

Abstract
Background
This study investigated the mechanism of miR-145 in targeting connective tissue growth factor (CTGF), which affects the proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) of ESCC cells.
Material/Methods
A total of 50 ESCC tissues and their corresponding normal adjacent esophageal tissue samples were collected. Then, miR-145 expression in both ESCC clinical specimens and cell lines was detected using quantitative real-time PCR. CTGF protein was detected using immunohistochemistry. Dual luciferase reporter gene assay was employed to assess the effect of miR-145 on the 3′UTR luciferase activity of CTGF. Eca109 cells were transfected with miR-145 mimics and CTGF siRNA, respectively, and changes in cellular proliferation, migration, and invasion were detected via MTT assay, wound-healing assay, and Transwell assay, respectively. Western blotting assay was used to detect the expression of marker genes related to EMT.
Results
MiR-145 was significantly down-regulated in ESCC tissues and cell lines compared with normal tissues and cell lines (P<0.05). We found significantly more positively expressed CTGF protein in ESCC tissues was than in normal adjacent esophageal tissues (P<0.01). Dual luciferase reporter gene assay showed that miR-145 can specifically bind with the 3′UTR of CTGF and significantly inhibit the luciferase activity by 55% (P<0.01). Up-regulation of miR-145 or down-regulation of CTGF can suppress the proliferation, migration, invasion, and EMT process of ESCC cells.
Conclusions
MiR-145 was significantly down-regulated in ESCC tissues and cell lines, while the protein expression of CTGF exhibited the opposite trend. MiR-145 inhibited the proliferation, migration, invasiveness, and the EMT process of ESCC cells through targeted regulation of CTGF expression.
MeSH Keywords: Cell Proliferation, Enoxaparin, Esophageal Diseases
Background
Esophageal cancer (EC) is a common cancer that is often diagnosed at an advanced stage. Patients with EC do not have specific symptoms until they reach a very late stage in which unfavorable prognostic outcomes usually occur [1]. Esophageal squamous cell carcinoma (ESCC) is defined as a major histological type of esophageal carcinoma and it has been ranked as the fourth most dangerous malignancy in China, with an unexpectedly high fatality rate [2]. Radical surgery alone or combined with radiotherapy or chemotherapy has substantially improved the prognosis of ESCC patients [3,4]. However, potential radiation hazards and limited effectiveness of surgery have motivated researchers to introduce alternative therapeutic approaches for managing patients with ESCC. Also, clarification of the molecular mechanisms in ESCC progression is believed to provide improvements in disease diagnosis.
As noncoding RNAs, microRNAs (miRNAs) contain 21–25 nucleotides and mature miRNAs play vital regulatory roles in cell proliferation, differentiation, and apoptosis [5]. Cell proliferation generally exerts bad influences in some diseases [6,7]. Regulating gene expression at the transcriptional level is the main function of miRNA; growing evidence indicates that miRNAs are abnormally expressed in various human cancers and they have significant effects on cancer progression and carcinogenesis [8–10]. As predicted by bioinformatic analyses, single miRNA has various targets; therefore, miRNAs are able to mediate numerous protein-coding genes and approximately one-third of human genes are regulated by miRNAs [11,12]. Among all miRNAs, down-regulated miR-145 has been observed in multiple types of cancers, including lung cancer [13], renal cell cancer [14], prostate cancer [15], bladder cancer [16], and colon cancer [17]. Moreover, miR-145 has been verified to inhibit tumor cell proliferation and invasion; therefore, it is considered as a presumptive tumor suppressor [18]. Studies also have confirmed that miR-145 plays an important tumor-suppressing role in ESCC [7,19–21]. The relationship between miR-145 and ESCC should be further studied.
CTGF is an immediate-early gene product of the CCN family and has certain appropriate expression levels under normal physiological conditions [22,23]. Imbalanced CTGF expression may trigger several pathological states, such as arthritis, fibrosis, and cancers [24]. Previous studies reported that CTGF as a downstream effector of the TGF-β pathway was over-expressed and indirectly exacerbated the cellular invasiveness and proliferation in ESCC [25,26]. Furthermore, connective tissue growth factor (CTGF) has been identified as a unique target of miR-145 in glioma cells [27], but such a relationship has not been confirmed between miR-145 and CTGF in ESCC.
Epithelial-mesenchymal transition (EMT) is a signal pathway that contains a key step involved in the evolution of tumor cell metastasis, including successive cell detaching, migrating, invading, dispersing, and residing [28]. EMT has been classified as a hallmark of tumor metastasis and it is associated with various transcriptional factors [29–31]. Tumor biology studies indicated that signaling pathways, including TGF-β, Notch, and Wnt, as well as growth factors such as FGF and PDGF may induce EMT [32]. Mechanism analysis suggested that CTGF can regulate EMT in several types of cancer [33,34].
Therefore, in the current study, the expression level of miR-145 and CTGF in ESCC and corresponding normal adjacent esophageal tissues were detected. We found that miR-145 can suppress cell proliferation, migration, invasion, and EMT process by directly targeting CTGF. The aims of this study were to further systematically clarify the potential function of miR-145 targeting CTGF in ESCC and promote the development of an effective diagnosis or therapeutic strategy for ESCC.
