio of monomeric to aggregated forms of A beta 40 and A beta 42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J. Biol. Chem. 2008;283(42):28176–28189. doi: 10.1074/jbc.M803159200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hsia A.Y., Masliah E., McConlogue L., et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc. Natl. Acad. Sci. USA. 1999;96(6):3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chapman P.F., White G.L., Jones M.W., et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 1999;2(3):271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 91.Walsh D.M., Klyubin I., Fadeeva J.V., et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 92.Jan A., Adolfsson O., Allaman I., et al. A beta 42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete A beta 42 Species. J. Biol. Chem. 2011;286(10):8585–8596. doi: 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goedert M., Spillantini M.G. A century of Alzheimer's disease. Science. 2006;314(5800):777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 94.Hamley I.W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012;112(10):5147–5192. doi: 10.1021/cr3000994. [DOI] [PubMed] [Google Scholar]
- 95.Butterfield S.M., Lashuel H.A. Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem. Int. Ed. 2010;49(33):5628–5654. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 96.Tanzi R.E., Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: A genetic perspective. Cell. 2005;120(4):545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 97.Campioni S., Mannini B., Zampagni M., et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010;6(2):140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- 98.Shankar G.M., Li S., Mehta T.H., et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mannini B., Mulvihill E., Sgromo C., et al. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 2014;9(10):2309–2317. doi: 10.1021/cb500505m. [DOI] [PubMed] [Google Scholar]
- 100.Meisl G., Yang X., Frohm B., Knowles T.P., Linse S. Quantitative analysis of intrinsic and extrinsic factors in the aggregation mechanism of Alzheimer-associated A beta-peptide. Sci. Rep. 2016;6:8728. doi: 10.1038/srep18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Szczepankiewicz O., Linse B., Meisl G., et al. N-Terminal Extensions Retard A beta 42 Fibril Formation but Allow Cross-Seeding and Coaggregation with A beta 42. J. Am. Chem. Soc. 2015;137(46):14673–14685. doi: 10.1021/jacs.5b07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Selkoe D.J. Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 2011;17(9):1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 103.Cummings J., Zhong K. Biomarker-driven therapeutic management of Alzheimer's disease: establishing the foundations. Clin. Pharmacol. Ther. 2014;95(1):67–77. doi: 10.1038/clpt.2013.205. [DOI] [PubMed] [Google Scholar]
- 104.Arosio P., Vendruscolo M., Dobson C.M., Knowles T.P. Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 2014;35(3):127–135. doi: 10.1016/j.tips.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 105.Martin S.R., Schilstra M.J. Circular Dichroism and Its Application to the Study of Biomolecules. In: John J.C., William Detrich H. III, editors. Methods in Cell Biology. Vol. 84. USA: Academic Press; 2008. pp. 263–293. [DOI] [PubMed] [Google Scholar]
- 106.Kelly S.M., Jess T.J., Price N.C. How to study proteins by circular dichroism. Biochim Biophys Acta (BBA) -. Proteins Proteomics. 2005;1751(2):119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 107.Chiti F., Webster P., Taddei N., et al. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA. 1999;96(7):3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Calero M., Gasset M. Featuring amyloids with Fourier transform infrared and circular dichroism spectroscopies. Methods Mol. Biol. 2012;849:53–68. doi: 10.1007/978-1-61779-551-0_5. [DOI] [PubMed] [Google Scholar]
