

Abstract
Sleep apnea (SA) is increasing in prevalence and is commonly comorbid with hypertension. Chronic intermittent hypoxia is used to model the arterial hypoxemia seen in SA, and through this paradigm, the mechanisms that underlie SA-induced hypertension are becoming clear. Cyclic hypoxic exposure during sleep chronically stimulates the carotid chemoreflexes, inducing sensory long-term facilitation, and drives sympathetic outflow from the hindbrain. The elevated sympathetic tone drives hypertension and renal sympathetic activity to the kidneys resulting in increased plasma renin activity and eventually angiotensin II (Ang II) peripherally. Upon waking, when respiration is normalized, the sympathetic activity does not diminish. This is partially because of adaptations leading to overactivation of the hindbrain regions controlling sympathetic outflow such as the nucleus tractus solitarius (NTS), and rostral ventrolateral medulla (RVLM). The sustained sympathetic activity is also due to enhanced synaptic signaling from the forebrain through the paraventricular nucleus (PVN). During the waking hours, when the chemoreceptors are not exposed to hypoxia, the forebrain circumventricular organs (CVOs) are stimulated by peripherally circulating Ang II from the elevated plasma renin activity. The CVOs and median preoptic nucleus chronically activate the PVN due to the Ang II signaling. All together, this leads to elevated nocturnal mean arterial pressure (MAP) as a response to hypoxemia, as well as inappropriately elevated diurnal MAP in response to maladaptations.
Keywords: Sleep apnea (SA), Sympathetic nerve activity (SNA), Hypertension, Chronic intermittent hypoxia (CIH)
Introduction
Sleep apnea (SA) commonly refers to a group of disorders that are characterized by periodic interruption of respiration or hypoventilatory breathing during sleep. Sufferers experience airway obstruction, hypoxia, hypercapnia, and sleep fragmentation from arousal during periods of insufficient oxygenation. The Wisconsin Sleep Cohort Study indicates that the number of individuals with sleep apnea has been steadily increasing. Depending on the subpopulation examined, the prevalence of moderate to severe SA has increased between 14 and 55 % since 1994 [1]. The Wisconsin Sleep Study also identified a variety of cardiovascular sequelae associated with SA including hypertension [2]. The hypertension associated with sleep apnea is sustained during the diurnal cycle and is accompanied by chronically elevated sympathetic nerve activity (SNA). The activation of the chemoreflex in sleep apnea leads to increased SNA which drives increased blood pressure [3]. Elevated SNA has been observed in SA patients in numerous studies, and even short bouts of voluntary apnea result in increased SNA [4–6].
A better understanding of the central nervous system (CNS) mechanisms that lead to chronic SNA and diurnal hypertension could be critical to understanding the pathophysiology of SA. While a number of reviews have comprehensively discussed the pathophysiology of sleep apnea [7], this work will focus on such CNS events related to hypertension in intermittent hypoxia which models the hypoxemia associated with SA.
Chronic Intermittent Hypoxia
Fletcher conceived the idea that exposing rodents to cyclical hypoxia would appropriately mimic the hypoxemia experienced by SA patients [8, 9]. Animals in this model increase their blood pressure in proportion to elevations reported in SA sufferers [10–12]. This hypertension is accompanied by increased muscle SNA during the hypoxic exposure and during normoxia in SA patients [3, 13]. The chronic intermittent hypoxia (CIH) model produces the same pathophysiological increase in blood pressure seen with SA by only mirroring the hypoxic exposures. In response to CIH, animal models exhibit elevated renal [14, 15] and splanchnic SNA [16, 17] along with increased phrenic nerve activity [16]. CIH also leads to enhanced sympathetic responses to chemoreceptor stimulation [18, 19]. Therefore, CIH alone is sufficient to produce the hypertension without any need for the obstruction or sleep fragmentation present in SA.
Many labs have adapted this protocol in order to understand the mechanisms that produce CIH hypertension and to gain insight into the pathophysiology of SA. Patients with SA exhibit different durations and severity of hypoxemia and so there are a variety of CIH models for SA. Eventually, this variation in condition will allow for comparison between the cycle time and severity of hypoxia in the disease process, but until enough studies are completed, the lack of consensus makes it difficult to determine what the defining characteristics of the disease process are and what is specific to the idiosyncratic CIH protocols. A more standardized CIH protocol may be beneficial in the short term to tease out the core features of the disease process.
Hypertension Initiation During CIH
The carotid bodies that lie in the bifurcation of the internal and external carotid arteries sense blood oxygenation. This tissue does not sense oxygen content directly, but rather sufficient pressure of oxygenation [20]. Adequate pressure of oxygen in the arteries results in carbon monoxide (CO) production by heme oxygenase-2 in the glomus cells, and CO suppresses the production of dihydrogen sulfide (H2S) through a PKG-mediated pathway [21]. This is reversed in hypoxia and H2S increases carotid body firing by inhibiting potassium channels and increasing calcium influx [22].
Carotid body stimulation during the hypoxia in CIH results in increased afferent signals via the glossopharyngeal nerve to the nucleus tractus solitarius (NTS). This signaling is essential for sympathetic activation in CIH and numerous studies have verified that carotid body denervation prevents CIH hypertension [8, 23]. The acute increase in SNA from CIH is a productive response to diminished oxygenation and helps to maintain adequate perfusion and oxygenation to essential organs such as the heart and the brain.