Material and Methods
Clinical specimens and cell lines
Fifty pairs of primary ESCC tissues and corresponding normal adjacent esophageal tissues were taken from patients (38 males and 12 females) undergoing esophagectomy at the Fifth Affiliated Hospital of Sun Yat-Sen University between January 2013 and July 2015. Ages of involved patients ranged from 47 to 76 years with a median age of 63. No patients underwent radiotherapy, chemotherapy, or hormone therapy before surgery. Tissue samples were immediately frozen in liquid nitrogen and stored at −80°C until the commencement of RNA extraction. Another part of tumor tissues was fixed with 10% formalin, embedded in paraffin, and analyzed using immunohistochemistry. Histological diagnoses of ESCC were conducted by a pathologist. Informed consent was obtained from all patients and the Research Ethics Committee of the Fifth Affiliated Hospital of Sun Yat-Sen University approved this study.
Human esophageal endothelial cell line HEEC was purchased from Sciencell Research Laboratories (Sciencell, USA) and cultured in Epithelial Cell Medium-2 (Sciencell) containing epithelial cell growth supplement-2 (ScienCell). All human ESCC cell lines used in this study, including EC1, Eca109, KYSE150, and KYSE180, were acquired from the Cell Center of the Shanghai Institute of Life Science (Chinese Academy of Science, Shanghai, China) and were cultured in RPMI1640 medium (Gibco, USA) containing 10% fetal bovine serum (FBS; Gibco), streptomycin (100 mg/ml; Gibco), and penicillin (100 units/ml; Gibco) at 37°C in an incubator with 5% CO2.
Cell transfection
Eca109 cells were transfected into 5 different groups: blank group, scramble group, miR-145 mimics group, miR-145 inhibitor group, and CTGF siRNA group. Eca109 cells in the blank group were transfected without any sequence. Eca109 cells in the other 4 groups were separately transfected with scramble miRNA mimics as the negative control, miR-145 mimics, miR-145 inhibitors, and CTGF siRNA, which were all synthesized from GenePharma (Shanghai, China). Transfection was carried out using Lipofectamine 2000 (Invitrogen, USA) and cells were cultured at 37°C in an incubator with 5% CO2. Complete medium was replaced after 6~8 h and then cell culture was continued. Cells were harvested for further experiments after 48-h transfection.
RNA extraction and RT-PCR
Total RNA extraction from human ESCC tissues and cells was conducted using TRIzol reagent kit (Invitrogen) strictly following the manufacturer’s instructions. Complementary DNA (cDNA) was acquired using the Omniscript reverse transcription kit (Qiagen, Germany). Real-time quantitative RT-PCR assay was conducted by ABI7500 quantitative PCR instrument (Applied Biosystems) for detecting the relative expression levels of miR-145 and CTGF mRNA. The primers of miR-145 and CTGF (purchased from Invitrogen) were set as: miR-145 forward, 5′-TCGGTCCAGTTTTCCCAG -3′ and reverse, 5′-AGTGCGTGTCGTGGAGTC-3′; CTGF sense, 5′-CCCAAGGACCCAAACCGTG-3′ and antisense, 5′-CTAATCATAGTTGGGTCTGGGC-3′. The relative expression quantity of miR-145 and CTGF mRNA were calculated using the 2−ΔΔCt method and they were normalized to the expression quantity of U6 snRNA.
Immunohistochemistry
CTGF protein expression in ESCC tissues was detected by the immunohistochemistry universal PV-9000 two-step method. In brief, formalin-fixed and paraffin-embedded tissues from 50 ESCC patients were cut into 4-μm slices. Then, tissue slices underwent conventional dewaxing, graded ethanol dehydration, antigen retrieval, and addition of 3% hydrogen peroxide to block endogenous peroxidase. Primary antibodies (mouse anti-human CTGF monoclonal antibody, Bioss, China) were applied to tissues slices at 4°C overnight. Then, tissues were incubated for 20 min at room temperature after the addition of polymerase adjuvants. Secondary antibody labeled with HRP (Bioss) was also applied and tissues were incubated for another 30 min at room temperature. Staining was performed by diaminobenzidine (DAB) and slices were counterstained using hemalum. Phosphate-buffered saline (PBS) instead of a primary antibody was caused as the negative control and a known positive antibody was set as the positive control. The integral calculation of CTGF complied with both the product of staining intensity (3 – brown; 2 – yellow; 1 – light yellow; 0 – colorless) and positive cell percentage (4, >75%; 3, 51–75%; 2, 26–50%; 1, 6–25%; 0, ≤5%). Cells were randomly selected from 5 high-power fields (× 400) in each slice and 100 cells were counted in each field. As suggested by the double types of scores, the integral levels of CTGF were evaluated as: negative (−), 0–4 points; weakly positive (+), 5–8 points, and strongly positive (++), 9–12 points. Two independent pathologists were responsible for analyzing the slices.
Dual-luciferase reporter assay
MiRNA targets were predicted using the TargetScan system (https://www.targetscan.org). We constructed luciferase reporter vectors for the wild-type and mutant-type of CTGF 3′ untranslated regions (UTR). Either miR-145 mimics or control were co-transfected with the constructed wild-type or mutant-type luciferase reporter vector into Eca109 cells using Lipofectamine 2000 (Invitrogen). The pGL3 control vector (Promega, USA) was transfected and served as the control. The luciferase activity was assessed using the Dual-Luciferase Reporter Assay System (Promega) after 48-h cell transfection.
