

Abstract
Spinal cord injury (SCI) is a highly debilitating pathology that has irreversible impacts and results in functional loss. We evaluated the anti-inflammatory and immunologic role of antibody-mediated depletion of B cells through the glycoengineered anti-muCD20 antibody (18B12) in an experimental model of spinal cord compression, in vivo and ex vivo. Intraperitoneal 18B12 was administered at a dose of 30 mg/kg, 1 h and 6 h after SCI, and mice were sacrificed 24 h after trauma. We demonstrated, in vivo, that 18B12 slowed severe hindlimb motor dysfunction (Basso Mouse Scale score) and neuronal death by histological evaluation in SCI mice, as well as decreased expression of nuclear factor-kB, inducible nitric oxide synthase, cytokines, and glial fibrillary acidic protein. Also, 18B12 reduced expression of microglia, just as it lowered the expression of B and T lymphocytes. Moreover, in spinal cord organotypic cultures, pretreatment with 18B12 significantly reduced nitric oxide expression and protected cells from cell death [3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay]. In this study, we showed that 18B12 treatment reduces the development of inflammation and tissue injury by alteration of the immune system associated with SCI. This study increases the current knowledge that B-cell depletion is able to exert immunomodulating actions in damaged spinal cords.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-016-0446-2) contains supplementary material, which is available to authorized users.
Key Words: Spinal cord injury, 18B12, CD20, Inflammation, Immune system, B cell
Introduction
Spinal cord injury (SCI) is a central nervous insult that causes mortality and morbidity worldwide, representing a major social and health problem [1]. SCI is damage of the spinal cord at the end of the spinal canal, characterized by neurological dysfunction, due to the progressive destruction of neuronal networks, resulting from tissue damage and subsequent cellular and biochemical reactions. The initial physical trauma is followed by a secondary degenerative process that leads to the destruction of axonal tracts that control motor, sensory, and autonomic functions. Experimental studies and clinical observations showed that spinal cord lesions are greatly worsened by a process defined as secondary cell death [2]. Post-traumatic inflammatory reaction may play a decisive role in the secondary injury processes after SCI [3]. In particular, secondary damage results in a large number of cellular and biochemical cascades, and in a local inflammatory response. The cellular mechanisms remain uncertain, but might comprise dendritic remodeling, axonal sprouting, and local neuronal circuit reorganization. SCI studies mainly focus on prevention of secondary mechanisms after SCI, and many pharmacological agents are still under trial [4]. Many pharmacologic therapies have been assessed for the treatment of SCI, although none has met with substantial success.
Despite progress, a definitive agent for treatment of SCI has not yet been provided; therefore, SCI remains a very complex medical and psychological challenge.
It is known that the immune system plays remarkable functional roles in regeneration after SCI. The central nervous system (CNS) was considered immune privileged because of its inability to set off an immune response and process antigens [5]; however, evidence has reconsidered the CNS as being an “immunologically quiescent” rather than an immune privileged site [6]. This quiescent state is strongly altered in the injured spinal cord, where circulating immune cells invade the environment, activate resident microglia and astrocytes, and express classic immune and inflammatory mediators [7]. The immune response that accompanies SCI contributes to both injury and reparative processes, causing infiltration of leukocytes and glial activation, oxidative stress, and tissue damage, and subsequently modulating locomotor recovery [8].
Human and animal studies have both demonstrated that trauma unleashes an endogenous repertoire of CNS-reactive lymphocytes. The level of T lymphocytes reactive to myelin proteins is amplified in the serum of patients with SCI [9]; collectively, these data indicate the functional activation of autoreactive T and B cells. SCI disrupts the balance between immune effectors and regulatory cells, and allows the antigen-specific CD4+ T cells to become activated and contribute to pathology. Mechanisms by which immune cells contribute to pathology include the production of proinflammatory cytokines and chemokines, and the promotion of a proinflammatory phenotype in macrophages [10]. Recent evidence suggests that leukocytes, especially lymphocytes and neutrophils, penetrate the injured spinal cord and are directly implicated in the pathogenesis and extension of SCI [11].
The immune response to SCI involves an intricate interplay of immune system cells, such as neutrophils, macrophages and microglia, and T and B lymphocytes [12]; it clearly demonstrated that trauma of moderate severity leads to a particularly robust B-cell response that produces pathogenic antibodies [13].
T cells can have direct effects on neurons or glia [14], or indirect effects on other CNS cells following the production of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukins (ILs), that play a major role in cytotoxic cell damage [15]. It is suggested that increased cytokine production at the injury site is correlated with the augmentation of neural injury [16]. However, production of autoantibodies and activation of numerous inflammatory pathways causes activated B-cell accumulation at the SCI region, correlated with the pathological events after SCI [17].
Current therapeutic strategies address divergent methods, including early immune blockade and vaccination with immune cells to prevent early tissue damage [18]. B-cell depletion as a neuroprotective treatment is a significantly viable option for SCI, as therapies that mark the B cell or obstruct the effects of pathogenic antibodies have demonstrated considerable promise in clinical trials for neurological autoimmune diseases, and rituximab has been approved for the treatment of rheumatoid arthritis [19, 20]. Because it has been demonstrated that anti-CD20 treatment eliminates pre-existing B-cell activity in human autoimmunity [21], the purpose of the current proposal has been to evaluate the effects of treatment with glycoengineered anti-CD20 antibody 18B12 in mice subjected to SCI [22], in an attempt to investigate the anti-inflammatory and immunological potential of this anti-CD20 drug. 18B12 has been glycoengineered to make a nonfucosylated Fc region that substantially enhances the ability to attach and recruit effector cells, and hence to an increased antibody-dependent cell-mediated cytotoxicity and antibody-dependent cell phagocytosis compared with nonglycoengineered fully fucosylated antibodies [23, 24].
In this study, we assessed the role of B-cell depletion through the glycoengineered muCD20 antibody 18B12 on inflammatory cytokines, such as TNF-α and ILs, and on glial cells, that are overexpressed after SCI [25]. As B cells have a role in the trafficking of T cells into the CNS [26], this study also aimed to provide information on immune receptors, such as CD19+ (present on B cells), CD4+ (a co-receptor that assists the T-cell receptor in communicating with an antigen-presenting cell), CD45R+ (a member of the protein tyrosine phosphatase family, expressed in activated and memory T lymphocytes), CD8β+ (co-receptor predominantly expressed on the surface of cytotoxic T cells), and forkhead box P3 transcription factor (Foxp3).
Materials and Methods
In Vivo Procedures
Animals
Male adult CD1 mice (25–30 g; Envigo, Udine, Italy) were housed in a controlled environment and supplied with standard rodent chow and water. Mice were housed in stainless steel cages in a room kept at 22 ± 1 °C with a 12-h light/12-h dark cycle. Mice were acclimatized to their environment for 1 week and had ad libitum access to tap water and rodent standard diet. The study was approved by the University of Messina review board for the care of animals. All animal experiments were carried out in agreement with Italian (DM 116192) and European Union regulations (OJ of EC L 358/1 12/18/1986).
