

Abstract
Development of tool molecules that inhibit Jumonji demethylases allows for the investigation of cancer-associated transcription. While scaffolds such as 2,4-pyridinedicarboxylic acid (2,4-PDCA) are potent inhibitors, they exhibit limited selectivity. To discover new inhibitors for the KDM4 demethylases, enzymes overexpressed in several cancers, we docked a library of 600 000 fragments into the high-resolution structure of KDM4A. Among the most interesting chemotypes were the 5-aminosalicylates, which docked in two distinct but overlapping orientations. Docking poses informed the design of covalently linked fragment compounds, which were further derivatized. This combined approach improved affinity by ∼3 log-orders to yield compound 35 (Ki = 43 nM). Several hybrid inhibitors were selective for KDM4C over the related enzymes FIH, KDM2A, and KDM6B while lacking selectivity against the KDM3 and KDM5 subfamilies. Cocrystal structures corroborated the docking predictions. This study extends the use of structure-based docking from fragment discovery to fragment linking optimization, yielding novel KDM4 inhibitors.
Graphical abstract

INTRODUCTION
Methylation of lysine residues in histone proteins profoundly impacts the regulation of cellular processes such as transcription, formation of heterochromatin, genomic imprinting, and X-chromosome inactivation. The discovery of lysine-specific demethylase 1 (LSD1)1 and Jumonji C (JmjC) domain-containing histone lysine demethylases (KDMs)2 uncovered the dynamic character of methyllysine modifications in regulating gene expression. The JmjC domain-containing protein family comprises 33 members in humans,3 of which 24 are classified as histone lysine demethylases.4 In the context of lysine demethylation, these enzymes use iron(II), α-ketoglutarate (α-KG), and molecular oxygen to hydroxylate methyl groups attached to the ε-amino group of lysines. Subsequent decomposition of the hemiaminal intermediate releases formaldehyde and a demethylated residue.2 Accordingly, these enzymes are considered “erasers” of histone marks.5
Aberrant lysine methylation, caused by mutation or misregulation of histone demethylases and histone methyltransferases, profoundly impacts cell physiology. Of particular interest is the hyperactivity of the KDM4 enzyme subfamily (also known as JMJD2). This subfamily consists of five enzymes, KDM4A–E,6 which erase methylation marks on several lysine residues in both histone and non-histone proteins (e.g., polycomb 2 protein (Pc2)).7–13 High expression of individual isoforms of KDM4 is thought to promote oncogenesis in human tumors, especially in prostate cancer8,14 but also in colon15 and some types of breast16 cancers.
The availability of selective chemical probes against individual Jumonji C demethylase subfamilies is critical for exploring their physiological and pathological roles. Since the discovery of Jumonji demethylases in 2006, several inhibitor classes have been identified (Figure S1A) (for reviews, see refs 4, 17–20). These molecules are largely pan-Jumonji inhibitors, often with only modest selectivity among subfamilies and activity against related oxygen-sensing enzymes,21–24 such as factor-inhibiting hypoxia-inducible factor (FIH)19 and prolyl hydroxylases.25 Despite recent advances in KDM inhibitor development, resulting in molecules such as 5-chloro-2-[(E)-2-[phenyl(pyridin-2-yl)methylidene]hydrazin-1-yl]pyridine (JIB-04)26 and methylstat,27 the challenge of identifying selective molecules remains.23 This is further highlighted by N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3H-3-benzazepin-3-yl)-4-pyrimidinyl]-β-alanine (GSK-J1),28 a dual specificity inhibitor that potently inhibits both the KDM6 and KDM5 subfamilies.28,29 Among the only class of molecules with excellent selectivity are the 8-mer peptide substrate analogs,30 but their peptidic nature limits their cellular permeability and utility as chemical probes. To develop new molecules, we employed a structure-based discovery approach that broadly samples chemical space as a method to identify novel and diverse chemotypes targeting the KDM4 demethylases.31–37
We began with molecular docking screens focused on a library of 600 000 commercially available fragments.38 This screen resulted in the identification of unique lead fragments that inhibited KDM4C with good ligand efficiency (LE) values. The docking poses informed subsequent fragment linking, leading to hybrid compounds with as much as 700-fold improvement in inhibition relative to the parent fragments. Further improvement in potency was achieved by iterative cycles of optimization through chemical synthesis, isozyme profiling, and docking. Importantly, these optimized compounds displayed excellent selectivity against FIH and had substantial selectivity over several other Jumonji demethylase subfamilies. Beyond the discovery of novel fragments, this work suggests that docking can inform the design of fused fragments. This docking approach and the corroborating X-ray structures provide atomic-resolution insight into the activity of this new family of demethylase inhibitors.
RESULTS
Fragment Docking and Initial Testing
Molecular docking was performed on the model structure of KDM4A, since the residues lining the active site cavity are conserved between KDM4A and KDM4C, and extensive high-resolution structural data were available for KDM4A. The ZINC fragment library, containing over 600 000 molecules, was docked into the model active site using DOCK3.6. Compounds in this library share common features: commercial availability, xLogP value of ≤3.5, molecular weight of ≤250 Da, and fewer than five rotatable bonds. Each fragment was docked in an average of 21 857 orientations and 42 conformations, resulting in approximately 1.5 × 1012 calculated complexes. Partial ligand desolvation was accounted for in the docking calculations using a context-dependent implementation of GB/SA solvation values from AMSOL.39
The docked fragments were scored for van der Waals interactions using the AMBER potential function40 and for electrostatic complementarity using a point charge model with precalculated maps generated by Poisson–Boltzmann calculations using Qnifft.40,41 Importantly, all candidate compounds selected were among the very top-ranking 0.1% of the docking-ranked library. The selection criteria following docking included the following: (1) formation of favorable interactions with the active site iron center, which was not an explicit parameter in the initial scoring function;42 (2) identification of novel chemotypes; (3) elimination of poses with high internal energies, which are not always captured by Omega program calculations used to generate docking conformations.39 Such postscreening criteria are widely used for “hit-picking”, both in docking43,44 and in high-throughput screening.45 On the basis of these criteria, 14 fragments were selected for testing in an antibody-based demethylation assay coupled with time-resolved FRET (TR-FRET) detection (Table 1), which revealed four compounds that inhibited KDM4C with an IC50 below 100 μM (LE values 0.32–0.36, Table 1, compounds 2–5). Three other compounds exhibited IC50 values in the 100–200 μM range (LE values of 0.3–0.41, Table 1, compounds 6–8). Finally, the remaining seven molecules had detectable inhibition, albeit above 200 μM (LE values of 0.2–0.31, Table 1, compounds 9–15). In this study, 50% of the tested candidate compounds demonstrated IC50 values lower than 200 μM and LE values of 0.3 or higher.
Table 1.
Fragment Docking Hits Tested for Inhibition of KDM4Ca
| ID | Compound | Docking Rank |
IC50(μM) 2μM αKG |
LE | ID | Compound | Docking Rank |
IC50(μM) 2μM αKG |
LE | ID | Compound | Docking Rank |
IC50(μM) 2μM αKG |
LE |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |

|
* | 0.188 | 0.76 | 6 |

|
387 | 126 | 0.41 | 11 |

|
377 | 676 | 0.31 |
| 2 |

|
339 | 18.6 | 0.36 | 7 |

|
187 | 165 | 0.32 | 12 |

|
360 | 1210 | 0.27 |
| 3 |

|
** | 54.7 | 0.32 | 8 |

|
106 | 176 | 0.30 | 13 |

|
337 | 2070 | 0.28 |
| 4 |

|
252 | 58.0 | 0.32 | 9 |

|
138 | 387 | 0.26 | 14 |

|
361 | 2320 | 0.21 |
| 5 |

|
128 | 98.2 | 0.34 | 10 |

|
109 | 507 | 0.25 | 15 |

|
426 | 2810 | 0.20 |
IC50 values determined by TR-FRET assay with 2 μM α-KG.
Control compound, 2,4-PDCA.
Compound 3 is the enantiomer of compound 2 and has no docking rank associated with it.
Among the eight fragments with the highest ligand efficiency, compounds 4, 5, 7 share a common 5-aminosalicylate scaffold (Figure 1, Table 1). The 5-aminosalicylate scaffold docking poses predicted coordination to the active site Fe(II) via the carboxylate and hydroxyl moieties. The 5-amino group was predicted not to participate in metal coordination but rather to form a hydrogen bond with Asp135 (Figure 1). KDM4C inhibition analysis by TR-FRET yielded IC50 values ranging from 58 to 165 μM for compounds with this scaffold (Table 1).
Figure 1.

5-Aminosalicylate docking hits. Poses of the 5-aminosalicylates identified in the initial docking screens. Fragments 4, 5, and 7 are predicted to bind the metal in a bivalent fashion through the carboxylate and hydroxyl groups, while the amide proton is predicted to hydrogen-bond with Asp135 of KDM4A.
Initial efforts to optimize the 5-aminosalicylate scaffold explored 80 analogs, which were either commercially available or de novo synthesized. Among these analogs, the smallest compound tested was N-acetyl-5-aminosalicylic acid, which showed no inhibition. However, 13 molecules (Table S2) showed well-behaved dose–repose curves and a narrow confidence interval for IC50 values (Table S5). Importantly, the 5-aminosalicylate chemotype, represented by compound 20, stabilized the enzyme in thermal shift assays and did not exhibit nonspecific inhibition (Figure S2, Table S1). Compound 21 (Table S2) was the most potent molecule in this series with an IC50 of 5.2 μM, showing a 10-fold increase in activity over starting fragment 4. However, once we arrived at these low micromolar compounds, we could not optimize them any further. In general these new analogs showed relatively flat SAR, and no derivative emerged with affinity better than low μM.
Controls for Colloidal Aggregation
Surprisingly, though the molecules developed and tested here were almost without exception polar and small (many of them fragments) a substantial number had steep concentration–response curves in inhibition assays, a harbinger for colloidal aggregation46,47 that is precedented for fragments.48 For two of the initial docking hits, compounds 4 and 5, we specifically controlled for this artifact by counterscreening them against the unrelated enzyme AmpC β-lactamase and testing them for particle formation by DLS; neither of the two inhibitors acted as aggregators in a relevant concentration range (Table S1). However, of the 80 5-aminosalicylate inhibitors optimized, 18 had steep Hill slopes of above 3. For these compounds the molecular weight varied between 192 and 333 Da and the median xLogP was 2.39. Compounds that had unfavorable concentration–response curves, often a sign of artifactual activity, often by aggregation (Table S1C), were not further pursued.
Optimization by Fragment Linking
As derivatization of the amino moiety of the 5-aminosalicylates scaffold did not substantially improve potency, we considered alternative approaches to improve ligand binding. Specifically, we observed that the 5-aminosalicylates could adopt two high-scoring docking poses in the active site, leading us to explore ligand optimization through fragment linking. While most highly ranked 5-aminosalicylates docked in a pose dominated by iron chelation via the carboxylate and alcohol moieties (compound 4, Figure 2A), a subset of 5-aminosalicylates paired the carboxylate with Lys206 and Tyr132 in the α-KG binding site of the enzyme (e.g., compound 29, Figure 2A). Intriguingly, on the basis of the docking poses, a linking strategy for the two fragments appeared feasible. Specifically, we envisioned that the hybrid molecule should retain the 1,5-bidentate coordination of iron, as in fragment 4. In addition, the hybrid scaffold should contain a carboxylate group to engage Lys206-Tyr132, as present in fragment 29 (Figure 2B). To evaluate if a hybrid molecule could exploit both sets of interactions, 12 scaffolds were created computationally (Table S3). Each molecule incorporated a 1,5-bidentate Fe(II) ligand in addition to a carboxylate (Table S3). Docking of these hybrid molecules revealed that scaffolds I, II, and III (Figure 2C, Table S3, entries 1, 2, 3) had favorable binding geometries and minimal internal strain. Among these, the most appealing was scaffold II, where the iron coordination is achieved by phenol and pyridine moieties. The position of the pyridine ring in this scaffold, as predicted by docking, is reminiscent of 2,4-pyridinedicarboxylic acid (2,4-PDCA) in its cocrystal structure with KDM4A (PDB code 2VD7),25 providing further encouragement for our fragment fusion strategy. Furthermore, this scaffold can be synthetically accessed by a cross-coupling reaction between two aromatic rings leading to the core compound 30 (Table 2) that can be further derivatized to explore the chemical space around the hybrid scaffold.
Figure 2.