Patients with SA exhibit higher baseline muscle sympathetic nerve activity (MSNA) and blood pressure during periods of normal oxygenation indicating that some part of the adaptive response remains after the end of the hypoxic challenge [13]. These same subjects have significantly lower blood pressure and MSNA in response to a 15-min inhalation of 100 % oxygen, but not room air [13]. The abnormally high MSNA during normal oxygenation indicates that the carotid chemoreceptors may be inappropriately active during normal oxygenation. The chronic activation of carotid chemoreceptors is present in cat and rodent CIH models as well [24–26]. Abnormal chemoreceptor activation may be due to sensory long-term facilitation (sLTF) in the glomus cell of the carotid body [24]. Glomus cells are the oxygen sensing cells in the carotid bodies and induce firing of the glossopharyngeal afferents in response to inadequate oxygenation. The carotid bodies also possess a renin-independent pathway capable of producing angiotensin II (Ang II) [27, 28]. Locally administered Ang II briefly increases carotid afferent firing when applied continuously and results in sLTF when applied in discrete intervals [29]. This same phenomenon is produced by application of serotonin (5-HT) as well [30, 31]. Both 5-HT and Ang II function by activating NADPH oxidase 2 (NOX 2) which produces reactive oxygen species (ROS). The presence of ROS is essential for sLTF to occur [30]. Repetitive stimulation of the glomus cells via Ang II or 5-HT during CIH causes sLTF and results in increased basal and chemoreflex stimulation which may help to maintain the elevated MAP seen in SA patients.
After a week of CIH, the chronic chemoreflex stimulation from hypoxemia leads to a rightward resetting of the arterial pressure and renal SNA (rSNA) baroreflex curve. This resetting occurs without any changes in sensitivity so the baroreflex is not inhibited after a 7-day exposure to CIH, but there is greater discharge at each pressure point [32]. In a separate study, animals exhibited exaggerated chemoreflex responses to acute hypoxic exposures with elevated sympathetic discharge after 2 weeks of CIH [17]. This elevated discharge was not coupled with an enhanced pressor response though. IV application of phenylephrine, an α-adrenergic agonist, revealed that the CIH animals had an attenuated vasoconstrictor response to phenylephrine. These same anesthetized CIH animals had a greater decrease in MAP relative to their normoxic counterparts after ganglionic blockade with mecamylamine providing evidence that the increased arterial pressure after CIH is not completely governed by sympathetic vasoconstriction [17]. At this same 2-week time point, another study found that there was not an increase in total peripheral resistance, but there was an increase in cardiac output in CIH-treated animals providing support for the hypothesis that CIH-induced hypertension is not solely a result of vasoconstriction [33•]. With CIH protocols of 30 days or longer, there is impaired baroreflex control of heart rate [34, 35]. Overall, the increased sympathetic outflow at the early stages of CIH may desensitize adrenergic receptors in the vasculature, and hypertension may be driven by increased cardiac output and baroreflex impairment at later time points.
Maintaining Hypertension—Hindbrain
Increased chemoreceptor activation stimulates the NTS which leads to the activation of the rostral ventrolateral medulla (RVLM) and increased renal [14, 15], splanchnic, and phrenic SNA [16, 17]. rSNA leads to increased plasma renin activity (PRA), and through the renin angiotensin system (RAS), elevated circulating Ang II peripherally [36].
Glutamate is the major neurotransmitter released by chemoreceptor afferents [37], and CIH has complex effects on glutamate neurotransmission in the NTS. For example, 10 days of CIH increases the excitability of second-order NTS neurons due to enhanced spontaneous neurotransmitter release, but this effect is offset by a decrease in amplitude of evoked excitatory postsynaptic currents (EPSCs) [38]. Consistent with this observation, it has recently been reported that the number of active synapses is reduced in the NTS after CIH [39]. Moreover, CIH may differentially affect the sensitivity of second-order NTS neurons to glutamate with AMPA-mediated responses being enhanced while NMDA responses are decreased [40]. These observations suggest that, while chemoreceptor sensitivity may be increased by CIH, compensatory mechanisms at the level of the NTS may reduce the overall impact of this effect on autonomic control.
In addition to chemoreceptor stimulation, hypoxia activates KATP currents in neurons and numerous other cell types causing hyperpolarization that may protect vulnerable tissue during adverse events that result in ATP depletion. After 7 days of CIH, acute hypoxic exposure to a NTS slice preparation results in less outward potassium current, and, therefore, less protection and more excitability in NTS neurons receiving carotid body inputs [41]. The KATP channels normally dampen excitability in the neurons, but CIH diminishes this potassium current in the NTS allowing for increased activation. This lack of potassium efflux could contribute to a more robust sympathetic response to hypoxemia.
Enhanced chemoreceptor activation during CIH also augments signaling in the pre-sympathetic hindbrain nuclei. Catecholamine-producing tyrosine hydroxylase (TH)-positive A2 neurons located in the NTS are one subtype which receives direct inputs from the chemoreceptors [42, 43]. The more caudal aspects of the NTS are responsible for modulation of sympathetic outflow in part through their connections with the RVLM. Knockdown of TH and, therefore, catecholamine production in A2 neurons reduces the sustained component of CIH hypertension [44]. ΔFosB staining in the paraventricular nucleus (PVN) of the hypothalamus after CIH was significantly reduced in the rats that received TH knockdown in the NTS. In these studies, knockdown of TH was achieved via an adenoviral vector (AAV) specific for neurons which demonstrates the importance of neural inputs in the NTS rather than glia for the progression of hypertension from CIH. This is reinforced by another study that acutely administered fluorocitrate into the NTS of CIH animals to inhibit glia and found no change in baseline, or post CIH sympathetic activity from the animals [45]. A more chronic inhibition of glial cells may unmask a role for the tripartite synapse in the regulation of CIH sympathetic control via purinergic signaling though. Glial release of ATP onto NTS neurons that project to the RVLM enhances synaptic transmission via the activation of P2X receptors [46]. Downstream from the NTS, purinergic inputs to the RVLM are augmented after a 10-day CIH protocol, and this effect is most likely mediated by an increase in P2X3 and P2X4 receptors [47].