Materials
All compounds were obtained from Sigma-Aldrich (Milan, Italy). For B-cell depletion, a glycoengineered muIgG2 version of the murine CD20 antibody 18B12 was applied and produced by transient transfection [22]. All chemicals were of the best commercial grade available. All stock solutions were prepared in nonpyrogenic saline (0.9 % NaCl; Baxter, Lurago D’erba, Italy). Tissue preparation was performed in aseptic conditions using sterile instruments.
SCI
We used the clip compression model as previously described (see Supplementary data 1) [27].
Experimental Groups
Mice were randomly allocated to the following groups: 1) sham + vehicle group (n = 10)—mice were subjected to the surgical procedures except for the aneurysm clip, which was not applied, and these mice were administered saline (administered i.p., 1 h and 6 h after SCI); 2) sham + 18B12 group (n = 10)—identical to sham + vehicle group except for the administration of 18B12 (at a dose of 30 mg/kg, i.p., 1 h and 6 h after SCI); 3) SCI + vehicle (n = 10)—mice were subjected to laminectomy and the aneurysm clip was applied; 4) SCI + 18B12 (n = 10)—mice were subjected to SCI and administered 18B12 (at a dose of 30 mg/kg, i.p., 1 h and 6 h after SCI).
The dose of 18B12 (30 mg/kg) used here was based on that used in a previous study [23]; the dose–response and time-course studies were previously done at our laboratory (data not shown).
To evaluate the various parameters, mice (n = 10 from each group) were sacrificed at 24 h after SCI.
In a separate set of experiments, another 10 animals in each group were observed until 10 days after SCI, to evaluate the motor score.
Tissue Processing and Light Microscopy
Spinal cord tissues from the perilesional zone were taken 24 h after trauma and processed, as previously described, for hematoxylin and eosin coloration (see Supplementary data 2) [28]. Representative images are shown. Quantitative assessments of histopathologic inflammation were made and were scored on a 9-point scale [29]. The scores from all sections of every spinal cord were averaged to give a final score for distinct mice. Each histological analyses was completed in a blinded fashion.
2.6 Grading of Motor Disturbance
The motor function of animals subjected to compression trauma was evaluated once a week for 6 weeks after injury. Recovery from motor disturbance was graded using the Basso Mouse Scale (BMS) open-field score as the BMS has been shown to be a valid locomotor rating scale for mice [30]. The BMS scale ranges from 0 (indicating complete paralysis) to 9 (indicating normal hindlimb function), and rates locomotion on such aspects of hindlimb function as weight support, stepping ability, coordination, and toe clearance.
Western Blot Analysis for IkB-α, Nuclear Factor Kappa B, Glial Fibrillary Acidic Protein, IL-1β, IL-12A, and Inducible Nitric Oxide Synthase
The levels of IkB-α, nuclear factor kappa B (NF-kB), glial fibrillary acidic protein (GFAP), IL-1β, IL-12A, inducible nitric oxide synthase (iNOS), lamin a/c, and β-actin were calculated, as previously described, in cytosolic and nuclear fractions from spinal cord tissue collected at the end of the experiment, with minor modifications (see Supplementary data 3) [31]. Bands were detected with a chemiluminescence system (Thermo, Waltham, Massachusetts, USA), visualized with the Chemi Doc XRS (Bio-Rad, USA) and analyzed by using Image Lab 3.0 software (Bio-Rad, Hercules, CA, USA). The expression levels of β-actin and lamin A/C served as an internal control for protein loading.
Immunohistochemical Localization of CD19, CD8β, CD4, CD45, CD11β, IL-1β, TNF-α, GFAP, and Foxp3
Immunohistochemical localization of CD19, CD8β, CD4, CD45, CD11β, IL-1β, TNF-α, GFAP, and Foxp3 was carried out as previously described (see Supplementary data 4) [28]. The immunohistochemical pictures were collected by a Zeiss microscope using Axio Vision software. For graphic exemplification of densitometric analysis, we measured the intensity of positive staining (brown staining) by computer-assisted color image analysis (QWin V3; Leica, Milton Keynes UK). The percentage area of immunoreactivity (determined by the number of positive pixels) was expressed as percentage of total tissue area (red staining). Replicates for each experimental condition and histochemical staining were obtained from each mouse in each experimental group. In sham-operated mice, the central areas of equivalent tissue sections were taken as reference points and a comparable number of optical fields were counted [32]. All histological examinations were carried out in a blinded fashion.
Immunofluorescence
Detection of GFAP, TNF-α, IL-1β, Iba-1, CD4, and Foxp3 was carried out as previously described (see Supplementary data 5) [28]. All images were digitalized at a resolution of 8 bits into an array of 2560 × 1920 pixels. Sections were viewed and photographed with a Leica DM2000 microscope. Optical sections of fluorescence samples were obtained using an Ar laser (458 nm), a HeNe laser (543 nm), and an ultraviolet laser (361–365 nm) at a 1-min, 2-s scanning speed with up to 8 averages; 1.5-μm sections were obtained using a pinhole of 250. Contrast and brightness were established by examining the most brightly labeled pixels and applying settings that allowed clear visualization of structural details while keeping the highest pixel intensities close to 200. The same settings were used for all images obtained from the other samples that had been processed in parallel. Digital images were cropped and figure montages prepared using Adobe Photoshop CS5 (Adobe Systems, Palo Alto, CA, USA).
Ex Vivo Procedures
Preparation of Spinal Cord Organotypic Slice Cultures
Spinal cord slice cultures were staged from mouse spinal cord at postnatal day 6, as previously described [33].
Treatments
Cell cultures were divided into the following groups, after 7 days of stabilization of growth: 1) control (Ctr)—undamaged spinal cord slices were cultured with normal culture medium and treated with vehicle only; 2) damage—spinal cord slices were cut sagittally with a blade, under microscopic control; 3) damage + 18B12—spinal cord slices were cut sagittally, as described, and 18B12 was applied at a concentration of 4.35 μg/μl and placed in culture medium 1 h before injury.
In all of the groups, the compounds were left in a culture medium for 24 h after injury. Spinal cord slices were then used for Western blot analysis and 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
Viability of Organotypic Cultures
At 24 h after mechanical damage, viability of organotypic cultures was assessed by using a mitochondria-dependent dye for live cells (tetrazolium dye; MTT), as previously described [34].
Measurement of Nitrite Levels
Total nitrite levels, as an indicator of nitric oxide (NO) synthesis, were measured in the supernatant as previously described [35].
Statistical Evaluation
All values in the figures and text are expressed as mean ± SEM of n observations. For the in vivo studies, n represents the number of animals studied. In the histology or immunohistochemistry experiments, the figures shown are representative of at least 3 experiments performed on different days. The results were analyzed by 1-way ANOVA followed by a Bonferroni post-hoc test for multiple comparisons. A p-value < 0.05 was considered significant. BMS scale data were analyzed by the Mann–Whitney test and considered significant when p < 0.05.
