

Abstract
Cardiomyopathy caused by lamin A/C gene mutations (LMNA cardiomyopathy) is characterized by increased myocardial fibrosis, which impairs left ventricular relaxation and predisposes to heart failure, and cardiac conduction abnormalities. While we previously discovered abnormally elevated extracellular signal-regulated kinase 1/2 (ERK1/2) activities in heart in LMNA cardiomyopathy, its role on the development of myocardial fibrosis remains unclear. We now showed that transforming growth factor (TGF)-β/Smad signaling participates in the activation of ERK1/2 signaling in LMNA cardiomyopathy. ERK1/2 acts on connective tissue growth factor (CTGF/CCN2) expression to mediate the myocardial fibrosis and left ventricular dysfunction. Studies in vivo demonstrate that inhibiting CTGF/CCN2 using a specific antibody decreases myocardial fibrosis and improves the left ventricular dysfunction. Together, these findings show that cardiac ERK1/2 activity is modulated in part by TGF-β/Smad signaling, leading to altered activation of CTGF/CCN2 to mediate fibrosis and alter cardiac function. This identifies a novel mechanism in the development of LMNA cardiomyopathy.
Introduction
One cause of dilated cardiomyopathy is dominant mutations in LMNA, the gene encoding A-type nuclear lamins, intermediate filament proteins of the nuclear envelope (1,2). Cardiomyopathy caused by LMNA mutations (i.e. LMNA cardiomyopathy) is characterized by increased myocardial fibrosis that impairs left ventricular relaxation and predisposes to heart failure, and cardiac conduction abnormalities (3–6). The onset of symptoms in LMNA cardiomyopathy, although variable, occurs most frequently in the third decade and the disease has a more aggressive course than most other inherited dilated cardiomyopathies (7). While sudden death from arrhythmias may be prevented by implantation of a pacemaker and/or defibrillator, the progressive heart failure eventually becomes resistant to treatment and heart transplantation is often the last therapeutic option (4).
We previously discovered abnormally elevated activity of extracellular signal-regulated kinase 1/2 (ERK1/2) in the hearts of LmnaH222P/H222P mice that develop cardiomyopathy (8). These mice recapitulate the cardiac disease in patients with LMNA mutations and therefore serve as a useful animal model. Inhibition of ERK1/2 signaling in LmnaH222P/H222P mice by an inhibitor of mitogen-activated protein kinase 1/2 (MEK1/2), the enzyme that activates ERK1/2, improved left ventricular ejection fraction and prolonged survival (9–11). MEK1/2 inhibitor treatment of these mice further slowed progression of myocardial fibrosis, which is prominent in humans with LMNA cardiomyopathy (10,11). Myocardial fibrosis consists of the replacement of functional cells with accumulation of collagen-rich extracellular matrix (ECM). Cardiomyocytes are tethered within the ECM, consisting of collagen, elastin, proteoglycans and glycoproteins. ECM provides a scaffold for myofiber alignment protects against sarcomere overstretching and plays a role in electrical behavior of the myocardium. Therefore, ECM stiffness and deposition of fibrous tissue have dramatic effects on heart function (12). Indeed, myocardial fibrosis contributes to diastolic and systolic dysfunctions and conduction defects in the heart (13,14).
Pro-fibrotic changes during cardiac remodeling are mainly driven by cytokines such as transforming growth factor-β (TGF-β) and the matricellular protein connective tissue growth factor (CTGF/CCN2) (15). TGF-β-dimers bind to type II receptors, which recruit and phosphorylate type I receptors such as activin receptor-like 5 (ALK5). Ligand binding to this type I receptor recruits and phosphorylates Smad2/3, which is then translocated to the nucleus and activates target gene transcription. We therefore assessed the modulation of TGF-β/Smad signaling implicated in activating fibrosis in LmnaH222P/H222P mice and its interaction with ERK1/2 signaling. We found that TGF-β/Smad signaling participates in the activation of ERK1/2 signaling in LMNA cardiomyopathy and that ERK1/2 acts on CTGF/CCN2 expression to mediate the myocardial fibrosis and left ventricular dysfunction.
Results
Myocardial fibrosis in LmnaH222P/H222P mice
Consistent with previous results (16), hearts of LmnaH222P/H222P mice at 20 weeks of age, when left ventricles function is altered (10), had an increase in fibrotic tissue compared with Lmna+/+ mice (WT mice) (Supplementary Material, Fig. S1). At 20 weeks of age, hearts from LmnaH222P/H222P mice also had significantly increased expression of mRNAs from the Col1a1 and Col1a2 encoding type I collagens of the ECM and Fn1, Scd1, Nid1 and Dcn1, respectively, encoding the basal lamina proteins fibronectin, syndecan, nidogen and decorin (Fig. 1). Our data confirmed that there is increased myocardial fibrosis in LmnaH222P/H222P mice compared with WT mice.
Figure 1.

Quantification of myocardial fibrosis in LmnaH222P/H222P mice. Cardiac expression of genes encoding proteins of the ECM in LmnaH222P/H222P mice. Bar diagrams represent relative mean mRNA (as well as standard errors of means) expression of Col1a1, Col1a2, Sdc1, Nid1, Fn1 and Dcn1 in hearts from WT mice (n = 4) and LmnaH222P/H222P mice (Lmna H222P) (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001.
TGF-β/Smad signaling is enhanced in hearts from LmnaH222P/H222P mice
Given that TGF-β signaling is an important mediator of fibrosis, we assessed its activity in hearts and isolated cardiomyocytes of LmnaH222P/H222P mice. We observed enhanced Tgf-b1 and Tgf-b2 mRNA expression in hearts from LmnaH222P/H222P mice at 20 weeks of age compared with hearts from WT mice (Fig. 2A). In hearts of LmnaH222P/H222P mice, Tgf-b1 and Tgf-b2 mRNAs were increased, respectively, by 2- and 20-fold compared with WT mice. We then further assessed the modulation of TGF-β signaling by studying the abundance of phosphorylated Smad2/3 (p-Smad2/3) in protein extracts from left ventricles and isolated ventricular cardiomyocytes from LmnaH222P/H222P and WT mice. There was a significant increase of cardiac p-Smad2/3 in LmnaH222P/H222P mouse heart (Fig. 2B). Immunofluorescence microscopy also revealed a nuclear localization of p-Smad2/3 in the heart from LmnaH222P/H222P mice (Fig. 2C), which was occurring to a lesser extend in cardiac endothelial cells (Fig. 2D). This increased nuclear translocation of p-Smad2/3 was confirmed by western blot analysis of fractionated cells (Fig. 2E). These results demonstrated abnormal activation of TGF-β signaling in hearts from LmnaH222P/H222P mice at an age when myocardial fibrosis occurs. These data extend a previous report showing activation of TGF-β/Smad2/3 signaling in failing hearts from LmnaH222P/H222P mice at 24 weeks of age (16).
Figure 2.