CIH also influences hindbrain circuits that couple respiratory and sympathetic outflow. After 10 bouts of brief hypoxic exposure, rats showed a marked increase in phrenic and splanchnic sympathetic activity [16]. Recording both of these variables simultaneously revealed that splanchnic and phrenic activity was coupled and that splanchnic discharge primarily occurred during the expiratory phase of respiration. A longer 10-day CIH exposure also resulted in an increased abdominal sympathetic activity that coupled with the expiration phase of respiration [48]. This respiratory coupling may be due in part to some C1 RVLM neurons that show increased excitability from respiratory neurons that are most likely expiratory [49].
While all of the studies presented so far have focused on increasing sympathetic outflow, there is also evidence for diminished parasympathetic control over heart rate. A 4-week model of CIH that includes hypercapnia along with hypoxia (CIHH) resulted in increased inhibitory neurotransmission to cardiac vagal neurons in the nucleus ambiguus and dorsal motor nucleus of the vagus [50]. This was coupled with a reduction in excitatory glutamatergic neurotransmission and indicates that parasympathetic outflow is reduced following CIH. The diminished parasympathetic outflow blunts the acute bradycardic response to CIHH but does not result in a sustained increase in heart rate, and overall may contribute to poor baroreflex sensitivity in longer-term CIH.
Maintaining Hypertension—PVN
The PVN is an important regulatory region for blood pressure control due to its parvocellular regions that influence SNA as well as the hypothalamic-pituitary-adrenal axis. In addition, its magnocellular region is in direct control of vasopressin which maintains body fluid regulation. The PVN receives inputs related to the chemoreflex [42, 51], and from forebrain regions that sense circulating Ang II such as the subfornical organ (SFO) as well as integrating centers such as the median preoptic nucleus (MnPO) [52, 53]. This pivotal location makes it an attractive target for intervention in the SA disease process. After 7 days of CIH, the transcription factor ΔFosB, a marker for neuronal activity, is significantly increased in the medial parvocellular, dorsal parvocellular, and lateral parvocellular subregions of the PVN [11]. Reducing the activity of the PVN acutely in an anesthetized preparation via microinjection of the GABA agonist muscimol produced greater decreases in MAP and lumbar SNA in 7-day CIH-exposed animals than in controls [54]. In agreement with the acute data, chronic bilateral infusion of muscimol over 14 days of CIH prevented the sustained hypertensive component from developing [55]. Together, these observations indicate that the PVN contributes to CIH hypertension.
Numerous studies have demonstrated a role for Ang II acting as a peptide neurotransmitter in hypertension [56–58]. CIH-induced neurogenic hypertension is also partially driven by Ang II. Chronic intracerebroventricular (ICV) infusions of losartan, an angiotensin type 1 (AT1) receptor blocker significantly attenuated the sustained component of CIH hypertension that occurs during the normoxic dark phase [59]. In addition, ICV losartan blocked increases in ΔFosB staining in the PVN as well as the RVLM, NTS, and MnPO [59]. Similarly, bilateral infusion of losartan directly in the PVN prevented an absolute increase in MAP after 14 days of CIH [55]. This beneficial effect on CIH hypertension was also present when AT2 or Ang 1–7 receptors were blocked, indicating that multiple different angiotensin peptide variants and receptors are contributing to the increase in firing from the PVN.
AT1 stimulation is partially responsible for the increase MAP in CIH so understanding the regulation of AT1 receptor expression may be critical to understanding the pathogenesis of CIH hypertension. Neuronal nitric oxide synthase (nNOS), which synthesizes the gaseous neurotransmitter nitric oxide, represses AT1 receptor expression in hypothalamic cell culture via a PKG-mediated mechanism [60]. A significant decrease in neurons expressing nNOS RNA and histological staining for nNOS was observed in the ventral parvocellular PVN of rats after 35 days CIH [61]. This study did not find a difference in the magnocellular region but also did not measure nNOS activity. In a separate study, a mouse model of 35 days of CIH resulted in a decrease of NO production in the dendrites of PVN neurons [62]. Decreased NO production was preceded by the internalization of NR1, an NMDA receptor subunit in neurons with nNOS-expressing dendrites on day 14 of CIH. This internalization reduces NMDA-mediated inward currents and may play a role in the decrease of nNOS expression and, therefore, NO production. The authors propose that the neurons examined in this study are potentially magnocellular since nNOS in the PVN is scarcely expressed in parvocellular pre-autonomic or GABAergic PVN neurons [63]. Conversely, nNOS in the PVN has been identified in the magnocellular neurosecretory neurons in numerous studies [64–66]. It has been shown that the dendrites from magnocellular neurons may be in close proximity to PVN pre-autonomic neurons and that their release of vasopressin can regulate the activity of PVN pre-autonomic neurons and sympathetic outflow [67]. It could be speculated that changes in NO release from magnocellular dendrites may influence AT1 receptor expression in PVN parvocellular neurons. It remains to be determined if this type of interaction between these two PVN cell types contributes to CIH hypertension.
Plasma vasopressin (AVP) is not increased after 35 days of CIH, and plasma osmolality is not perturbed after 7 days of CIH [11, 62]. However, recent studies indicate that AVP in PVN parvocellular neurons may contribute to increased sympathetic drive. Among the parvocellular neurons in the PVN that express vasopressin, there is a subset that project caudally to the RVLM [68–70]. After 8 days of CIH, the number of RVLM neurons positive for the AVP receptor V1A significantly increases. Functionally, when the PVN is disinhibited by the GABA antagonist bicuculine, there is increased sympathetic drive from the RLVM which can be stopped with a V1A blocker. Animals that were exposed to 8 days of CIH required significantly more pharmacological V1A blocker to normalize blood pressure. These results suggest that a vasopressin projection from the PVN to the RVLM may regulate sympathetic outflow during CIH and that CIH is associated with an increase in the excitatory drive from this pathway.