- 109.Khrapunov S. CD spectroscopy has intrinsic limitations for protein secondary structure analysis. Anal. Biochem. 2009;389(2):174–176. doi: 10.1016/j.ab.2009.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rogers DR. Screening for amyloid with thioflavin-t fluorescent method. 1965. [DOI] [PubMed]
- 111.Krebs M.R., Bromley E.H., Donald A.M. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J. Struct. Biol. 2005;149(1):30–37. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 112.Biancalana M., Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta (BBA) -. Proteins Proteomics. 2010;1804(7):1405–1412. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Michaels T.C., Lazell H.W., Arosio P., Knowles T.P. Dynamics of protein aggregation and oligomer formation governed by secondary nucleation. J. Chem. Phys. 2015;143(5):054901. doi: 10.1063/1.4927655. [DOI] [PubMed] [Google Scholar]
- 114.Meisl G., Kirkegaard J.B., Arosio P., et al. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 2016;11(2):252–272. doi: 10.1038/nprot.2016.010. [DOI] [PubMed] [Google Scholar]
- 115.Habchi J., Arosio P., Perni M., et al. An anti-cancer drug suppresses the primary nucleation reaction and delays the production of the toxic Ab42 aggregates linked with Alzheimer’s disease. Sci Adv. 2016;2(2):e1501244. doi: 10.1126/sciadv.1501244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wolfe L.S., Calabrese M.F., Nath A., Blaho D.V., Miranker A.D., Xiong Y. Protein-induced photophysical changes to the amyloid indicator dye thioflavin T. Proc. Natl. Acad. Sci. USA. 2010;107(39):16863–16868. doi: 10.1073/pnas.1002867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.LeVine Iii H. [18] Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 118.Sanchez M.M., Moghadam S., Naik P., Martin K.J., Salehi A. Hippocampal network alterations in Alzheimer's disease and down syndrome: from structure to therapy. Adv. Alzheimer Dis. 2011;26(Suppl. 3):29–47. doi: 10.3233/JAD-2011-0050. [DOI] [PubMed] [Google Scholar]
- 119.Sanchez L., Madurga S., Pukala T., et al. A beta 40 and A beta 42 amyloid fibrils exhibit distinct molecular recycling properties. J. Am. Chem. Soc. 2011;133(17):6505–6508. doi: 10.1021/ja1117123. [DOI] [PubMed] [Google Scholar]
- 120.Serra-Vidal B., Pujadas L., Rossi D., Soriano E., Madurga S., Carulla N. Hydrogen/Deuterium exchange-protected oligomers populated during a beta fibril formation correlate with neuronal cell death. ACS Chem. Biol. 2014;9(11):2678–2685. doi: 10.1021/cb500621x. [DOI] [PubMed] [Google Scholar]
- 121.Marcoux J., Robinson Carol V. Twenty years of gas phase structural biology. Structure. 2013;21(9):1541–1550. doi: 10.1016/j.str.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 122.Bernstein S.L., Dupuis N.F., Lazo N.D., et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 2009;1(4):326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kloniecki M., Jablonowska A., Poznanski J., et al. Ion mobility separation coupled with MS detects two structural states of Alzheimer's disease A beta 1-40 peptide oligomers. J. Mol. Biol. 2011;407(1):110–124. doi: 10.1016/j.jmb.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 124.Bernstein S.L., Wyttenbach T., Baumketner A., et al. Amyloid beta-protein: Monomer structure and early aggregation states of A beta 42 and its Pro(19) alloform. J. Am. Chem. Soc. 2005;127(7):2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 125.Pujol-Pina R., Vilaprinyo-Pascual S., Mazzucato R., et al. SDS-PAGE analysis of A beta oligomers is disserving research into Alzheimer's disease: appealing for ESI-IM-MS. Sci. Rep. 2015:5. doi: 10.1038/srep14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lanucara F., Holman S.W., Gray C.J., Eyers C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014;6(4):281–294. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- 127.Kumosinski T.F., Unruh J.J. Quantitation of the global secondary structure of globular proteins by FTIR spectroscopy: Comparison with X-ray crystallographic structure. Talanta. 1996;43(2):199–219. doi: 10.1016/0039-9140(95)01726-7. [DOI] [PubMed] [Google Scholar]