Activity of Tgf-β signaling in LmnaH222P/H222P mice. (A) Expression of genes encoding proteins of the Tgf-β signaling in heart from LmnaH222P/H222P mice. Bar graphs indicate the expression of Tgf-b1 and Tgf-b2 from WT mice (n = 3) and LmnaH222P/H222P mice (Lmna H222P) (n = 8). *P < 0.05, **P < 0.01. (B) Immunoblots showing phosphorylated Smad2/3 (p-smad2/3) and total smad2/3 in protein extracts of hearts (upper panel) and isolated cardiomyocytes (lower panel) from WT and LmnaH222P/H222P mice (Lmna H222P). Each lane contains protein extracts from a different mouse. (C) Micrographs showing p-smad2/3 labeling (upper part, Scale bar: 25 μm) of cross sections of hearts from WT compared with LmnaH222P/H222P mice (Lmna H222P). Nuclei counter-stained with 4′,6-diamidino-2-phenylindole (dapi) are also shown. (D) Micrographs showing p-smad2/3 and CD31 staining (lower part, Scale bar: 25 μm) of cross sections of hearts from WT compared with LmnaH222P/H222P mice (Lmna H222P). Nuclei counter-stained with 4′,6-diamidino-2-phenylindole (dapi) are also shown. (E) Immunoblots showing p-smad2/3 and total smad2/3 in nuclear and cytosol extracts from hearts from WT and LmnaH222P/H222P mice (Lmna H222P). Each lane contains protein extracts from a different mouse.
Inhibiting TGF-β/Smad signaling improves cardiac dysfunction
Given the enhanced TGF-β/Smad signaling in hearts of LmnaH222P/H222P mice, we hypothesized that reducing this activity would attenuate myocardial fibrosis and improve cardiac function. We inhibited ALK5, a receptor for TGF-β that recruits and phosphorylates Smad2/3 (17), with daily intra-peritoneal injections of SB-431542 (5 mg/kg) starting at 16 weeks of age (Fig. 3A). SB-431542 specifically targets the TGF-β signaling (18,19). After 4 weeks of treatment, mice were analyzed by echocardiography and sacrificed for biochemical and histological analysis. SB-431542 treatment reduced Smad2/3 phosphorylation in hearts of LmnaH222P/H222P mice compared with DMSO placebo treatment (Fig. 3B). After the treatment with SB-431542, myocardial fibrosis was reduced in treated LmnaH222P/H222P mice compared with placebo-treated mice (Fig. 3C). SB-431542 also reduced the levels of Col1a1 and Col1a2 mRNAs in heart (Fig. 3C). M-mode echocardiography showed that left ventricular end diastolic and end systolic diameters in LmnaH222P/H222P mice treated with SB-431542 were significantly smaller and fractional shortening was significantly increased compared with the placebo-treated mice (Fig. 3D and Table 1). SB-431542 also reduced the mRNA levels of NppA and NppB, genes that encode natriuretic peptide precursors that are synthesized in response to left ventricular dilatation, as well as Myl4, the gene encoding the light chain of myosin (Fig. 3E). Hence, TGF-β/Smad signaling inhibition that reduces fibrosis concomitantly improves left ventricular function in LmnaH222P/H222P mice.
Figure 3.

Pharmacological inhibition of Tgf-β signaling improves left-ventricular function. (A) Schematic representation of the treatment protocol of LmnaH222P/H222P mice with SB-431542. (B) Representative immunoblots using antibodies against p-smad2/3 and total smad2/3 to probe proteins extracted from hearts from LmnaH222P/H222P mice (Lmna H222P) treated with placebo or SB-431542. Each lane contains protein extracts from a different mouse. (C) Mason trichrome staining of cross sections of hearts from LmnaH222P/H222P mice (Lmna H222P) treated with placebo or SB-431542. Scale bar: 50 μm. Bar diagrams indicate the expression of Col1a1 and Col1a2 from LmnaH222P/H222P mice (Lmna H222P) treated with placebo (n = 3) or SB-431542 (n = 5). *P < 0.05. (D) Graph showing mean left ventricle end-diastolic diameter (LVEDD), mean left ventricle end-systolic diameter (LVESD) and fraction shortening (FS) in 20-week-old male LmnaH222P/H222P mice (Lmna H222P) treated with placebo (n = 15) or SB-431542 (n = 9). Values for each individual mouse receiving placebo or SB-431542 as well as standard errors of means (bars) are shown. *P < 0.05, **P < 0.01. (E) Relative expression of mRNAs from genes encoding myosin light chain (Myl4), atrial natriuretic peptide A (NppA) and brain natriuretic peptide B (NppB) in LmnaH222P/H222P mice (Lmna H222P) treated with placebo (n = 3) or SB-431542 (n = 5). Values shown are means ± standard errors. **P < 0.01.
Table 1.
Quantification of left ventricular function in Lmna H222P mice treated with SB-431542
| Genotype | Treatment | n | Heart rate (beats/min) | LVEDD (mm) | LVESD (mm) | FS (%) |
|---|---|---|---|---|---|---|
| Lmna WT | None | 10 | 532.0 ± 21.1 | 4.2 ± 0.2 | 2.4 ± 0.1 | 42.9 ± 1.5 |
| Lmna H222P | DMSO | 15 | 500.7 ± 4.6 | 4.4 ± 0.3 | 3.6 ± 0.4 | 18.9 ± 5.3 |
| Lmna H222P | SB-431542 | 9 | 496.3 ± 13.5 | 4.0 ± 0.3* | 3.0 ± 0.5** | 26.2 ± 5.0 ** |
*P < 0.05.
**P < 0.005 between DMSO-treated and SB-431542-treated mice.
Decreased ERK1/2 activation lowers myocardial fibrosis
We previously showed that reducing ERK1/2 signaling in LmnaH222P/H222P mice, either genetically or using chemical inhibitors, reduces myocardial fibrosis (9–11,20). We confirmed this using PD325901, an inhibitor of MEK1/2, to reduce ERK1/2 signaling in LmnaH222P/H222P mice from 16 to 20 weeks of age. Treating mice for 4 weeks with PD325901 reduced ERK1/2 signaling, reduced left ventricular diameters and improved function (Supplementary Material, Fig. S2A, Supplementary Material, Table S1). Inhibition of ERK1/2 signaling further decreased the degree of fibrosis in hearts from LmnaH222P/H222P mice (Supplementary Material, Fig S2B). Immunohistochemistry also revealed decreased nuclear translocation of p-Smad2/3 when LmnaH222P/H222P mice were treated with PD325901, compared with untreated or DMSO-treated animals (Fig. 4). In PD325901-treated LmnaH222P/H222P mice, Col1a1 and Col1a2 mRNAs were significantly decreased compared with DMSO-treated mice (Supplementary Material, Fig. S2C). We similarly assessed expression of Col1a1 and Col1a2 mRNAs in LmnaH222P/H222P/Erk1−/− mice lacking ERK1 (20). The level of Col1a2 mRNA was significantly decreased in hearts from these mice compared with LmnaH222P/H222P/Erk1+/+ mice (Supplementary Material, Fig. S2C). These results suggest that ERK1/2 hyperactivation contributes to the enhancement of myocardial fibrosis in the hearts of LmnaH222P/H222P mice.
Figure 4.

Pharmacological inhibition of ERK1/2 signaling acts on Tgf-β signaling in LmnaH222P/H222P mice. Micrographs showing p-smad2/3 labeling (immunohistochemistry, Scale bar: 50 μm) of cross sections of hearts from WT compared with LmnaH222P/H222P mice (Lmna H222P) treated with placebo or PD325901. Nuclei are counter-stained with 4′,6-diamidino-2-phenylindole (dapi).
We next assessed the impact of inhibition of TGF-β signaling on ERK1/2 activity in LmnaH222P/H222P mice. After treatment with SB-431542 for 4 weeks, expression of p-ERK1/2 was lower in protein extracts from hearts of LmnaH222P/H222P mice compared with placebo-treated mice (Fig. 5A). Smad2/3 phosphorylation paralleled activation of ERK1/2 in hearts of LmnaH222P/H222P mice at 12 and 24 weeks of age (Fig. 5B). This was confirmed in C2C12 cells stimulated with TGF-β. When cells were treated for 6 h with 2 nm TGF-β1, an increase of both p-Smad2/3 and p-ERK1/2 was observed (Fig. 5C). This demonstrated the role played by TGF-β on ERK1/2 signaling in LMNA cardiomyopathy.