In addition to increasing sympathetic outflow, the PVN contributes to diminishing parasympathetic discharge as well. A subpopulation of parvocellular neurons in the PVN projects to the cardiac vagal neurons (CVN), and after exposure to 4 weeks of CIHH, these parasympathetic control centers exhibit reduced excitatory glutamatergic input after photo-stimulation of the PVN [71•]. The CVNs neurons also showed a reduction in EPSC frequency and amplitude after photo-stimulation of PVN. This supports the increased inhibitory input to the CVNs and demonstrates a role for the PVN in diminished parasympathetic tone after SA.
Maintaining Hypertension—Forebrain
Both SA and CIH increase PRA and RAS [36, 72]. Increased peripherally circulating Ang II can bind to CVOs in the forebrain and contribute to the pathophysiological increase in sympathetic tone. After 7 days of CIH, there is increased ΔFosB staining in the SFO, organum vasculosum lamina terminalis (OVLT), and MnPO [11]. These observations are consistent with the hypothesis that circulating Ang II is activating the circumventricular organs, SFO and OVLT, which in turn stimulate the MnPO to drive sympathetic tone though its connections with the PVN. The electrolytic lesion of the AV3V region that contains MnPO, OVLT, and SFO afferents prevents the sustained increase in MAP during waking, but does not affect the MAP increase during intermittent hypoxia [73••].
Supporting the idea that peripheral Ang II plays a role in CIH-induced hypertension, knockdown of AT1a receptors in the SFO via an AAV prevents the sustained component of hypertension from CIH [36]. In addition to the functional effect, the knockdown decreased the expression of ΔFosB in the downstream regions of the MnPO and PVN of rats. This suggests that SFO stimulation is necessary to drive the increase MAP during the waking hours and ΔFosB expression in both the MnPO and the PVN.
Both the MnPO and SFO have projections to the PVN, and both increase ΔFosB in response to CIH. The redundant connections to the PVN from these regions to the PVN have not been explored in CIH and the contribution of each nucleus separately is not well defined. ICV administration of losartan decreased ΔFosB expression in the MnPO along with numerous other autonomic nuclei. ΔFosB is an AP-1 transcription factor that signals for increased neuronal activity and transcriptionally modulates the expression of numerous genes. A dominant negative inhibition of ΔFosB in the MnPO via a virus expressing ΔJunD also prevented sustained hypertension from CIH [73••]. This decrease in MAP along with a reduction in the number of ΔFosB-positive cells in the PVN, RVLM, and NTS demonstrates an essential role for the MnPO in CIH-induced hypertension. Furthermore, several genes were identified as regulated by ΔFosB in the MnPO such as angiotensin converting enzyme (ACE), ACE 2, angiotensin type 1a receptor (unpublished observation), and nNOS. Future experiments will examine how these ΔFosB-regulated genes contribute to CIH hypertension. It could be that, as described above for the NTS, some of these changes in gene expression contribute to the development of CIH hypertension (ACE & AT1a) while others represent homeostatic changes (ACE2 and nNOS) that reduce the magnitude of CIH hypertension.
Summary
Utilizing CIH as a model of the hypoxemia associated with SA has allowed for unprecedented exploration into the early mechanisms that could underlie SA-induced hypertension. Initially, hypertension may arise from repeated chemoreflex stimulation of the sympathetic nervous system (Fig. 1). This point is supported by the observations that removal of the carotid body prevents or eliminates the hypertension. This repeated exposure to hypoxemia acutely leads to increased blood pressure and through rSNA increases PRA. At the same time, the glomus cells of the carotid body could be contributing to the generation of sLTF due to increased ROS and Ang II. The combination of increased peripheral Ang II and sensitized chemoreception by both may be required to sustain elevated blood pressure and increased SNA during the normoxic waking hours. During the waking hours, the hindbrain continues to be overactive and this leads to increased SNA. Augmented signaling from the carotid bodies during normal oxygenation along with changes in excitability and synaptic sensitively in the NTS could lead to increased activation of the RVLM, and through A2 projections of the NTS, the PVN (Fig. 1).
Fig. 1.
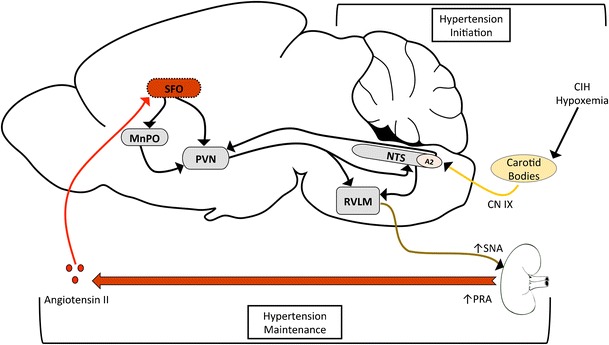
Hypertension Initiation—The hypoxemia from CIH or sleep apnea drives the activation of the carotid bodies. This in turn activates the NTS and the RVLM to increase sympathetic tone. In addition, the sympathetic outflow to the kidneys increases plasma renin activity (PRA) and results in increased circulating angiotensin II (Ang II) during both the hypoxic period and later during the normoxic period. Hypertension Maintenance—Circulating Ang II activates SFO which synapses and activates MnPO. Both of these nuclei stimulate the PVN. The NTS also has afferent connections to PVN that could converge with excitation from the SFO and MnPO. Together with enhanced activity from the NTS due to altered chemoreceptor function, these forebrain mechanisms could contribute to the maintenance of the hypertension during the normoxic periods of CIH
Peripheral Ang II may play an additional role in supporting sustained elevations in blood pressure and SNA associated with CIH. Circulating Ang II stimulates the SFO, and possibly the OVLT, which in turn stimulates the MnPO leading to increased expression of ΔFosB (Fig. 1). ΔFosB-mediated changes in gene expression in the MnPO are necessary for the sustained component of CIH hypertension that occurs during normoxia. Ang II signaling is likely an important component of these central pathways as ICV Losartan administration prevents the sustained component CIH hypertension. Furthermore, three of the candidate ΔFosB-regulated genes in the MnPO are part of the brain RAS such as ACE and the At1a receptor. Enhanced angiotensin signaling at the level of the MnPO could create a feed forward loop that contributes to sympathetic drive through the PVN, where information from the lamina terminalis could be integrated with chemoreceptor information from the hindbrain (Fig. 1). The evidence described above indicates that experimental manipulation effecting either the lamina terminalis, the PVN, or the hindbrain is sufficient to abrogate CIH hypertension suggesting that the entire network may be required for the sustained component of CIH hypertension that is similar to diurnal hypertension in SA patients.