- 128.Barth A. Infrared spectroscopy of proteins. Biochim Biophys Acta (BBA) -. Bioenergetics. 2007;1767(9):1073–1101. doi: 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 129.Sarroukh R., Goormaghtigh E., Ruysschaert J-M., Raussens V. ATR-FTIR: A “rejuvenated” tool to investigate amyloid proteins. Biochim Biophys Acta (BBA) -. Biomembranes. 2013;1828(10):2328–2338. doi: 10.1016/j.bbamem.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 130.Sassi P., Giugliarelli R., Paolantoni M., et al. Unfolding and aggregation of lysozyme: A thermodynamic and kinetic study by FTIR spectroscopy. Biophys. Chem. 2011;158(1):46–53. doi: 10.1016/j.bpc.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 131.Kong J., Yu S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. (Shanghai) 2007;39(8):549–559. doi: 10.1111/j.1745-7270.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- 132.Haris P., Severcan F. FTIR spectroscopic characterization of protein structure in aqueous and non-aqueous media. J. Mol. Catal., B Enzym. 1999;7(1-4):207–221. [Google Scholar]
- 133.Shivu B., Seshadri S., Li J., Oberg K.A., Uversky V.N., Fink A.L. Distinct β-sheet structure in protein aggregates determined by ATR-FTIR Spectroscopy. Biochemistry. 2013;52(31):5176–5183. doi: 10.1021/bi400625v. [DOI] [PubMed] [Google Scholar]
- 134.Khurana R., Fink A.L. Do Parallel β-helix proteins have a unique fourier transform infrared spectrum? Biophys. J. 2000;78(2):994–1000. doi: 10.1016/S0006-3495(00)76657-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stuart B.H. Infrared Spectroscopy. Fundam Appl. 2005;25:1–208. [Google Scholar]
- 136.Cappella B., Dietler G. Force-distance curves by atomic force microscopy. Surf. Sci. Rep. 1999;34(1-3):1–104. [Google Scholar]
- 137.Ruggeri F.S., Byrne C., Khemtemourian L., Ducouret G., Dietler G., Jacquot Y. Concentration-dependent and surface-assisted self-assembly properties of a bioactive estrogen receptor alpha-derived peptide. J. Pept. Sci. 2015;21(2):95–104. doi: 10.1002/psc.2730. [DOI] [PubMed] [Google Scholar]
- 138.Müller T., Ruggeri F.S., Kulik A.J., et al. Nanoscale spatially resolved infrared spectra from single microdroplets. Lab Chip. 2014;14(7):1315–1319. doi: 10.1039/c3lc51219c. [DOI] [PubMed] [Google Scholar]
- 139.Kulik A., Ruggeri F.S., Gruszecki W.I., Dietler G. Nanoscale infrared spectroscopy of light harvesting proteins, amyloid structures and collagen fibres. Microsc. Anal. (Am. Ed.) 2014;28(4):11–15. [Google Scholar]
- 140.Born M., Wolf E. Principles of optics: electromagnetic theory of propagation, interference and diffraction of light. UK: Cambridge University Press; 1999. [Google Scholar]
- 141.Binnig G., Rohrer H. Scanning tunneling microscopy. IBM J. Res. Develop. 2000;44(1-2):279–293. [Google Scholar]
- 142.Allison D.P., Mortensen N.P., Sullivan C.J., Doktycz M.J. Atomic force microscopy of biological samples. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010;2(6):618–634. doi: 10.1002/wnan.104. [DOI] [PubMed] [Google Scholar]
- 143.Hendrik H. Atomic force microscopy and spectroscopy. Dynamic Force Spectroscopy and Biomolecular Recognition. USA: CRC Press; 2012. pp. 51–91. [Google Scholar]
- 144.Garcia R., Perez R. Dynamic atomic force microscopy methods. Surf. Sci. Rep. 2002;47(6-8):197–301. [Google Scholar]
- 145.de Pablo PJ, Carrion-Vazquez M. Imaging biological samples with atomic force microscopy. 2014. [DOI] [PubMed]
- 146.Kuznetsov Y.G., McPherson A. Atomic force microscopy in imaging of viruses and virus-infected cells. Microbiol. Mol. Biol. Rev. 2011;75(2):268–285. doi: 10.1128/MMBR.00041-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alonso-Sarduy L., Longo G., Dietler G., Kasas S. Time-Lapse AFM imaging of DNA conformational changes induced by daunorubicin. Nano Lett. 2013;13(11):5679–5684. doi: 10.1021/nl403361f. [DOI] [PubMed] [Google Scholar]