Docking-based fragment linking. (A) Superimposition of the two major 5-aminosalicylate poses (compound 4, orange; compound 29, magenta). (B) Structures of 5-aminosalicylate fragments used to create hybrid scaffolds. Key interacting groups are highlighted with a box colored to match the fragments. Iron coordinating atoms are marked by asterisk, and possible ring positions are highlighted. (C) Among 12 virtual hybrid scaffolds that were docked (Table S3), three had good scores and maintained the hypothesized network of interactions. (D) Overlay of a docking pose of 46 (orange) and a crystal structure of 30 (green), a representative hybrid scaffold synthesized in this study.
Table 2.
Synthesis and Structure-Activity Relationships of the Acylated Hybrid Scaffoldsa

| ||||||||
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) values for KDM4C using TR-FRET assay
| ||||||||
| ID | Compound | 2 μM αKG 50 μM αKG | ID | Compound | 2 μM αKG 50 μM αKG |
ID | Compound | 2 μM αKG 50 μM αKG |
| 30 |

|
8.3 N.D. |
35 |

|
0.064 2.6 |
40 |

|
0.14 1.5 |
| 31 |

|
310 N.D. |
36 |

|
0.065 5.0 |
41 |

|
1.02 N.D. |
| 32 |

|
N.I. | 37 |

|
0.43 17 |
42 |

|
0.14 7.4 |
| 33 |

|
4.6 N.D. |
38 |

|
0.17 3.4 |
43 |

|
6.0 N.D. |
| 34 |

|
0.46 N.D. |
39 |

|
0.069 3.7 |
44 |

|
1.2 40 |
Top: synthetic route to N-acyl hybrid derivatives: (a) CombiPhos-Pd6 mixed catalyst, Cs2CO3, 20:1 acetonitrile/water, 100 °C, 24 h; (b) NaI, HBr, 110 °C, 4 days; (c) typically, (i) NHS, R-COCl, MeCN, 20 °C, 1 h; (ii) compound 30, 1:1:1 MeCN/H2O/MeOH, 20 °C, 16 h. Bottom: IC50 values for selected hybrid derivatives tested for inhibition of KDM4C using TR-FRET with the indicated concentration of α-KG. ND = not determined, NI = not inhibitory.
Hybrid Scaffold Structure–Activity Relationship
The core biaryl hybrid scaffold was generated via Suzuki–Miyaura coupling followed by simultaneous nitro group reduction and deprotection of the methyl ester and methyl ether groups using HBr/NaI at reflux (Table 2). Acylation of the resultant amino group yielded the N-acylated hybrid analogs (Table 2). Several hybrid derivatives strongly inhibited KDM4C: compounds 35, 36, and 39 exhibited IC50 values below 70 nM using the TR-FRET assay. A subset of the hybrid compounds was tested under stringent competition with 50 μM α-KG to differentiate relative IC50 values among the most potent derivatives. Under these conditions, the tested molecules demonstrated a range of low micromolar values at half-maximal KDM4C inhibition (Table 2).
The FDH-coupled assay was used for reciprocal (Lineweaver–Burk) plot analysis of selected potent acylated hybrid derivatives, 35 and 42. The Ki constants were calculated based on the Morrison equation using the experimentally determined KM of 5.1 μM for α-KG (Figure S3C). The established nonselective α-KG competitive inhibitor, 2,4-PDCA (1), exhibited a characteristic competitive reciprocal plot profile and a Ki of 2 nM (Figure 3C, Table 3). Compounds 35 and 42 displayed Ki values of 43 and 680 nM, respectively (Figure 3, Table 3). Importantly, these inhibition constants allow for direct comparison of the compound potency irrespective of the assay conditions. While these two compounds bind competitively with respect to α-KG, they do not show competition with the peptide substrate (Table 3).
Figure 3.

Lineweaver–Burke plots. Plots of 1/[α-KG] vs 1/Vi for a range of concentrations of (A) 42, (B) 35, and (C) 1: ● = 19 μM inhibitor, ■ = 6 μM inhibitor, ◆ = 2 μM inhibitor, ▲ = 0.7 μM inhibitor, ▼ = 0 μM inhibitor.
Table 3.
Competition Experiments Performed with a Range of α-KG and Peptide Concentrationsa
| ID | Compound | Ki(μM) Std. Error | LE | IC50(μM) for concentrations [αKG], (peptide) (μM) | ||
|---|---|---|---|---|---|---|
| [50],[50] | [50],[250] | [5],[50] | ||||
| 1 |

|
0.0022 (±0.0006) |
0.98 | 1.5 | 1.9 | 0.53 |
| 35 |

|
0.0432 (±0.0140) |
0.39 | 4.8 | 5.5 | 1.1 |
| 42 |

|
0.6829 (±0,1625 |
0.38 | 14.2 | 14.9 | 3.3 |
Compounds 1, 35, and 42 were tested over a range of concentrations of α-KG and peptide to determine inhibitory constants (Ki) and competition with respect to both substrates. The KM for H3K9me3 peptide is 76 μM,76 and the determined KM for α-KG is 5.1 μM (Figure S3C). Measurements were performed in triplicates.
The potency improvement conferred by the hybrid scaffold over the lead 5-aminosalicylates is illustrated by direct comparison of the corresponding derivatized compounds (Table 4). Relative to the 5-aminosalicylate compounds, the potencies of the hybrid analogs improved substantially. The greatest increase in potency was observed for 42 vs 5 with IC50 values of 0.14 and 98 μM, respectively (∼700-fold improvement, Table 4). Several other corresponding pairs of compounds, such as 4 vs 45 and 28 vs 46, showed improvements in affinity. Naturally, there was an exception where the hybrid scaffold did not improve activity (21 vs 47, Table 4).
Table 4.
Potency Comparison between 5-Aminosalicylates and the Corresponding Hybrid Moleculesa
| ID | Compound | IC50(μM), 2 μM αKG | ID | Compound | IC50(μM), 2 μM αKG |
|---|---|---|---|---|---|
| 5 |

|
98 | 42 |

|
0.14 |
|
| |||||
| 4 |

|
58 | 45 |

|
0.77 |
|
| |||||
| 28 |

|
15 | 46 |

|
0.56 |
|
| |||||
| 21 |

|
5.2 | 47 |

|
2.5 |
Selected 5-aminosalicylate and hybrid derivatives tested for inhibition of KDM4C using TR-FRET with 2 μM α-KG.
Selectivity Counterscreens
The in vitro selectivity profiles of derivatives 35, 36, 40, 42, and 44 (Table 5) were determined using representative members from distinct phylogenetic groups within the JmjC domain-containing enzyme family (Figure 4A) and were screened using the MALDI or AlphaScreen assays. We tested each inhibitor against the H3K4 demethylases KDM5A (JARID1A, RBP2) and KDM5B (JARID1B, PLU-1), the H3K27 demethylase KDM6B (JMJD3), the H3K36me2 demethylase KDM2A (FBXL11), the H3K9me1/2 demethylase KDM3A (JMJD1A), and the asparagine hydroxylase FIH. While the parent hybrid scaffold 30 exhibited 6-fold selectivity for KDM4C over FIH (Figure 4B), this selectivity was improved for all N-acyl derivatives, with compound 35 demonstrating >85-fold selectivity against FIH (IC50 > 500 μM) (Table 5). Given the important physiological role of FIH in regulating broad transcriptional networks, the excellent selectivity of the hybrid analogs for KDM4C over FIH inhibition is critical. All tested derivatives exhibited 6-fold or greater selectivity for KDM4C vs the H3K36me2 demethylase KDM2A, with the best discrimination (24-fold, Table 5) achieved by 42. Moreover, in this series compound 42 was also the most selective inhibitor of KDM4C over the H3K27 demethylase KDM6B (>26-fold). The tested hybrid compounds demonstrated poor discrimination of KDM4C over the H3K4 demethylases KDM5A and KDM5B and the H3K9me1/2 demethylase KDM3A. As expected, these molecules do not discriminate among the members of the KDM4 family (Table 5).
Table 5.
Selectivity Profile of Selected Inhibitorsa

|
IC50 values for selected JmjC proteins (μM) [fold selectivity over KDM4C] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| MALDI Assays | AlphaScreen Assays | |||||||||
|
| ||||||||||
| ID | Hybrid core compound | KDM4C | FIH | KDM5A | KDM4C | KDM2A | KDM6B | KDM5B | KDM3A | KDM4D |
| 1 |

|
0.62 | 8 6 [14] |
0.76 [1] |
1.2 | 12.8 [11] |
44.6 [37] |
2.7 [2] |
3.3 [3] |
2.4 [2] |
| 44 |
|
63 | >500* [>8] |
240 [4] |
15 | >100″ [>6] |
>100∧ [>6] |
72 [5] |
33 [2] |
14 [1] |
| 42 |
|
47 | >500* [>11] |
180 [4] |
3.8 | 91 [24] |
>100∧ [>26] |
12 [3] |
11 [3] |
4.2 [1] |
| 36 |

|
16 | ~473** [31] |
23 [1] |
5.5 | 76 [14] |
31 [6] |
2.7 [−2] |
5.7 [1] |
9.2 [2] |
| 35 |

|
5.9 | >500* [>85] |
45 [4] |
12 | 89 [7] |
27 [2] |
19 Ml | 2.3 [−5] |
13 [1] |
| 40 |
|
7.3 | >500* [>68] |
52 [7] |
9.9 | 89 [9] |
16 [2] |
22 [2] |
9 [1] |
11 [1] |
Selectivity of inhibitors for KDM4C compared to FIH and KDM5A was determined by MALDI assay with 100 μM α-KG. Selectivity against other JmjC domain containing proteins was determined by AlphaScreen, wherein α-KG concentrations used are at approximately the KM for each of the enzymes. No inhibition was observed at maximum concentrations of compounds tested for each of the enzymes:
KDM2A, [inhibitor] = 100 μM;
KDM6B, [inhibitor] = 100 μM;
FIH, [inhibitor] = 500 μM. The reported IC50 are therefore an underestimate.
IC50 value was inferred by extrapolating the IC50 curve (see Figure S3B).
Figure 4.

Jumonji family enzymes and hybrid compound selectivity against FIH. (A) Phylogenetic diagram based on the catalytic domains of JmjC proteins, with KDM4C highlighted in red, and enzymes used in the counterscreening experiments in this study highlighted in blue. (B) Selectivity profile obtained using the MALDI assay with 100 μM α-KG against KDM4C (red) and FIH (green) for hybrid scaffold 30 indicating an IC50 of 135 and 828 μM, respectively, and (C) its acyl derivative 44 showing an IC50 of 63 μM for KDM4C (red) and no inhibition for FIH (green).
Crystallography
To investigate the agreement between docking predictions and experimentally observed orientations and to enable future elaboration of the hybrid scaffold, we turned to X-ray crystallography. Crystal structures of KDM4A, a representative KDM4 enzyme that is well suited for crystallographic studies,23,25,49–51 were determined in complex with hybrid compounds 30, 35, 36, 40, 42, 43, and 44 (Figure 5A–G). The resolution of the structures ranged from 2.0 to 2.39 Å (Figure 5A–G), and other statistics can be found in Supporting Information Table S4. Unambiguous positions for the ligands in the structures were identified in unbiased difference density maps (Fo – Fc for compound 36 contoured at 2.5σ (Figure 5H) and contoured at 3σ (Figure S4G)), and the inhibitors placement refined well (2Fo – Fc map for compound 36, contoured at 1σ (Figure S4G, and composite omit map contoured at 1σ (Figure 5I)).
Figure 5.