MTT assay
Cell proliferation was evaluated by MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide] assays. Briefly, transfected cells washed twice using PBS were cultured until a density of 80% was reached. Cells were digested with trypsin into cell suspensions and then the number of cells was counted using a cell counter. Eca109 cells were inoculated into 96-well plates with 3×103~6×103 cells/well and a total of 6 wells were replicated. RPMI-1640 medium was added as a zero well in unseeded cell wells. Cells were detected after they were transfected for 24, 48, 72, and 96 h, respectively. MTT (20 μl, 5 mg/ml, Sigma) was placed into each well and the culture process was sustained for 4 h at 37°C in an incubator with 5% CO2. Subsequently, dimethyl sulfoxide (150 μl) was added into each well and cells were gently shaken for about 10 min to dissolve the crystals. Samples were detected using a microplate reader (SpectraMAX Plus, Molecular Devices, Sunnyvale, CA) at a wavelength of 490 nm.
Wound-healing assay
Cell migration status was detected by the wound-healing assay. Briefly, Eca109 cells were placed in 6-well plates with a density of 5×105 cells/well. When cells were cultured to the level of 90% confluence, monolayer cells were scratch-wounded using a sterilized 1000-μl pipette tip. Then cells were washed with PBS 3 times to remove the detached cells, then cells were cultured for another 48 h. Migration distances of cells were imaged and monitored using an inverted microscope (Carl Zeiss AG, Germany) with 3 randomly selected fields in the wounded region at 0 h, 24 h, and 48 h. The percentage of wound width was calculated as: the wound width at 0, 24, or 48 h/the original wound width measured at 0 h.
Transwell migration and invasion assays
Transwell invasion assay was performed using 24-well transwell plates (8-μm pores) and a coating of Matrigel (BD Biosciences, US) was introduced to measure the invasion status of Eca109 cells. Transwell migration assay was performed without the Matrigel coating. Briefly, cells were trypsinized and resuspended in serum-free medium after 48-h transfection and 2×104 cells were placed in the upper transwell chamber. Then, 0.5 ml complete RPMI 1640 medium was added in the lower chamber as the chemoattractant. After cells were incubated for 24 h at 37°C, non-migratory or non-invasive cells in the top chamber were removed with cotton swabs. Migrated or invaded cells at the bottom of the membrane were fixed with methanol and then stained with crystal violet. The number of migrated or invaded cells was counted to assess the migration and invasion status of cells.
Western blotting assay
The expression levels of CTGF, E-cadherin, N-cadherin, fibronectin, and vimentin were examined by Western blotting assay. Cellular proteins were extracted after 48-h transfection. The BCA approach was used to detect the protein density. An equal amount of proteins for each group was loaded, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto polyvinylidene fluoride membranes, and blocked with 5% skim milk. Membranes were incubated with primary antibodies (CTGF, E-cadherin, N-cadherin, fibronectin, and vimentin) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (CST, American), respectively, at 4°C overnight. Membranes were washed with TBST 3 times (10 min each) and incubated with horseradish-peroxidase-linked secondary antibodies at room temperature for 1 h. After that, membranes were washed again with TBST another 3 times (10 min each) and signal detection was carried out using Super ECL Plus Detection Reagent (Applygen Technologies Inc., China).
Statistical analysis
Statistical analyses were performed using SPSS 19.0 software. Significant differences in continuous data (mean ±SD) were estimated using the analysis of variance (ANOVA). Differences between 2 groups were analyzed by unpaired t tests. Results from the immunohistochemistry of CTGF protein were analyzed by the rank sum test. Two-sided P value <0.05 was defined as the cut-off value for evaluating statistical significance.
Results
MiR-145 was down-regulated in human ESCC cell lines and clinical specimens
Quantitative real-time PCR was used to evaluate the expression level of miR-145 in 50 ESCC tissues and matched normal adjacent esophageal tissues (Figure 1A) as well as in ESCC cell lines (Figure 1B). MiR-145 expression in ESCC tissues was significantly down-regulated compared to that in normal adjacent esophageal tissues (P<0.05). MiR-145 expression was also significantly down-regulated in ESCC cell lines in comparison to normal esophageal endothelial cell line HEEC (P<0.05), while the lowest level of miR-145 expression was observed in Eca109 cell lines. Therefore, Eca109 cell lines were used to perform subsequent experiments.
Figure 1.

The relative expression of miR-145 was detected by quantitative real-time PCR in ESCC clinical specimens and cell lines. (A) The relative expression of miR-145 in ESCC tissues and corresponding normal adjacent esophageal tissues; (B) The relative expression of miR-145 in human esophageal endothelial cell line HEEC and ESCC cell lines. The results were from 3 independent experiments. Data are expressed as mean ±SD. * P<0.05, compared with the control group.
CTGF expression in ESCC clinical specimens
The protein expression of CTGF in ESCC tissues and their corresponding normal adjacent esophageal tissues was detected by immunohistochemistry. Positively expressed CTGF was mainly located in cell cytoplasm with yellow granules (Figure 2). Positively expressed CTGF proteins in ESCC tissues significantly exceeded those in normal adjacent esophageal tissues (P<0.01) (Table 1).