Figure 5.

Tgf-β signaling acts on ERK1/2 signaling in LmnaH222P/H222P mice. (A) Immunoblots showing pERK1/2 and ERK1/2 in protein extracts of hearts from LmnaH222P/H222P mice (Lmna H222P) treated with placebo or SB-431542. Each lane contains protein extracts from a different mouse. The bar graph shows means ± standard errors values of the ratio pERK1/2 normalized to ERK1/2 from scan band densities of three immunoblots from n = 4 different mice per groups. *P < 0.05. (B) Immunoblots showing p-smad2/3, pERK1/2 and ERK1/2 in protein extracts of hearts from LmnaH222P/H222P mice (Lmna H222P) at 12 and 24 weeks of age. Each lane contains protein extracts from a different mouse. (C) Immunoblots showing p-smad2/3, pERK1/2 and ERK1/2 in protein extracts of C2C12 cells treated with or without (−) TGF-β1 for 6 h (2 nM).
Cardiac fibrosis is mediated by CTGF/CCN2
As ERK1/2 has been shown to mediate up-regulation of CTGF/CCN2, which may be involved in myocardial fibrosis, we sought to determine if CTGF/CCN2 expression is altered in hearts of LmnaH222P/H222P mice. CTGF/CCN2 expression was measured in hearts at 20 weeks of age. Compared with WT mice, hearts and isolated cardiomyocytes from LmnaH222P/H222P mice exhibited increased CTGF/CCN2 protein and mRNA expression (Fig. 6A and B). We next assessed the expression of Ctgf mRNA level in hearts of LmnaH222P/H222P mice between 4 weeks of age and 16 weeks of age concurrent with the progression of the left ventricular dysfunction. A significant increase of Ctgf mRNA was observed in hearts of LmnaH222P/H222P mice as early as 12 weeks of age, when compared with hearts from WT mice (Fig. 6C). This increase corresponded to the beginning of fibrosis, as we did not observe any increase of Col1a1 mRNA up to 12 weeks of age and of Col1a2 mRNA up to 16 weeks of age (Fig. 6C). To test whether the changes we observed in hearts of LmnaH222P/H222P mice also occur in humans, we examined CTGF/CCN2 levels in left ventricular tissue from human subjects with cardiomyopathy caused by mutations in LMNA. Compared with controls, elevated CTGF/CCN2 expression was observed in left ventricles tissue of human subjects with LMNA cardiomyopathy (Fig. 6D). These observations established that enhanced CTGF/CCN2 expression is a feature of cardiac remodeling in LMNA cardiomyopathy.
Figure 6.

Abnormal CTGF expression in hearts of LmnaH222P/H222P mice and human subjects with LMNA cardiomyopathy. (A) Immunoblots showing CTGF and GAPDH protein expression in hearts from WT and LmnaH222P/H222P mice. Each lane contains protein extracts from a different mouse. The bar graph represents mRNA relative expression (means ± standard errors of means) of Ctgf in hearts from WT (n = 3) and LmnaH222P/H222P (Lmna H222P) (n = 3) mice. **P < 0.01. (B) Immunoblots showing CTGF and GAPDH protein expression in isolated cardiomyocytes from WT and LmnaH222P/H222P mice. Each lane contains protein extracts from cardiomyocytes isolated from a different mouse. The bar graph represents mRNA relative expression (means ± standard errors of means) of Ctgf in cardiomyocytes from WT (n = 3) and LmnaH222P/H222P (Lmna H222P) (n = 3) mice. ***P < 0.001. (C) Validation of differential mRNA expression of Ctgf, Col1a1 and Col1a2 in hearts from WT and LmnaH222P/H222P mice. RNA was obtained from hearts of mice at 4, 8, 12, 16 weeks of age. White bars show relative RNA expression levels in hearts of WT mice and black bars in hearts of LmnaH222P/H222P mice. Values are means ± standard errors for n = 4 samples per group. *P < 0.05, ***P < 0.001. (D) Immunoblots showing CTGF and GAPDH in protein extracts of heart tissue from three human control (LMNA WT) and three individuals with dilated cardiomyopathy caused by LMNA mutations (LMNA mut).
CTGF/CCN2 expression influences myocardial fibrosis and left ventricular function
We next hypothesized that reducing CTGF/CCN2 activity would reduce myocardial fibrosis and improve left ventricular function. To test this hypothesis, an inhibitory human monoclonal antibody against CTGF (FG-3019) was administered to LmnaH222P/H222P mice for 2 weeks, starting at 16 weeks of age. As a control group, LmnaH222P/H222P mice were injected with non-specific human immunoglobulin G (Fig. 7A). There was less myocardial fibrosis in hearts from LmnaH222P/H222P mice treated with FG-3019 relative to mice treated with control human immunoglobulin G (Fig. 7B). Consistently, there was a decreased Col1a1 mRNA expression in hearts from LmnaH222P/H222P mice treated with FG-3019 compared with mice treated with control human immunoglobulin G (Fig. 7B). Two weeks of treatment with FG-3019 significantly improved both the left ventricular end systolic diameter and fractional shortening (Fig. 7C and Table 2). There was also a significant decrease in Myh7 and NppA mRNA expression after the treatment (Fig. 7D).
Figure 7.

Pharmacological inhibition of CTGF/CCN2 improves left-ventricular function in LmnaH222P/H222P mice. (A) Schematic representation of the treatment protocol of LmnaH222P/H222P mice with FG-3019. (B) Sirius Red staining of cross sections of hearts from LmnaH222P/H222P mice (Lmna H222P) treated with IgG or FG-3019. Scale bar: 50 μm. Left panel bar graph shows the quantification of myocardial fibrosis (means ± standard errors of means) from LmnaH222P/H222P mice (Lmna H222P) treated with immunoglobulin G (IgG) or FG-3019. *P < 0.05. Right panel shows expression of Col1a1 gene (means ± standard errors of means) in heart from LmnaH222P/H222P mice treated with IgG (n = 3) or FG-3019 (n = 3). *P < 0.05. (C) Graph showing mean LVEDD, mean LVESD and FS in 18-week old male LmnaH222P/H222P mice (Lmna H222P) treated with IgG (n = 6) or FG-3019 (n = 6). Values for each individual mouse receiving placebo or FG-3019 as well as standard errors (bars) are shown. (D) Expression of Myh7 and NppA genes in heart from LmnaH222P/H222P mice (Lmna H222P) treated with IgG (n = 3) or FG-3019 (n = 3). *P < 0.05, n.s. non-significant.
Table 2.
. Quantification of left ventricular function in Lmna H222P mice treated with FG3019
| 2 weeks treatment | ||||||
|---|---|---|---|---|---|---|
| Genotype | Treatment | n | Heart rate (beats/min) | LVEDD (mm) | LVESD (mm) | FS (%) |
| Lmna WT | None | 4 | 350.2 ± 34.5 | 3.7 ± 0.4 | 2.1 ± 0.3 | 43.4 ± 1.6 |
| Lmna H222P | Human IgG | 6 | 317.3 ± 51.5 | 3.9 ± 0.2 | 2.6 ± 0.2 | 34.4 ± 4.1 |
| Lmna H222P | FG3019 | 6 | 329.5 ± 65.9 | 3.7 ± 0.2* | 2.2 ± 0.2** | 39.3 ± 4.5* |
*P < 0.05.