Crystal structures of hybrid compounds in complex with KDM4A. Seven cocrystal structures were obtained with KDM4A and the hybrid molecules (A) 43, (B) 30, (C) 44, (D) 42, (E) 35, (F) 40, and (G) 36 to a resolution of 2.39, 2.00, 2.20, 2.15, 2.27, 2.16, and 2.28 Å, respectively. Interacting residues are shown as sticks. (H) Omit map (green) for compound 36 contoured at 2.5σ. (I) 2Fo – Fc composite omit map (gray) contoured at 1σ showing residues 5 Å around compound 36. (J) As a representative structure, compound 36 (wheat) is superimposed with the docked 5-aminosalicylate compound 4 (orange) and the corresponding docked hybrid compound 45 (green). (K) Hydrogen bond network with compound 36. (L) Stacking interactions with compound 36; the hydrophobic centers are indicated by a green sphere.
In each of the seven complexes, the hybrid core of the compounds superimposes well with the docked pose (rmsd range from 0.45 to 0.77 Å, represented by 42 and 43, respectively), forming nearly identical key interactions with the metal and α-KG pocket (Figure 5J). As predicted by the docking poses, in all seven structures the inhibitor carboxylate salt-bridges with Lys206 Nε (2.6 Å) and hydrogen-bonds with Tyr132 OH (2.6 Å) (Figure 5K), mimicking the interactions observed between the carboxylic acid of the α-KG cofactor in the active site52,53(PDB code 2OX0). Similarly, in both the docking and the crystal structures, the inhibitor pyridine ring stacks with Phe185 (Figure 5L), while the pyridine nitrogen, as expected, chelates the active site metal (Mn(II) was used as a mimic of oxygen sensitive Fe(II)) (Figure 5K), an interaction analogous to the interaction between the metal and previously identified inhibitors such as 2,4-PDCA (PDB code 2VD7)25 and 4′-[(2-aminoethyl)carbamoyl]-2,2′-bipyridine-4-carboxylic acid (PDB code 3PDQ).51 The second interaction formed with the metal is with the hydroxyl moiety (2.1 Å) of the N-acylamino substituted phenol ring (Figure 5K). As in the docked structures, in the crystal structures this coordination forms an octahedral geometry, with an angle of 171° between the inhibitor’s hydroxyl group, the metal, and the Nε of His276; Glu190, His188, and a water molecule provide the remaining three metal coordinations. Finally, as anticipated by docking, the phenol ring of the hybrid molecule is sandwiched between the hydroxyl moiety of Tyr177 and the side chain of Lys241, while the pyridine ring is positioned between Phe185 and the aromatic ring of Tyr177 (Figure 5L). We note that in several of the structures there is unexplained electron density that superimposes well with the position occupied by the trimethylated Nε of the lysine peptide substrate. This electron density is approximately 4 Å from the phenol ring and may be modeled as a DMSO molecule that could make π–σ stacking interactions with the inhibitors (Figure 5L).
The one substantial difference between the docking poses and the crystallographic results is in the position of the exocyclic amide substituent, common to the five compounds crystallized (Figure 5C–G). Whereas this difference has little effect on the overall placement of the core scaffold in the site (Figure 5J), the details of the hydrogen-bonding to the enzyme change. In the docking predictions, the amide proton is predicted to hydrogen bond directly with Asp135. While a hydrogen bond between this amide and the protein is observed crystallographically, in some of the complexes (for example, 35, 40, and 42) the nitrogen engages both Tyr177 and Asp135 through a bridging water molecule (Figure 5D–F). In contrast, in the crystal structures of compounds 36 and 44, the Tyr177 and Asp135 form a water-mediated hydrogen bond with the oxygen atom of the exocyclic amide of the inhibitor (Figure 5C,G). Compound 40 is the largest compound for which a structure was solved; however, poor density is observed for its acyl substituent, which occupies different orientations in each crystallographic monomer (Figure S4F). The acyl moieties of these inhibitors reach the peptide binding pocket and mostly occupy the area in which Ser10, Thr11, and Gly12 of the histone H3 substrate bind (Figure S5).52 For example, the oxygen atom of the isoxazolyl moiety of compound 36 forms a hydrogen bond with the side chain nitrogen of Asn86 (Figure 5K), consistent with docking poses of 5-aminosalicylate fragments (fragment 4, Figure 5J).
DISCUSSION
In this study we applied fragment-based docking screens to identify novel KDM4 inhibitor chemotypes. Subsequent fragment optimization (typically requiring numerous iterations of structure determination, modeling, and synthesis) was streamlined by the use of docked geometries to inform fragment linking and the design of a hybrid scaffold. While fragment linking is considered more difficult than fragment elaboration,54 it has been successfully implemented.55–57 Typically, fragment linking is guided by experimental binding geometries, either from NMR or from crystallography;58 this work establishes the use of docking geometries to effectively guide fragment fusion. The success of the strategy here (supported by the 2 log-orders of affinity gained by the fused molecules and the correspondence of the docking predictions to the subsequent crystallographic results) support the use of docking not only to prioritize initial hits for testing but also to guide their optimization. This is further supported by earlier studies that suggest that docked fragments can pose in orientations that accurately represent experimental structures59–62 and that docking can prioritize among multiple binding modes sometimes suggested by experimental structures.63 A detailed analysis of representative hybrid salicylate compounds revealed a competitive binding mode with respect to α-KG that is supported by seven crystal structures (Figure 5A–G), with Ki values of 43 nM for 35 and 680 nM for 42 (Table 3). Acyl derivatives of the hybrid scaffold exhibited increased inhibition of KDM4 demethylases in addition to increased selectivity versus the asparagine hydroxylase FIH (Figure 4B,C, Table 5). Substantial selectivity was achieved against the H3K36 demethylase KDM2A and the H3K27 demethylase KDM6B, for example, compound 42 showed levels of selectivity ≥24-fold against these enzymes. However, selectivity versus the KDM5 and KDM3 subfamilies remains limited for all tested compounds.
Due to the presence of the pyridyl-4-carboxylate moiety, our inhibitors resemble some of the known Jumonji inhibitor scaffolds64 that also use this moiety, such as 2,4-PDCA derivatives,25 bipyridyl derivatives,51 and 8-hydroxyquinoline derivatives23 (Figure S1A). Indeed, since its discovery as a collagen prolyl hydroxylase inhibitor in the 1980s,22 the 2,4-PDCA scaffold and its derivatives have been used for the inhibition of Fe(II) α-KG dependent dioxygenases. However, in contrast to previously reported KDM4 demethylase inhibitors, a unique feature of the molecules described in this study is the coordination geometry of the iron chelating atoms. While other iron-coordinating demethylase inhibitors are usually 1,4-bidentate ligands (Figure S1A), molecules that resulted from this study are 1,5-bidentate ligands. This ligand architecture imposes an out of plane rotation of the two aromatic rings, with a median dihedral angle between the two planes of 36°, as determined in cocrystal structures of KDM4A with the inhibitors (Figure 5). The observation that the hybrid ligands can adopt different dihedral angles between the two aromatic rings, which accommodate the interactions between the acyl substituent and the enzyme, suggests flexibility of the hybrid inhibitors. Importantly, the identification of these novel hybrid molecules was only possible through fragment discovery of the 5-aminosalicylate chemotype. The favorable ligand efficiency and the small size of the 5-aminosalicylate chemotype allowed us to grow this fragment to gain potency.
Two key observations emerge from this study. First, against a soluble enzyme with little ligand precedence, a structure-based docking screen of 600 000 fragments revealed new inhibitors. All 14 of the docking-prioritized molecules demonstrated KDM4C inhibition. Half of the candidate inhibitor fragments showed IC50 values better than 200 μM and LE values of 0.3 or better. Whereas structure-based docking has been shown to be effective in fragment prioritization, this was often against model enzymes, like β-lactamase59,60,63 for which there was substantial ligand precedence; this is much less true for KDM4C, a target of active biological interest for which such ligand precedence is much reduced. Second, and more ambitiously, this study represents the first successful use of docking poses to guide a fragment-linking and synthetic elaboration strategy. This approach was born out of the affinity maturation of the early compounds, and the subsequent crystallography verified the docking predictions.
An important caveat to consider is that we do not suggest that docking can replace the cycles of structure determination and synthesis that are widely practiced in the field of fragment discovery and optimization; indeed, this study itself uses such cycles. We used docking to guide the fusion and optimization because we were unable to determine the cocomplex structures of the initial 5-aminosalicylate fragment hits, which bind in the 50–200 μM range. Thus, we do not advocate docking as a first strategy to guide fragment fusion and optimization. However, when determining initial fragment structures is difficult, this study suggests that docking may be a viable alternative.
In summary, this study has revealed how discovery of the novel midmicromolar 5-aminosalicylate series of inhibitors has been used for fragment linking to yield a potent hybrid scaffold. Optimization of this new scaffold has led to nanomolar hybrid inhibitors of the KDM4 family of epigenetic erasers. This work sets the stage for evaluation of these molecules in cellular assays and for further elaboration of these molecules to improve their selectivity with the ultimate goal of using them as potent and selective chemical probes of KDM4 family demethylase function in physiology and disease.
EXPERIMENTAL SECTION
Docking and Fragment Identification for Testing
For docking we considered the demethylase domain of KDM4A and KDM4C enzymes to be identical, as the residues lining the active site cavity are conserved. Hence 20 chains from the structures of KDM4A (PDB codes 2GP5, 2GP3, 2OQ7, 2OQ6, 2Q8C, 2VD7, 2YBK, 2WWJ, 3NJY, 3PDQ) and KDM4C (PDB code 2XML) were superimposed (Figure S1B). Chain B of structure 3PDQ was chosen as the docking model (Figure S1C) because it was the highest resolution (1.99 Å) structure among the five inhibitor complexes that were used for pose recapitulation. Additionally, this chain had a well-defined rim of the active site, encompassing loop residues Leu153 to Thr173 that were unstructured in other models.
Chain B of 3PDQ was prepared by replacing the Ni(II) metal in the active site with the catalytically relevant Fe(II) metal. Docking was calibrated by screening 40 known inhibitors (extracted from PDB codes 2VD7, 2WWJ, 2YBK, 3NJY, and 3PDQ and selected compounds from literature with IC50 of 0.6–8.2 μM)23,25,27,49–51,65 vs 10 200 physically matched decoys. Results were evaluated based on known inhibitor enrichment and pose recapitulation. In the optimization, we explored representing the Fe(II) metal charge as +1.1, +1.2, +1.3, and +1.4 electrons vs +2, thereby displacing the extra charge from the Fe(II) to the metal chelating residues His188, His276, and Glu190, as previously described.42,66,67 The hydrogen placement on the enzyme was optimized to interact with known inhibitors. A good log AUC enrichment of 20.28 was obtained with Fe(II) at a charge of +1.3, with the charge on His188 and His276 increased by +0.25 each and the charge on Glu190 increased by +0.2 (the net charge of the iron system was unchanged). In DOCK3.6 spheres are typically calculated to represent the negative image of the binding site, on which ligand atoms are placed in calculating initial poses.68 Here, these spheres were calculated automatically69 and a few spheres found to be distant from the metal center were manually removed. For the docking screen that led to the 5-aminosalicylates, the partial atomic charge on the phenolic oxygen and hydrogen of Tyr132 was changed by 0.4 electron in reciprocal directions (the overall charge was again unchanged), a technique we have used previously to slightly compensate for the polar component of ligand recognition.70 A ligand and receptor bin size of 0.4, with an overlap of 0.1 was used to dock fragments from the ZINC database38 (http://zinc.docking.org/subsets/fragment-like).
KDM4C Time-Resolved FRET Assay
3-Fold serial dilutions of compound stocks in DMSO were added to 5 μL of assay buffer (50 mM HEPES, pH 7.0, 0.01 v/v % Tween-20, 0.01 m/v % BSA) supplemented with 10 μM iron(II) ammonium sulfate hexahydrate, 4 μM or 100 μM α-KG, 900 nM histone H3 (residues 1–21) lysine 9 trimethylated peptide with biotin tag (Anaspec), and 200 μM ascorbic acid in a 384-well white opaque OptiPlate (PerkinElmer). The reaction was started by adding 5 μL of 20 nM KDM4C (residues 1–352; see Supporting Information methods for cloning and purification) in assay buffer, then sealed and incubated for 45 min at ambient temperature. The final concentration of DMSO was 2%. The reaction was quenched by addition of 10 μL of detection mix containing 2 nM europium-conjugated anti-H3K9(me2) antibody (PerkinElmer), 100 nM Ulight-streptavadin conjugate (PerkinElmer), 2 mM EDTA, pH 8.0, and 1× LANCE detection buffer (PerkinElmer) in water. The quenched reaction was covered and incubated for 1 h in the dark and then analyzed by a SpectraMax M5e plate reader using TR-FRET mode with an excitation wavelength of 320 nm, emission wavelengths of 665 and 615 nm, 50 μs delay, 500 μs integration, and 100 reads per well. Signal was calculated as E665/E615. Values were plotted in GraphPad Prism and fit by nonlinear regression to calculate IC50 values.
Crystallography
The paralog KDM4A was expressed, purified, and used for crystallography as described.52 Typically, protein solution at 1.8 mg/mL containing 500 μM MnCl2 and 400 μM compound was incubated for 2 h on ice and spun for 5 min at 10 000 rcf at 4 °C to remove precipitate. The supernatant was collected and concentrated to 14–16 mg/mL. Crystals were grown by vapor diffusion by mixing 100 nL of protein solution with 50 nL of reservoir solution at 4 °C. Crystals appeared in conditions containing 22–30% PEG3350, 0.1 M Bis-Tris, pH 5.5–7.5, or 0.1 M Tris, pH 8.5, 0.15–0.25 M ammonium sulfate. Crystals were cryoprotected by addition of 25% ethylene glycol based on crystallization solution and flash-cooled in N2. Data were collected at beamlines Diamond-I04-1 or I02 and I04. Data sets were processed using Xia2.71 Iterative model building with COOT and refinement with PHENIX and BUSTER resulted in final models.
FDH-Coupled Assay for Generation of Lineweaver–Burk Plots
The following components were added to a black 96-well round-bottom Microfluor 1 plate (Thermo Scientific): reaction buffer (10 mM HEPES, pH 7.9, 50 mM NaCl, 0.01 v/v % Tween-20), enzymes, cofactors, additives (vide infra), and 3-fold compound dilutions from DMSO stocks (1 v/v % final DMSO concentration) to a final volume of 90 μL. Each well was then mixed thoroughly by manual pipetting, followed by the immediate addition of 10 μL of ARK(Me3)STGGK peptide substrate. Typical final concentrations are 50 μM iron(II) ammonium sulfate hexahydrate, 500 μM ascorbate, 2 mM NAD+, 0.0252 U formaldehyde dehydrogenase (FDH) per reaction, 1 μM KDM4C (residues 1–352), 50 μM peptide substrate, and variable concentration of α-KG (2.5–50 μM). The reaction was monitored by measuring the change in fluorescence intensity over time on a SpectraMax M5e plate reader with an excitation wavelength of 350 nm and emission wavelength of 460 nm.
Determination of Ki Values
Ki values were determined by FDH-coupled assay under identical conditions to those described above except that the inhibitor was incubated for 15 min in reaction mixture containing 500 nM enzyme concentration and lacking the peptide and α-KG. Following incubation, the reaction was initiated by addition of α-KG (10–100 μM) and peptide substrate.
The rate of the first 2 min of the reaction was calculated by a linear fit, normalized to the fluorescence intensity of 1 μM NADH. Values were plotted in GraphPad Prism. Owing to the high concentration of enzyme, the Morrison equation for tight-binding inhibition was used to globally fit data derived from testing inhibition in the presence of a range of concentrations of α-KG:72
with the form of for competitive inhibitors,
Counterscreening with MALDI Assay
General methods for counter screening of inhibitors against FIH and KDM5A can be found in the methods section in Supporting Information.
Counterscreening of KDM4C with AlphaScreen Assay
Antibody based AlphaScreen assays were used to detect demethylated peptide product73 to counterscreen the KDM4C inhibitors against KDM2A, KDM3A, KDM4D, KDM5B, KDM6B. Details of enzyme purification and assay conditions are described in the methods section in Supporting Information.
Controls for Colloidal Aggregation
Two of the initial docking fragment hits, compounds 4 and 5, were tested for colloidal aggregation in an AmpC β-lactamase counterscreen and by dynamic light scattering (DLS).74,75 Synthesized inhibitors that had steep concentration–response curves were dropped from further consideration, as we were concerned that they were likely aggregating and inhibiting nonspecifically.
Synthesis of Hybrid Scaffold. 2-(5-Amino-2-hydroxyphenyl)-isonicotinic Acid (30)
1.28 mmol of methyl 2-bromoisonicotinate (277 mg) (Combi-Blocks), 0.85 mmol of 2-methoxy-5-nitrophenylboronic acid pinacol ester (237 mg) (Frontier Scientific), Cs2CO3 (1.63 g), 0.1 mmol of CombiPhos-Pd6 (50 mg) (CombiPhos Catalysts), and 10 mL of DMF were combined in a 15 mL pressure vial and stirred for 20 h at 100 °C. The mixture was then diluted in 75 mL of water, acidified to pH 2 with 2 N HCl, and extracted three times with 50 mL of EtOAc. The combined organic phase was washed with 5% citric acid, followed by 50 mL of saturated NaCl, which included 1 mL of 2 N HCl, and concentrated under reduced pressure. The crude mixture was transferred to a new 15 mL pressure vial. 6 mL of 48% aqueous HBr (Sigma) and 4 mmol NaI (600 mg) were added, and the mixture was stirred at 100 °C for 4 days. The mixture was then diluted in 50 mL of water and washed once with 50 mL of EtOAc, and the aqueous fraction was concentrated under reduced pressure. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–10 min, 10% B; 10–89 min, 10–40% B; 89–90 min, 40–100% B; 90–100 min, 100% B) was followed by freeze-drying and yielded 38 mg of product (0.165 mmol, 19%). LCMS [M + H]+ m/z = 231.54. 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.85 (d, J = 5.1 Hz, 1H), 8.52 (d, J = 0.8 Hz, 1H), 8.07 (d, J = 2.1 Hz, 1H), 7.92−7.79 (m, 1H), 7.38−7.27 (m, 1H), 7.10 (dd, J = 8.7, 0.8 Hz, 1H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 165.9, 158.0, 156.4, 156.0, 148.7, 139.8, 125.2, 121.5, 120.8, 120.3, 118.7.
Typical Reductive Amination
Hybrid scaffold 30 (1 equiv) and aldehyde (2 equiv) were combined in 0.02 M ethanol and refluxed for 2 h. NaBH4 (4 equiv) was then added and the resulting mixture was refluxed for an additional 30 min. This mixture was diluted in water and purified by HPLC.
Typical Acylation
Acid chloride (5 equiv) and N-hydroxysuccinimide (10 equiv) were combined in 0.15 M acetonitrile and stirred at rt for 1 h. 30 (1 equiv) was added in an equal volume of water/acetonitrile/methanol (1:1:1) and stirred at rt for 16 h. This mixture was diluted in water and purified by HPLC.
General Methods for Synthesis of Compounds
All reagents and solvents were purchased as the highest available grade from Sigma-Aldrich and used without further purification, unless otherwise indicated. Reverse-phase high performance liquid chromatography (RP-HPLC) was performed with a Varian ProStar solvent delivery system equipped with a Phenomenex Luna 10 μm C18(2) 100 Å column. Separation was achieved using a gradient of acetonitrile or methanol in water with 0.1% TFA, at a flow rate of 15 mL/min. Compounds were purified by RP-HPLC to >95% purity, as assessed by UPLC–MS peak integration and proton NMR. 1H NMR data were recorded with a Varian Innova 400 MHz spectrometer. 13C NMR data were recorded either with a Varian Innova 400 MHz spectrometer or a 500 MHz Bruker AVANCE DRX500 spectrometer equipped with an actively shielded Z-gradient QCI cryoprobe (H-P/C/N-D). 13C spectra were analyzed following multipoint baseline correction. Mass spectrometry (ESI-MS) was performed using a Waters Acquity UPLC/ESI-TQD equipped with a 2.1 mm × 50 mm Acquity UPLC BEH C18 column.
-
1