Perspectives
All of the pathophysiological processes described above could lead to potential avenues for the future treatment of hypertension associated with SA. Alternative treatment options are needed for patients who do not experience any therapeutic effect on blood pressure from CPAP. Even among individuals who adhere to CPAP for 4 h per night, at least 25 % do not show a therapeutic benefit reducing hypertension [74, 75]. This leaves a significant number of individuals at risk for cardiovascular sequelae despite some relief from the hypoxemia. A novel analysis of circulating miRNA in plasma has identified potential biomarkers that may help to identify these non-responders so that they can be medicated appropriately along with CPAP [76••].
A lack of response to CPAP makes intuitive sense in light of the processes examined. Treating the hypoxemia with CPAP after a prolonged disease period only alleviates the pathophysiological mechanisms that occur acutely during the apneic period. The forebrain and hindbrain mechanisms altered by long-term exposure to hypoxia may continue to drive the high blood pressure until neural adaptations have been reversed. Angiotensin receptor blockers (ARB) are a prime candidate to treat hypertension associated with SA through the brain. Losartan [77, 78] and Candesartan [79] are among the ARBs that have functional evidence that they cross the blood brain barrier. Despite this evidence, there has been no detection of the active form of these ARBs in the brain, and in animal models central administration of losartan produces a more robust and consistent decrease in blood pressure compared to peripheral administration [59, 80]. A more blood brain barrier permeable ARB could be an important addition to the therapeutic tools available for SA induced hypertension. A number of the mechanisms that have been identified in animal models of CIH appear to be sufficient to establish and sustain hypertension, but SA is increasingly associated with treatment resistant hypertension [81]. For most SA patients, single modality therapies may be sufficient for treatment, but long-term sufferers with resistant hypertension may benefit from targeting multiple mechanisms to significantly reduce MAP.
Unfortunately, around 80 % of those who are afflicted with sleep apnea remain undiagnosed [82]. An essential step for the pathophysiology outlined here to be effectively treated, and treated early, is to diagnose SA. Utilizing a similar circulating biomarker approach in the blood, there could be a cheaper, simpler way to identify those suffering from sleep apnea. A circulating biomarker approach would be a useful tool in identifying individuals who are sleep apneic during their routine blood draws and examinations rather than through self or partner identification. This would not only provide information on who should undergo polysomnography, but also would allow for some degree of intervention in underserved populations that may not be able to afford a sleep study. It is possible that these biomarkers could be related to the mechanisms that sustain hypertension during normoxia.
Conclusions
Overall, CIH has proven to be an extremely useful model to dissect the neurological pathogenesis of sleep apnea. Further studies utilizing this design will provide insight into the onset and maintenance of the disease. This will allow for treatments that are not palliative, but actively ameliorating the hypertension.
Acknowledgments
Dr. Cunningham reports the corresponding author is funded by NIH grants P01 HL088052 and R01 HL119458.
Compliance with Ethical Standard
Conflict of Interest
Drs. Shell, Faulk, and Cunningham declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
This article is part of the Topical Collection on Hypertension and the Brain
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Peppard PE, et al. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177:1006–14. doi: 10.1093/aje/kws342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peppard PE, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342(19):1378–84. doi: 10.1056/NEJM200005113421901. [DOI] [PubMed] [Google Scholar]
- 3.Somers VK, et al. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96:1897–1904. doi: 10.1172/JCI118235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cutler MJ, et al. Hypoxia-mediated prolonged elevation of sympathetic nerve activity after periods of intermittent hypoxic apnea. J Appl Physiol (Bethesda, Md : 1985) 2004;96:754–61. doi: 10.1152/japplphysiol.00506.2003. [DOI] [PubMed] [Google Scholar]
- 5.Smith ML, et al. Role of hypoxemia in sleep apnea-induced sympathoexcitation. J Auton Nerv Syst. 1996;56:184–190. doi: 10.1016/0165-1838(95)00062-3. [DOI] [PubMed] [Google Scholar]
- 6.Leuenberger UA, et al. Effects of intermittent hypoxia on sympathetic activity and blood pressure in humans. Auton Neurosci. 2005;121(1–2):87–93. doi: 10.1016/j.autneu.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Dempsey JA, et al. Pathophysiology of sleep apnea. Physiol Rev. 2010;90(1):47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fletcher EC, et al. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol (Bethesda, Md : 1985) 1992;72:1978–84. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- 9.Fletcher EC, et al. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension. 1992;19:555–61. doi: 10.1161/01.HYP.19.6.555. [DOI] [PubMed] [Google Scholar]
- 10.Foster GE, et al. Intermittent hypoxia increases arterial blood pressure in humans through a Renin-Angiotensin system-dependent mechanism. Hypertension. 2010;56(3):369–77. doi: 10.1161/HYPERTENSIONAHA.110.152108. [DOI] [PubMed] [Google Scholar]