**P < 0.005 between IgG-treated and FG3019-treated mice.
ERK1/2 signaling modulates CTGF/CCN2 expression
Given the enhanced ERK1/2 signaling that is observable in hearts of LmnaH222P/H222P mice as early as 4 weeks of age (21), which is before detectable myocardial fibrosis, we hypothesized that ERK1/2 may drive expression of CTGF/CCN2. Treatment of 16 week-old LmnaH222P/H222P mice with a MEK1/2 inhibitor (PD325901) for 1 month significantly decreased Ctgf mRNA in heart compared to that in DMSO-treated mice (Fig. 8A). Similarly, Ctgf mRNA was significantly decreased in hearts of LmnaH222P/H222P/Erk1−/− mice lacking ERK1 compared with hearts of LmnaH222P/H222P/Erk1+/+ mice (Fig. 8A). To further demonstrate the link between ERK1/2 signaling and enhanced expression of CTGF/CCN2, we used C2C12 cells. When we treated C2C12 cells with different doses of PD325901 (10, 5 and 1 µM), we observed decreased expression of CTGF/CCN2 (Fig. 8B). Conversely, the overexpression of ERK2 triggered an increase in CTGF/CCN2 expression (Fig. 8B).
Figure 8.

Effect of ERK1/2 on CTGF/CCN2 in hearts of LmnaH222P/H222P mice and C2C12 cells. (A) Expression of Ctgf gene in heart from LmnaH222P/H222P mice (Lmna H222P) treated with PD325901 (left panel) and LmnaH222P/H222P mice crossed with Erk1−/− mice (right panel). Bar diagrams indicate the expression of Ctgf (means ± standard errors of means) from LmnaH222P/H222P mice treated with placebo (n = 8) or PD325901 (n = 8) and from LmnaH222P/H222P/Erk1+/+ mice (n = 4) or LmnaH222P/H222P/Erk1−/− mice (n = 3). *P < 0.05, **P < 0.01. (B) C2C12 were treated with DMSO or PD325901 (10, 5 or 1 μM) or transfected with plasmid encoding ERK2-WT (5, 10 nM). Immunoblots showing phosphorylated ERK1/2 (p-ERK1/2), total ERK1/2, GFP and CTGF in protein extracts of C2C12 cells treated or transfected. (C) C2C12 cells stably expressing FLAG-tagged H222P-lamin A (C2-H222P) were transfected with the plasmid encoding the promoter of CTGF alone or in association with ERK2-WT (5, 10 nM) or ERK2-DN (5 nM). C2-H222P transfected with the plasmid encoding the promoter of CTGF were also treated with DMSO or PD325901 (10, 5 or 1 μM). After 48 h, luciferase activities induced by expression of CTGF were measured in cell lysates and normalized to β-gal activities obtained from a protein encoded by a co-transfected plasmid. Results are means ± standard errors of means of three experiments. *P < 0.05, n.s. non-significant (compared to C2-H222P transfected with the plasmid encoding the promoter of CTGF alone).
To test whether expression of the lamin A H222P variant was correlated with CTGF/CCN2 activation, we assessed CTGF/CCN2 promoter activity in stably transfected C2C12 cells over-expressing wild type or H222P lamin A (22). CTGF/CCN2 promoter activity was increased in transfected cells expressing lamin A H222P compared with cells expressing wild type lamin A (Fig. 8C). These results demonstrated a correlation between H222P lamin A expression and CTGF/CCN2 expression, recapitulating the expression pattern in hearts from LmnaH222P/H222P mice. We next assessed if the modulation of ERK1/2 signaling caused by H222P lamin A altered CTGF/CCN2 expression. Treatment of transfected C2C12 cells expressing H222P lamin A with PD325901 (10, 5 and 1 µM) showed a dose-dependent decrease in activity of the CTGF/CCN2 promoter (Fig. 8C). To address whether ERK1/2 activation drove the enhanced CTGF/CCN2 expression, ERK2 was overexpressed in C2C12 cells expressing lamin A H222P. CTGF/CCN2 expression increased in proportion to the amount of transfected ERK2 while cells transfected with a dominant negative variant of ERK2 did not exhibit elevated CTGF/CCN2 expression (Fig. 8C). These data demonstrate that cardiac CTGF/CCN2 expression in LmnaH222P/H222P mice was ERK1/2-dependent.
Crosstalk between ERK1/2 and TGF-β signaling in the hearts from LmnaH222P/H222P mice
Feedback control could exist between ERK1/2 and TGF-β/Smad signaling. To address this possibility, we examined expression of p-Smad2/3 in hearts of LmnaH222P/H222P mice treated with MEK1/2 inhibitor. The amount of p-Smad2/3 from left ventricular tissue was lowered in hearts of PD325901-treated LmnaH222P/H222P mice compared with hearts of placebo-treated LmnaH222P/H222P mice (Supplementary Material, Fig. S3A). To determine if CTGF affects TGF-β/Smad or ERK1/2 signaling, the hearts of LmnaH222P/H222P mice treated with FG-3019 were examined. FG-3019 had no effect on Smad2/3 signaling in these mice (Supplementary Material, Fig. S3B). However, compared to mice treated with control human immunoglobulin G, FG-3019 appeared to decrease the extent of ERK1/2 activation (Supplementary Material, Fig. S3B). These results suggested that in hearts of LmnaH222P/H222P mice TGF-β signaling mediates the activation of ERK1/2, which modulates myocardial fibrosis through up-regulation of CTGF (Fig. 9).
Figure 9.
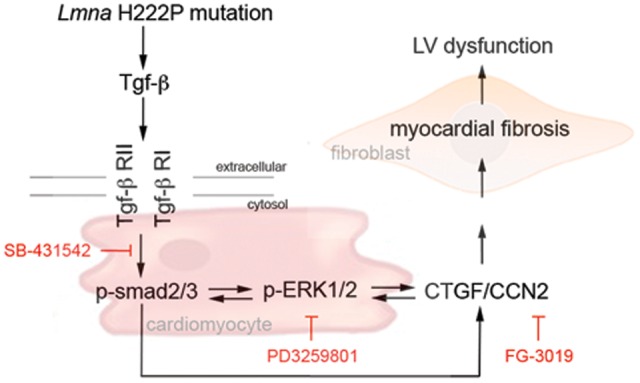
Proposed mechanism of myocardial fibrosis and cardiac dysfunction caused by Lmna mutation. Schematic representation of the sequential events of our hypothesis leading to left ventricular dysfunction in LMNA cardiomyopathy.
Discussion
Cardiac fibrosis exacerbates the clinical progression of heart failure (23,24). Therefore, early expression of mediators of fibrosis may contribute to the progressive development of cardiac dysfunction in primary cardiomyopathy. In this study, LmnaH222P/H222P mice were used to explore mechanisms of cardiac fibrosis in dilated cardiomyopathy. At 20 weeks, LmnaH222P/H222P mice had elevated levels of mRNA for ECM proteins, elevated expression of TGF-β and activation of TGF-β/Smad signaling. Up-regulation of TGF-β/Smad signaling was observed as early as 12 weeks of age in LmnaH222P/H222P mice, which is a time that precedes development of both cardiac fibrosis and the onset of overt cardiac dysfunction. Inhibition of TGF-β/Smad signaling suppressed cardiac fibrosis and attenuated cardiac dysfunction. These observations indicate that TGF-β is a mediator of the cardiomyopathy that develops as a result of LMNA mutations.