Purchased from Acros.
-
2
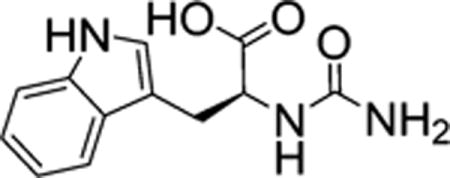
Molecular weight: 246.26. L-tryptophan (6 mmol, 1.226 g), urea (10.3 mmol, 0.661 g), and 1.8 mL of a 3.3 N aqueous solution of NaOH were combined in a 50 mL beaker and mixed. Mixture was microwaved (1100 W) on high for 4 min along with a 500 mL beaker full of water. Mixture was cooled to 0 °C, and 3 mL of a 2 N aqueous solution of HCl was added. The precipitate was isolated by filtration and washed with water. Crude yield was 848.5 mg (3.43 mmol, 57%). 100 mg of crude material was HPLC purified (0.1% TFA, acetonitrile/water gradient of 0–5 min, 20% B; 5–30 min, 20–100% B; 30–60 min, 100% B) followed by freeze-drying and yielded 92.9 mg of solid (0.38 mmol, 53%). LCMS [ES]− m/z = 246.08. 1H NMR (400 MHz, DMSO-d6, ppm) δ 12.51 (s, 1H), 10.85 (s, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.09 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 6.97 (ddd, J = 7.9, 7.1, 1.0 Hz, 1H), 6.11 (d, J = 8.1 Hz, 1H), 5.61 (s, 2H), 4.46−4.32 (m, 1H), 3.10 (dd, J = 14.6, 5.3 Hz, 1H), 3.01 (dd, J = 14.7, 7.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 174.4, 158.2, 136.1, 127.4, 123.6, 120.9, 118.4, 118.3, 111.3, 109.7, 53.1, 27.9.
-
3

Molecular weight: 246.26. D-Tryptophan (3 mmol, 0.614 g), urea (4.99 mmol, 0.300 g), and 0.9 mL of a 3.3 N aqueous solution of NaOH were combined in a 50 mL beaker and mixed. The mixture was microwaved (1100 W) on high for 2 min along with a 500 mL beaker full of water. The mixture was then cooled to 0 °C. 1.5 mL of a 2 N aqueous solution of HCl was then added and the precipitate was isolated by filtration and washed with water, yielding 942.7 mg of solid (3.82 mmol, 128%). LCMS [ES]− m/z = 248.63. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.83 (s, 1H), 7.51 (d, J = 8.2 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.09 (d, J = 2.3 Hz, 1H), 7.04 (t, J = 7.0 Hz, 1H), 7.00−6.91 (m, 1H), 6.08 (d, J = 8.1 Hz, 1H), 5.58 (s, 2H), 5.39 (s, 1H), 4.31 (s, 1H), 3.10 (dd, J = 14.6, 5.5 Hz, 1H), 3.00 (dd, J = 14.5, 6.7 Hz, 1H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 174.4, 158.2, 136.1, 127.4, 123.6, 120.9, 118.4, 118.3, 111.3, 109.7, 53.1, 27.9.
-
4

Molecular weight: 247.25. 5-Aminosalicylate (1 mmol, 153 mg), 1-methyl-1H-pyrazole-5-carbaldehyde (0.5 mmol, 49.2 μL), and 10 mL of EtOH were combined in a flame-dried round-bottom flask fitted with a reflux condenser, under an argon atmosphere. The mixture was stirred for 2 h at 100 °C. Sodium borohydride (1 mmol, 38 mg) was added, and mixture was stirred for another 15 h at ambient temperature. 20 mL of saturated NH4Cl was used to quench the reaction, and the product was extracted into 2 × 50 mL of EtOAc. Organic layers were combined and washed with saturated NaCl, dried over Na2SO4, filtered, and concentrated under reduced pressure, yielding 218.8 mg of crude product (0.88 mmol, 177%). 22.2 mg of crude product was HPLC purified (0.1% TFA, acetonitrile/water gradient of 0–5 min, 15% B; 5–25 min, 15–80% B; 25–35 min, 80–100% B; 35–50 min, 100% B) and the purified product freeze-dried. Final yield was 12.3 mg of solid (0.050 mmol, 98%). LCMS [M − H]− m/z = 246.45. 1H NMR (400 MHz, acetonitrile-d3, ppm) δ 7.71 (s, 1H), 7.13 (d, J = 2.8 Hz, 1H), 6.98 (dd, J = 8.9, 2.8 Hz, 1H), 6.83 (d, J = 8.9 Hz, 1H), 6.42 (s, 1H), 4.38 (s, 2H), 3.95 (s, 3H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 171.8, 158.5, 158.2, 137.3, 117.7, 116.5, 114.2, 112.7, 106.0, 36.4, 36.3.
-
5

Purchased from Enamine, Ltd.
-
6

Molecular weight: 188.22. 2-Aminoisobutyric acid (2 mmol, 206 mg), 1-isocyanatopropane (2.1 mmol, 200 uL), and 3 mL of 0.33 N NaOH were combined in a 10 mL flask and stirred at ambient temperature for 2 days. Precipitate was filtered and the solution was acidified to pH 2.0 with 2 N HCl, extracted with EtOAc, and concentrate under reduced pressure. The compound was then HPLC purified (0.1% TFA, acetonitrile/water gradient of 0–5 min, 20% B; 5–30 min, 20–100% B; 30–60 min, 100% B) followed by freeze-drying, yielding 67 mg of solid (0.36 mmol, 18%). LCMS [M + H]+ m/z = 189.11. 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.23 (s, 1H), 3.29 (t, J = 7.0 Hz, 2H), 1.50 (p, J = 7.2 Hz, 2H), 1.27 (s, 6H), 0.80 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 177.56, 155.50, 57.63, 24.69, 20.92, 10.96.
-
7

Molecular weight: 240.28. 5-Aminosalicylic acid (1 mmol, 153 mg), ethyl isocyanate (1 mmol, 90 uL), and 2 mL of acetonitrile were combined in a 50 mL flask and refluxed for 1.5 h. The mixture was then cooled to ambient temperature, 20 mL of DMF was added, and the mixture was refluxed for an additional 75 min. The mixture was again cooled to ambient temperature and concentrated under reduced pressure. Crude material was suspended in 20 mL of EtOAc and extracted with 10 mL of saturated aqueous NaHCO3. The aqueous layer was washed with 2 × 20 mL EtOAc, and the combined EtOAc washes were re-extracted with 2 × 10 mL saturated aqueous NaHCO3. The aqueous layer was acidified with 60 mL of 2 N HCl and then extracted with 3 × 20 mL of EtOAc. Organic fractions were combined and washed with 50 mL of brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The product was HPLC purified (0.1% TFA, acetonitrile/water gradient of 0–10 min, 20% B; 10–25 min, 20–80% B; 25–30 min, 80% B; 35–60 min, 100% B), followed by freeze-drying, yielding 156.3 mg of solid (0.65 mmol, 65%). LCMS [M − H]− m/z = 239.4. 1H NMR (400 MHz, DMSO-d6, ppm) δ 11.12 (s, 1H), 9.25 (s, 1H), 7.77−7.53 (m, 1H), 7.45 (d, J = 8.8 Hz, 1H), 6.91 (d, J = 8.8 Hz, 1H), 3.45 (s, 2H), 1.09 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 180.7, 171.6, 158.3, 132.8, 130.6, 126.0, 117.2, 112.5, 56.0, 18.6, 14.3.
-
8

Molecular weight: 234.20. Citric acid (5.04 mmol, 968 mg) was added portionwise to 1.4 mL of H2SO4 and stirred for 40 min at ambient temperature, followed by stirring for 70 min at 70 °C. The mixture was cooled to 0 °C, and 5-methylbenzene-1,3-diol (3.97 mmol, 492 mg) was added portionwise over 15 min. The mixture was stirred at 0 °C for 2 h and then poured onto 30 g of ice. The filtrate was collected, washed with 4 × 5 mL of water, and eluted with 5 × 5 mL of saturated aqueous NaHCO3. The filtrate was acidified with 30 mL of 2 N HCl and extracted with 2 × 30 mL of EtOAc. EtOAc fractions were combined and washed with 2 × 50 mL of brine, dried over Na2SO4, and concentrated under reduced pressure, yielding 152.2 mg of solid (0.65 mmol, 16%). LCMS [M − H]− m/z = 233.50. 1H NMR (400 MHz, CDCl3, ppm) δ 6.94 (dd, J = 2.1, 1.2 Hz, 1H), 6.81 (s, 1H), 6.15 (t, J = 1.8 Hz, 1H), 4.03 (d, J = 1.7 Hz, 2H), 2.49−2.44 (m, 3H). 13C NMR (126 MHz, CDCl3, ppm) δ 216.5, 162.5, 159.9, 153.2, 149.6, 146.2, 144.3, 113.0, 112.3, 110.9, 102.3, 33.1, 22.5.
-
9

Molecular weight: 252.27. 1-(2-Methoxy-2-oxoethyl)pyrazole-3-carboxylic acid (Aurora) (0.16 mmol, 30 mg), HATU (0.187 mmol, 71 mg), DIPEA (0.172 mmol, 30 μL), N-methylpiperazine (0.27 mmol, 30 μL), and dry DMF (0.7 mL) were added to a 5 mL flask and stirred at ambient temperature overnight. The reaction was concentrated under reduced pressure, then partitioned between water (1 mL) and ethyl acetate (2 mL). The aqueous layer was washed twice with ethyl acetate (2 mL). The aqueous layer was HPLC purified (0.1% TFA, acetonitrile/water gradient of 0–5 min, 0% B; 5–30 min, 0–30% B; 30–60 min, 100% B) followed by freeze-drying, yielding 19.0 mg of solid (0.070 mmol, 46%). LCMS [M + H]+ m/z = 253. 1H NMR (400 MHz, D2O, ppm) δ 7.80 (dd, J = 2.4, 0.9 Hz, 1H), 6.72 (dd, J = 2.3, 1.0 Hz, 1H), 5.14 (d, J = 0.7 Hz, 2H), 4.79−4.68 (m, 2H), 3.66−3.57 (m, 2H), 3.37−3.17 (m, 2H), 2.10−1.98 (s, 3H). 13C NMR (100 MHz, D2O, ppm) δ 171.8, 164.9, 145.0, 133.9, 108.4, 53.3, 53.0, 43.0, 39.7.
-
10

Molecular weight: 241.24. Methyl 2-chloroisonicotinate (0.76 mmol, 130 mg), 2-acetylphenylboronic acid (1.39 mmol, 229 mg) (Alpha Aesar), 2.3 mL of aqueous 2 M K2CO3, and 3 mL of 1,4-dioxane were combined in a 50 mL flask and bubbled with argon for 10 min. Pd(PPh3)4 (0.073 mmol, 84 mg) was added, and the mixture was stirred for 12 h at reflux. The mixture was cooled to ambient temperature, diluted with 30 mL of EtOAc, and the organic layer was washed with 2 × 10 mL of saturated aqueous NaHCO3. The aqueous layers were combined, acidified with 2 N HCl, extracted with 2 × 15 mL of n-BuOH, concentrated under reduced pressure, and finally HPLC purified (0.1% TFA, acetonitrile/water gradient of 0–10 min, 20% B; 10–25 min, 20–80% B; 25–30 min, 80% B; 35–60 min, 100% B). The product was freeze-dried, yielding 73.8 mg of solid (0.31 mmol, 40%). LCMS [M − H]− m/z = 240.5. 1H NMR (400 MHz, D2O, ppm) δ 9.19 (d, J = 6.4 Hz, 1H), 8.72 (s, 1H), 8.30−8.22 (m, 2H), 7.91−7.77 (m, 3H), 2.09 (d, J = 0.9 Hz, 3H). 13C NMR (126 MHz, D2O, ppm) δ 166.7, 151.0, 150.3, 143.3, 139.1, 134.4, 131.6, 128.2, 125.2, 123.9, 123.4, 120.2, 101.6, 25.7.
-
11