Figure 2.

Representative immunohistochemistry images of clinical specimens detected by immunohistochemical universal PV-9000 two-step method (×400).
Table 1.
The expression of CTGF protein in ESCC and normal adjacent esophageal tissues.
| Groups | n | CTGF | Z-value | P-value | ||
|---|---|---|---|---|---|---|
| − | + | ++ | ||||
| ESCC | 50 | 10 | 16 | 24 | −6.773 | <0.01 |
| Normal | 50 | 42 | 8 | 0 | ||
ESCC – esophageal squamous cell carcinoma; P<0.01, compared with the normal group.
Targeting CTGF by miR-145
A putative conserved binding site of miR-145 at nucleotide position 31–37 of human CTGF 3′UTR was predicted by the TargetScan database. Perfect base pairing was observed between the seed sequence of mature miR-145 and the 3′UTR of CTGF mRNA (Figure 3A). Dual luciferase reporter gene assays revealed that miR-145 weakened the luciferase activity of CTGF wild-type by 55% (P<0.01). However, miR-145 did not significantly affect the luciferase activity of CTGF with mutant-type 3′UTR (Figure 3B). As suggested by RT-PCR and Western blot assays, CTGF mRNA and protein expressions were significantly inhibited in the miR-145 mimics group and stimulated in the miR-145 inhibitors group compares to the reference scramble group (P<0.05) (Figure 3C, 3D). Collectively, the above findings indicated that CTGF was a direct target of miR-145.
Figure 3.

CTGF is a target gene of miR-145. (A) Binding of miR-145 with CTGF 3′-UTR predicted by TargetScan; (B) Dual luciferase reporter gene assay revealed that miR-145 significantly decreased the luciferase activity of CTGF wild-type (wt) 3′UTR; (C) CTGF mRNA levels in Eca109 cells transfected with miR-145 mimics, miR-145 inhibitor or scramble sequence were examined by qRT-PCR; (D) CTGF proteins were detected by Western blotting using GAPDH as a loading control. The results were from 3 experiments. Data are expressed as mean±SD. ** P<0.01 vs. corresponding control; * P<0.05 vs. corresponding control.
MiR-145 targeted CTGF for suppressing ESCC cell proliferation
As suggested by MTT assays, significantly constrained proliferation of Eca109 cells was observed when cells were transfected with miR-145 mimics and CTGF siRNA for 48 h compared with the scramble group and blank group (all P<0.05). Furthermore, this trend appeared to become more significant over time. In contrast, no significant difference in the proliferation status of Eca109 cells was observed between the miR-145 mimics and CTGF siRNA group (P>0.05). No notable difference in cell survival ability was detected between the blank and scramble group at each time point (all P>0.05) (Figure 4).
Figure 4.
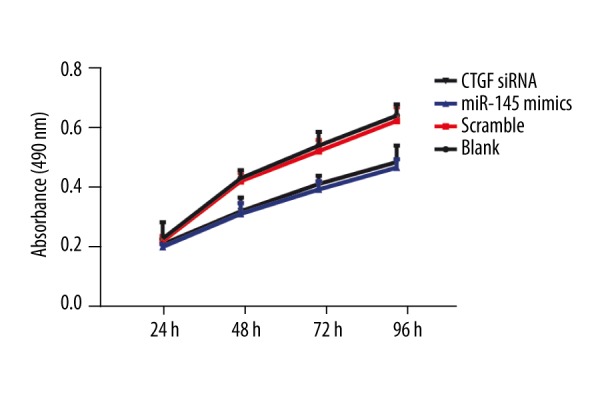
The proliferation of Eca109 cells was repressed by miR-145. A490 values of cells were measured by MTT assay. The results (mean ±SD) were from 6 independent experiments.
MiR-145 inhibited ESCC cell migration
As suggested by the transwell migration assay, there were significantly fewer migrated cells in the miR-145 mimics (65.5±5.8) and CTGF siRNA (69.3±4.6) group than in the blank (117.2±7.5) and scramble groups (112.6±6.2) (all P<0.05), while the difference between the miR-145 mimics group and CTGF siRNA group was insignificant (P>0.05) (Figure 5A). The wound healing assay also found that the percentage of wound width in the blank and scramble group were significantly less than those in the miR-145 mimics and CTGF siRNA group, both at 24 h and 48 h (all P<0.05) (Figure 5B). Therefore, up-regulation of miR-145 or down-regulation of CTGF can restrict the migration ability of Eca109 cells.
Figure 5.

The migration of Eca109 cells was suppressed by miR-145. (A) Transwell migration assay. (B) Wound healing assay. Photomicrographs were taken at 0, 24, and 48 h after cell wounding. The results (mean ±SD) were from 6 independent experiments. * P<0.05 vs. the blank and scramble group.
MiR-145 inhibited ESCC cell invasion
As shown in Figure 6, transwell matrigel invasion assay indicated that the number of invaded cells in the blank and scramble groups were 78.5±4.9 and 75.6±5.2, respectively, with no significant difference (P > 0.05). Invaded Eca109 cells transfected with miR-145 mimics (42.8±5.5) and CTGF siRNA (45.2±4.2) were notable fewer than those in the blank and scramble group (all P<0.05), while the difference between the miR-145 mimics and CTGF siRNA groups was insignificant (P>0.05). These data indicate that up-regulation of miR-145 or down-regulation of CTGF can suppress the invasive ability of Eca109 cells.