The mechanism by which LMNA mutations lead to TGF-β expression and activation remains to be elucidated. However, our study demonstrated some of the pathways by which TGF-β signaling promotes fibrosis. The canonical TGF-β/Smad signaling pathway involves phosphorylation of Smad2 and/or Smad3, which facilitates their interaction with Smad4 and nuclear translocation to induce transcriptional activation of target genes (25,26). LmnaH222P/H222P mice exhibited elevated cardiac p-Smad2/3, its enrichment in the nucleus and a concomitant depletion in the cytosol, which are all characteristic of canonical TGF-β/Smad signaling. TGF-β also has several non-canonical functions, one of which involves activation of the ERK1/2 signaling pathway (27). We have previously reported that ERK1/2 is activated in the hearts of LmnaH222P/H222P mice (8) and that reducing its activity ameliorates development of cardiomyopathy (9–11,20). We have demonstrated that at least one path to ERK1/2 activation in the hearts of LmnaH222P/H222P mice is via TGF-β/Smad signaling, since the ALK5 kinase inhibitor SB-431542 decreased activated ERK1/2. This is in line with a recent finding by Huang et al. (28), showing that ERK1/2 inhibition resulted in significant decreased of Smad2/3-dependent myocardial fibrosis. The same group also showed that another member of ALK (i.e. ALK7) attenuates cardiac fibrosis by blocking ERK1/2 signaling (28).
One of the well-known TGF-β target genes is CTGF. TGF-β can induce CTGF/CCN2 production in cardiac fibroblasts and cardiomyocytes (29), endothelial cells (30,31) and pericytes (32). The regulation of CTGF/CCN2 expression is complex (33). A TGF-β response element located in the promoter region of the CTGF/CCN2 gene was first reported by Grotendorst et al. (34) and regulation by Smad2/3 binding to the promoter was subsequently confirmed (35). Our data demonstrate that cardiac CTGF/CCN2 expression is elevated in LmnaH222P/H222P mice, as early as 12 weeks after birth. Inhibition of ERK1/2 activation or genetic depletion of ERK1/2 decreased the expression of CTGF/CCN2 mRNA. This confirmed that CTGF/CCN2 mRNA expression is mediated in part by ERK1/2 signaling (36–38). CTGF/CCN2 was reported to be essential for the sustained fibrotic activity of TGF-β (39) and to promote fibrosis in multiple organs (40–43). There are some reports showing that CTGF/CCN2 expression is increased in cardiac fibrosis (44) and failing hearts (23,45,46). However, CTGF/CCN2’s mechanism of action is incompletely understood (47). While it is broadly recognized that TGF-β induces the expression of CTGF/CCN2 (34), there are also data indicating that CTGF/CCN2 promotes the expression of TGF-β (45,48–50). Thus, CTGF/CCN2 appears to promote TGF-β/Smad signaling by inducing expression of TGF-β in a positive feedback loop. This may involve TGFβ-induced ERK1/2 signaling, since inhibition of ERK1/2 strongly reduced CTGF/CCN2 expression and transcription from the CTGF/CCN2 promoter.
The effect of CTGF/CCN2 overexpression in cardiac fibroblasts, which are the major cell type responsible for collagen synthesis in the heart, has not yet been examined. Genetic manipulation of CTGF/CCN2 in cardiomyocytes has resulted in a variety of apparently contradictory effects. The first group to generate transgenic mice reported that overexpression of CTGF/CCN2 in cardiomyocytes did not lead to cardiac fibrosis but the mice developed age-dependent cardiomyopathy that was concordant with cardiac hypertrophy (51). Soon after, Yoon et al. (52) generated transgenic mice overexpressing CTGF/CCN2 in cardiomyocytes. These mice had no cardiac hypertrophy but increased fibrosis upon pressure overload (52). A contradictory report by others showed that overexpressing CTGF/CCN2 in cardiomyocytes lead to mice with smaller myocytes, enhanced collagen and fibronectin expression and greater blood vessel density but normal left ventricular function (45). These mice were protected against ischemia/reperfusion injury (45), myocardial infarction (53) and pressure overload (54). Most recently, transgenic mice were generated that only overexpressed CTGF in cardiomyocytes (55). After a month of CTGF/CCN2 expression, these mice exhibited no signs of cardiac fibrosis or hypertrophy. It remains unclear if the cause of the discrepant results between these studies is the result of the promoters used to drive CTGF/CCN2 expression, the background mouse strains or some other reasons.
In the context of the heart, fibrosis can cause increased tissue stiffness, cardiomyocyte atrophy, arrhythmias and hypoxia. Although targeting TGF-β has shown promise as an anti-fibrotic therapy, it also functions in other biological processes (56). Hence, broad targeting of TGF-β/Smad signaling may be problematic. Inhibiting CTGF/CCN2 may therefore be a more specific target for blocking fibrosis. Similarly, it may be a more specific anti-fibrotic therapy than the anti-protozoal drug, halofuginone, which was reported to attenuate cardiac fibrosis and dysfunction in the mdx mouse model via inhibition of TGF-β/Smad signaling (57,58). While some of the data from transgenic mouse models could be interpreted as indicating that inhibition of CTGF/CCN2 might be detrimental to cardiac structure and function, this has not been observed when CTGF/CCN2 was pharmacologically inhibited with antibodies. In a cardiac allograft rejection model, FG-3019 administration for 3 weeks decreased fibrosis in the transplanted heart and suppressed cardiac hypertrophy (59). In a rat model of bronchopulmonary dysplasia, inhibition of CTGF/CCN2 with FG-3149, a mouse chimera of FG-3019, inhibited hyperoxia-induced increases of right ventricular systolic pressure and prevented right ventricular hypertrophy (60). And in a transverse aortic constriction-induced mouse model of pressure overload, FG-3149 inhibited cardiac hypertrophy, improved cardiac function and exhibited a trend toward reduced fibrosis that was not significant for the cohort size used (61). Treating mdx mice with FG-3019 leads also to an improvement in tissue morphology, less fibrosis, a decrease in damage and an improvement in strength of skeletal muscle (62). These data suggest that CTGF/CCN2 function in the fibrotic process and inhibiting it prevents or reverses fibrosis. In our study, 2 weeks of FG-3019 administration inhibited collagen deposition. Hence, inhibiting CTGF/CCN2 could be beneficial for patients with LMNA cardiomyopathy by reducing cardiac fibrosis.
Established management of LMNA cardiomyopathy included the use of angiotensin-converting enzyme (ACE) inhibitors or the combination of an ACE inhibitor with β-adrenergic receptor blockade (63). Treatment with ACE inhibitors improves cardiac function in patients and mice with dilated cardiomyopathy; however, ACE inhibition does not attenuate cardiac fibrosis (64,65). Modulation of ECM function may prove beneficial in attenuating disease progression, as shown by the success of anti-fibrotic therapy in the mdx mouse (57,58). Future studies will better define the role of CTGF/CCN2 in LMNA cardiomyopathy, as well as determine if CTGF/CCN2 can be a therapeutic target more generally in dilated cardiomyopathy.
Materials and Methods
Animals
LmnaH222P/H222P mice (16) were fed chow and housed in a disease-free barrier facility with 12 h/12 h light/dark cycles. For treatments, PD0325901, SB-431542 and FG-3019 were delivered (intraperitoneal injection) daily at a dose of 1, 5 and 25 mg/kg, respectively. All animal experiments were carried our according to Ministère de l’Éducation Nationale de l’Enseignement Supérieur et de la Recherche at Center for Research in Myology and The Institutional Animal Care and Use Committee at Columbia University Medical Center for the care and use of experimental animals.