- 11.Knight WD, et al. Chronic intermittent hypoxia increases blood pressure and expression of FosB/ FosB in central autonomic regions. AJP: Regul Integr Comp Physiol. 2011;301:R131–R139. doi: 10.1152/ajpregu.00830.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pankow W, et al. Continuous positive airway pressure lowers blood pressure in hypertensive patients with obstructive sleep apnea. Somnologie. 2003;7(1):17–22. doi: 10.1046/j.1439-054X.2003.02195.x. [DOI] [Google Scholar]
- 13.Narkiewicz K, et al. Contribution of tonic chemoreflex activation to sympathetic activity and blood pressure in patients with obstructive sleep apnea. Circulation. 1998;97:943–945. doi: 10.1161/01.CIR.97.10.943. [DOI] [PubMed] [Google Scholar]
- 14.Lim K, et al. Differential activation of renal sympathetic burst amplitude and frequency during hypoxia, stress and baroreflexes with chronic angiotensin treatment. Exp Physiol. 2015;100(10):1132–44. doi: 10.1113/EP085312. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto K, Lalley P, Mifflin S. Acute intermittent optogenetic stimulation of nucleus tractus solitarius neurons induces sympathetic long-term facilitation. Am J Physiol Regul Integr Comp Physiol. 2015;308(4):R266–75. doi: 10.1152/ajpregu.00381.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dick TE, et al. Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol. 2007;92:87–97. doi: 10.1113/expphysiol.2006.035758. [DOI] [PubMed] [Google Scholar]
- 17.Silva AQ, Schreihofer AM. Altered sympathetic reflexes and vascular reactivity in rats after exposure to chronic intermittent hypoxia. J Physiol. 2011;589(Pt 6):1463–76. doi: 10.1113/jphysiol.2010.200691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenberg HE, et al. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol. 1999;86(1):298–305. doi: 10.1152/jappl.1999.86.1.298. [DOI] [PubMed] [Google Scholar]
- 19.Huang J, et al. Sympathetic response to chemostimulation in conscious rats exposed to chronic intermittent hypoxia. Resp Physiol Neurobiol. 2009;166:102–106. doi: 10.1016/j.resp.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez C, et al. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74(4):829–98. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- 21.Yuan G, et al. Protein kinase G-regulated production of H2S governs oxygen sensing. Sci Signal. 2015;8(373):ra37. doi: 10.1126/scisignal.2005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prabhakhar NR, Joyner MJ. Tasting arterial blood: what do the carotid chemoreceptors sense? Front Physiol. 2014;5:524. doi: 10.3389/fphys.2014.00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lesske J, et al. Hypertension caused by chronic intermittent hypoxia—influence of chemoreceptors and sympathetic nervous system. J Hypertens. 1997;15:1593–603. doi: 10.1097/00004872-199715120-00060. [DOI] [PubMed] [Google Scholar]
- 24.Peng Y-JJ, et al. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rey S, et al. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560(Pt 2):577–86. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcus NJ, et al. Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: role of the angiotensin II type 1 receptor. Respir Physiol Neurobiol. 2010;171(1):36–45. doi: 10.1016/j.resp.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam SY, Leung PS. A locally generated angiotensin system in rat carotid body. Regul Pept. 2002;107(1–3):97–103. doi: 10.1016/S0167-0115(02)00068-X. [DOI] [PubMed] [Google Scholar]
- 28.Leung PS, Lam SY, Fung ML. Chronic hypoxia upregulates the expression and function of AT(1) receptor in rat carotid body. J Endocrinol. 2000;167(3):517–24. doi: 10.1677/joe.0.1670517. [DOI] [PubMed] [Google Scholar]
- 29.Peng Y-JJ, et al. Angiotensin II evokes sensory long-term facilitation of the carotid body via NADPH oxidase. J Appl Physiol (Bethesda, Md: 1985) 2011;111:964–970. doi: 10.1152/japplphysiol.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng YJ, et al. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci. 2009;29(15):4903–10. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng YJ, et al. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol. 2006;576(Pt 1):289–95. doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto K, et al. Resetting of the sympathetic baroreflex is associated with the onset of hypertension during chronic intermittent hypoxia. Auton Neurosci. 2013;173:22–27. doi: 10.1016/j.autneu.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.•.Lucking EF, O’Halloran KD, Jones JFX. Increased cardiac output contributes to the development of chronic intermittent hypoxia-induced hypertension. Exp Physiol. 2014;99:1312–1324. doi: 10.1113/expphysiol.2014.080556. [DOI] [PubMed] [Google Scholar]
- 34.Gu H, et al. Selective impairment of central mediation of baroreflex in anesthetized young adult Fischer 344 rats after chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2007;293(5):H2809–18. doi: 10.1152/ajpheart.00358.2007. [DOI] [PubMed] [Google Scholar]
- 35.Yan B, et al. Attenuation of heart rate control and neural degeneration in nucleus ambiguus following chronic intermittent hypoxia in young adult Fischer 344 rats. Neuroscience. 2008;153(3):709–20. doi: 10.1016/j.neuroscience.2008.01.066. [DOI] [PubMed] [Google Scholar]