Results
18B12 Reduces the Severity of SCI
The severity of trauma in the perilesional area, assessed by the presence of edema, as well as estimation of alterations of white matter, was evaluated by hematoxylin and eosin staining 24 h after trauma. Significant damage to the spinal cord was observed in tissue from mice subjected to SCI (Fig. 1b; see histological score, Fig. 1d) when compared with sham-operated mice (Fig. 1a; see histological score, Fig. 1d). Notably, a significant protection against SCI was shown in 18B12-treated mice (Fig. 1c; see histological score, Fig. 1d). To evaluate whether histological damage to the spinal cord was associated with a loss of motor function, was used the BMS open-field score. Motor function was not altered in sham mice. SCI mice showed significant deficits in hindlimb movement (Fig. 1e), starting with the first evaluation performed 1 week after trauma. In the 18B12-treated mice group, the neurological score was significantly enhanced after acute administration compared with the SCI vehicle mice (Fig. 1e).
Fig. 1.

Effect of 18B12 treatment on histological alterations and behavioral disturbance after spinal cord injury (SCI). An important injury to the spinal cord was observed in (b) SCI mice compared with (a) sham and (c) 18B12-treated mice. The severity of tissue damage was enhanced in tissue sections obtained from (b) injured mice and from (c) 18B12-treated mice. (d) Histological score was prepared by an independent observer. (e) The motor function of mice subjected to trauma was evaluated up to 6 weeks after damage. Recovery from motor disturbance was graded using the Basso Mouse Scale. Each data are expressed as mean ± SEM of 10 mice for each group. ***p < 0.001 vs sham; ### p < 0.001 vs SCI. (a, b) Hematoxylin and eosin ND = not detectable
Effect of 18B12 on IkB-α Degradation, NF-kB p65 Nuclear Localization, and iNOS Expression
To analyze the molecular mechanisms by which 18B12 treatment may attenuate the development of SCI, we assessed by Western blot analysis the classical NF-kB pathway, evaluating IkB-α levels and NF-kB nuclear localization. Also, to determine the levels of NO produced during SCI, iNOS expression was evaluated by Western blotting analysis in the spinal cord sections 24 h after SCI. A basal level of IkB-α was detected in the spinal cord sections from sham-operated mice (Fig. 2a), whereas SCI substantially reduced IkB-α levels (Fig. 2a). 18B12 prevented the SCI-induced IkB-α degradation and restored IkB-α to levels similar to those in sham-operated mice (Fig. 2a). In addition, SCI mice resulted in a significant increase in nuclear NF-kB p65 levels compared with sham-operated mice (Fig. 2b). 18B12 treatment significantly decreased NF-kB p65 expression (Fig. 2b). Moreover, an increased expression of iNOS was observed in mice subject to SCI compared with the control group (Fig. 2c), while treatment with 18B12 reduced iNOS expression significantly (Fig. 2c).
Fig. 2.

Effect of 18B12 treatment on nuclear factor kappa B (NF-kB) pathways and inducible nitric oxide synthase (iNOS) expression. Western blot analysis for IkB-α, NF-kB, and iNOS proteins in the spinal cord were detected at 24 h after SCI. (a) IkB-α levels were reduced substantially in 18B12 tissues compared with spinal cord injury (SCI) mice. (b) NF-kB p65 levels in the SCI mice were increased significantly compared to sham mice. Low levels of NF-kB p65 were found in 18B12-treated animals. (c) A substantial increase in expression of iNOS was found in SCI mice compared with control; 18B12 considerably reduced iNOS expression. Data were normalized on the basis of β-actin and Lamin A/C levels. A representative blot of lysates obtained from 5 animals/group is shown and densitometry analysis is reported for all animals. Data are expressed as mean ± SEM of 10 mice from each group. *p < 0.1 vs sham; # p < 0.1 vs SCI
Effect of 18B12 on IL-12A and IL-1β Expression
To test whether 18B12 modulates inflammatory development through the regulation of the secretion of cytokines, we analyzed spinal cord expression and levels of the proinflammatory cytokines IL-12A and IL-1β. A substantial increase in IL-12A and IL-1β expression was found in the spinal cord tissues collected 24 hours after SCI (Fig. 3b, d–f). 18B12 treatment reduced SCI expression of IL-1β in injured mice and lowered the expression of IL-12A (p < 0.01) (Fig. c–f).
Fig. 3.

Effect of 18B12 treatment on cytokine expression. (e, f) Homogenized spinal cord sections were evaluated by Western blot analysis. Injury caused a significant increase in interleukin (IL)-1β and IL-12A expression compared with the control group. Treatment with 18B12 significantly reduced cytokine levels. Spinal cord sections were also processed for immunohistologic staining for IL-1β. (a, d) No positive staining for IL-1β was found in the spinal cord tissue from sham-operated mice. (b, d)A substantial increase in IL-1β expression was found in spinal cord tissues from spinal cord injury (SCI) mice 24 h after SCI. (c, d) 18B12 treatment significantly attenuated IL-1β levels in the spinal cord. Data were normalized on the basis of β-actin levels. A representative blot of lysates obtained from 5 animals/group is shown and densitometry analysis of all animals is reported. Data are expressed as mean ± SEM of 10 mice from each group. **p < 0.01 and *p < 0.1 vs sham; # p < 0.1 and ## p < 0.1 vs SCI ND = not detectable
Effect of 18B12 on Astrocyte Activation and TNF-α Expression in Spinal Cord Tissue
It has been known that after SCI astrocytes become reactive as a response to damage. In this study, using GFAP staining, we mean a glial response mediated by damage, which reflects that astrocytes become activated, as indicated by staining for GFAP cells in injured animals compared with sham animals (Fig. 4e and d, respectively; see densitometric analysis, Fig. 4g). Instead, treatment with 18B12 decreased the number of reactive astrocytes (GFAP-activated astrocytes) (Fig. 4f; see densitometric analysis, Fig. 4g). To test whether 18B12 regulates the inflammatory process through the secretion of proinflammatory cytokines, we analyzed the spinal cord levels of TNF-α by immunohistochemical staining. There was no staining for TNF-α in spinal cord obtained from sham mice (Fig. 4a; see densitometric analysis Fig. 4g). A substantial alteration in TNF-α expression was found in the spinal cord tissues collected from mice at 24 h after SCI (Figs. 4b, b1; see densitometric analysis, Fig. 4g). Spinal cord expression of TNF-α was significantly attenuated in 18B12-treated SCI mice compared with SCI animals (Fig. 4c, c1; see densitometric analysis, Fig. 4g).
Fig. 4.