Human heart tissue
Sections of explanted hearts from human subjects with LMNA mutations were obtained from Myobank-AFM de l’lnstitut de Myologie (Paris, France). Myobank-AFM received the authorizations from the French Ministry of Health and from the Comity for Protection of Patient to share tissues and cells of human origin for scientific purposes, ensuring the donors the maintenance of anonymity, respect of their volition, and consent according to the legislation. Control human heart samples were obtained from the National Disease Research Interchange (Philadelphia, PA, USA); information regarding donor confidentiality and consent can be found at http://www.ndriresource.org.
Histology
Hearts from LmnaH222P/H222P mice were fixed in 4% formaldehyde for 48 h, embedded in paraffin, sectioned at 5 µm and stained with Masson trichrome. Representative stained sections were photographed using a Microphot SA (Nikon) light microscope attached to a Spot RT Slide camera (Diagnostic Instruments). Images were processed using Adobe Photoshop CS (Adobe Systems).
Immunohistochemistry and immunofluorescence microscopy
Immunofluorescence staining was performed on frozen 8 μm thick sections of transversal cardiac muscles by fixing them in 3.7% formaldehyde in PBS for 15 min, then blocking in 5% fetal goat serum in PBS/Triton X-100 for 1 h. Sections were incubated in blocking solution with anti-p-smad2/3 antibody (Cell Signaling Technology), CD31 (Abcam) or Col1a1 (Santa Cruz) antibody overnight at 4 °C followed by PBS washing and incubation with Texas red–conjugated goat anti-mouse IgG secondary antibody (Invitrogen) and counterstained with 0.1 μg/ml DAPI (Sigma-Aldrich).
Quantitative polymerase chain reaction analysis
Total RNA was extracted using the Rneasy isolation kit (Qiagen). cDNA was synthesized using Superscript first strand synthesis system according to the manufacturer’s instructions (Invitrogen) on total RNA. For each replicate in each experiment, RNA from tissue samples of different animals was used. Primers for Col1a1, Col1a2, Fn1, Sdc1, Nid1, Dcn1, Tgf-b1, Tgf-b2 and Ctgf were designed using Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Real-time quantitative polymerase chain reaction (qPCR) reactions containing HotStart-IT SYBR green qPCR Master Mix (Affymetrix), 200 nM of each primer and 0.2 µl of template in a 25 µl reaction volume were amplified using the ABI 7300 Real-Time PCR System (Applied Biosystems). Relative levels of mRNA expression calculated using the ΔΔCT method were normalized by comparison to housekeeping mRNA.
Protein extraction and immunoblotting
Mouse heart tissue and cultured cells were homogenized in sample extraction buffer (Cell Signaling) then separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes and blotted with primary antibodies against Smad2/3 (Santa Cruz Biotechnology), phosphorylated Smad2/3 (Cell Signaling), CTGF (Santa Cruz), ERK1/2 (Santa Cruz Biotechnology), phosphorylated ERK1/2 (Cell Signaling) and GFP, GAPDH (Ambion). Secondary antibodies were horseradish peroxidate–conjugated (GE Healthcare). Recognized proteins were visualized by enhanced chemiluminescence.
Isolation of mouse cardiomyocytes
Wild type and LmnaH222P/H222P mice were anesthetized with pentofurane and ventricular cardiomyocytes were isolated as described previously (8). Briefly, hearts were removed and the aorta cannulated. After Ca2+-free buffer was perfused for 2 min, 0.25 mg/ml collagenase I/II solution was perfused through the coronary arteries for 6 min with 12.5 mm Ca2+. Left ventricular tissue was teased apart and pipetted to release individual cells. After enzymatic dispersion, Ca2+ concentration in the buffer containing bovine serum albumin was elevated in three steps up to 500 mm.
Luciferase assays
C2C12 cells (American Type Culture Collection) were maintained in a humidified 37 °C/5% CO2 incubator and subcultured at 80% confluency. Unmodified and stable C2C12 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Invitrogen). Stable C2C12 cells were plated at 1 × 106 cells/9.5 cm2 DMEM medium supplemented with 10% FBS and 1% penicillin-streptomycin. Twenty-four hours after plating, the cells were co-transfected with 500 ng of the pGL3-CTGF-Luc, 50 ng of the pGFP-ERK2-DN and 50–100 ng of pGFP-ERK2-WT in OptiMEM medium using Lipofectamine Reagent (Invitrogen). Forty-eight hours after transfection, the luciferase substrate was applied using Luciferase Assay system (Promega). For cell normalization, CellTiterGlo® Luminescent Cell Viability Assay (Promega) was used.
Thansthoracic echocardiography
LmnaH222P/H222P mice were anesthetized with 1.5% isoflurane in O2 and placed on a heating pad (37 °C). Echocardiography was performed using a Visualsonics Vevo 770 ultrasound with a 30 MHz transducer applied to the chest wall. Cardiac ventricular dimensions and fractional shortening were measured in 2D mode and M-mode three times for the number of animals indicated. ‘Blinded’ echocardiographers, unaware of the treatment, performed the examinations.
Statistics
Values for real-time qPCR were compared using an unpaired Student t-test. Comparisons of echocardiographic parameters between PD0325901 and SB-431542-treated and placebo-treated LmnaH222P/H222P mice were performed using a Welch t-test; to validate these results, a non-parametric test (Mann–Whitney) was performed and concordance checked. Statistical analyses were performed using GraphPad Prism software.
Supplementary Material
Supplementary Material is available at HMG online.
Acknowledgements
The authors gratefully acknowledge Professor P. Stork for providing ERK constructs. The authors also gratefully acknowledge Professor B. Kone for providing pGL3-CTGF constructs. This study is supported by funds from the US National Institutes of Health grant [AR048997 to H.J.W.] and from the Institut National de la Sante et de la Recherche Médicale; the Université Pierre et Marie Curie-Paris 6, the Centre National de la Recherche Scientifique and the Association Française contre les Myopathies.
Conflict of Interest statement. K.E.L. is a FibroGen employee. All other authors have declared no conflicts of interest.
Funding
This work was supported by funds from the US National Institutes of Health grant [AR048997 to H.J.W.] and from the Institut National de la Santé et de la Recherche Médicale; the Université Pierre et Marie Curie-Paris 6, the Centre National de la Recherche Scientifique and the Association Française contre les Myopathies.