- 36.Saxena A, et al. Angiotensin II type 1a receptors in subfornical organ contribute towards chronic intermittent hypoxia-associated sustained increase in mean arterial pressure. Am J Physiol Heart Circ Physiol. 2015;308(5):H435–46. doi: 10.1152/ajpheart.00747.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kline DD. Plasticity in glutamatergic NTS neurotransmission. Respir Physiol Neurobiol. 2008;164(1–2):105–11. doi: 10.1016/j.resp.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kline DD, Ramirez-Navarro A, Kunze DL. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: evidence for homeostatic plasticity. J Neurosci. 2007;27(17):4663–73. doi: 10.1523/JNEUROSCI.4946-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Almado CE, Machado BH, Leao RM. Chronic intermittent hypoxia depresses afferent neurotransmission in NTS neurons by a reduction in the number of active synapses. J Neurosci. 2012;32(47):16736–46. doi: 10.1523/JNEUROSCI.2654-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Paula PM, Tolstykh G, Mifflin S. Chronic intermittent hypoxia alters NMDA and AMPA-evoked currents in NTS neurons receiving carotid body chemoreceptor inputs. Am J Physiol Regul Integr Comp Physiol. 2007;292(6):R2259–65. doi: 10.1152/ajpregu.00760.2006. [DOI] [PubMed] [Google Scholar]
- 41.Zhang W, et al. Chronic sustained and intermittent hypoxia reduce function of ATP-sensitive potassium channels in nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol. 2008;295(5):R1555–62. doi: 10.1152/ajpregu.90390.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.King TL, et al. Acute systemic hypoxia activates hypothalamic paraventricular nucleus-projecting catecholaminergic neurons in the caudal ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol. 2013;305(10):R1112–23. doi: 10.1152/ajpregu.00280.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King TL, et al. Catecholaminergic neurons projecting to the paraventricular nucleus of the hypothalamus are essential for cardiorespiratory adjustments to hypoxia. Am J Physiol Regul Integr Comp Physiol. 2015;309(7):R721–31. doi: 10.1152/ajpregu.00540.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bathina CS, et al. Knockdown of tyrosine hydroxylase in the nucleus of the solitary tract reduces elevated blood pressure during chronic intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2013;305:R1031–9. doi: 10.1152/ajpregu.00260.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa KM, Moraes DJ, Machado BH. Acute inhibition of glial cells in the NTS does not affect respiratory and sympathetic activities in rats exposed to chronic intermittent hypoxia. Brain Res. 2013;1496:36–48. doi: 10.1016/j.brainres.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Accorsi-Mendonca D, et al. Glial cells modulate the synaptic transmission of NTS neurons sending projections to ventral medulla of Wistar rats. Physiol Rep. 2013;1(4):e00080. doi: 10.1002/phy2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoccal DB, Huidobro-Toro JP, Machado BH. Chronic intermittent hypoxia augments sympatho-excitatory response to ATP but not to l-glutamate in the RVLM of rats. Auton Neurosci. 2011;165:156–62. doi: 10.1016/j.autneu.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 48.Zoccal DB, et al. Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol. 2008;586:3253–3265. doi: 10.1113/jphysiol.2008.154187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moraes DJ, et al. Electrophysiological properties of rostral ventrolateral medulla presympathetic neurons modulated by the respiratory network in rats. J Neurosci Off J Soc Neurosci. 2013;33:19223–37. doi: 10.1523/JNEUROSCI.3041-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dyavanapalli J, et al. Chronic intermittent hypoxia-hypercapnia blunts heart rate responses and alters neurotransmission to cardiac vagal neurons. J Physiol. 2014;592(Pt 13):2799–811. doi: 10.1113/jphysiol.2014.273482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reddy MK, Patel KP, Schultz HD. Differential role of the paraventricular nucleus of the hypothalamus in modulating the sympathoexcitatory component of peripheral and central chemoreflexes. Am J Physiol Regul Integr Comp Physiol. 2005;289(3):R789–97. doi: 10.1152/ajpregu.00222.2005. [DOI] [PubMed] [Google Scholar]
- 52.Ferguson AV, Bains JS. Electrophysiology of the circumventricular organs. Front Neuroendocrinol. 1996;17(4):440–75. doi: 10.1006/frne.1996.0012. [DOI] [PubMed] [Google Scholar]
- 53.Llewellyn T, et al. Median preoptic nucleus and subfornical organ drive renal sympathetic nerve activity via a glutamatergic mechanism within the paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol. 2012;302(4):R424–R432. doi: 10.1152/ajpregu.00403.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharpe AL, et al. Chronic intermittent hypoxia increases sympathetic control of blood pressure: role of neuronal activity in the hypothalamic paraventricular nucleus. Am J Physiol Heart Circ Physiol. 2013;305(12):H1772–80. doi: 10.1152/ajpheart.00592.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Da Silva AQG, Fontes MAP, Kanagy NL. Chronic infusion of angiotensin receptor antagonists in the hypothalamic paraventricular nucleus prevents hypertension in a rat model of sleep apnea. Brain Res. 2011;1368:231–238. doi: 10.1016/j.brainres.2010.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology. 2008;23(4):187–193. doi: 10.1152/physiol.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arnold AC, Gallagher PE, Diz DI. Brain renin-angiotensin system in the nexus of hypertension and aging. Hypertens Res. 2013;36(1):5–13. doi: 10.1038/hr.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Kloet AD, et al. Role of neurons and glia in the CNS actions of the renin-angiotensin system in cardiovascular control. Am J Physiol Regul Integr Comp Physiol. 2015;309(5):R444–58. doi: 10.1152/ajpregu.00078.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knight WD, et al. Central losartan attenuates increases in arterial pressure and expression of FosB/ΔFosB along the autonomic axis associated with chronic intermittent hypoxia. American journal of physiology. Regul Integr Comp Physiol. 