Purchased from Enamine, Ltd.
-
12

Molecular weight: 228.63. 4-Chloropicolinic acid (0.52 mmol, 81 mg), L-alanine methyl ester HCl (0.58 mmol, 81 mg), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide HCl (0.55 mmol, 105 mg), hydroxybenzotriazole (0.57 mmol, 77 mg), diisopropylethylamine (0.80 mmol, 140 uL), and 1 mL DMF were combined in a 10 mL Schlenk flask and stirred at ambient temperature for 18 h. The mixture was concentrated under reduced pressure and the crude material dissolved in 5 mL of EtOAc. The solution was subsequently washed with 2 × 5 mL of 5% aqueous citric acid; 2 × 5 mL of 5% aqueous NaHCO3; 2 × 5 mL of water; followed by 10 mL of brine. The organic layer was dried over Na2SO4, concentrated under reduced pressure, and purified by flash chromatography (eluting with EtOAc/hexanes (20/80)), yielding 72 mg of crude intermediate. 44 mg of this material was dissolved in 1.2 mL of 6 N HCl and 1.2 mL of 1,4-dioxane in a 10 mL flask and stirred at 60 °C for 9 h. Volatiles were removed under reduced pressure. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 25% B; 5–30 min, 25–100% B; 30–60 min, 100% B) was followed by freeze-drying, yielding 21.7 mg of solid (0.095 mmol, 53%). LCMS [M − H]− m/z = 227.43. 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.90 (s, 1H), 8.66 (d, J = 5.3 Hz, 1H), 8.04 (s, 1H), 7.80 (dd, J = 5.3, 0.9 Hz, 1H), 4.60−4.40 (m, 1H), 1.43 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 173.6, 162.25, 151.2, 150.2, 144.7, 126.7, 122.0, 47.9, 17.2.
-
13

Purchased from Oakwood.
-
14

Purchased from Otava.
-
15

Molecular weight: 230.22. Quinoline-2-carbonyl chloride (1.07 mmol, 205 mg), glycine methyl ester HCl (1.06 mmol, 134 mg), and 20 mL of CH2Cl2 were combined in a 100 mL Schlenk flask and stirred. Into this solution, Et3N (24.4 mmol, 3.4 mL) was added dropwise over 5 min. The mixture was stirred at ambient temperature for 2 days. The reaction mixture was then washed with 2 × 20 mL of water, dried over Na2SO4, concentrated under reduced pressure, and purified by flash chromatography (eluting in EtOAc/hexanes (25/75)), yielding 147 mg of crude intermediate. 70 mg of this material was dissolved in 1.5 mL of 6 N HCl and 1.5 mL of 1,4-dioxane and stirred at 60 °C for 1 h. Volatiles were removed under reduced pressure. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 30% B; 5–30 min, 30–100% B; 30–60 min, 100% B), followed by freeze-drying, yielded 54.4 mg of solid (0.24 mmol, 46%). LCMS [M + H]+ m/z = 231.57. 1H NMR (400 MHz, DMSO-d6, ppm) δ 12.74 (s, 1H), 9.13 (t, J = 6.1 Hz, 1H), 8.59 (dd, J = 8.5, 0.7 Hz, 1H), 8.19−8.08 (m, 3H), 7.89 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.74 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 4.05 (d, J = 6.1 Hz, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 171.1, 164.2, 149.6, 146.0, 138.0, 130.7, 129.2, 128.9, 128.2, 128.2, 118.6, 41.2.
-
16

Molecular weight: 287.27. 3-Methoxybenzoyl chloride (0.13 mmol, 22 mg), N-hydroxysuccinamide (0.26 mmol, 30 mg), diisopropylethylamine (0.5 mmol, 87 μL), and 1.5 mL of acetonitrile were combined and stirred at ambient temperature for 30 min. Subsequently 5-aminosalicylate (0.13 mmol, 20 mg) was added and the mixture was stirred at ambient temperature for 5 h. The reaction was acidified with 2 M HCl, extracted with EtOAc, and concentrated under reduced pressure. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 20% B; 5–25 min, 20–100% B), followed by freeze-drying, yielded 13.7 mg of white solid (0.048 mmol, 37%). LCMS [M + H]+ m/z = 288. 1H NMR (400 MHz, DMSO-d6, ppm) δ 11.06 (s, 1H), 10.19 (s, 1H), 8.27 (d, J = 2.7 Hz, 1H), 7.88 (dd, J = 8.9, 2.7 Hz, 1H), 7.58−7.47 (m, 1H), 7.44 (t, J = 7.9 Hz, 1H), 7.15 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 6.97 (d, J = 8.9 Hz, 1H), 4.15 (s, 1H), 3.84 (s, 3H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 171.9, 165.1, 159.4, 157.6, 136.3, 131.0, 129.8, 128.9, 122.1, 120.0, 117.5, 117.3, 113.0, 112.6, 55.5.
-
17

Molecular weight: 315.36. 1-Adamantanecarbonyl chloride (0.13 mmol, 26 mg), N-hydroxysuccinamide (0.26 mmol, 30 mg), diisopropylethylamine (0.5 mmol, 87 μL), and 1.5 mL of acetonitrile were combined and stirred at ambient temperature for 30 min. Then 5-aminosalicylate (0.13 mmol, 20 mg) was added and the mixture was stirred at ambient temperature for 5 h. The reaction was acidified with 2 M HCl, extracted with EtOAc, and concentrated under reduced pressure. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 50% B; 5–30 min, 50–100% B), followed by freeze-drying, yielded 1.0 mg of white solid (0.0032 mmol, 2.5%). LCMS [M + H]+ m/z = 316. 1H NMR (400 MHz, DMSO-d6, ppm) δ 9.06 (s, 1H), 7.96 (d, J = 2.8 Hz, 1H), 7.68 (dd, J = 8.9, 2.8 Hz, 1H), 6.74 (d, J = 8.8 Hz, 1H), 2.02 (s, 2H), 1.91 (s, 3H), 1.70 (s, 3H).
-
18

Molecular weight: 257.28. 0.25 mmol paraformaldehyde (0.25 mmol, 7.5 mg), N-benzyl-5-aminosalicylate (0.12 mmol, 29 mg), NaBH3CN (0.32 mmol, 20 mg), and 2 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was heated for 2 h at 100 °C and subsequently cooled to ambient temperature and acidified with 2 M HCl. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 10% B; 5–30 min, 10–80% B), followed by freeze-drying, yielded 11.9 mg of white solid (0.046 mmol, 39%). LCMS [M + H]+ m/z = 258. 1H NMR (400 MHz, DMSO-d6, ppm) δ 11.08 (s, 1H), 7.47−7.07 (m, 7H), 6.90 (d, J = 7.7 Hz, 1H), 4.55 (s, 2H), 3.01 (s, 3H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 171.5, 158.6, 158.2, 128.5, 128.4, 127.6, 124.3, 118.0, 117.0, 114.1, 112.9, 58.3, 40.6.
-
19

Molecular weight: 365.38. 4-(4-Methoxyphenoxy)benzaldehyde (0.25 mmol, 57 mg), 5-aminosalicylate (0.12 mmol, 18 mg), NaCNBH3 (0.32 mmol, 20 mg), and 2 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was heated for 2 h at 100 °C, then cooled to ambient temperature and acidified with 2 M HCl. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 30% B; 5–30 min, 30–100% B), followed by freeze-drying, yielded 17.2 mg of yellow solid (0.047 mmol, 23%). LCMS [M + H]+ m/z = 366. 1H NMR (400 MHz, DMSO-d6, ppm) δ 7.34 (dd, J = 8.4, 3.0 Hz, 1H), 7.17 (s, 1H), 7.04 (d, J = 8.7 Hz, 1H), 6.99−6.92 (m, 2H), 6.89 (dd, J = 8.6, 3.3 Hz, 1H), 6.81 (dd, J = 8.9, 3.3 Hz, 1H), 4.25 (s, 2H), 3.74 (d, J = 3.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 171.7, 157.2, 155.6, 154.7, 154.6, 149.5, 129.6, 123.9, 120.7, 120.6, 117.7, 117.3, 115.1, 112.8, 55.4, 48.5.
-
20

Molecular weight: 257.28. 2-Methylbenzaldehyde (0.25 mmol, 30 mg), 5-aminosalicylate (0.12 mmol, 18 mg), NaCNBH3 (0.32 mmol, 20 mg), and 2 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was heated for 2 h at 100 °C, then cooled to ambient temperature and acidified with 2 M HCl. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 10% B; 5–30 min, 10–80% B), followed by freeze-drying, yielded 36.2 mg of white solid (0.14 mmol, 70%). LCMS [M + H]+ m/z = 258.28. 1H NMR (400 MHz, DMSO-d6, ppm) δ 7.34−7.27 (m, 1H), 7.20−7.11 (m, 4H), 7.03 (dd, J = 8.9, 2.7 Hz, 1H), 6.81 (d, J = 8.9 Hz, 1H), 4.23 (s, 2H), 2.31 (s, 3H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 171.8, 154.4, 138.7, 136.3, 136.0, 130.1, 128.1, 127.2, 125.8, 123.5, 117.7, 114.0, 112.8, 46.8, 18.7.
-
21

Molecular weight: 322.15. 5-Aminosalicylate (0.2 mmol, 31 mg), 4-bromobenzaldehyde (0.2 mmol, 37 mg), and 2 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was heated for 1 h at 100 °C, then sodium borohydride (0.4 mmol, 15 mg) was added and the mixture was maintained at 100 °C for 3 h. The reaction was acidified with 2 M HCl. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 10% B; 5–20 min, 10–35% B; 20–30 min, 35–75% B), followed by freeze-drying, yielded 35.5 mg of orange solid (0.11 mmol, 55%). LCMS [M + H]+ m/z = 322.13, 324.11. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.50 (s, 1H), 7.51 (s, 1H), 7.49 (s, 1H), 7.32 (s, 1H), 7.29 (s, 1H), 6.92 (d, J = 2.9 Hz, 1H), 6.85 (dd, J = 8.8, 3.0 Hz, 1H), 6.72 (d, J = 8.9 Hz, 1H), 4.19 (s, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 172.0, 152.8, 141.0, 139.7, 131.1 (2C), 129.4 (2C), 121.9, 119.6, 117.5, 112.5, 111.5, 46.5.
-
22

Molecular weight: 273.28. 4-Methoxybenzaldehyde (0.25 mmol, 30.4 μL), 5-aminosalicylate (0.12 mmol, 18 mg), NaCNBH3 (0.32 mmol, 20 mg), and 2 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was heated for 2 h at 100 °C, then cooled to ambient temperature and acidified with 2 M HCl. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 10% B; 5–30 min, 10–80% B), followed by freeze-drying, yielded 46.9 mg of white solid (0.17 mmol, 86%). LCMS [M − H]− m/z = 272.19. 1H NMR (400 MHz, DMSO-d6, ppm) δ 7.56 (s, 1H), 7.32 (dd, J = 8.5, 3.4 Hz, 3H), 7.00−6.87 (m, 3H), 4.36 (d, J = 3.0 Hz, 2H), 3.73 (d, J = 3.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 171.3, 159.3, 158.9, 158.1, 130.8, 127.5, 126.3, 120.6, 118.2, 113.9, 113.5, 55.2, 51.6.
-
23

Molecular weight: 282.29. 5-Aminosalicylate (0.2 mmol, 31 mg), 1H-indole-7-carbaldehyde (0.2 mmol, 29 mg) (Acros), and 2 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was heated for 1 h at 100 °C, then sodium borohydride (0.4 mmol, 15 mg) was added and the mixture was maintained at 100 °C for 3 h. The reaction was acidified with 2 M HCl. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 10% B; 5–30 min, 10–80% B), followed by freeze-drying, yielded 14.3 mg of yellow solid (0.051 mmol, 25%). LCMS [M − H]− m/z = 281.21. 1H NMR (400 MHz, DMSO-d6, ppm) δ 11.24 (s, 1H), 7.51 (d, J = 7.8 Hz, 2H), 7.42 (s, 1H), 7.41−7.38 (m, 2H), 7.23 (d, J = 7.9 Hz, 2H), 7.16 (d, J = 7.0 Hz, 2H), 6.99 (t, J = 7.6 Hz, 2H), 6.88 (d, J = 8.7 Hz, 1H), 6.48 (dd, J = 2.9, 1.5 Hz, 1H), 4.61 (s, 2H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 171.3, 162.0, 134.2, 127.8, 125.2, 121.0, 120.7, 119.6, 118.6, 117.6, 117.2, 114.3, 112.7, 101.3, 93.8, 60.6.
-
24

Molecular weight 322.15. 5-Aminosalicylate (0.2 mmol, 31 mg), 3-bromobenzaldehyde (0.2 mmol, 23.3 μL), and 2 mL of EtOH were added in a round-bottom flask fitted with a reflux condenser. The mixture was heated for 1 h at 100 °C, then sodium borohydride (0.4 mmol, 15 mg) was added and the mixture was maintained at 100 °C for 3 h. The reaction was acidified with 2 M HCl. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 10% B; 5–20 min, 10–35% B; 20–30 min, 35–75% B), followed by freeze-drying, yielded 23.7 mg of orange solid (0.074 mmol, 37%). LCMS [M + H]+ m/z = 322.09, 324.08. 1H NMR (400 MHz, DMSO-d6, ppm) δ 7.66 (s,1H), 7.50 (d, J = 7.9 Hz, 1H), 7.46−7.39 (m, 1H), 7.39−7.22 (m, 2H), 7.05 (d, J = 27.5 Hz, 1H), 6.76 (dd, J = 8.6, 3.2 Hz, 1H), 4.37 (s, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 171.9, 130.6, 130.5, 130.1, 129.7, 126.6, 121.7, 121.7, 117.6, 112.6, 47.2.
-
25