Figure 6.
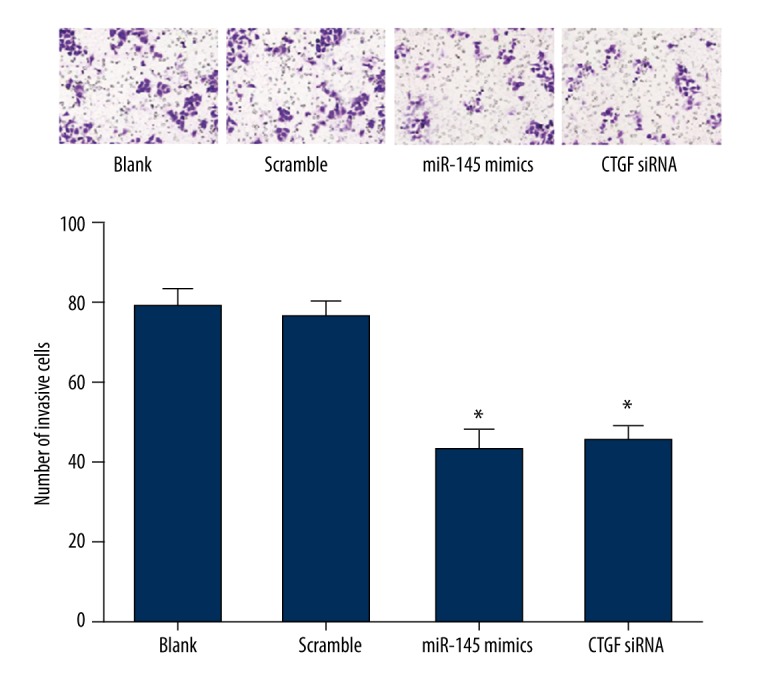
The invasion of Eca109 cells were inhibited by miR-145. The invasion ability of cells at 48 h after transfection was detected by a transwell invasion assay. The results (mean ±SD) were from 6 independent experiments. * P<0.05 vs. the blank and scramble group.
MiR-145 repressed EMT in ESCC cells
In order to understand how miR-145 and CTGF siRNA inhibited the migration and invasion of Eca109 cells, the protein expressions of EMT markers were examined via Western blot analysis. The results indicated that N-cadherin, which is a mesenchymal marker and supposed to be up-regulated during EMT, was decreased in Eca109 cells transfected with miR-145 mimics or CTGF siRNA when compared with the blank and scramble groups. Moreover, fibronectin and vimentin, another mesenchymal marker, was also down-regulated in Eca109 cells when transfected with miR-145 mimics or CTGF siRNA. However, E-cadherin, which is an epithelial marker and should be down-regulated during EMT, was promoted in Eca109 cells transfected with miR-145 mimics or CTGF siRNA when compared to the blank and scramble groups (Figure 7). These findings indicate that miR-145 repressed the migration and invasion of Eca109 cells through inhibiting the EMT process.
Figure 7.
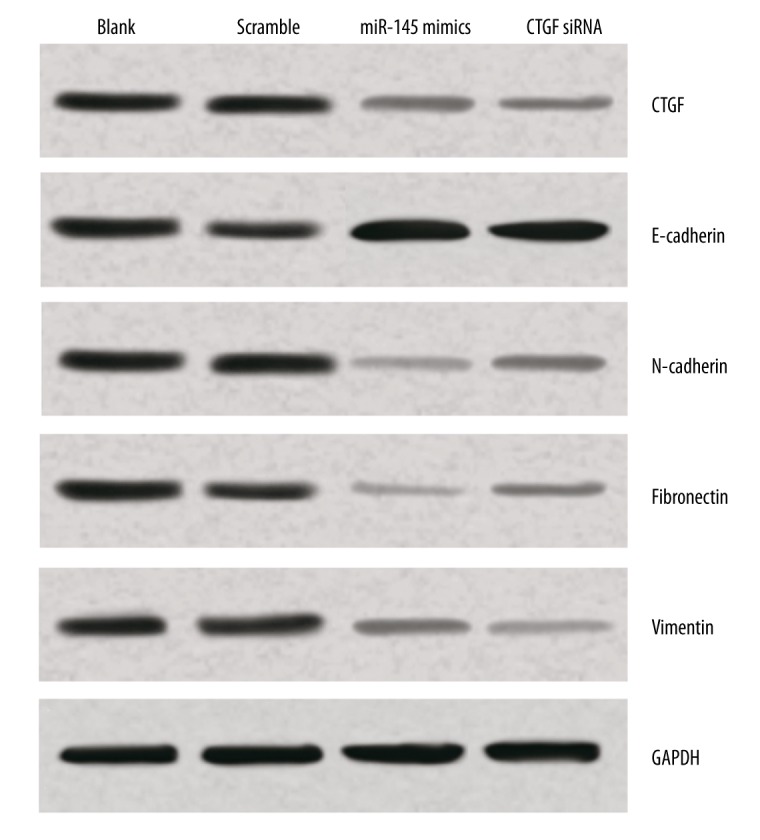
The effect of miR-145 on EMT by targeting CTGF in Eca109 cells. Results from Western blot analysis showed that over-expression of miR-145 and down-regulation of CTGF were associated with increased E-cadherin expression and supressed N-cadherin, fibronectin, and vimentin expression.