- 148.Sweers K.K., Segers-Nolten I.M., Bennink M.L., Subramaniam V. Structural model for α-synuclein fibrils derived from high resolution imaging and nanomechanical studies using atomic force microscopy. Soft Matter. 2012;8(27):7215. [Google Scholar]
- 149.Adamcik J., Jung J.M., Flakowski J., De Los Rios P., Dietler G., Mezzenga R. Understanding amyloid aggregation by statistical analysis of atomic force microscopy images. Nat. Nanotechnol. 2010;5(6):423–428. doi: 10.1038/nnano.2010.59. [DOI] [PubMed] [Google Scholar]
- 150.Khalaf O., Fauvet B., Oueslati A., et al. The H50Q mutation enhances alpha-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 2014;289(32):21856–21876. doi: 10.1074/jbc.M114.553297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim H., Cho M., Kumar A., et al. Structural properties of pore-forming oligomers of α-synuclein. J. Am. Chem. Soc. 2009;131(47):17482–17489. doi: 10.1021/ja9077599. [DOI] [PubMed] [Google Scholar]
- 152.Hoyer W., Cherny D., Subramaniam V., Jovin T.M. Rapid self-assembly of α-synuclein observed by in situ atomic force microscopy. J. Mol. Biol. 2004;340(1):127–139. doi: 10.1016/j.jmb.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 153.Ansaloni A., Wang Z-M., Jeong J.S., Ruggeri F.S., Dietler G., Lashuel H.A. One-Pot Semisynthesis of Exon 1 of the Huntingtin Protein: New tools for elucidating the role of posttranslational modifications in the pathogenesis of Huntington's disease. Angew. Chem. Int. Ed. 2014;53(7):1928–1933. doi: 10.1002/anie.201307510. [DOI] [PubMed] [Google Scholar]
- 154.Conway K.A., Lee S.J., Rochet J.C., Ding T.T., Williamson R.E., Lansbury P.T. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA. 2000;97(2):571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Poirier M.A., Li H., Macosko J., Cai S., Amzel M., Ross C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 2002;277(43):41032–41037. doi: 10.1074/jbc.M205809200. [DOI] [PubMed] [Google Scholar]
- 156.Conway K.A., Harper J.D., Lansbury P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998;4(11):1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 157.Eanes E.D., Glenner G.G. X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 1968;16(11):673. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- 158.Greenwald J., Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18(10):1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 159.Makin O.S., Atkins E., Sikorski P., Johansson J., Serpell L.C. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA. 2005;102(2):315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Khurana R., Ionescu-Zanetti C., Pope M., et al. A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophys. J. 2003;85(2):1135–1144. doi: 10.1016/S0006-3495(03)74550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fändrich M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell. Mol. Life Sci. 2007;64(16):2066–2078. doi: 10.1007/s00018-007-7110-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fandrich M., Meinhardt J., Grigorieff N. Structural polymorphism of Alzheimer Abeta and other amyloid fibrils. Prion. 2009;3(2):89–93. doi: 10.4161/pri.3.2.8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Volpatti L.R., Vendruscolo M., Dobson C.M., Knowles T.P. A clear view of polymorphism, twist, and chirality in amyloid fibril formation. ACS Nano. 2013;7(12):10443–10448. doi: 10.1021/nn406121w. [DOI] [PubMed] [Google Scholar]
- 164.Usov I., Adamcik J., Mezzenga R. Polymorphism complexity and handedness inversion in serum albumin amyloid fibrils. ACS Nano. 2013;7(12):10465–10474. doi: 10.1021/nn404886k. [DOI] [PubMed] [Google Scholar]