Molecular weight 277.70. 5-Aminosalicylate (0.2 mmol, 31 mg), 2-chlorobenzaldehyde (0.2 mmol, 22.5 μL), and 2 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was heated for 1 h at 100 °C, then sodium borohydride (0.4 mmol, 15 mg) was added and the mixture was maintained at 100 °C for 3 h. The reaction was acidified with 2 M HCl. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 10% B; 5–10 min, 10–35% B; 10–30 min, 35–80% B), followed by freeze-drying, yielded 39.2 mg of white solid (0.14 mmol, 70%). LCMS [M + H]+ m/z = 276.13, 278.13. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.53 (s, 1H), 7.48−7.38 (m, 2H), 7.34−7.25 (m, 2H), 6.95 (d, J = 2.9 Hz, 1H), 6.88 (dd, J = 8.8, 3.0 Hz, 1H), 6.76 (dd, J = 8.8, 0.4 Hz, 1H), 4.30 (s, 2H).
-
26

Molecular weight: 366.16. 5-Aminosalicylate (0.065 mmol, 10 mg), 6-bromo-1,3-benzodioxole-5-carboxaldehyde (0.13 mmol, 30 mg), 1 mL of EtOH, and 100 μL of acetic acid were combined in a round-bottom flask fitted with a reflux condenser. The mixture was stirred for 2 h at 100 °C, then sodium borohydride (0.26 mmol, 10 mg) was added and the mixture was stirred at 100 °C for another 30 min. HPLC purification (0.1% TFA, methanol/water gradient of 0–8 min, 5% B; 8–22 min, 5–75% B; 22–34 min, 100% B), followed by freeze-drying, yielded 9.7 mg of product (0.027 mmol, 41%). LCMS [M − H]− m/z = 364.00, 365.95. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.53 (s, 1H), 7.21 (d, J = 0.5 Hz, 1H), 6.94 (d, J = 4.6 Hz, 2H), 6.85 (d, J = 2.9 Hz, 1H), 6.77 (s, 1H), 6.03 (s, 2H), 4.15 (s, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 171.9, 153.3, 147.3, 147.2, 131.5, 122.1, 117.6, 112.8, 112.6, 112.3, 112.3, 108.7, 102.0, 101.9, 47.5.
-
27

Molecular weight: 321.71. 5-Aminosalicylate (0.065 mmol, 10 mg), 6-chloro-1,3-benzodioxole-5-carboxaldehyde (0.13 mmol, 24 mg), 1 mL of EtOH, and 100 μL of acetic acid were combined in a round-bottom flask fitted with a reflux condenser. The mixture was stirred for 2 h at 100 °C, then sodium borohydride (0.26 mmol, 10 mg) was added and mixture was stirred for 30 min at 100 °C. HPLC purification (0.1% TFA, methanol/water gradient of 0–8 min, 5% B; 8–22 min, 5–75% B; 22–34 min, 100% B) was followed by freeze-drying, yielding 10.1 mg of product (0.031 mmol, 48%). LCMS [M − H]− m/z = 320.08. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.55 (s, 1H), 7.07 (s, 1H), 6.96 (d, J = 2.9 Hz, 1H), 6.94 (s, 1H), 6.87 (dd, J = 8.8, 2.9 Hz, 1H), 6.76 (d, J = 8.9 Hz, 1H), 6.03 (d, J = 0.4 Hz, 2H), 4.19 (s, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 171.9, 153.3, 147.0, 146.7, 140.1, 129.9, 123.8, 122.2, 117.6, 112.6, 111.9, 109.5, 108.5, 101.9, 45.0.
-
28

Molecular weight: 287.27. 5-Aminosalicylate (0.4 mmol, 61 mg), 2,3-(methylenedioxy)benzaldehyde (0.4 mmol, 45.8 μL) (Combi-Blocks), and 4 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was stirred for 30 min at 100 °C, then sodium borohydride (0.8 mol, 30 mg) was added and stirring was continued for 30 min at 100 °C. HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–10 min, 20% B; 10–40 min, 20–100% B; 40–50 min, 100% B) was followed by freeze-drying, yielding 85.3 mg of product (0.30 mmol, 74%). LCMS [M − H]− m/z = 286.15. 1H NMR (400 MHz, DMSO-d6, ppm) δ 7.31 (s, 1H), 7.12−7.05 (m, 1H), 6.98−6.73 (m, 5H), 6.01 (d, J = 2.4 Hz, 2H), 4.31 (s, 2H), 4.18 (s, 1H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 172.0, 153.5, 146.7, 145.1, 122.5, 121.5, 121.4, 121.2, 120.9, 117.6, 112.6, 107.3, 107.1, 100.8, 57.2, 41.8.
-
29

Purchased from ChemBridge.
-
30

Molecular weight: 230.22. See section “Synthesis of Hybrid Scaffold. 2-(5-Amino-2-hydroxyphenyl)isonicotinic Acid (30)”.
-
31

Molecular weight: 244.25. This compound was purified as a side product of the reaction to produce compound 30. LCMS [M + H]+ m/z = 245.19. 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.86 (dd, J = 5.0, 0.9 Hz, 1H), 8.38 (dd, J = 1.6, 0.9 Hz, 1H), 7.78 (dd, J = 5.0, 1.6 Hz, 1H), 7.76 (d, J = 2.7 Hz, 1H), 7.31 (dd, J = 8.8, 2.7 Hz, 1H), 7.25 (d, J = 8.9 Hz, 1H), 3.88 (s, 3H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 166.3, 154.9, 154.7, 150.4, 138.3, 127.9, 123.6, 123.3, 123.2, 121.4, 113.5, 56.3.
-
32

Purchased from Sigma.
-
33

Molecular weight: 350.32. 2-(Acetoxy)benzoyl chloride (0.071 mmol, 14 mg), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial. The mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and the mixture was stirred at ambient temperature for 16 h. The intermediate was isolated by HPLC (0.1% TFA, MeOH/water gradient of 0–5 min, 15% B; 5–10 min, 15–40% B; 10–35 min, 40–80% B), followed by concentration at reduced pressure. The concentrate was transferred to a sealed pressure vial with 0.5 mL of 2 M NaOH and heated to 100 °C for 1 h. The reaction was quenched with 2 M HCl. The final product was HPLC purified (0.1% TFA, MeOH/water gradient of 0–5 min, 15% B; 5–10 min, 15–50% B; 10–35 min, 50–100% B), followed by freeze-drying, yielding 0.9 mg of yellow solid (0.003 mmol, 20%). LCMS [M + H]+ m/z = 351.26. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.35 (s, 1H), 8.84 (d, J = 5.4 Hz, 1H), 8.50 (s, 1H), 8.37 (s, 1H), 8.00 (d, J = 7.9 Hz, 1H), 7.84 (d, J = 5.3 Hz, 1H), 7.72 (dd, J = 8.2, 1.5 Hz, 1H), 7.44 (s, 1H), 7.00 (s, 1H), 6.99 (d, J = 2.8 Hz, 1H), 6.97 (s, 1H).
-
34

Molecular weight: 350.32. 3-(Acetoxy)benzoyl chloride (0.071 mmol, 14 mg), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and stirring was continued at ambient temperature for 16 h. The reaction mixture was then transferred to a sealed pressure vial with 0.5 mL of 2 M NaOH and heated to 100 °C for 1 h. The reaction was quenched with 2 M HCl. HPLC purification (0.1% TFA, MeOH/water gradient of 0–5 min, 15% B; 5–10 min, 15–50% B; 10–35 min, 50–100% B), followed by freeze-drying, yielded 1.9 mg of yellow solid (0.005 mmol, 32%). LCMS [M + H]+ m/z = 351.16. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.11 (s, 1H), 9.74 (s, 1H), 8.83 (dd, J = 5.3, 1.1 Hz, 1H), 8.45 (d, J = 16.9 Hz, 1H), 7.88−7.80 (m, 1H), 7.45−7.39 (m, 1H), 7.38−7.27 (m, 2H), 6.97 (ddt, J = 4.9, 2.6, 1.3 Hz, 2H), 6.95 (d, J = 1.2 Hz, 1H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 165.9, 165.2, 157.4, 154.7, 148.2, 140.1, 136.4, 131.3, 129.4, 124.5, 121.1, 119.3. 119.1, 118.8, 118.4, 118.1, 117.7, 114.4.
-
35

Molecular weight: 350.32. 4-(Acetoxy)benzoyl chloride (0.071 mmol, 14 mg) and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and the mixture was stirred at ambient temperature for 16 h. The reaction mixture was then transferred to a sealed pressure vial with 0.5 mL of 2 M NaOH and heated to 100 °C for 1 h. The reaction was quenched with 2 M HCl. HPLC purification (0.1% TFA, MeOH/water gradient of 0–5 min, 15% B; 5–10 min, 15–50% B; 10–35 min, 50–100% B), followed by freeze-drying, yielded 2.0 mg of yellow solid (0.006 mmol, 34%). LCMS [M + H]+ m/z = 351.18. 1H NMR (400 MHz, DMSO-d6, ppm) δ 12.99 (s, 1H), 10.08 (s, 1H), 9.95 (s, 1H), 8.83 (dd, J = 5.1, 0.8 Hz, 1H), 8.53−8.35 (m, 2H), 7.87 (d, J = 8.7 Hz, 2H), 7.85−7.80 (m, 2H), 6.95 (d, J = 8.8 Hz, 1H), 6.87 (d, J = 8.7 Hz, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 165.9, 164.8, 160.4, 157.4, 154.5, 148.1, 140.1, 131.5, 129.5 (2C), 125.4, 124.5, 121.1, 119.2, 118.9, 118.7, 117.7, 114.9 (2C).
-
36

Molecular weight: 339.30. 5-Methylisoxazole-3-carboxyl chloride (0.071 mmol, 10 mg) (Oakwood Research), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and the mixture was stirred at ambient temperature for an additional 16 h. HPLC purification (0.1% TFA, MeOH/water gradient of 0–10 min, 15% B; 10–15 min, 15–40% B; 15–35 min, 40–80% B) was followed by freeze-drying, yielding 1.5 mg of yellow solid (0.004 mmol, 40%). LCMS [M + H]+ m/z = 340.16. 1H NMR (400 MHz, DMSO-d6, ppm) δ 13.16 (s, 1H), 10.57 (s, 1H), 8.82 (d, J = 5.1 Hz, 1H), 8.49 (d, J = 2.5 Hz, 1H), 8.46 (s, 1H), 7.85 (d, J = 2.5 Hz, 1H), 7.85−7.81 (m, 2H), 6.97 (d, J = 8.8 Hz, 1H), 6.67 (d, J = 1.1 Hz, 1H), 2.54 (s, 3H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 171.4, 165.9, 159.3, 157.1, 157.0, 155.3, 148.0, 130.1, 124.4, 121.3, 119.3, 119.3, 118.8, 117.8, 101.6, 11.9.
-
37

Molecular weight: 348.35. Phenylacetyl chloride (0.071 mmol, 9.4 μL) (Sigma), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and stirring was continued at ambient temperature for 16 h. HPLC purification (0.1% TFA, MeOH/water gradient of 0–10 min, 15% B; 10–15 min, 15–50% B; 15–35 min, 50–90% B) was followed by freeze-drying, yielding 0.3 mg of yellow solid (0.001 mmol, 8%). LCMS [M + H]+ m/z = 349.18. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.11 (s, 1H), 8.81 (d, J = 5.2 Hz, 1H), 8.40 (s, 1H), 8.28 (d, J = 2.7 Hz, 1H), 7.84−7.75 (m, 1H), 7.62−7.52 (m, 1H), 7.34 (d, J = 6.7 Hz, 3H), 7.21 (s, 1H), 7.08 (s, 1H), 7.00−6.86 (m, 2H), 3.62 (s, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 168.7, 165.9, 157.2, 154.3, 148.3, 139.8, 136.1, 131.4, 129.1 (2C), 128.3 (2C), 126.5, 123.2, 121.1, 119.4, 119.1, 117.9, 117.9, 43.4.
-
38

Molecular weight: 427.25. 2-(3-Bromophenyl)acetyl chloride (0.71 mmol, 17 mg), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and the mixture was stirred at ambient temperature for 16 h. HPLC purification (0.1% TFA, MeOH/water gradient of 0–10 min, 15% B; 10–15 min, 15–50% B; 15–35 min, 50–90% B), followed by freeze-drying, yielded 0.6 mg of yellow solid (0.001 mmol, 13%). LCMS [M + H]+ m/z = 427.09, 429.05. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.13 (d, J = 1.5 Hz, 1H), 8.81 (dd, J = 5.3, 1.3 Hz, 1H), 8.41 (s, 1H), 8.27 (s, 1H), 7.84−7.77 (m, 1H), 7.62−7.51 (m, 2H), 7.50−7.42 (m, 1H), 7.39−7.25 (m, 2H), 6.92 (dd, J = 8.8, 1.5 Hz, 1H), 3.65 (d, J = 1.6 Hz, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 219.0, 168.1, 165.9, 157.1, 154.3, 148.3, 139.9, 138.7, 131.9, 131.2, 130.5, 129.4, 128.3, 123.2, 121.5, 121.1, 119.4, 118.0, 117.9, 42.6.
-
39

Molecular weight: 378.38. 2-(4-Methoxyphenyl)acetyl chloride (0.071 mmol, 10.9 μL), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and the mixture was stirred at ambient temperature for 16 h. HPLC purification (0.1% TFA, MeOH/water gradient of 0–10 min, 15% B; 10–15 min, 15–50% B; 15–35 min, 50–90% B) was followed by freeze-drying, yielding 1.1 mg of yellow solid (0.003 mmol, 29%). LCMS [M + H]+ m/z = 379. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.05 (s, 1H), 8.87−8.76 (m, 1H), 8.41 (s, 1H), 8.27 (s, 1H), 7.82 (d, J = 5.3 Hz, 1H), 7.58 (d, J = 8.5 Hz, 1H), 7.29−7.24 (m, 2H), 6.94−6.86 (m, 2H), 3.73 (d, J = 1.9 Hz, 3H), 3.54 (d, J = 1.3 Hz, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 169.1, 165.9, 158.0, 157.2, 154.2, 148.2, 131.4, 130.1 (2C), 128.0, 123.2, 121.1, 119.4, 119.0, 117.8, 113.8 (2C), 55.0, 42.5.
-
40