Discussion
Many studies have demonstrated that miRNAs regulate the progression of various tumors [35], and miR-145 plays a particularly crucial role in many cancers, including glioma [27], breast cancer [36], renal cell carcinoma [37], and prostate cancer [38]. Our results show that miR-145 is dramatically down-regulated in ESCC cell lines and ESCC tissues and these conclusions are similar to those from previous studies [7,39]. CTGF is a member of the CCN family and has been reported to participate in cell proliferation, adhesion, and tumor angiogenesis [22]. Several studies have reported that CTGF was not only up-regulated in ESCC, but also stimulated the development and progression of ESCC [25,26]. Results from immunohistochemistry confirmed that CTCF protein levels in ESCC tissues were significantly elevated compared to those in normal adjacent esophageal tissues (P<0.01).
Generally, miRNAs mediate a series of biological processes via different target sites and they also regulate the expression of their downstream targeting mRNAs [40,41]. Many studies demonstrated that miR-145 influence tumor development by mediating different downstream targeting mRNAs, including c-Myc in breast cancer [42] and human lung cancer [43], FSCN1 in prostate cancer [44], ANGPT2 and NEDD9 in renal cell carcinoma [37], and Sox2 in human choriocarcinoma cells [45]. In this study, we predicted that CTGF was a target gene of miR-145 using the TargetScan database. Furthermore, dual luciferase reporter gene assay enabled us to prove that miR-145 is able to directly bind with the 3′UTR of CTGF mRNA. Moreover, our study demonstrated that miR-145 mimics inhibited the mRNA and protein expression level of CTGF in Eca109 cells, and miR-145 inhibitors can significantly elevate CTGF expression in Eca109 cells. Thus, our results provide evidence that gene and transcription levels of CTGF are modulated by miR-145. Accordingly, Lee et al. reported that miR-145 regulates cell migration in glioma by targeting CTGF [27].
MiRNAs are known for their multifunctional roles in cell differentiation, apoptosis, proliferation, and metabolism [46,47]. To determine the regulatory roles of miR-145 that target CTGF in ESCC, Eca109 cells were separately transfected with miR-145 mimics and CTGF siRNA and changes in cellular proliferation, migration, and invasion were assessed. Our findings reveal that up-regulated miR-145 significantly inhibits the proliferation, migration, and invasion of Eca-109 cells. Consistent with our findings, it was reported that over-expression of miR-145 inhibits the proliferation and invasion of human choriocarcinoma [45] and prostate cancer [44]. In addition, we discovered that the proliferation, migration, and invasion of Eca-109 cells were restricted by the knockdown of CTCF. Deng et al. reported that knockdown of CTGF in ESCC cells significantly inhibit cell growth, colony formation, and tumorigenicity in vivo [26]. These results suggest that miR-145 and CTGF may have significant effects on ESCC pathogenesis.
EMT not only triggers the invasion and metastasis of tumors, but also endows cancer cells with immortalized proliferation [48]. Importantly, many studies have validated that miRNAs play a key role in tumor metastasis by regulating EMT. For instance, previous studies demonstrated that over-expression of miR-145 can repress EMT in both prostate cancer [38,49] and breast cancer [36]. Our experiments indicated that miR-145 mimics inhibit the expression of several mesenchymal markers, including N-cadherin, fibronectin, and vimentin, and stimulate the expression of E-cadherin, which is a typical epithelial cell maker. Additionally, siRNA-mediated knockdown of CTGF specifically suppressed N-cadherin, fibronectin, and vimentin expression, and this is associated with increased E-cadherin expression. Results from our study are consistent with previous reports concluding that CTGF contributes to changes in epithelial cells both in vivo and in vitro, and CTGF promotes EMT in neuroblastoma cells [50,51]. Hence, all of these results suggest that miR-145, which targets CTGF, inhibits ESCC cell migration and invasion through the suppression of EMT.
The present study has several imitations. Although we demonstrated the relationship between miR-145 and CTGF in ESCC, our study had a relatively small sample size due to limited resources. In addition, only the Eca109 cell line was used to perform experiments of cell transfection. The molecular mechanism of miR-145 and CTGF in ESCC progression needs further study.
Conclusions
In conclusion, miR-145 expression exhibited down-regulation in ESCC cell lines and ESCC tissues. Our experiments further demonstrated that both over-expression of miR-145 and knockdown of CTGF inhibited the proliferation, migration, and invasion of ESCC cells through their influence on EMT. Therefore, miR-145 and CTGF can potentially be considered as therapeutic targets or diagnostic biomarkers of ESCC.
Acknowledgements
We thank Dr Xian-Pin Yi and Dr Yu-Jing Lin (Department of Pathology, Fifth Affiliated Hospital of Sun Yat-Sen University), and Dr Ling-Ling Li (Central Lab, Fifth Affiliated Hospital of Sun Yat-Sen University) for their academic help.