References
- 1.Bonne G., Di Barletta M.R., Varnous S., Becane H.M., Hammouda E.H., Merlini L., Muntoni F., Greenberg C.R., Gary F., Urtizberea J.A. et al. (1999) Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet., 21, 285–288. [DOI] [PubMed] [Google Scholar]
- 2.Fatkin D., MacRae C., Sasaki T., Wolff M.R., Porcu M., Frenneaux M., Atherton J., Vidaillet H.J., Jr, Spudich S., De Girolami U. et al. (1999) Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med., 341, 1715–1724. [DOI] [PubMed] [Google Scholar]
- 3.Meune C., Van Berlo J.H., Anselme F., Bonne G., Pinto Y.M., Duboc D. (2006) Primary prevention of sudden death in patients with lamin A/C gene mutations. N. Engl. J. Med., 354, 209–210. [DOI] [PubMed] [Google Scholar]
- 4.Ben Yaou R., Gueneau L., Demay L., Stora S., Chikhaoui K., Richard P., Bonne G. (2006) Heart involvement in lamin A/C related diseases. Arch. Mal. Coeur Vaiss, 99, 848–855. [PubMed] [Google Scholar]
- 5.Fontana M., Barison A., Botto N., Panchetti L., Ricci G., Milanesi M., Poletti R., Positano V., Siciliano G., Passino C. et al. (2013) CMR-verified interstitial myocardial fibrosis as a marker of subclinical cardiac involvement in LMNA mutation carriers. JACC Cardiovasc. Imaging, 6, 124–126. [DOI] [PubMed] [Google Scholar]
- 6.Holmstrom M., Kivisto S., Helio T., Jurkko R., Kaartinen M., Antial M., Reissell E., Kuusisto J., Karjjainen S., Peukurinen K. et al. (2011) Late gadolinium enhanced cardiovascular magnetic resonance of lamin A/C gene mutation related diolated cardiomyopathy. J. Cardiovasc. Magn. Reson., 13, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu J.T., Muchir A., Nagy P.L., Worman H.J. (2011) LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis. Model. Mech., 4, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muchir A., Pavlidis P., Decostre V., Herron A.J., Arimura T., Bonne G., Worman H.J. (2007) Activation of MAPK pathway links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest., 117, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muchir A., Shan J., Bonne G., Lehnart S.E., Worman H.J. (2009) Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet., 19, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu W., Muchir A., Shan J., Bonne G., Worman H.J. (2011) Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation, 123, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muchir A., Reilly S.A., Wu W., Iwata S., Homma S., Bonne G., Worman H.J. (2012) Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc. Res., 93, 311–319. [DOI] [PubMed] [Google Scholar]
- 12.Weber K.T., Sun Y., Bhattacharya S.K., Ahokas R.A., Gerling I.C. (2013) Myofibroblast-mediated mechanisms of pathological remodeling of the heart. Nat. Rev. Cardiol., 10, 15–26. [DOI] [PubMed] [Google Scholar]
- 13.van Tintelen J.P., Tio R.A., Kerstjens-Frederikse W.S., van Berlo J.H., Boven L.G., Suurmeijer A.J., White S.J., den Dunnen J.T., te Meerman G.J., Vos Y.J. et al. (2007) Severe myocardial fibrosis caused by a deletion of the 5’ end of the lamin A/C gene. J. Am. Coll. Cardiol., 49, 2430–2439. [DOI] [PubMed] [Google Scholar]
- 14.Raman S.V., Sparks E.A., Baker P.M., McCarthy B., Wooley C.F. (2007) Mid-myocardial fibrosis by cardiac magnetic resonance in patients with lamin A/C cardiomyopathy: possible substrate for diastolic dysfunction. J. Cardiovasc. Magn. Reson., 9, 907–913. [DOI] [PubMed] [Google Scholar]
- 15.Dobaczewski M., Chen W., Frangogiannis N.G. (2011) Transforming growth (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol., 51, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arimura T., Helbling-Leclerc A., Massart C., Varnous S., Niel F., Lacene E., Fromes Y., Toussaint M., Mura A.M., Keller D.I. et al. (2005) Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet., 14, 155–169. [DOI] [PubMed] [Google Scholar]
- 17.Weiss A., Attisano L. (2013) The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol., 2, 47–63. [DOI] [PubMed] [Google Scholar]
- 18.Inman G.J., Nicolàs F.J., Callhan J.F., Harling J.D., Gsater L.M., Reith A.D., Laping N.J., Hill C.S. (2002) SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol., 62, 65–74. [DOI] [PubMed] [Google Scholar]
- 19.Ogunjimi A.A., Zeqiraj E., Ceccarelli D.F., Sicheri F., Wrana J.L., David L. (2012) Structural basis for specificity of TGF family receptor small molecule inhibitors. Cell Signal, 24, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu W., Iwata S., Homma S., Worman H.J., Muchir A. (2014) Depletion of extracellular signal-regulated kinase 1 in mice with cardiomyopathy caused by lamin A/C gene mutation partially prevents pathology before isoenzyme activation. Hum. Mol. Genet, 23, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi J.C., Muchir A., Wu W., Iwata S., Homma S., Morrow J.P., Worman H.J. (2012) Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med., 4, 144ra102.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi J.C., Wu W., Muchir A., Iwata S., Homma S., Worman H.J. (2012) Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J. Biol. Chem., 287, 40513–40524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koshman Y.E., Patel N., Chu M., Iyengar R., Kim T., Ersahin C., Lewis W., Heroux A., Samarel A.M. (2013) Regulation of connective tissue growth factor gene expression and fibrosis in human heart failure. J. Cardiac Fail., 19, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segura A.M., Frazier O.H., Buja L.M. (2014) Fibrosis and heart failure. Heart Fail. Rev., 19, 173–185. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y., Massague J. (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell, 13, 685–700. [DOI] [PubMed] [Google Scholar]
- 26.Derynck R., Zhang Y.E. (2003) Smad-dependent and smad-independent pathways in TGF-beta family signaling. Nature, 425, 577–584. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y.E. (2009) Non-Smad pathways in TGF-beta signaling. Cell Res., 19, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang H., Tang Y., Wu G., Mei Y., Liu W., Liu X., Wan N., Liu Y., Huang C. (2015) ALK7 protects against pathological cardiac hypertrophy in mice. Cardiovasc. Res., 108, 50–61. [DOI] [PubMed] [Google Scholar]
- 29.Chen M.M., Lam A., Abraham J.A., Schreiner G.F., oly A.H. (2000) CTGF expression is induced by TGF-beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J. Mol. Cell. Cardiol., 32, 1805–1819. [DOI] [PubMed] [Google Scholar]
- 30.Sohn M., Tan Y., Wang B., Klein R.L., Trojanowska M., Jaffa A.A. (2006) Mechanisms of low-density lipoprotein-induced expression of connective tissue growth factor in human aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol., 290, H1624–H1634. [DOI] [PubMed] [Google Scholar]
- 31.Rosin N.L., Falkenham A., Sopel M.J., Lee T.D., Legare J.F. (2013) Regulation and role of connective tissue growth factor in AngII-induced myocardial fibrosis. Am. J. Pathol., 182, 714–726. [DOI] [PubMed] [Google Scholar]
- 32.Van Geest R.J., Klaassen I., Vogels I.M., Van Noorden C.J., Schlingemann R.O. (2010) Differential TGF-{beta} signaling in retinal vascular cells: a role in diabetic retinopathy? Invest. Ophthalmol. Vis. Sci., 51, 1857–1865. [DOI] [PubMed] [Google Scholar]
- 33.Oliver N., Sternlicht M., Gerritsen K., Goldschmeding R. (2010) Could aging human skin use a connective tissue growth factor boost to increase collagen content? J. Invest. Dermatol., 130, 338–341. [DOI] [PubMed] [Google Scholar]
- 34.Grotendorst G.R., Okochi H., Hayashi N. (1996) A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth. Diff. 7, 469–480. [PubMed] [Google Scholar]
- 35.Holmes A., Abraham D.J., Sa S., Shiwen X., Black C.M., Leask A. (2001) CTGF and SMADs maintenance of schleroderma phenotype is independent of SMAD signaling. J. Biol. Chem., 276, 10594–10601. [DOI] [PubMed] [Google Scholar]
