

Summary
Many infections are caused by pathogens that are similar, but not identical, to previously encountered viruses, bacteria, or vaccines. In such re-infections, pathogens introduce known antigens, which are recognized by memory T cells and new antigens that activate naive T cells. How preexisting memory T cells impact the repertoire of T cells responding to new antigens is still largely unknown. We demonstrate that even a minimum epitope overlap between infections strongly increases the activation threshold and narrows the diversity of T cells recruited in response to new antigens. Thus, minimal cross-reactivity between infections can significantly impact the outcome of a subsequent immune response. Interestingly, we found that non-transferrable memory T cells are most effective in raising the activation threshold. Our findings have implications for designing vaccines and suggest that vaccines meant to target low-affinity T cells are less effective when they contain a strong CD8 T cell epitope that has previously been encountered.
Keywords: T cell activation threshold, cytotoxic T cells, secondary immune responses, strength of TCR stimulation
Graphical Abstract
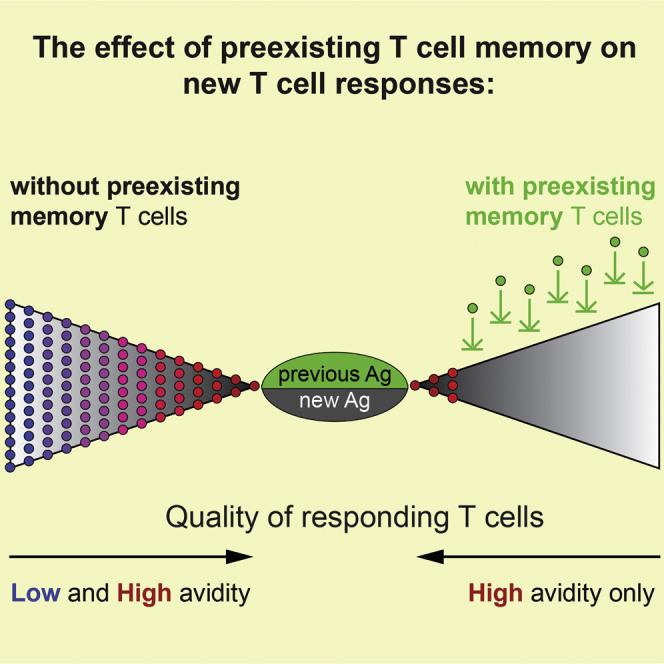
Highlights
-
•
The T cell activation threshold differs between naive and immune individuals
-
•
A minimum epitope overlap between infections causes a threshold change
-
•
Shared epitopes particularly interfere with generation of low-affinity T cell responses
-
•
The threshold is more effectively raised by endogenous than transferred T cells
Oberle et al. show that even minimal antigen overlap between infections diminishes the breadth of the T cell repertoire and increases the T cell activation threshold. We provide evidence for the type of memory T cell population that mediates this threshold change, and we discuss the implications of our findings for vaccine design.
Introduction
Throughout life, we are constantly challenged by pathogens, and re-exposure to previously encountered viruses or bacteria happens frequently. Homologous re-infections are often effectively controlled by neutralizing antibodies, but pathogens that express altered serological epitopes (heterosubtypic re-infection) can bypass antibody-mediated immunity and cause a secondary infection (Corti and Lanzavecchia, 2013). The immunological conditions found in such infections differ in many respects from primary infections. Right at the beginning of such a re-infection, the T cell repertoire may already contain large numbers of pathogen-specific T cells, and some of these can immediately exert effector function (Zhang and Bevan, 2011). Moreover, preexisting pathogen-specific but insufficiently neutralizing antibodies could strongly alter infection and replication kinetics (Beura et al., 2016) of a pathogen, and this may significantly alter tissue tropism (Rothman, 2011). Such changes in host immunity are not limited to the adaptive immune response. It has been shown that cells of the innate arm of the immune system can respond more vigorously (Sun et al., 2009), and memory CD8 T cells can even contribute to controlling pathogen load in the early phase of infection in a non-cognate, innate-like fashion (Chu et al., 2013). Thus, pathogen spreading, antigen presentation, T cell activation kinetics, and levels of inflammation and tissue destruction can significantly vary between primary and secondary infections.