Footnotes
Source of support: Departmental sources
References
- 1.Eslick GD. Epidemiology of esophageal cancer. Gastroenterol Clin North Am. 2009;38:17–25. vii. doi: 10.1016/j.gtc.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, et al. Global cancer statistics. Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Boonstra JJ, Kok TC, Wijnhoven BP, et al. Chemotherapy followed by surgery versus surgery alone in patients with resectable oesophageal squamous cell carcinoma: Long-term results of a randomized controlled trial. BMC Cancer. 2011;11:181. doi: 10.1186/1471-2407-11-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crehange G, Bonnetain F, Peignaux K, et al. Preoperative radiochemotherapy for resectable localised oesophageal cancer: A controversial strategy. Crit Rev Oncol Hematol. 2010;75:235–42. doi: 10.1016/j.critrevonc.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Gu J, Wang Y, Wu X. MicroRNA in the pathogenesis and prognosis of esophageal cancer. Curr Pharm Des. 2013;19:1292–300. doi: 10.2174/138161213804805775. [DOI] [PubMed] [Google Scholar]
- 6.Guo R, Li W, Liu B, et al. Resveratrol protects vascular smooth muscle cells against high glucose-induced oxidative stress and cell proliferation in vitro. Med Sci Monit Basic Res. 2014;20:82–92. doi: 10.12659/MSMBR.890858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang F, Xia J, Wang N, Zong H. miR-145 inhibits proliferation and invasion of esophageal squamous cell carcinoma in part by targeting c-Myc. Onkologie. 2013;36:754–58. doi: 10.1159/000356978. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 9.Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959–74. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- 10.Murray MJ, Saini HK, van Dongen S, et al. The two most common histological subtypes of malignant germ cell tumour are distinguished by global microRNA profiles, associated with differential transcription factor expression. Mol Cancer. 2010;9:290. doi: 10.1186/1476-4598-9-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Qu Y, Duan J, et al. Integrated analysis of the miRNA, gene and pathway regulatory network in gastric cancer. Oncol Rep. 2016;35:1135–46. doi: 10.3892/or.2015.4451. [DOI] [PubMed] [Google Scholar]
- 12.Xie X, Lu J, Kulbokas EJ, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–45. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Ma X, Zhang B, et al. Prognostic role of microRNA-210 in various carcinomas: A systematic review and meta-analysis. Dis Markers. 2014;2014:106197. doi: 10.1155/2014/106197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doberstein K, Steinmeyer N, Hartmetz AK, et al. MicroRNA-145 targets the metalloprotease ADAM17 and is suppressed in renal cell carcinoma patients. Neoplasia. 2013;15:218–30. doi: 10.1593/neo.121222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larne O, Hagman Z, Lilja H, et al. miR-145 suppress the androgen receptor in prostate cancer cells and correlates to prostate cancer prognosis. Carcinogenesis. 2015;36:858–66. doi: 10.1093/carcin/bgv063. [DOI] [PubMed] [Google Scholar]
- 16.Chiyomaru T, Enokida H, Tatarano S, et al. miR-145 and miR-133a function as tumour suppressors and directly regulate FSCN1 expression in bladder cancer. Br J Cancer. 2010;102:883–91. doi: 10.1038/sj.bjc.6605570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Guo H, Zhang H, et al. Putative tumor suppressor miR-145 inhibits colon cancer cell growth by targeting oncogene Friend leukemia virus integration 1 gene. Cancer. 2011;117:86–95. doi: 10.1002/cncr.25522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sachdeva M, Zhu S, Wu F, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA. 2009;106:3207–12. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derouet MF, Liu G, Darling GE. MiR-145 expression accelerates esophageal adenocarcinoma progression by enhancing cell invasion and anoikis resistance. PLoS One. 2014;9:e115589. doi: 10.1371/journal.pone.0115589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabrizi M, Khalili M, Vasei M, et al. Evaluating the miR-302b and miR-145 expression in formalin-fixed paraffin-embedded samples of esophageal squamous cell carcinoma. Arch Iran Med. 2015;18:173–78. [PubMed] [Google Scholar]
- 21.Kano M, Seki N, Kikkawa N, et al. miR-145, miR-133a and miR-133b: Tumor-suppressive miRNAs target FSCN1 in esophageal squamous cell carcinoma. Int J Cancer. 2010;127:2804–14. doi: 10.1002/ijc.25284. [DOI] [PubMed] [Google Scholar]
- 22.Holbourn KP, Acharya KR, Perbal B. The CCN family of proteins: Structure-function relationships. Trends Biochem Sci. 2008;33:461–73. doi: 10.1016/j.tibs.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brigstock DR. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: Lessons from transgenic animals. J Cell Commun Signal. 2010;4:1–4. doi: 10.1007/s12079-009-0071-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19:133–44. doi: 10.1016/j.cytogfr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Xie JJ, Xu LY, Wu JY, et al. Involvement of CYR61 and CTGF in the fascin-mediated proliferation and invasiveness of esophageal squamous cell carcinomas cells. Am J Pathol. 2010;176:939–51. doi: 10.2353/ajpath.2010.090118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng YZ, Chen PP, Wang Y, et al. Connective tissue growth factor is overexpressed in esophageal squamous cell carcinoma and promotes tumorigenicity through beta-catenin-T-cell factor/Lef signaling. J Biol Chem. 2007;282:36571–81. doi: 10.1074/jbc.M704141200. [DOI] [PubMed] [Google Scholar]