Molecular weight: 378.38. 2-(3-Methoxyphenyl)acetyl chloride (0.071 mmol, 11.1 μL), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and the mixture was stirred at ambient temperature for 16 h. HPLC purification (0.1% TFA, MeOH/water gradient of 0–5 min, 15% B; 5–10 min, 15–50% B; 10–35 min, 50–100% B) was followed by freeze-drying, yielding 1.6 mg of yellow solid (0.004 mmol, 25%). LCMS [M + H]+ m/z = 379.20. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.09 (s, 1H), 8.81 (d, J = 5.2 Hz, 1H), 8.41 (d, J = 1.3 Hz, 1H), 8.27 (d, J = 2.6 Hz, 1H), 7.81 (dd, J = 5.2, 1.4 Hz, 1H), 7.59 (dd, J = 8.9, 2.5 Hz, 1H), 7.23 (d, J = 8.1 Hz, 1H), 6.94−6.91 (m, 2H), 6.91 (s, 1H), 6.85−6.79 (m, 1H), 3.75 (s, 3H), 3.59 (s, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 169.0, 166.3, 159.7, 157.6, 154.8, 138.0, 131.8, 129.8, 123.7, 121.8, 121.6, 119.8, 119.5, 118.3, 118.3, 115.4, 112.4, 55.5, 43.9.
-
41

Molecular weight: 325.28. Isoxazole-5-carbonyl chloride (0.071 mmol, 9.3 mg) (Maybridge), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and the mixture was stirred at ambient temperature for 1 h. HPLC purification (0.1% TFA, MeOH/water gradient of 0–5 min, 15% B; 5–10 min, 15–40% B; 10–35 min, 40–100% B), followed by freeze-drying, yielded 1.0 mg of orange solid (0.004 mmol, 33%). LCMS [M + H]+ m/z = 326.18. 1H NMR (400 MHz, DMSO-d6, ppm) δ 10.72 (s, 1H), 8.84 (dd, J = 5.2, 0.8 Hz, 1H), 8.82 (d, J = 1.9 Hz, 1H), 8.48 (dd, J = 1.5, 0.9 Hz, 1H), 8.43 (d, J = 2.6 Hz, 1H), 7.86−7.81 (m, 2H), 7.24 (d, J = 1.9 Hz, 1H), 7.00 (d, J = 8.9 Hz, 1H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 165.9, 162.8, 157.1, 155.4, 153.8, 151.9, 148.3, 140.0, 129.8, 124.6, 121.3, 119.8, 119.5, 119.1, 118.0, 106.6.
-
42

Molecular weight: 302.28. 2-Methoxyacetyl chloride (0.071 mmol, 6.5 μL), N-hydroxysuccinimide (0.13 mmol, 15 mg), and 0.5 mL of acetonitrile were added to a glass vial, and the mixture was stirred at ambient temperature for 1 h. Compound 30 (0.013 mmol, 3 mg) in 0.5 mL of 1:1:1 water/acetonitrile/methanol was then added, and the mixture was stirred at ambient temperature for 1 h. HPLC purification (0.1% TFA, MeOH/water gradient of 0–10 min, 15% B; 10–15 min, 15–40% B; 15–35 min, 40–80% B), followed by freeze-drying, yielded 1.5 mg of yellow solid (0.005 mmol, 50%). LCMS [M + H]+ m/z = 303.22. 1H NMR (400 MHz, DMSO-d6, ppm) δ 9.70 (s, 1H), 8.82 (d, J = 5.2 Hz, 1H), 8.45 (s, 1H), 8.34 (d, J = 2.5 Hz, 1H), 7.83 (dd, J = 5.2, 1.5 Hz, 1H), 7.74 (dd, J = 9.1, 2.3 Hz, 1H), 6.92 (d, J = 8.8 Hz, 1H), 3.99 (s, 2H), 3.40 (s, 3H). 13C NMR (126 MHz, DMSO- d6, ppm) δ 167.7, 165.9, 157.3, 154.7, 148.1, 140.0, 130.5, 124.0, 121.1, 119.3, 118.7, 118.7, 117.7, 71.8, 58.7.
-
43

Molecular weight: 240.21. Methyl 2-bromo-4-pyridinecarboxylate (0.19 mmol, 30 mg), 5-cyano-2-hydroxyphenylboronic acid (0.28 mmol, 61 mg) (Combi-Blocks), cesium carbonate (0.70 mmol, 228 mg), Pd6 mixed catalyst (0.014 mmol, 7 mg) (CombiPhos), and 2 mL of DMF were added to a sealed pressure vial. The mixture was then heated to 120 °C for 5 h. Upon cooling to ambient temperature, the mixture was acidified with 2 M HCl, extracted with EtOAc, washed with brine, and concentrated. The product was isolated by HPLC purification (0.1% TFA, acetonitrile/water gradient of 0–5 min, 10% B; 5–20 min, 10–40% B; 20–30 min, 40–100% B), followed by freeze-drying, yielding 3.0 mg of gray solid (0.013 mmol, 7%), and the unhydrolyzed methyl ester was not collected. LCMS [M + H]+ m/z = 241.18. 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.85 (d, J = 5.1 Hz, 1H), 8.68 (s, 1H), 8.62 (d, J = 2.1 Hz, 1H), 7.88 (dd, J = 5.2, 0.9 Hz, 1H), 7.76 (dd, J = 8.5, 2.1 Hz, 1H), 7.12 (d, J = 8.5 Hz, 1H). 13C NMR (100 MHz, DMSO-d6, ppm) δ 166.0, 162.2, 155.7, 148.4, 140.4, 139.9, 135.1, 133.3, 122.2, 121.2, 120.9, 119.1, 101.9.
-
44

Molecular weight: 272.26. Compound 30 (2 mg, 0.009 mmol), acetic anhydride (5 μL, 0.053 mmol, Sigma), and 250 μL of water were added to a glass vial. The reaction was stirred at ambient temperature for 30 min and then diluted in HPLC solvent. HPLC purification (0.1% TFA, MeOH/water gradient of 0–5 min, 15% B; 5–10 min, 15–50% B; 10–35 min, 50–90% B) was followed by freeze-drying, yielding 2.2 mg of yellow solid (0.008 mmol, 90%). LCMS [M + H]+ m/z = 273.57. 1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 8.82 (dd, J = 5.2, 0.8 Hz, 1H), 8.41 (dd, J = 1.5, 0.9 Hz, 1H), 8.23 (d, J = 2.6 Hz, 1H), 7.82 (dd, J = 5.1, 1.4 Hz, 1H), 7.57 (dd, J = 8.8, 2.6 Hz, 1H), 6.91 (d, J = 8.8 Hz, 1H), 2.03 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 167.9, 165.9, 157.2, 154.1, 148.2, 139.9, 131.5, 123.3, 121.1, 119.4, 118.9, 117.9, 117.8, 23.9.
-
45

Molecular weight: 324.33. Compound 30 (0.017 mmol, 4 mg), 1-methyl-1H-pyrazole-5-carbaldehyde (0.034 mmol, 3.3 μL), and 1 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was stirred for 2 h at 100 °C, then sodium borohydride (0.068 mmol, 2.6 mg) was added and the mixture was stirred for 30 min at 100 °C. HPLC purification (0.1% TFA, methanol/water gradient of 0–8 min, 5% B; 5–22 min, 5–75% B; 22–34 min, 100% B), followed by freeze-drying, yielded 1.8 mg of product (0.00356 mmol, 33%). LCMS [M + H]+ m/z = 325.16. 1H NMR (400 MHz, CD3OD, ppm) δ 8.79 (dd, J = 5.3, 0.9 Hz, 1H), 8.53 (t, J = 1.2 Hz, 1H), 7.98 (dd, J = 5.3, 1.4 Hz, 1H), 7.76 (d, J = 2.7 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.23 (dd, J = 8.8, 2.7 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 6.51 (d, J = 2.0 Hz, 1H), 4.75 (s, 2H), 3.70 (s, 3H). 13C NMR (126 MHz, CD3OD, ppm) δ 165.8, 157.2, 155.8, 147.0, 140.7, 138.0, 137.2, 122.4, 121.4, 119.7, 119.3, 119.0, 116.3, 107.0, 42.4, 35.2.
-
46

Molecular weight: 364.35. Compound 30 (0.038 mmol, 9 mg), 2,3-(methylenedioxy)benzaldehyde (0.075 mmol, 8.6 μL) (Combi-Blocks), 2 mL of EtOH, and 100 μL of glacial acetic acid were combined in a round-bottom flask fitted with a reflux condenser. The mixture was stirred for 3 h at ambient temperature, then sodium borohydride (0.15 mmol, 6 mg) was added and the mixture was stirred for an additional 30 min at ambient temperature. The reaction was quenched with ∼10 drops of 2 N HCl. HPLC purification (0.1% TFA, methanol/water gradient of 0–8 min, 5% B; 5–22 min, 5–75% B; 22–34 min, 100% B) was followed by freeze-drying, yielding 6.4 mg of product (0.018 mmol, 47%). LCMS [M − H]− m/z = 363.14. 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.79 (dd, J = 5.2, 0.8 Hz, 1H), 8.43 (d, J = 1.4 Hz, 1H), 7.80 (dd, J = 5.2, 1.4 Hz, 1H), 7.47 (s, 1H), 7.04−6.74 (m, 5H), 6.03 (s, 2H), 4.32 (s, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 166.0, 158.2, 157.9, 157.5, 148.1, 146.8, 145.3, 139.9, 121.9, 121.6, 120.9, 119.3, 118.5, 117.4, 115.1, 107.6, 100.8.
-
47