A particular feature of heterosubtypic re-infections is that many antigens are shared in the primary and secondary infection, but the second pathogen expresses epitopes, which are new to the immune system. Such a situation of preexisting partial immunity to a pathogen can also occur in a primary infection when a pathogen happens to share an epitope with a previously encountered unrelated pathogen—a phenomenon known as heterologous immunity (Welsh et al., 2010, Che et al., 2015). Though highly relevant for understanding immunity in humans, interferences between past and present infections (or vaccinations) are rarely investigated under well-defined conditions. Thus, we still have very limited insight into how existing immunity impacts T cell activation, expansion, and conversion into effector and memory T cells. Similarly, pre-existing partial immunity can significantly alter the outcome of vaccination (Frahm et al., 2012). It is also particularly important to consider that pre-existing immunity does not inevitably result in better immune protection. Instead, certain re-infections are known to be accompanied by enhanced pathology, as occurs when a Dengue-virus-immune individual is exposed to a different virus serovar (Rothman, 2011). Similarly, certain vaccination trials are suspected to have augmented the severity of subsequent infections (Blanco et al., 2010, Moore et al., 2008). The mechanisms underlying such disease enhancement are not clearly resolved, and it is not known how frequently this may occur. All of these points strongly underline the need to investigate immune responses in immune or partially immune individuals.
Here, we utilize well-controlled experimental systems to dissect and characterize how different levels of pre-existing immunity influence T cell responses. We observed that T cell activation thresholds differ substantially between primary and secondary infections. We report that only the highest-affinity ligands induce T cell expansion in heterosubtypic re-infections, whereas minor reductions in the levels of TCR stimulation fail to induce T cell expansion. Importantly, we show that neither a shortened inflammatory response nor a globally altered antigen presentation pattern are responsible for raising the threshold. Instead, we found that even a single shared epitope recognized by previously formed memory CD8+ T cells is sufficient to increase the T cell activation threshold of naive T cells. We noted that this elevated threshold is mediated by a non-transferrable endogenous memory T cell population. Together our data provide relevant insights into T cell differentiation mechanisms in secondary heterosubtypic infections and for the design of vaccines that are given multiple times.
Results
Only Very High-Affinity T Cells Expand in Secondary Infections
We previously reported that low-affinity T cells differentiate into effector and memory cells in primary infections (Zehn et al., 2009). We established this using recombinant Listeria strains, which express full-length Ovalbumin [Ova] that contains either the original high-affinity, H-2Kb-restricted OT-1 ligand SIINFEKL (Ova357–364) or different altered peptide ligands (APLs) A2 > Y3 > Q4 > T4 APL (listed in decreasing OT-1 stimulatory potency). We then became interested in addressing the stimulation requirements in secondary heterosubtypic re-infections. To do so, we infected naive or wild-type Listeria (Lm-WT)-experienced mice with Lm-Ova- or APL-expressing Listeria (see Figure 1A). With this experimental setup, we mimic a frequently occurring situation where an individual is immune to some but not all antigens in a secondary infection. We observed that the high-affinity OT-1 ligand N4 induces similar expansion in mice with or without a previous Lm-WT immunization. In contrast, OT-1 expansion in response to Listeria expressing the slightly weaker SAINFEKL (A2) or SIYNFEKL (Y3) APL was decreased in Lm-WT immune mice (Figure 1B) and significantly impaired in response to Q4 or T4. Similar exclusions of low-affinity T cells were observed when we transferred OT-1 T cells before the primary Lm-WT infection (Figure S1A) and when memory OT-1 T cells were used instead of naive OT-1 T cells (Figure S1B). Notably, both N4 and T4 induce similar carboxyfluorescein succinimidyl ester (CFSE) dilution in primary infections, but only N4 induces robust proliferation in immune mice (Figure 1C). Some OT-1 T cells proliferate in response to T4 in Listeria-immune mice, but this did not increase total OT-1 T cells (data not shown). We therefore conclude that low-affinity stimulation leads to an abortive T cell response in heterosubtypic re-infections.
Figure 1.

Only High-Affinity T Cells Expand in Listeria-Immune Mice
(A) Schematic illustration of the Experimental Procedures. Naive controls and C57BL/6 mice infected with wild-type Listeria (Lm-WT) received 104 naive OT-1 4 weeks later and the indicated Listeria strains.
(B) Frequency of OT-1 T cells among peripheral blood CD8+ T cells 6 days after infection. Numbers indicate the ratio of OT-1 T cells in naive and Lm-WT immune hosts.
(C) Naive or Lm-WT immune mice grafted with 2.5 × 105 CFSE-labeled OT-1 cells and infected with Lm-N4 or Lm-T4. CFSE dilution among splenic OT-1 (filled histogram) was measured 78 hr later. Dashed lines are unlabeled host CD8+ T cells. Data are representative of three (B) or two (C) independent experiments with three to five mice per group.
(D) Naive or Lm-WT infected Rip-mOva mice were 4 weeks later engrafted with low-affinity H-2Kb/Ova-specific OT-3 TCR transgenic T cells and then infected with Lm-N4. Blood glucose levels were determined 8 days later. Statistical analysis are unpaired Student’s t tests with ∗∗p < 0.01 and ∗∗∗∗p < 0.0001.