- 165.Usov I., Mezzenga R. Correlation between nanomechanics and polymorphic conformations in amyloid fibrils. ACS Nano. 2014;8(11):11035–11041. doi: 10.1021/nn503530a. [DOI] [PubMed] [Google Scholar]
- 166.Usov I., Adamcik J., Mezzenga R. Polymorphism in bovine serum albumin fibrils: morphology and statistical analysis. Faraday Discuss. 2013;2013(166):151–162. doi: 10.1039/c3fd00083d. [DOI] [PubMed] [Google Scholar]
- 167.vandenAkker CC, Engel MFM, Velikov KP, Bonn M, Koenderink GH. Morphology and persistence length of amyloid fibrils are correlated to peptide molecular structure. J. Am. Chem. Soc. 2011;133(45):18030–18033. doi: 10.1021/ja206513r. [DOI] [PubMed] [Google Scholar]
- 168.Volpatti L.R., Knowles T.P. Polymer physics inspired approaches for the study of the mechanical properties of amyloid fibrils. J. Polym. Sci., B, Polym. Phys. 2014;52(4):281–292. [Google Scholar]
- 169.Gittes F., Mickey B., Nettleton J., Howard J. Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. J. Cell Biol. 1993;120(4):923–934. doi: 10.1083/jcb.120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Knowles T.P., Fitzpatrick A.W., Meehan S., et al. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318(5858):1900–1903. doi: 10.1126/science.1150057. [DOI] [PubMed] [Google Scholar]
- 171.Harper J.D., Wong S.S., Lieber C.M., Lansbury P.T. Assembly of A beta amyloid protofibrils: An in vitro model for a possible early event in Alzheimer's disease. Biochemistry. 1999;38(28):8972–8980. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- 172.Lovas S., Zhang Y., Yu J., Lyubchenko Y.L. Molecular mechanism of misfolding and aggregation of Aβ(13-23). J. Phys. Chem. 2013;117(20):6175–6186. doi: 10.1021/jp402938p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Antzutkin O.N. Amyloidosis of Alzheimer's Aβ peptides: solid-state nuclear magnetic resonance, electron paramagnetic resonance, transmission electron microscopy, scanning transmission electron microscopy and atomic force microscopy studies. Magn. Reson. Chem. 2004;42(2):231–246. doi: 10.1002/mrc.1341. [DOI] [PubMed] [Google Scholar]
- 174.Kowalewski T., Holtzman D.M. In situ atomic force microscopy study of Alzheimer’s β-amyloid peptide on different substrates: New insights into mechanism of β-sheet formation. Proc. Natl. Acad. Sci. USA. 1999;96(7):3688–3693. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chromy B.A., Nowak R.J., Lambert M.P., et al. Self-assembly of Aβ1-42 into globular Neurotoxins†. Biochemistry. 2003;42(44):12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 176.Liu R., Yuan B., Emadi S., et al. Single chain variable fragments against β-amyloid (Aβ) can inhibit Aβ aggregation and prevent Aβ-induced neurotoxicity†. Biochemistry. 2004;43(22):6959–6967. doi: 10.1021/bi049933o. [DOI] [PubMed] [Google Scholar]
- 177.Legleiter J., Czilli D.L., Gitter B., DeMattos R.B., Holtzman D.M., Kowalewski T. Effect of different anti-Aβ antibodies on aβ fibrillogenesis as assessed by atomic force microscopy. J. Mol. Biol. 2004;335(4):997–1006. doi: 10.1016/j.jmb.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 178.McConney M.E., Singamaneni S., Tsukruk V.V. Probing Soft Matter with the Atomic Force Microscopies: Imaging and Force Spectroscopy. Polym. Rev. (Phila. Pa.) 2010;50(3):235–286. [Google Scholar]
- 179.Florin E.L., Rief M., Lehmann H., et al. Sensing specific molecular interactions with the atomic force microscope. Biosens. Bioelectron. 1995;10(9-10):895–901. [Google Scholar]
- 180.Kellermayer M.S., Grama L., Karsai A., et al. Reversible mechanical unzipping of amyloid beta-fibrils. J. Biol. Chem. 2005;280(9):8464–8470. doi: 10.1074/jbc.M411556200. [DOI] [PubMed] [Google Scholar]
- 181.Karsai A., Martonfalvi Z., Nagy A., Grama L., Penke B., Kellermayer M.S. Mechanical manipulation of Alzheimer's amyloid beta 1-42 fibrils. J. Struct. Biol. 2006;155(2):316–326. doi: 10.1016/j.jsb.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 182.Dunstan D.E., Hamilton-Brown P., Asimakis P., Ducker W., Bertolini J. Shear-induced structure and mechanics of [small beta]-lactoglobulin amyloid fibrils. Soft Matter. 2009;5(24):5020–5028. [Google Scholar]