- 27.Moon MH, Jeong JK, Lee YJ, et al. SIRT1, a class III histone deacetylase, regulates TNF-alpha-induced inflammation in human chondrocytes. Osteoarthritis Cartilage. 2013;21:470–80. doi: 10.1016/j.joca.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 28.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–28. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15:169–90. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radisky ES, Radisky DC. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:201–12. doi: 10.1007/s10911-010-9177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai Z, Wang Q, Zhou Y, et al. Epidermal growth factor-induced epithelial-mesenchymal transition in human esophageal carcinoma cells – a model for the study of metastasis. Cancer Lett. 2010;296:88–95. doi: 10.1016/j.canlet.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 32.Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512–20. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tao L, Chen Y, Kong J, et al. Lack of association between the promoter -667T>C (rs2233679) polymorphism and cancer risk: Evidence from meta-analysis. Biomed Rep. 2014;2:223–28. doi: 10.3892/br.2014.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobson A, Cunningham JL. Connective tissue growth factor in tumor pathogenesis. Fibrogenesis Tissue Repair. 2012;5:S8. doi: 10.1186/1755-1536-5-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 36.Hu J, Guo H, Li H, et al. MiR-145 regulates epithelial to mesenchymal transition of breast cancer cells by targeting Oct4. PLoS One. 2012;7:e45965. doi: 10.1371/journal.pone.0045965. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Lu R, Ji Z, Li X, et al. miR-145 functions as tumor suppressor and targets two oncogenes, ANGPT2 and NEDD9, in renal cell carcinoma. J Cancer Res Clin Oncol. 2014;140:387–97. doi: 10.1007/s00432-013-1577-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren D, Wang M, Guo W, et al. Double-negative feedback loop between ZEB2 and miR-145 regulates epithelial-mesenchymal transition and stem cell properties in prostate cancer cells. Cell Tissue Res. 2014;358:763–78. doi: 10.1007/s00441-014-2001-y. [DOI] [PubMed] [Google Scholar]
- 39.Wu BL, Xu LY, Du ZP, et al. MiRNA profile in esophageal squamous cell carcinoma: Down regulation of miR-143 and miR-145. World J Gastroenterol. 2011;17:79–88. doi: 10.3748/wjg.v17.i1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritchie W, Rasko JE, Flamant S. MicroRNA target prediction and validation. Adv Exp Med Biol. 2013;774:39–53. doi: 10.1007/978-94-007-5590-1_3. [DOI] [PubMed] [Google Scholar]
- 41.Zheng L, Zhang Y, Liu Y, et al. MiR-106b induces cell radioresistance via the PTEN/PI3K/AKT pathways and p21 in colorectal cancer. J Transl Med. 2015;13:252. doi: 10.1186/s12967-015-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim SJ, Oh JS, Shin JY, et al. Development of microRNA-145 for therapeutic application in breast cancer. J Control Release. 2011;155:427–34. doi: 10.1016/j.jconrel.2011.06.026. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, Zeng H, Guo Y, et al. miRNA-145 inhibits non-small cell lung cancer cell proliferation by targeting c-Myc. J Exp Clin Cancer Res. 2010;29:151. doi: 10.1186/1756-9966-29-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuse M, Nohata N, Kojima S, et al. Restoration of miR-145 expression suppresses cell proliferation, migration and invasion in prostate cancer by targeting FSCN1. Int J Oncol. 2011;38:1093–101. doi: 10.3892/ijo.2011.919. [DOI] [PubMed] [Google Scholar]
- 45.Xu F, Wang H, Zhang X, et al. Cell proliferation and invasion ability of human choriocarcinoma cells lessened due to inhibition of Sox2 expression by microRNA-145. Exp Ther Med. 2013;5:77–84. doi: 10.3892/etm.2012.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J. 2011;278:1598–609. doi: 10.1111/j.1742-4658.2011.08089.x. [DOI] [PubMed] [Google Scholar]
- 47.Huang Y, Shen XJ, Zou Q, et al. Biological functions of microRNAs: A review. J Physiol Biochem. 2011;67:129–39. doi: 10.1007/s13105-010-0050-6. [DOI] [PubMed] [Google Scholar]
- 48.Berx G, Raspe E, Christofori G, et al. Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin Exp Metastasis. 2007;24:587–97. doi: 10.1007/s10585-007-9114-6. [DOI] [PubMed] [Google Scholar]
- 49.Guo W, Ren D, Chen X, et al. HEF1 promotes epithelial mesenchymal transition and bone invasion in prostate cancer under the regulation of microRNA-145. J Cell Biochem. 2013;114:1606–15. doi: 10.1002/jcb.24502. [DOI] [PubMed] [Google Scholar]
- 50.Sonnylal S, Xu S, Jones H, et al. Connective tissue growth factor causes EMT-like cell fate changes in vivo and in vitro. J Cell Sci. 2013;126:2164–75. doi: 10.1242/jcs.111302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Q, Xu Z, An Q, et al. TAZ promotes epithelial to mesenchymal transition via the upregulation of connective tissue growth factor expression in neuroblastoma cells. Mol Med Rep. 2015;11:982–88. doi: 10.3892/mmr.2014.2818. [DOI] [PMC free article] [PubMed] [Google Scholar]