Effects of 18B12 treatment on astrocyte activation and tumor necrosis factor (TNF)-α expression in spinal cord tissue. Spinal cord sections were processed to determine the immunohistologic staining for glial fibrillary acidic protein (GFAP) and TNF-α expression. (a, g) No positive staining for TNF-α was found in the spinal cord tissue from the sham group. (b, g) TNF-α expression increased in spinal cord tissues from spinal cord injury (SCI) mice 24 h after SCI. (c, g) 18B12 treatment significantly attenuated TNF-α levels in the spinal cord. (d, g) Moreover, sham animals never expressed GFAP, but (e, g) the number of GFAP+ cells was significantly increased after induction of SCI. (f, g) 18B12 significantly decreased the activation of astrocyte GFAP+ cells. The figure is representative of at least 3 experiments performed on different experimental days. Data are expressed as percentage of total tissue area. Data are mean ± SEM of 10 mice from each group. ### p < 0.001 vs sham; ***p < 0.01 vs SCI ND = not detectable
Moreover, to confirm astrogliosis and TNF-α and IL-1β expression, we performed immunofluorescence staining. Sections of spinal cord were double stained with antibodies against GFAP (green), Iba1 (green), TNF-α (red), and IL-1β (red). Spinal cord sections revealed amplified astrogliosis (GFAP + cells) in SCI mice (Figs. 5d, f and 6d, f) such as activation of the microglia (Figs. 5o, q and 6o, q). GFAP immunoreactivity and microglia activation were significantly reduced in SCI-treated mice (Figs. 5g, i, r, t and 6g, i, r, t). The yellow spots indicate the colocalization between TNF-α and GFAP (Fig. 5f, i) and the colocalization between IL-1β and GFAP (Fig. 6f, i), as well as between TNF-α and Iba1 (Fig. 5q, t), and IL-1β and Iba1 (Fig. 6q, t). Images are representative of triplicate experiments.
Fig. 5.

Effects of 18B12 treatment on colocalization of glial fibrillary acidic protein (GFAP)/tumor necrosis factor (TNF)-α and Iba1/TNF-α after spinal cord injury (SCI). (a, c, l, n) Results are shown for sham-operated mice. (d, f, o, q) Mice with SCI and (g, i, r, t) mice with SCI treated with 18B12. Spinal cord sections were double stained with antibodies against GFAP (a, d, j; green), Iba1 (l, o, r; green), and TNF-α (b, e, h, m, p, s; red). (d, o) Spinal cord sections revealed increased astrogliosis (GFAP + cells) in SCI mice. (g, r) GFAP immunoreactivity was reduced in 18B12-treated mice.(i, t) Yellow spots indicate colocalizations and revealed a high colocalization between GFAP/TNF-α and Iba1/TNF-α double staining
Fig. 6.

Effects of 18B12 treatment on colocalization of glial fibrillary acidic protein (GFAP)/interleukin (IL)-1β and Iba1/IL-1β after spinal cord injury (SCI). (a, c, l, n) Results are shown for sham-operated mice, (d, f, o, q) mice with SCI, and (g, i, r, t) mice with SCI treated with 18B12. Spinal cord sections were double stained with antibodies against (a, d, j; green) GFAP, (l, o, r; green) Iba1, and (b, e, h, m, p, s; red) IL-1β. (d, o) Spinal cord sections revealed increased astrogliosis (GFAP+ cells) in SCI mice. (g, r) GFAP immunoreactivity was reduced in 18B12-treated mice.(i, t) Yellow spots indicate colocalizations and revealed high colocalization between GFAP/IL-1β and Iba1/IL-1β double staining
Effect of 18B12 on Immune Response
To determine whether 18B12 treatment affected the immune response in the acute phase after SCI, we evaluated B and T lymphocyte markers in the spinal cord tissues collected 24 h after SCI. We studied B-cell infiltration (by anti-CD19 staining) and other leukocyte infiltration via CD11β staining (a marker involved in the innate immune system and microglia activation). We found that expression of CD19+ B lymphocytes was considerably higher in SCI mice (Fig. 7b, g) than in the sham group (Fig. 7a, g). Also, spinal cord sections from control mice did not stain for CD11β (Fig. 7d, g) whereas SCI sections exhibited positive staining for CD11β expression (Fig. 7e, g). 18B12 treatment reduced CD19 and CD11 β-cell infiltration (Fig. 7c, f, g).
Fig. 7.

Effects of 18B12 treatment on CD19 and CD11β. (a, d, g) Spinal cord sections from sham-operated mice did not stain for CD19 or CD11β. (b, e, g) Sections obtained from vehicle-treated animals after spinal cord injury (SCI) demonstrate positive staining for CD19 and for CD11β, mainly localized in inflammatory cells. (c, f, g) 18B12 treatment reduced the degree of positive staining both for B lymphocytes and for CD11β. The figure is representative of at least 3 experiments carried out on different experimental days. Data are expressed as percentage of total tissue area. Data are mean ± SEM of 10 mice from each group.**p < 0.01 vs sham; # p < 0.1 vs SCI
In seeking to provide direct evidence that regulatory T cells (Tregs; CD8β+, CD4+, and CD45+) are required for CNS maintenance of the day-to-day need to cope with traumatic events, we evaluated their expression. We found that T lymphocytes (CD8β+, CD4+, and CD45+) were considerably more expressed in SCI mice (Fig. 8d, e, f, l, m, n) than in the sham group (Fig. 8a–c, l, m, n). 18B12 treatment reduced positive staining for CD8β+ (Fig. 8g, l), for CD4+ (Fig. 8h, m), and for CD45+ (Fig. 8i, n).
Fig. 8.

Effects of 18B12 treatment on T lymphocytes. (a, l) Spinal cord sections from sham-operated mice expressed basal levels for CD8β, (b, m) for CD4, and (c, n) for CD45. (d, e, f, g, h, i, l, m, n) 18B12 treatment reduced the amount of positive staining for T lymphocytes with respect to spinal cord injury (SCI) mice. The figure is representative of at least 3 experiments performed on different experimental days. Data are expressed as percentage of total tissue area. Data are mean ± SEM of 10 mice from each group.**p < 0.01 vs sham; # p < 0.1 vs SCI
In addition, as Foxp3+ cells are regarded as an important component of the immune system, we analyzed Foxp3 expression by immunohistochemistry. We found that Foxp3 was considerably more expressed in SCI mice (Fig. 9m; see densitometric analysis, Fig. 9o) than in the control group (Fig. 9l; see densitometric analysis, Fig. 9o). 18B12 treatment significantly reduced positive staining for Foxp3 (Fig. 9n; see densitometric analysis, Fig. 9o). Also, as Foxp3 is a critical component of CD4+ Treg development and function, we performed immunofluorescence staining for both CD4 and Foxp3. Spinal cord sections were double stained with antibodies against CD4+ (green) and Foxp3+ (red). Spinal cord sections revealed a significant increase in cells expressing the Treg markers CD4+ and Foxp3+ in the SCI group (Fig. 9b, e, h) compared with the sham group (Fig. 9a, d, h). Foxp3 and CD4 immunoreactivity were considerably reduced in SCI-treated mice (Fig. 9c, f, i).
Fig. 9.