- 36.Phanish M.K., Wahab N.A., Hendry B.M., Dockrell M.E. (2005) TGF-beta1-induced connective tissue growth factor (CCN2) expression in human renal proximal tubule epithelial cells requires Ras/MEK/ERK and Smad signaling. Nephron Exp. Nephrol., 100, e156–e165. [DOI] [PubMed] [Google Scholar]
- 37.Pickles M., Leask A. (2007) Analysis of CCN2 promoter activity in PANC-1 cells: regulation by ras/MEK/ERK. J. Cell. Commun. Signal, 1, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Secker G.A., Shortt A.J., Sampson E., Schwartz Q.P., Schultz G.S., Daniels J.T. (2008) TGFbeta stimulated re-epithelialisation is regulated by CTGF and Ras/MEK/ERK signaling. Exp. Cell Res., 314, 131–142. [DOI] [PubMed] [Google Scholar]
- 39.Mori T., Kawara S., Shinozaki M., Hayashi N., Kakinuma T., Igarashi A., Takigawa M., Nakanishi T., Takehara K. (1999) Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: a mouse fibrosis model. J. Cell. Physiol., 181, 153–159. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q., Usinger W., Nichols B., Gray J., Xu L., Seeley T.W., Brenner M., Guo G., Zhang W., Oliver N., Lin A., Yeowell D. (2011) Cooperative interaction of CTGF and TGF-β in animal models of fibrotic disease. Fibrogenesis Tissue Repair, 4, 4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li G., Xie Q., Shi Y., Li D., Zhang M., Jiang S., Zhou H., Lu H., Jin Y. (2006) Inhibition of connective tissue growth factor by siRNA prevents liver fibrosis in rats. J. Gene Med., 8, 889–900. [DOI] [PubMed] [Google Scholar]
- 42.Guha M., Xu Z.G., Tung D., Lanting L., Natarajan R. (2007) Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. Faseb J., 21, 3355–3368. [DOI] [PubMed] [Google Scholar]
- 43.Brigstock D.R. (2009) Strategies for blocking the fibrogenic actions of connective tissue growth factor (CCN2): From pharmacological inhibition in vitro to targeted siRNA therapy in vivo. J. Cell. Commun. Signal, 3, 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohnishi H., Oka T., Kusachi S., Nakanishi T., Takeda K., Nakahama M., Doi M., Murakami T., Ninomiya Y., Takigawa M. et al. (1998) Increased expression of connective tissue growth factor in the infarct zone of experimentally induced myocardial infarction in rats. J. Mol. Cell. Cardiol, 30, 2411–2422. [DOI] [PubMed] [Google Scholar]
- 45.Ahmed M.S., Gravning J., Martinov V.N., von Lueder T.G., Edvardsen T., Czibik G., Moe I.T., Vinge L.E., Øie E., Valen G. et al. (2011) Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol, 300, H1291–H1302. [DOI] [PubMed] [Google Scholar]
- 46.Au C.G., Butler T.L., Sherwood M.C., Egan J.R., North K.N., Winlaw D.S. (2011) Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiaomyopathy. Int. J. Exp. Path, 92, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lipson K.E., Wong C., Teng Y., Spong S. (2012) CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair, 5, S24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakerakanti S.S., Bujor A.M., Trojanowska M. (2011) CCN2 is required for the TGF-β induced activation of Smad1-Erk1/2 signaling network. PLoS One, 6, e21977.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kunzmann S., Seher A., Kramer B.W., Schenk R., Schutze N., Jakob F., Sebald W., Speer C.P. (2008) Connective tissue growth factor does not affect transforming growth factor-beta 1-induced Smad3 phosphorylation and T lymphocyte proliferation inhibition. Int. Arch. Allergy Immunol, 147, 152–160. [DOI] [PubMed] [Google Scholar]
- 50.Yang H., Huang Y., Chen X., Liu J., Lu Y., Bu L., Xia L., Xiao W., Chen M., Nie Q. et al. (2010) The role of CTGF in the diabetic rat retina and its relationship with VEGF and TGF-β(2), elucidated by treatment with CTGF siRNA. Acta Ophthalmol., 88, 652–659. [DOI] [PubMed] [Google Scholar]
- 51.Panek A.N., Posch M.G., Alenina N., Ghadge S.K., Erdmann B., Popova E., Perrot A., Geier C., Dietz R., Morano I. et al. (2009) Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS One, 4, e6743.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoon P.O., Lee M.A., Cha H., Jeong M.H., Kim J., Jang S.P., Choi B.Y., Jeong D., Yang D.K., Hajjar R.J. et al. (2010) The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J. Mol. Cell. Cardiol., 49, 294–303. [DOI] [PubMed] [Google Scholar]
- 53.Gravning J., Ørn S., Kaasbøll O.J., Martinov V.N., Manhenke C., Dickstein K., Edvardsen T., Attramadal H., Ahmed M.S. (2012) Myocardial connective tissue growth factor (CCN2/CTGF) attenuates left ventricular remodeling after myocardial infarction. PLoS One, 7, e52120.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gravning J., Ahmed M.S., von Lueder T.G., Edvardsen T., Attramadal H. (2013) CCN2/CTGF attenuates myocardial hypertrophy and cardiac dysfunction upon chronic pressure-overload. Int. J. Cardiol., 168, 2049–2056. [DOI] [PubMed] [Google Scholar]
- 55.Accornero F., van Berlo J.H., Correll R.N., Elrod J.W., Sargent M.A., York A., Rabinowitz J.E., Leask A., Molkentin J.D. (2015) Genetic analysis of connective tissue growth factor as an effector of transforming growth factor β signaling and cardiac remodeling. Mol. Cell. Biol., 35, 2154–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaefer C.J., Ruhrmund D.W., Pan L., Seiwert S.D., Kossen K. (2011) Antifibrotic activities of pirgenidone in animal models. Eur. Respir. Rev., 20, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huebner K.D., Jassal D.S., Halevy O., Pines M., Anderson J.E. (2008) Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. Am. J. Physiol. Heart Circ. Physiol., 294, H1550–H1561. [DOI] [PubMed] [Google Scholar]
- 58.Turgeman T., Hagai Y., Huebner K., Jassal D.S., Anderson J.E., Genin O., Nagler A., Halevy O., Pines M. (2008) Prevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginone. Neuromuscul. Disord., 18, 857–868. [DOI] [PubMed] [Google Scholar]
- 59.Booth A.J., Csencsits-Smith K., Wood S.C., Lu G., Lipson K.E., Bishop D.K. (2010) Connective tissue growth factor promotes fibrosis downstream of TGFbeta and IL-6 in chronic cardiac allograft rejection. Am. J. Transplant., 10, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alapati D., Rong M., Chen S., Hehre D., Rodriguez M.M., Lipson K.E., Wu S. (2011) Connective tissue growth factor antibody therapy attenuates hyperoxia-induced lung injury in neonatal rats. Am. J. Respir. Cell. Mol. Biol., 45, 1169–1177. [DOI] [PubMed] [Google Scholar]
- 61.Szabó Z.I., Magga J., Alakoski T., Ulvila J., Piuhola J., Vainio L., Kivirikko K.I., Vuolteenaho O., Ruskoaho H., Lipson K.E. et al. (2014) Connective tissue growth factor inhibition attenuates left ventricular remodeling and dysfunction in pressure overload-induced heart failure. Hypertension, 63, 1235–1240. [DOI] [PubMed] [Google Scholar]
- 62.Morales M.G., Gutierrez J., Cabello-Verrugio C., Cabrera D., Lipson K.E., Golschmeding R., Brandan E. (2013) Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum. Mol. Genet., 15, 4938–4951. [DOI] [PubMed] [Google Scholar]
- 63.Bushby K., Muntoni F., Bourke J.P. (2003) 107th ENMC international workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. Neuromuscul. Disord., 13, 166–172. [DOI] [PubMed] [Google Scholar]
- 64.Jefferies J.L., Eidem B.W., Belmont J.W., Craigen W.J., Ware S.M., Fernbach S.D., Neish S.R., Smith E.O., Towbin J.A. (2005) Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation, 112, 2799–2804. [DOI] [PubMed] [Google Scholar]
- 65.Bauer R., Straub V., Blain A., Bushby K., MacGowan G.A. (2009) Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur. J. Heart Fail., 11, 463–471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.