Molecular weight: 399.24. Compound 30 (0.02 mmol, 4.6 mg), 4-bromobenzaldehyde (0.08 mmol, 15 mg), and 1 mL of EtOH were combined in a round-bottom flask fitted with a reflux condenser. The mixture was stirred for 2 h at 100 °C, then sodium borohydride (0.08 mmol, 3 mg) was added and the mixture was stirred for 30 min at 100 °C. HPLC purification (0.1% TFA, methanol/water gradient of 0–8 min, 5% B; 5–22 min, 5–75% B; 22–34 min, 100% B), followed by freeze-drying, yielded 3.2 mg of product (0.008 mmol, 40%). LCMS [M + H]+ m/z = 399.13, 401.11. 1H NMR (400 MHz, DMSO-d6, ppm) δ 8.79 (dd, J = 5.1, 0.8 Hz, 1H), 8.41 (s, 1H), 7.79 (dd, J = 5.2, 1.4 Hz, 1H), 7.55 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.26−6.95 (m, 1H), 6.83 (s, 1H), 4.36 (s, 2H). 13C NMR (126 MHz, DMSO-d6, ppm) δ 171.9, 166.4, 158.6, 158.4, 157.9, 148.6, 140.3, 131.7 (2C), 130.8 (2C), 121.4, 120.1, 118.8, 118.1, 115.7, 23.0.
Supplementary Material
Acknowledgments
The authors thank Dr. Wendell Lim and Dr. Denise Chan for the kind gift of expression plasmid and of FIH gene, respectively, Hayarpi Torosyan for help with aggregation assays, and Dr. Anat Levit, Dr. Trent E. Balius, Lindsey Pack, and Christina Fitzsimmons for valuable comments on the manuscript. This work was supported by Grant GM59957 (for docking) and Grant GM71630 (for colloidal aggregation controls) (to B.K.S.), Searle Scholar award, V Foundation Scholar awards, and UCSF Prostate Cancer Developmental Research Program (to D.G.F.) and by Arthritis Research UK (program Grant number 20522), the NIHR Oxford Biomedical Research Unit and the Rosetrees Trust (to U.O.). The Structural Genomics Consortium is a registered charity (No. 1097737) that receives funds from Abbvie, Bayer Healthcare, Boehringer Ingelheim, the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Eli Lilly and Company, Genome Canada, GlaxoSmithKline, the Ontario Ministry of Economic Development and Innovation, Janssen, the Novartis Research Foundation, Pfizer, Takeda, and the Wellcome Trust.
ABBREVIATIONS USED
- 2,4-PDCA
2,4-pyridinedicarboxylic acid
- LSD1
lysine-specific demethylase 1
- JmjC
Jumonji C domain
- α-KG
α-ketoglutarate
- FIH
factor-inhibiting hypoxia-inducible factor
- LE
ligand efficiency
- xLogP
predicted octanol–water partitioning coefficient
- H3
histone H3
- KDM
lysine-specific demethylase
- NHS
N-hydroxysuccinimide
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.5b01527.
Experimental procedure for cloning, expression for all enzymes, MALDI and AlphaScreen counterscreening; IC50 for selected 5-aminosalicylates series of inhibitors; structures of hybrid scaffolds that were virtually screened; crystallography statistics for compounds 43, 30, 44, 42, 35, 40, and 36; omit maps and 2Fo − Fc maps for all compounds; figures showing docking targets used, docking binding site used, and superimposition of hybrid compounds with structure 2OQ6 showing H3 histone peptide; counterscreening confirming specific inhibition and binding of compounds through an aggregation test, DLS test, and enzymes Tm stabilization curves (PDF)
Accession Codes
Atomic coordinates and structure factors for the KDM4A complexes have been deposited in the Protein Data Bank as entries 5A7N in complex with compound 43, 5A7O with 42, 5A7P with 36, 5A7Q with 30, 5A7S with 44, 5A7W with 35, and 5A80 with 40.
Notes
The authors declare no competing financial interest.
References
- 1.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 3.Johansson C, Tumber A, Che K, Cain P, Nowak R, Gileadi C, Oppermann U. The roles of Jumonji-type oxygenases in human disease. Epigenomics. 2014;6:89–120. doi: 10.2217/epi.13.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Labbe RM, Holowatyj A, Yang ZQ. Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. Am J Transl Res. 2013;6:1–15. [PMC free article] [PubMed] [Google Scholar]
- 5.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 6.Katoh M, Katoh M. Identification and characterization of JMJD2 family genes in silico. Int J Oncol. 2004;24:1623–1628. [PubMed] [Google Scholar]
- 7.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 8.Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 9.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 10.Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20:1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun. 2007;359:742–746. doi: 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- 12.Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-containing JMJD2/KDM4 Proteins. J Biol Chem. 2009;284:8395–8405. doi: 10.1074/jbc.M807818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, Dorrestein PC, Rosenfeld MG. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. 2011;147:773–788. doi: 10.1016/j.cell.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu CH, Wang LY, Hsu KC, Chen CC, Cheng HH, Wang SM, Wu CM, Chen TJ, Li LT, Liu R, Hung CL, Yang JM, Kung HJ, Wang WC. KDM4B as a target for prostate cancer: structural analysis and selective inhibition by a novel inhibitor. J Med Chem. 2014;57:5975–5985. doi: 10.1021/jm500249n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Zheng P, Liu Y, Ji T, Liu X, Yao S, Cheng X, Li Y, Chen L, Xiao Z, Zhou J, Li J. An epigenetic role for PRL-3 as a regulator of H3K9 methylation in colorectal cancer. Gut. 2013;62:571–581. doi: 10.1136/gutjnl-2011-301059. [DOI] [PubMed] [Google Scholar]
- 16.Yang J, Jubb AM, Pike L, Buffa FM, Turley H, Baban D, Leek R, Gatter KC, Ragoussis J, Harris AL. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010;70:6456–6466. doi: 10.1158/0008-5472.CAN-10-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walport LJ, Hopkinson RJ, Schofield CJ. Mechanisms of human histone and nucleic acid demethylases. Curr Opin Chem Biol. 2012;16:525–534. doi: 10.1016/j.cbpa.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 18.McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20:659–672. doi: 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 20.Thinnes CC, England KS, Kawamura A, Chowdhury R, Schofield CJ, Hopkinson RJ. Targeting histone lysine demethylases – Progress, challenges, and the future. Biochim Biophys Acta, Gene Regul Mech. 2014;1839:1416–1432. doi: 10.1016/j.bbagrm.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thalhammer A, Mecinovic J, Loenarz C, Tumber A, Rose NR, Heightman TD, Schofield CJ. Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates. Org Biomol Chem. 2011;9:127–135. doi: 10.1039/c0ob00592d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majamaa K, Hanauske-Abel HM, Gunzler V, Kivirikko KI. The 2-oxoglutarate binding site of prolyl 4-hydroxylase. Identification of distinct subsites and evidence for 2-oxoglutarate decarboxylation in a ligand reaction at the enzyme-bound ferrousion. Eur J Biochem. 1984;138:239–245. doi: 10.1111/j.1432-1033.1984.tb07907.x. [DOI] [PubMed] [Google Scholar]
- 23.King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, Klose RJ, Oppermann U, Jadhav A, Heightman TD, Maloney DJ, Schofield CJ, Simeonov A. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One. 2010;5:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopkinson RJ, Tumber A, Yapp C, Chowdhury R, Aik W, Che KH, Li XS, Kristensen JBL, King ONF, Chan MC, Yeoh KK, Choi H, Walport LJ, Thinnes CC, Bush JT, Lejeune C, Rydzik AM, Rose NR, Bagg EA, McDonough MA, Krojer TJ, Yue WW, Ng SS, Olsen L, Brennan PE, Oppermann U, Muller S, Klose RJ, Ratcliffe PJ, Schofield CJ, Kawamura A. 5-Carboxy-8-hydroxyquinoline is a broad spectrum 2-oxoglutarate oxygenase inhibitor which causes iron translocation. Chem Sci. 2013;4:3110–3117. doi: 10.1039/C3SC51122G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rose NR, Ng SS, Mecinovic J, Lienard BM, Bello SH, Sun Z, McDonough MA, Oppermann U, Schofield CJ. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J Med Chem. 2008;51:7053–7056. doi: 10.1021/jm800936s. [DOI] [PubMed] [Google Scholar]
- 26.Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, Ruiz J, Bruick R, Pena-Llopis S, Xu J, Babinski DJ, Frantz DE, Brekken RA, Quinn AM, Simeonov A, Easmon J, Martinez ED. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035. doi: 10.1038/ncomms3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo X, Liu Y, Kubicek S, Myllyharju J, Tumber A, Ng S, Che KH, Podoll J, Heightman TD, Oppermann U, Schreiber SL, Wang X. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J Am Chem Soc. 2011;133:9451–9456. doi: 10.1021/ja201597b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinemann B, Nielsen JM, Hudlebusch HR, Lees MJ, Larsen DV, Boesen T, Labelle M, Gerlach LO, Birk P, Helin K. Inhibition of demethylases by GSK-J1/J4. Nature. 2014;514:E1–2. doi: 10.1038/nature13688. [DOI] [PubMed] [Google Scholar]
- 30.Woon EC, Tumber A, Kawamura A, Hillringhaus L, Ge W, Rose NR, Ma JH, Chan MC, Walport LJ, Che KH, Ng SS, Marsden BD, Oppermann U, McDonough MA, Schofield CJ. Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of JmjC histone demethylases. Angew Chem, Int Ed. 2012;51:1631–1634. doi: 10.1002/anie.201107833. [DOI] [PubMed] [Google Scholar]
- 31.Murray CW, Blundell TL. Structural biology in fragment-based drug design. Curr Opin Struct Biol. 2010;20:497–507. doi: 10.1016/j.sbi.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Hamada S, Kim TD, Suzuki T, Itoh Y, Tsumoto H, Nakagawa H, Janknecht R, Miyata N. Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg Med Chem Lett. 2009;19:2852–2855. doi: 10.1016/j.bmcl.2009.03.098. [DOI] [PubMed] [Google Scholar]
- 33.Hann MM, Leach AR, Harper G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J Chem Inf Model. 2001;41:856–864. doi: 10.1021/ci000403i. [DOI] [PubMed] [Google Scholar]
- 34.Hann MM, Keseru GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat Rev Drug Discovery. 2012;11:355–365. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- 35.Murray CW, Carr MG, Callaghan O, Chessari G, Congreve M, Cowan S, Coyle JE, Downham R, Figueroa E, Frederickson M, Graham B, McMenamin R, O’Brien MA, Patel S, Phillips TR, Williams G, Woodhead AJ, Woolford AJ. Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency. J Med Chem. 2010;53:5942–5955. doi: 10.1021/jm100059d. [DOI] [PubMed] [Google Scholar]
- 36.Bembenek SD, Tounge BA, Reynolds CH. Ligand efficiency and fragment-based drug discovery. Drug Discovery Today. 2009;14:278–283. doi: 10.1016/j.drudis.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 37.Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 38.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mysinger MM, Shoichet BK. Rapid context-dependent ligand desolvation in molecular docking. J Chem Inf Model. 2010;50:1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 40.Meng EC, Shoichet BK, Kuntz ID. Automated Docking with Grid-Based Energy Evaluation. J Comput Chem. 1992;13:505–524. [Google Scholar]
- 41.Gilson MK, Honig B. The inclusion of electrostatic hydration energies in molecular mechanics calculations. J Comput-Aided Mol Des. 1991;5:5–20. doi: 10.1007/BF00173467. [DOI] [PubMed] [Google Scholar]
- 42.Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry. 2005;44:12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- 43.Babaoglu K, Simeonov A, Irwin JJ, Nelson ME, Feng B, Thomas CJ, Cancian L, Costi MP, Maltby DA, Jadhav A, Inglese J, Austin CP, Shoichet BK. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J Med Chem. 2008;51:2502–2511. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, Heveker N, Shoichet BK, Volkman BF. Structure-based ligand discovery for the protein-protein interface of chemokine receptor CXCR4. Proc Natl Acad Sci U S A. 2012;109:5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng ZW, Gillespie P, Weisel M, So SS, So WV, Kondru R, Narayanan A, Hermann JC. A Crowd-Based Process and Tool for HTS Hit Triage. Mol Inf. 2013;32:337–345. doi: 10.1002/minf.201200154. [DOI] [PubMed] [Google Scholar]
- 46.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 47.Shoichet BK. Screening in a spirit haunted world. Drug Discovery Today. 2006;11:607–615. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giannetti AM, Koch BD, Browner MF. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J Med Chem. 2008;51:574–580. doi: 10.1021/jm700952v. [DOI] [PubMed] [Google Scholar]
- 49.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rose NR, Woon EC, Kingham GL, King ON, Mecinovic J, Clifton IJ, Ng SS, Talib-Hardy J, Oppermann U, McDonough MA, Schofield CJ. Selective inhibitors of the JMJD2 histone demethylases: combined nondenaturing mass spectrometric screening and crystallographic approaches. J Med Chem. 2010;53:1810–1818. doi: 10.1021/jm901680b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang KH, King ON, Tumber A, Woon EC, Heightman TD, McDonough MA, Schofield CJ, Rose NR. Inhibition of histone demethylases by 4-carboxy-2,2′-bipyridyl compounds. ChemMedChem. 2011;6:759–764. doi: 10.1002/cmdc.201100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BM, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- 53.Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, Hagman J, Hansen K, Shi Y, Zhang G. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 54.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 55.Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barker JJ, Barker O, Courtney SM, Gardiner M, Hesterkamp T, Ichihara O, Mather O, Montalbetti CA, Muller A, Varasi M, Whittaker M, Yarnold CJ. Discovery of a novel Hsp90 inhibitor by fragment linking. ChemMedChem. 2010;5:1697–1700. doi: 10.1002/cmdc.201000219. [DOI] [PubMed] [Google Scholar]
- 57.Villemagne B, Flipo M, Blondiaux N, Crauste C, Malaquin S, Leroux F, Piveteau C, Villeret V, Brodin P, Villoutreix BO, Sperandio O, Soror SH, Wohlkonig A, Wintjens R, Deprez B, Baulard AR, Willand N. Ligand Efficiency Driven Design of New Inhibitors of Mycobacterium tuberculosis Transcriptional Repressor EthR Using Fragment Growing, Merging, and Linking Approaches. J Med Chem. 2014;57:4876–4888. doi: 10.1021/jm500422b. [DOI] [PubMed] [Google Scholar]
- 58.Erlanson DA. Introduction to fragment-based drug discovery. Top Curr Chem. 2011;317:1–32. doi: 10.1007/128_2011_180. [DOI] [PubMed] [Google Scholar]
- 59.Teotico DG, Babaoglu K, Rocklin GJ, Ferreira RS, Giannetti AM, Shoichet BK. Docking for fragment inhibitors of AmpC beta-lactamase. Proc Natl Acad Sci U S A. 2009;106:7455–7460. doi: 10.1073/pnas.0813029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Y, Shoichet BK. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat Chem Biol. 2009;5:358–364. doi: 10.1038/nchembio.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tiefenbrunn T, Forli S, Baksh MM, Chang MW, Happer M, Lin YC, Perryman AL, Rhee JK, Torbett BE, Olson AJ, Elder JH, Finn MG, Stout CD. Small Molecule Regulation of Protein Conformation by Binding in the Flap of HIV Protease. ACS Chem Biol. 2013;8:1223–1231. doi: 10.1021/cb300611p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nichols DA, Renslo AR, Chen Y. Fragment-based inhibitor discovery against beta-lactamase. Future Med Chem. 2014;6:413–427. doi: 10.4155/fmc.14.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barelier S, Eidam O, Fish I, Hollander J, Figaroa F, Nachane R, Irwin JJ, Shoichet BK, Siegal G. Increasing chemical space coverage by combining empirical and computational fragment screens. ACS Chem Biol. 2014;9:1528–1535. doi: 10.1021/cb5001636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki T, Miyata N. Lysine demethylases inhibitors. J Med Chem. 2011;54:8236–8250. doi: 10.1021/jm201048w. [DOI] [PubMed] [Google Scholar]
- 65.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang SC, Chen X, Chau I, Mangano TJ, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hermann JC, Ghanem E, Li YC, Raushel FM, Irwin JJ, Shoichet BK. Predicting substrates by docking high-energy intermediates to enzyme structures. J Am Chem Soc. 2006;128:15882–15891. doi: 10.1021/ja065860f. [DOI] [PubMed] [Google Scholar]
- 67.Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM. Structure-based activity prediction for an enzyme of unknown function. Nature. 2007;448:775–779. doi: 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–88. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 69.Irwin JJ, Shoichet BK, Mysinger MM, Huang N, Colizzi F, Wassam P, Cao Y. Automated docking screens: a feasibility study. J Med Chem. 2009;52:5712–5720. doi: 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure. 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 71.Winter G, Lobley CM, Prince SM. Decision making in xia2. Acta Crystallogr, Sect D: Biol Crystallogr. 2013;69:1260–1273. doi: 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Copeland RA. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. 2nd. Wiley; New York: 2000. p. xvi.p. 397. [Google Scholar]
- 73.Kawamura A, Tumber A, Rose NR, King ON, Daniel M, Oppermann U, Heightman TD, Schofield C. Development of homogeneous luminescence assays for histone demethylase catalysis and binding. Anal Biochem. 2010;404:86–93. doi: 10.1016/j.ab.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 75.Irwin JJ, Duan D, Torosyan H, Doak AK, Ziebart KT, Sterling T, Tumanian G, Shoichet BK. An Aggregation Advisor for Ligand Discovery. J Med Chem. 2015;58:7076–7087. doi: 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cascella B, Mirica LM. Kinetic analysis of iron-dependent histone demethylases: alpha-ketoglutarate substrate inhibition and potential relevance to the regulation of histone demethylation in cancer cells. Biochemistry. 2012;51:8699–8701. doi: 10.1021/bi3012466. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.