Effects of 18B12 treatment on regulatory T cell (Treg) expression. (l, o) Spinal cord sections from sham-operated mice expressed basal levels for forkhead box P3 transcription factor (Foxp3). 18B12 treatment reduced the degree of positive staining for (n, o) Foxp3 with respect to (m, o) spinal cord injury (SCI) mice. The figure is representative of at least 3 experiments performed on different experimental days. Data are expressed as percentage of total tissue area. Data are mean ± SEM of 10 mice from each group.**p < 0.01 vs sham. ## p < 0.01 vs SCI. Also, spinal cord sections were double stained with antibodies against CD4 (green) and Foxp3 (red). (b, e) Increased Treg cell expression was revealed in SCI mice, (c, f) while Foxp3 and CD4 immunoreactivity was reduced in 18B12-treated mice.Yellow arrows indicate CD4 and Foxp3 expression
Effect of 18B12 on Cell Viability in Spinal Cord Slices and on Nitrite (NO2–) Concentration
Slices were cultured for up to 7 days. During this time, slices conserved their morphology and structure with clear differentiation of white and gray matter (data not shown). Viable cells within the slices, detected using MTT tetrazolium dye, were observed under light microscopy. The grade of cell death was assessed in each slice 24 h after injury. Spinal cord organotypic slice cultures were treated with 18B12. Mechanical damage induced a significant reduction in viability compared with the uninjured group (Fig. 10a). We clearly showed that pretreatment with 18B12 significantly reduced cell death compared with the damage group (Fig. 10a). Moreover, this effect was confirmed by nitrite (NO2 –) concentration; we assessed the levels of nitrite released into the culture medium by Griess reagent. The untreated control group released low levels of NO2 –; instead, injury significantly enhanced the levels of NO2 –production (Fig. 10b). 18B12 treatment decreased damage-induced NO production in the medium in a concentration-dependent manner (Fig. 10b).
Fig. 10.

Percentage of cell death and nitrite levels in spinal cord injury slices. Viable cells, identified using 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, were visualized under light microscopy. The level of cell death was evaluated in each slice at 7 days. Cell death in injured groups was considerably higher compared with the control group. (a) Cell death was significantly attenuated by 18B12 pretreatment. ***p <0.001 vs control (CTR); ### p < 0.001 vs damage. (b) Indeed, an augmented formation of nitrite levels was observed in the damage group, while the pretreatment with 18B12 decreased injury-induced nitric oxide (NO) production. *** p < 0.001 vs CTR; # p < 0.1 vs damage
Discussion
SCI is one of the most disturbing of all traumatic conditions that can be experienced by patients. Over the last few years, much research has been performed on elucidating the mechanisms of SCI that, with its pathological sequelae, result in permanent neurological deficit. The importance of the immune response in functional recovery after traumatic SCI has been investigated in adult mammals under various conditions in different experimental model systems. The inflammatory response in the acutely injured cord is the main defense mechanism, which is designed to eliminate invading pathogens and tissue debris [36]. It has been proposed that the immune system can have both beneficial and adverse impacts on regeneration after trauma [37]. Results from clinical trials testing the beneficial effects of B cell reduction in various CNS autoimmune conditions cautiously support the approach [38]. Because the involvement of various components of the immune system in the regeneration after SCI has been studied in various experimental paradigms [39], in the present work, we evaluated the anti-inflammatory and immunologic role of B-cell depletion using the muCD20 antibody 18B12 in an experimental model of compression injury of the mouse spinal cord.
In this study, we used a clip compression model because only compression injury produces an outcome similar to that found in human SCI [40, 41]. SCI results in a necrotic district of cavitation that progressively increases, promoted by acute inflammation, apoptosis, and glial scarring. The acute loss of motor neurons and degeneration of white matter tracts results most often in irreversible motor dysfunction [42]. In this work, we clearly demonstrated that 18B12 treatment, administered 1 h and 6 h postinjury, attenuates the degree of secondary inflammation and reduced the loss of motor function associated with histological damage. This treatment is based on the assumption that glucocorticoids, given within the first 8 h of injury and continued for 24–48 h, are regarded as part of the standard treatment regimen [3, 43]. Many studies suggest that activation of the transcription factor NF-kB plays a central role in the generation of mediators or proteins involved in secondary inflammation associated with SCI [44]. As has been demonstrated, we confirmed that 18B12 inhibits constitutive NF-kB signaling pathways by reducing the phosphorylation of the components of the NF-kB pathway [45]. In this study, we report that SCI is associated with important IkB-α degradation and augmented nuclear expression of p65 in spinal cord tissues 24 h after injury. Treatment with 18B12 significantly reduced IkB-α degradation and NF-kB localization. NF-kB is one of the pivotal regulators of proinflammatory gene expression and induces transcription of proinflammatory cytokines, chemokines, and adhesion molecules [46]. NF-kB is greatly activated at sites of inflammation in various immunological diseases and this activation is accompanied by enhanced recruitment of inflammatory cells and production of proinflammatory mediators like iNOS, IL-1β, IL-12A, and TNF-α. In addition, B lymphocytes were noted at the injury site within the first week of SCI and subsequent secretion of cytokines plays a major role in cytotoxic cell damage. In particular, IL-12 directs human T-cell differentiation towards more mature cells, delineating an important role of IL-12A in the activation of damage [47]. However, the activation of numerous inflammatory pathways determines activated B-cell accumulation at the SCI region and is undoubtedly correlated with pathological events after SCI. This mechanism was similar to neuroinflammatory cell damage, which is a significant mechanism for autoimmune neurological diseases [48]. Therefore, the inhibition of the production of proinflammatory mediators by 18B12 described in this study can most probably be attributed to the inhibitory effect of the activation of NF-kB. Moreover, preceding studies have shown that TNF-α activates iNOS and causes the generation of the NO radical from L-arginine [49]. NO generation is a common aspect of immune cells, including neutrophils, monocytes, macrophages, and NK cells, including endothelial cells. This study demonstrates that 18B12 attenuates the expression of iNOS in the tissue from SCI-treated mice when compared with control group mice. In this study, we have found that the damage induced by SCI is associated with intense immunostaining for TNF-α and IL-1β. Trauma was noted to increase expression of TNF-α and IL-1 in neuronal and glial cells of spinal cord; we clearly observed that 18B12 treatment significantly prevents the appearance of staining of TNF-α and IL-1β.
It is known that microglia and astrocytes play a special role in sustaining the immune response [50], so we evaluated the effect of 18B12 on microglial and astrocytic markers. Microglial Iba1 and astrocytic GFAP expression are usually used as markers of glial cells, though their use as markers of cellular activation has been increasingly questioned. Augmented levels of GFAP have been associated with increased neurological disease activity and, in particular, with disability [51, 52]. Astrocytes respond to various neurodegenerative insults rapidly, causing vigorous astrogliosis [53], so we questioned the use of GFAP, demonstrating that 18B12 has effects on astrocytes and potentially on their activation state. Also, as microglia become activated within minutes to hours after SCI and are transformed into macrophages [54], using CD11β that regulates activation of microglia, in this work we studied microgliosis, demonstrating that CD11β and Iba1 immunoreactivity was significantly increased in SCI mice 24 h after the trauma, whereas the treatment with 18B12 reduced its expression.