- 183.Perkins T.T., Smith D.E., Larson R.G., Chu S. Stretching of a single tethered polymer in a uniform-flow. Science. 1995;268(5207):83–87. doi: 10.1126/science.7701345. [DOI] [PubMed] [Google Scholar]
- 184.Wiggins P.A., Phillips R., Nelson P.C. Exact theory of kinkable elastic polymers. Phys Rev E Stat Nortin Soft Matter Phyp. 2005;71(2 pt 1):021909. doi: 10.1103/PhysRevE.71.021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mostaert A.S., Higgins M.J., Fukuma T., Rindi F., Jarvis S.P. Nanoscale mechanical characterisation of amyloid fibrils discovered in a natural adhesive. J. Biol. Phys. 2006;32(5):393–401. doi: 10.1007/s10867-006-9023-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Anika S.M., Suzanne P.J. Beneficial characteristics of mechanically functional amyloid fibrils evolutionarily preserved in natural adhesives. Nanotechnology. 2007;18(4):044010. [Google Scholar]
- 187.Schleeger M. Amyloids: From molecular structure to mechanical properties. Polymer (Guildf.) 2013;54(10):2473–2488. [Google Scholar]
- 188.Sweers K., Werf K., Bennink M., Subramaniam V., Werf K. Nanomechanical properties of α-synuclein amyloid fibrils: a comparative study by nanoindentation, harmonic force microscopy, and Peakforce QNM. Nanoscale Res. Lett. 2011;6(1):270. doi: 10.1186/1556-276X-6-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ramos J.R., Pabijan J., Garcia R., Lekka M. The softening of human bladder cancer cells happens at an early stage of the malignancy process. Beilstein J. Nanotechnol. 2014;5:447–457. doi: 10.3762/bjnano.5.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kasas S., Longo G., Dietler G. Mechanical properties of biological specimens explored by atomic force microscopy. J. Phys. D Appl. Phys. 2013;46(13) [Google Scholar]
- 191.Adamcik J., Berquand A., Mezzenga R. Single-step direct measurement of amyloid fibrils stiffness by peak force quantitative nanomechanical atomic force microscopy. Appl. Phys. Lett. 2011;98(19):193701. [Google Scholar]
- 192.Adamcik J., Lara C., Usov I., et al. Measurement of intrinsic properties of amyloid fibrils by the peak force QNM method. Nanoscale. 2012;4(15):4426. doi: 10.1039/c2nr30768e. [DOI] [PubMed] [Google Scholar]
- 193.Lara C, Gourdin-Bertin S, Adamcik J, Bolisetty S, Mezzenga R. Self-assembly of ovalbumin into amyloid and non-amyloid fibrils. 2012. [DOI] [PubMed]
- 194.Ruggeri F.S., Adamcik J., Jeong J.S., Lashuel H.A., Mezzenga R., Dietler G. Influence of the β-sheet content on the mechanical properties of aggregates during amyloid fibrillization. Angew. Chem. Int. Ed. Engl. 2015;54(8):2462–2466. doi: 10.1002/anie.201409050. [DOI] [PubMed] [Google Scholar]
- 195.Jackson M., Mantsch H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Critic Rev Biochem. 1995;30(2):95–120. doi: 10.3109/10409239509085140. [DOI] [PubMed] [Google Scholar]
- 196.Carr G.L. Resolution limits for infrared microspectroscopy explored with synchrotron radiation. Rev. Sci. Instrum. 2001;72(3):1613–1619. [Google Scholar]
- 197.Dazzi A., Prater C.B., Hu Q., Chase D.B., Rabolt J.F., Marcott C. AFM-IR: Combining atomic force microscopy and infrared spectroscopy for nanoscale chemical characterization. Appl. Spectrosc. 2012;66(12):1365–1384. doi: 10.1366/12-06804. [DOI] [PubMed] [Google Scholar]
- 198.Dazzi A., Glotin F., Carminati R. Theory of infrared nanospectro-scopy by photothermal induced resonance. J. Appl. Phys. 2010;107(12) [Google Scholar]
- 199.Lahiri B., Holland G., Centrone A. Chemical imaging beyond the diffraction limit: experimental validation of the PTIR technique. Small. 2013;9(3):439–445. doi: 10.1002/smll.201200788. [DOI] [PubMed] [Google Scholar]