2013;305:R1051–8. doi: 10.1152/ajpregu.00541.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharma NM, et al. Nitric oxide inhibits the expression of AT1 receptors in neurons. Am J Physiol Cell Physiol. 2012;302(8):C1162–73. doi: 10.1152/ajpcell.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang J, et al. Chronic intermittent hypoxia modulates nNOS mRNA and protein expression in the rat hypothalamus. Respir Physiol Neurobiol. 2007;158:30–38. doi: 10.1016/j.resp.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 62.Coleman CG, et al. Chronic intermittent hypoxia induces NMDA receptor-dependent plasticity and suppresses nitric oxide signaling in the mouse hypothalamic paraventricular nucleus. J Neurosci. 2010;30:12103–12. doi: 10.1523/JNEUROSCI.3367-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watkins ND, Cork SC, Pyner S. An immunohistochemical investigation of the relationship between neuronal nitric oxide synthase, GABA and presympathetic paraventricular neurons in the hypothalamus. Neuroscience. 2009;159(3):1079–88. doi: 10.1016/j.neuroscience.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 64.Hatakeyama S, et al. Nitric oxide synthase-containing magnocellular neurons of the rat hypothalamus synthesize oxytocin and vasopressin and express Fos following stress stimuli. J Chem Neuroanat. 1996;11(4):243–56. doi: 10.1016/S0891-0618(96)00166-4. [DOI] [PubMed] [Google Scholar]
- 65.Sanchez F, et al. Coexistence of NADPH-diaphorase with vasopressin and oxytocin in the hypothalamic magnocellular neurosecretory nuclei of the rat. Cell Tissue Res. 1994;276(1):31–4. doi: 10.1007/BF00354781. [DOI] [PubMed] [Google Scholar]
- 66.Calka J, Block CH. Relationship of vasopressin with NADPH-diaphorase in the hypothalamo-neurohypophysial system. Brain Res Bull. 1993;32(3):207–10. doi: 10.1016/0361-9230(93)90177-D. [DOI] [PubMed] [Google Scholar]
- 67.Son SJ, et al. Dendritic peptide release mediates interpopulation crosstalk between neurosecretory and preautonomic networks. Neuron. 2013;78(6):1036–1049. doi: 10.1016/j.neuron.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kc P, et al. Increased vasopressin transmission from the paraventricular nucleus to the rostral medulla augments cardiorespiratory outflow in chronic intermittent hypoxia-conditioned rats. J Physiol. 2010;588:725–40. doi: 10.1113/jphysiol.2009.184580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prabha K, et al. Chronic intermittent hypoxia-induced augmented cardiorespiratory outflow mediated by vasopressin-V(1)A receptor signaling in the medulla. Adv Exp Med Biol. 2011;701:319–25. doi: 10.1007/978-1-4419-7756-4_43. [DOI] [PubMed] [Google Scholar]
- 70.Stern JE. Nitric oxide and homeostatic control: an intercellular signalling molecule contributing to autonomic and neuroendocrine integration? Prog Biophys Mol Biol. 2004;84(2–3):197–215. doi: 10.1016/j.pbiomolbio.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 71.•.Dergacheva O, et al. Chronic intermittent hypoxia and hypercapnia inhibit the hypothalamic paraventricular nucleus neurotransmission to parasympathetic cardiac neurons in the brain stem. Hypertension. 2014;64(3):597–603. doi: 10.1161/HYPERTENSIONAHA.114.03603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fletcher EC, Bao G, Li R. Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension. 1999;34:309–314. doi: 10.1161/01.HYP.34.2.309. [DOI] [PubMed] [Google Scholar]
- 73.••.Cunningham JT, et al. An essential role for ΔFosB in the median preoptic nucleus in the sustained hypertensive effects of chronic intermittent hypoxia. Hypertension. 2012;60:179–187. doi: 10.1161/HYPERTENSIONAHA.112.193789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barbe F, et al. Pressure on the incidence of hypertension. Jama. 2013;307:2161–2168. doi: 10.1001/jama.2012.4366. [DOI] [PubMed] [Google Scholar]
- 75.Martínez-García M-A, et al. Effect of CPAP on blood pressure in patients with obstructive sleep apnea and resistant hypertension. Jama. 2013;310:2407. doi: 10.1001/jama.2013.281250. [DOI] [PubMed] [Google Scholar]
- 76.••.Sánchez-de-la-Torre M, et al. Precision medicine in patients with resistant hypertension and obstructive sleep apnea. J Am Coll Cardiol. 2015;66:1023–1032. doi: 10.1016/j.jacc.2015.06.1315. [DOI] [PubMed] [Google Scholar]
- 77.Pediconi D, et al. Effects of losartan and irbesartan administration on brain angiotensinogen mRNA levels. Eur J Pharmacol. 2005;528(1–3):79–87. doi: 10.1016/j.ejphar.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 78.Polidori C, et al. Functional evidence for the ability of angiotensin AT1 receptor antagonists to cross the blood–brain barrier in rats. Eur J Pharmacol. 1996;307(3):259–67. doi: 10.1016/0014-2999(96)00270-1. [DOI] [PubMed] [Google Scholar]
- 79.Nishimura Y, et al. Chronic peripheral administration of the angiotensin II AT(1) receptor antagonist candesartan blocks brain AT(1) receptors. Brain Res. 2000;871(1):29–38. doi: 10.1016/S0006-8993(00)02377-5. [DOI] [PubMed] [Google Scholar]
- 80.Pelisch N, et al. Systemic candesartan reduces brain angiotensin II via downregulation of brain renin-angiotensin system. Hypertens Res. 2010;33(2):161–4. doi: 10.1038/hr.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muxfeldt ES, et al. Prevalence and associated factors of obstructive sleep apnea in patients with resistant hypertension. Am J Hypertens. 2014;27(8):1069–78. doi: 10.1093/ajh/hpu023. [DOI] [PubMed] [Google Scholar]
- 82.Finkel KJ, et al. Prevalence of undiagnosed obstructive sleep apnea among adult surgical patients in an academic medical center. Sleep Med. 2009;10(7):753–8. doi: 10.1016/j.sleep.2008.08.007. [DOI] [PubMed] [Google Scholar]