Because SCI has been identified to trigger a systemic, neurogenic immune syndrome [55], we analyzed fluctuations of immune cell populations and proved that SCI induces immune suppression. Regulatory B and T cells control CNS autoimmune disease, independently of each other [56]. The role of B cells and autoimmunity as factors that contribute to neurological outcomes following SCI is poorly understood. The alteration of the local cytokine environment during SCI favors B-cell autoimmunity [57], and B cells produce pathogenic antibodies that impair lesion repair [58], resulting in worse neurological outcome. Therapies that target the B cell or obstruct the effects of pathogenic antibodies have demonstrated to be effective [20, 59].
Thus, in this study we analyzed the role of 18B12 on CD19+ B lymphocytes, confirming the observation that 18B12 effectively reduced B cells in inflamed tissues and lymphoid organs [60]. Previous studies have indicated that 18B12 may affect T-cell responses through downregulation of its activation. This effect was attributed to decreased antigen presenting function of B cells after 18B12 treatment [61].
Experimental SCI also induced depletion of CD45+ T lymphocytes. An invasion of T lymphocytes occurs early after injury, forming clusters around B lymphocyte infiltrates that persist during the chronic phase [62]. This study showed that CD45+, CD4+, and CD8+ T cell and Foxp3+ expression decreased in response to 18B12, compared with their presence in damage. Thus, we have confirmed that 18B12 provides significant protection in SCI-induced autoimmunity at the sites of inflammation. CD4+Foxp3+ Tregs have been considered critical in controlling immune system homeostasis [63], and their derangement is frequently associated with autoimmunity. Identification of Tregs is, however, difficult because most markers, including Foxp3, are expressed by recently activated T cells [64]. Therefore, we confirmed the presence of proliferating Foxp3+ and CD4+ cells in SCI mice and showed a significant decrease owing to 18B12 treatment in SCI-treated mice; however, we did not observe a Foxp3+CD4+ colocalization.
In this study, to confirm our in vivo observation, we also tested the efficacy of 18B12 using spinal cord organotypic slice cultures. We showed a significant reduction in cell death following 18B12 pretreatment. Cell death and neuron degeneration, which occur after the injury, are a consequence of an increase in reactive oxygen species and NO production. NO is a unique molecule involved in a variety of physiological processes in the CNS [65]. The effects of NO on spinal cord damage are correlated with several factors, such as concentration of NO production, activity of different synthase isoforms, cellular source of production, and time of release [35]. In this study, we demonstrated increased NO expression after injury, reduced by 18B12 pretreatment.
Conclusion
Research to evaluate possible new molecules that participate in neuroprotection has become more important, particularly with regard to protecting nervous tissue from inflammatory and immunological events in neuroinflammatory and neurodegenerative disease. In this study, we clearly showed that B-cell depletion by 18B12 treatment reduces the increase of inflammation and tissue injury, such as the alteration of the immune system associated with SCI. Moreover, this study enhances current understanding that B-cell depletion exerts immunomodulating actions, offering the possibility of pursuing new therapeutic strategies involving therapeutic CD20 antibodies such as rituximab or obinutuzumab as a new approach in the treatment of inflammatory and immunological effects associated with SCI.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 1224 kb)
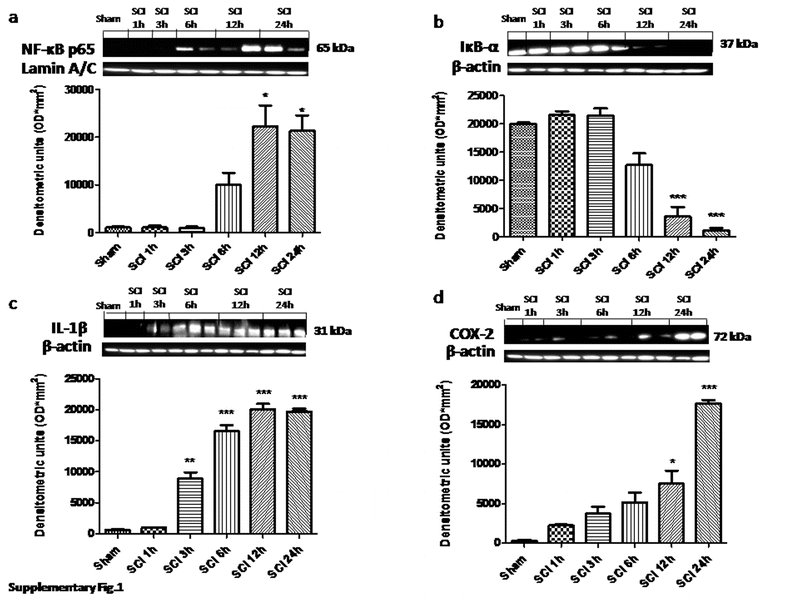{kind=link}
Evaluation of proinflammatory mediators at different time points after injury (GIF 90 kb)
Acknowledgments
We thank Antonietta Medici for her excellent technical assistance during this study, and Miss Valentina Malvagni for editorial assistance with the manuscript. 18B12 was provided by Christian Kleina and Marina Bacac, Roche Innovation Center Zurich, Roche Pharmaceutical Research and Early Development.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Compliance With Ethical Standards
Conflict of Interest
No competing financial interests exist.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-016-0446-2) contains supplementary material, which is available to authorized users.