- 200.Janik E., Bednarska J., Zubik M., et al. Molecular architecture of plant thylakoids under physiological and light stress conditions: A study of lipid-Light-Harvesting complex II model membranes. Plant Cell. 2013;25(6):2155–2170. doi: 10.1105/tpc.113.113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lu F., Jin M., Belkin M.A. Tip-enhanced infrared nanospectroscopy via molecular expansion force detection. Nat. Photonics. 2014;8(4):307–312. [Google Scholar]
- 202.Mayet C., Dazzi A., Prazeres R., Allot F., Glotin F., Ortega J.M. Sub-100 nm IR spectromicroscopy of living cells. Opt. Lett. 2008;33(14):1611–1613. doi: 10.1364/ol.33.001611. [DOI] [PubMed] [Google Scholar]
- 203.Dazzi A., Prazeres R., Glotin F., Ortega J.M., Al-Sawaftah M., de Frutos M. Chemical mapping of the distribution of viruses into infected bacteria with a photothermal method. Ultramicroscopy. 2008;108(7):635–641. doi: 10.1016/j.ultramic.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 204.Mayet C., Dazzi A., Prazeres R., Ortega J.M., Jaillard D. In situ identification and imaging of bacterial polymer nanogranules by infrared nanospectroscopy. Analyst (Lond.) 2010;135(10):2540–2545. doi: 10.1039/c0an00290a. [DOI] [PubMed] [Google Scholar]
- 205.Mayet C., Deniset-Besseau A., Prazeres R., Ortega J.M., Dazzi A. Analysis of bacterial polyhydroxybutyrate production by multimodal nanoimaging. Biotechnol. Adv. 2013;31(3):369–374. doi: 10.1016/j.biotechadv.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 206.Policar C., Waern J.B., Plamont M.A., et al. Subcellular IR imaging of a metal-carbonyl moiety using photothermally induced resonance. Angew. Chem. Int. Ed. Engl. 2011;50(4):860–864. doi: 10.1002/anie.201003161. [DOI] [PubMed] [Google Scholar]
- 207.Gourion-Arsiquaud S., Marcott C., Hu Q., Boskey A.L. Studying variations in bone composition at nano-scale resolution: a preliminary report. Calcif. Tissue Int. 2014;95(5):413–418. doi: 10.1007/s00223-014-9909-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Baldassarre L., Giliberti V., Rosa A., et al. Mapping the amide I absorption in single bacteria and mammalian cells with resonant infrared nanospectroscopy. Nanotechnology. 2016;27(7):075101. doi: 10.1088/0957-4484/27/7/075101. [DOI] [PubMed] [Google Scholar]
- 209.Marcott C., Lo M., Kjoller K., Domanov Y., Balooch G., Luengo G.S. Nanoscale infrared (IR) spectroscopy and imaging of structural lipids in human stratum corneum using an atomic force microscope to directly detect absorbed light from a tunable IR laser source. Exp. Dermatol. 2013;22(6):419–421. doi: 10.1111/exd.12144. [DOI] [PubMed] [Google Scholar]
- 210.Marcott C., Lo M., Kjoller K., et al. Localization of human hair structural lipids using nanoscale infrared spectroscopy and imaging. Appl. Spectrosc. 2014;68(5):564–569. doi: 10.1366/13-07328. [DOI] [PubMed] [Google Scholar]
- 211.Deniset-Besseau A., Prater C.B., Virolle M.J., Dazzi A. Monitoring triacylglycerols accumulation by atomic force microscopy based infrared spectroscopy in streptomyces species for biodiesel applications. J. Phys. Chem. Lett. 2014;5(4):654–658. doi: 10.1021/jz402393a. [DOI] [PubMed] [Google Scholar]
- 212.Janik E., Bednarska J., Zubik M., et al. Molecular architecture of plant thylakoids under physiological and light stress conditions: a study of lipid-light-harvesting complex II model membranes. Plant Cell. 2013;25(6):2155–2170. doi: 10.1105/tpc.113.113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Gruszecki W.I., Kulik A.J., Janik E., et al. Nanoscale resolution in infrared imaging of protein-containing lipid membranes. Nanoscale. 2015;7(35):14659–14662. doi: 10.1039/c5nr03090k. [DOI] [PubMed] [Google Scholar]
- 214.Muller T., Ruggeri F.S., Kulik A.J., et al. Nanoscale spatially resolved infrared spectra from single microdroplets. Lab Chip. 2014;14(7):1315–1319. doi: 10.1039/c3lc51219c. [DOI] [PubMed] [Google Scholar]