References
- 1.Whiteneck GG, Charlifue SW, Frankel HL, et al. Mortality, morbidity, and psychosocial outcomes of persons spinal cord injured more than 20 years ago. Paraplegia. 1992;30:617–630. doi: 10.1038/sc.1992.124. [DOI] [PubMed] [Google Scholar]
- 2.Bartholdi D, Schwab ME. Methylprednisolone inhibits early inflammatory processes but not ischemic cell death after experimental spinal cord lesion in the rat. Brain Res. 1995;672:177–186. doi: 10.1016/0006-8993(94)01410-J. [DOI] [PubMed] [Google Scholar]
- 3.Genovese T, Esposito E, Mazzon E, et al. PPAR-alpha modulate the anti-inflammatory effect of glucocorticoids in the secondary damage in experimental spinal cord trauma. Pharmacol Res. 2009;59:338–350. doi: 10.1016/j.phrs.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 4.Dumont AS, Dumont RJ, Oskouian RJ. Will improved understanding of the pathophysiological mechanisms involved in acute spinal cord injury improve the potential for therapeutic intervention? Curr Opin Neurol. 2002;15:713–720. doi: 10.1097/00019052-200212000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Cserr HF, Knopf PM. Cervical lymphatics, the blood–brain barrier and the immunoreactivity of the brain: a new view. Immunol Today. 1992;13:507–512. doi: 10.1016/0167-5699(92)90027-5. [DOI] [PubMed] [Google Scholar]
- 6.Prins RM, Liau LM. Immunology and immunotherapy in neurosurgical disease. Neurosurgery. 2003;53:144–152. doi: 10.1227/01.NEU.0000068865.34216.3A. [DOI] [PubMed] [Google Scholar]
- 7.Stoll G, Jander S, Schroeter M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol. 2002;513:87–113. doi: 10.1007/978-1-4615-0123-7_3. [DOI] [PubMed] [Google Scholar]
- 8.Trivedi A, Olivas AD, Noble-Haeusslein LJ. Inflammation and spinal cord injury: infiltrating leukocytes as determinants of injury and repair processes. Clin Neurosci Res. 2006;6:283–292. doi: 10.1016/j.cnr.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zajarias-Fainsod D, Carrillo-Ruiz J, Mestre H, Grijalva I, Madrazo I, Ibarra A. Autoreactivity against myelin basic protein in patients with chronic paraplegia. Eur Spine J. 2012;21:964–970. doi: 10.1007/s00586-011-2060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones TB. Lymphocytes and autoimmunity after spinal cord injury. Exp Neurol. 2014;258:78–90. doi: 10.1016/j.expneurol.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Oropallo MA, Goenka R, Cancro MP. Spinal cord injury impacts B cell production, homeostasis, and activation. Semin Immunol. 2014;26:421–427. doi: 10.1016/j.smim.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 12.Wu B, Matic D, Djogo N, Szpotowicz E, Schachner M, Jakovcevski I. Improved regeneration after spinal cord injury in mice lacking functional T- and B-lymphocytes. Exp Neurol. 2012;237:274–285. doi: 10.1016/j.expneurol.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 13.Ankeny DP, Guan Z, Popovich PG. B cells produce pathogenic antibodies and impair recovery after spinal cord injury in mice. J Clin Invest. 2009;119:2990–2999. doi: 10.1172/JCI39780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nitsch R, Pohl EE, Smorodchenko A, Infante-Duarte C, Aktas O, Zipp F. Direct impact of T cells on neurons revealed by two-photon microscopy in living brain tissue. J Neurosci. 2004;24:2458–2464. doi: 10.1523/JNEUROSCI.4703-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones TB, Hart RP, Popovich PG. Molecular control of physiological and pathological T-cell recruitment after mouse spinal cord injury. J Neurosci. 2005;25:6576–6583. doi: 10.1523/JNEUROSCI.0305-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ankeny DP, Popovich PG. B cells and autoantibodies: complex roles in CNS injury. Trends Immunol. 2010;31:332–338. doi: 10.1016/j.it.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu H, Zhang GX, Ciric B, Rostami A. IDO: a double-edged sword for T(H)1/T(H)2 regulation. Immunol Lett. 2008;121:1–6. doi: 10.1016/j.imlet.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang DW, McKerracher L, Braun PE, David S. A therapeutic vaccine approach to stimulate axon regeneration in the adult mammalian spinal cord. Neuron. 1999;24:639–647. doi: 10.1016/S0896-6273(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 19.Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neurol. 2008;4:557–567. doi: 10.1038/ncpneuro0901. [DOI] [PubMed] [Google Scholar]
- 20.Gold R, Stangel M, Dalakas MC. Drug Insight: the use of intravenous immunoglobulin in neurology--therapeutic considerations and practical issues. Nat Clin Pract Neurol. 2007;3:36–44. doi: 10.1038/ncpneuro0376. [DOI] [PubMed] [Google Scholar]
- 21.Lehmann-Horn K, Schleich E, Hertzenberg D, et al. Anti-CD20 B-cell depletion enhances monocyte reactivity in neuroimmunological disorders. J Neuroinflammation. 2010;8:146. doi: 10.1186/1742-2094-8-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. 2007;179:3351–3361. doi: 10.4049/jimmunol.179.5.3351. [DOI] [PubMed] [Google Scholar]
- 23.Mossner E, Brunker P, Moser S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115:4393–4402. doi: 10.1182/blood-2009-06-225979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrara C, Stuart F, Sondermann P, Brunker P, Umana P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem. 2006;281:5032–5036. doi: 10.1074/jbc.M510171200. [DOI] [PubMed] [Google Scholar]
- 25.Gal P, Kravcukova P, Mokry M, Kluchova D. Chemokines as possible targets in modulation of the secondary damage after acute spinal cord injury: a review. Cell Mol Neurobiol. 2009;29:1025–1035. doi: 10.1007/s10571-009-9392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anthony DC, Dickens AM, Seneca N, et al. Anti-CD20 inhibits T cell-mediated pathology and microgliosis in the rat brain. Ann Clin Transl Neurol. 2014;1:659–669. doi: 10.1002/acn3.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Impellizzeri D, Esposito E, Mazzon E, et al. The effects of a polyphenol present in olive oil, oleuropein aglycone, in an experimental model of spinal cord injury in mice. Biochem Pharmacol. 2012;83:1413–1426. doi: 10.1016/j.bcp.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Siracusa R, Paterniti I, Bruschetta G, et al. The association of palmitoylethanolamide with luteolin decreases autophagy in spinal cord injury. Mol Neurobiol 2015 Jul 5 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Retracted]
- 29.Qiao F, Atkinson C, Kindy MS, et al. The alternative and terminal pathways of complement mediate post-traumatic spinal cord inflammation and injury. Am J Pathol. 2010;177:3061–3070. doi: 10.2353/ajpath.2010.100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- 31.Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, Yezierski RP. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci. 1998;18:3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shea TB. Technical report. An inexpensive densitometric analysis system using a Macintosh computer and a desktop scanner. Biotechniques. 1994;16:1126–1128. [PubMed] [Google Scholar]
- 33.Esposito E, Paterniti I, Meli R, Bramanti P, Cuzzocrea S. GW0742, a high-affinity PPAR-delta agonist, mediates protection in an organotypic model of spinal cord damage. Spine (Phila Pa 1976) 2012;37:E73–E78. doi: 10.1097/BRS.0b013e3182276d88. [DOI] [PubMed] [Google Scholar]
- 34.Abe K, Matsuki N. Measurement of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction activity and lactate dehydrogenase release using MTT. Neurosci Res. 2000;38:325–329. doi: 10.1016/S0168-0102(00)00188-7. [DOI] [PubMed] [Google Scholar]
- 35.Paterniti I, Impellizzeri D, Di Paola R, Navarra M, Cuzzocrea S, Esposito E. A new co-ultramicronized composite including palmitoylethanolamide and luteolin to prevent neuroinflammation in spinal cord injury. J Neuroinflammation. 2013;10:91. doi: 10.1186/1742-2094-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beattie MS. Inflammation and apoptosis: linked therapeutic targets in spinal cord injury. Trends Mol Med. 2004;10:580–583. doi: 10.1016/j.molmed.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Bechmann I. Failed central nervous system regeneration: a downside of immune privilege? Neuromolecular Med. 2005;7:217–228. doi: 10.1385/NMM:7:3:217. [DOI] [PubMed] [Google Scholar]
- 38.Jagessar SA, Heijmans N, Bauer J, et al. B-cell depletion abrogates T cell-mediated demyelination in an antibody-nondependent common marmoset experimental autoimmune encephalomyelitis model. J Neuropathol Exp Neurol. 2012;71:716–728. doi: 10.1097/NEN.0b013e3182622691. [DOI] [PubMed] [Google Scholar]
- 39.Ankeny DP, Popovich PG. Mechanisms and implications of adaptive immune responses after traumatic spinal cord injury. Neuroscience. 2009;158:1112–1121. doi: 10.1016/j.neuroscience.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDonough A, Martinez-Cerdeno V. Endogenous proliferation after spinal cord injury in animal models. Stem Cells Int. 2012;2012:387513. doi: 10.1155/2012/387513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonough A, Monterrubio A, Ariza J, Martinez-Cerdeno V. Calibrated forceps model of spinal cord compression injury. J Vis Exp. 2015;24:98. doi: 10.3791/52318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paterniti I, Impellizzeri D, Di Paola R, et al. Docosahexaenoic acid attenuates the early inflammatory response following spinal cord injury in mice: in-vivo and in-vitro studies. J Neuroinflammation. 2014;11:6. doi: 10.1186/1742-2094-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bracken MB, Shepard MJ, Holford TR, et al. Methylprednisolone or tirilazad mesylate administration after acute spinal cord injury: 1-year follow up. Results of the third National Acute Spinal Cord Injury randomized controlled trial. J Neurosurg. 1998;89:699–706. doi: 10.3171/jns.1998.89.5.0699. [DOI] [PubMed] [Google Scholar]
- 44.La Rosa G, Cardali S, Genovese T, et al. Inhibition of the nuclear factor-kappaB activation with pyrrolidine dithiocarbamate attenuating inflammation and oxidative stress after experimental spinal cord trauma in rats. J Neurosurg Spine. 2004;1:311–321. doi: 10.3171/spi.2004.1.3.0311. [DOI] [PubMed] [Google Scholar]
- 45.Baritaki S, Militello L, Malaponte G, Spandidos DA, Salcedo M, Bonavida B. The anti-CD20 mAb LFB-R603 interrupts the dysregulated NF-kappaB/Snail/RKIP/PTEN resistance loop in B-NHL cells: role in sensitization to TRAIL apoptosis. Int J Oncol. 2011;38:1683–1694. doi: 10.3892/ijo.2011.984. [DOI] [PubMed] [Google Scholar]
- 46.Baeuerle PA, Baichwal VR. NF-kappa B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv Immunol. 1997;65:111–137. doi: 10.1016/S0065-2776(08)60742-7. [DOI] [PubMed] [Google Scholar]
- 47.Lehmann D, Spanholtz J, Sturtzel C, et al. IL-12 directs further maturation of ex vivo differentiated NK cells with improved therapeutic potential. PLoS One. 2014;9:e87131. doi: 10.1371/journal.pone.0087131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alter A, Duddy M, Hebert S, et al. Determinants of human B cell migration across brain endothelial cells. J Immunol. 2003;170:4497–4505. doi: 10.4049/jimmunol.170.9.4497. [DOI] [PubMed] [Google Scholar]
- 49.Paterniti I, Genovese T, Mazzon E, et al. Liver X receptor agonist treatment regulates inflammatory response after spinal cord trauma. J Neurochem. 2010;112:611–624. doi: 10.1111/j.1471-4159.2009.06471.x. [DOI] [PubMed] [Google Scholar]
- 50.De Luca C, Savarese L, Colangelo AM, et al. Astrocytes and microglia-mediated immune response in maladaptive plasticity is differently modulated by NGF in the ventral horn of the spinal cord following peripheral nerve injury. Cell Mol Neurobiol. 2016;36:37–46. doi: 10.1007/s10571-015-0218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Axelsson M, Malmestrom C, Nilsson S, Haghighi S, Rosengren L, Lycke J. Glial fibrillary acidic protein: a potential biomarker for progression in multiple sclerosis. J Neurol. 2011;258:882–888. doi: 10.1007/s00415-010-5863-2. [DOI] [PubMed] [Google Scholar]
- 52.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4:229–237. doi: 10.1111/j.1750-3639.1994.tb00838.x. [DOI] [PubMed] [Google Scholar]
- 54.Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol. 2002;61:623–633. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- 55.Riegger T, Conrad S, Schluesener HJ, et al. Immune depression syndrome following human spinal cord injury (SCI): a pilot study. Neuroscience. 2009;158:1194–1199. doi: 10.1016/j.neuroscience.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 56.Matsushita T. Regulatory B cell and autoimmune disease. Nihon Rinsho Meneki Gakkai Kaishi. 2010;33:234–241. doi: 10.2177/jsci.33.234. [DOI] [PubMed] [Google Scholar]
- 57.Scott GN, DuHadaway J, Pigott E, et al. The immunoregulatory enzyme IDO paradoxically drives B cell-mediated autoimmunity. J Immunol. 2009;182:7509–7517. doi: 10.4049/jimmunol.0804328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dekaban GA, Thawer S. Pathogenic antibodies are active participants in spinal cord injury. J Clin Invest. 2009;119:2881–2884. doi: 10.1172/JCI40839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waubant E. Spotlight on anti-CD20. Int MS J. 2008;15:19–25. [PubMed] [Google Scholar]
- 60.Tipton TR, Roghanian A, Oldham RJ, et al. Antigenic modulation limits the effector cell mechanisms employed by type I anti-CD20 monoclonal antibodies. Blood. 2015;125:1901–1909. doi: 10.1182/blood-2014-07-588376. [DOI] [PubMed] [Google Scholar]
- 61.Merli M, Ferrario A, Maffioli M, Arcaini L, Passamonti F. Investigational therapies targeting lymphocyte antigens for the treatment of non-Hodgkin's lymphoma. Expert Opin Investig Drugs. 2015;24:897–912. doi: 10.1517/13543784.2015.1038342. [DOI] [PubMed] [Google Scholar]
- 62.Ankeny DP, Lucin KM, Sanders VM, McGaughy VM, Popovich PG. Spinal cord injury triggers systemic autoimmunity: evidence for chronic B lymphocyte activation and lupus-like autoantibody synthesis. J Neurochem. 2006;99:1073–1087. doi: 10.1111/j.1471-4159.2006.04147.x. [DOI] [PubMed] [Google Scholar]
- 63.Li X, Meng Y, Plotnikoff NP, et al. Methionine enkephalin (MENK) inhibits tumor growth through regulating CD4 + Foxp3+ regulatory T cells (Tregs) in mice. Cancer Biol Ther. 2015;16:450–459. doi: 10.1080/15384047.2014.1003006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Esposito M, Ruffini F, Bergami A, et al. IL-17- and IFN-gamma-secreting Foxp3+ T cells infiltrate the target tissue in experimental autoimmunity. J Immunol. 2010;185:7467–7473. doi: 10.4049/jimmunol.1001519. [DOI] [PubMed] [Google Scholar]
- 65.Conti A, Miscusi M, Cardali S, et al. Nitric oxide in the injured spinal cord: synthases cross-talk, oxidative stress and inflammation. Brain Res Rev. 2007;54:205–218. doi: 10.1016/j.brainresrev.2007.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1224 kb)
Evaluation of proinflammatory mediators at different time points after injury (GIF 90 kb)