To illustrate the relevance of our observation, we transferred low-affinity H-2Kb/Ova-specific OT-3 TCR transgenic T cells (Enouz et al., 2012) into naive or Lm-WT immune Rip-mOva mice. The Lm-WT immune mice were then infected with the same challenge as in Figure 1A, whereas the naive Rip-mOva mice received a priming dose. Using this setup, only naive mice showed diabetic blood glucose levels above 300 mg/dL (Figure 1D).
T Cell Activation Threshold in Primary Infections
Next, we wanted to define the difference in the affinity range of T cells responding in primary versus heterosubtypic infections. We previously showed that the low-affinity V4 ligand activates OT-1 T cells (Zehn et al., 2009), even though it has a ∼1,000-fold lower EC50 than the N4 ligand (Figure 2A). Interestingly, the D4 APL has an EC50 value that is 10,000-fold below N4. To test the OT-1 T cell response to this very low-affinity ligand, we generated a recombinant Lm-D4 strain. We confirmed that the Lm-D4 strain is infectious and comparable to Lm-N4 (Figure S2A). Unexpectedly, even Lm-D4 induced rapid initial OT-1 proliferation (Figure 2B). D4 activated OT-1 T cells secrete TNF and IFNγ (Figure 2C) and produce granzyme B (Figure 2D), and they differentiate into memory T cells that can undergo secondary expansion (Figure 2E). We illustrated this by transferring OT-1 cells into mice that were either infected with Lm-D4 or with Lm-WT. Four weeks later, we challenged the mice with Vesicular stomatits virus expressing Ova (VSV-Ova). We noticed higher numbers of OT-1 cells in mice primed by D4 in comparison to Lm-WT-primed mice that do not stimulate OT-1 T cells (Figure 2E). Thus, the ability of D4 to activate OT-1 T cells indicated that the signal 1 (TCR stimulation) activation requirement in primary infections is very low.
Figure 2.

Even Extremely Low-Affinity Stimulation Induces Normal Activation and Memory Generation
(A) Dose response curves graphing peptide concentration against the fraction of maximum numbers of IFNγ producing T cells were obtained by briefly stimulating an OT-1 T cell line in vitro with the indicated peptides. Note that only a fraction of the OT-1 T cells produced IFNγ after very low-affinity ligand stimulation. Data were therefore normalized to better compare the EC50.
(B) C57BL/6 mice transferred with 2 × 105 CFSE-labeled OT-1 cells and infected the indicated Listeria strains were analyzed for splenic CFSE dilution at 76 hr post infection.
(C and D) Naive mice received 105 OT-1 T cells and an Lm-D4 or Lm-N4 infection. Splenocytes were 4.5 days later and briefly re-stimulated with SIINFEKL peptide and stained intracellularly for IFNγ, TNF (C), and GrzB (D). (C) shows representative OT-1 and (D) CD8+ gated flow cytometry plots.
(E) Naive mice received 2 × 104 OT-1 and the indicated Listeria strains and OT-1 frequency among peripheral blood CD8+ T cells was determined 28 days later. The mice were then infected with VSV-N4, and the OT-1 frequency was measured 4 days later in the blood.
(F) CFSE+ OT-1-containing mice were infected with Lm-N4, Lm-Cpα1, Lm-βcat, or Lm-E1 and analyzed as described in (B).
The data are representative for four (A) or three independent experiments with three to five mice per group (B, C, and D) or two experiments with two to five mice per group (E and F). Statistical analysis are unpaired Student’s t tests, ∗∗∗∗p < 0.0001.
In contrast, the difference between the normal N4 and the A2 or Y3 APL is rather small (Figure 2A). Nonetheless, this level already strongly impairs T cell expansion in secondary or cross-reactive infections. We therefore conclude that there is a substantial stimulation threshold difference between primary and secondary infections. To further illustrate the significance of this difference, we marked the known boundaries for ligands which induce positive and negative selection of OT-1 T cells in Figure 2A, whereby T4 is the threshold ligand between positive and negative selection (Figure 2A) (Daniels et al., 2006).
G4 is known to induce positive selection in fetal thymic organ cultures (Yachi et al., 2006). Given that D4 with a potency much below G4 supports effector T cell differentiation, we wondered whether even the presumed endogenous positive selecting ligands for OT-1, ISFKFDHL from the F-Actin capping protein (cpα1) and RTYTYEKL from β-catenin (Hogquist et al., 1997, Santori et al., 2002) would drive OT-1 expansion when expressed by Listeria, but this was not the case (Figure 2F). We confirmed that the lack of an OT-1 response is not due to a lack of bacterial replication and that an Lm-βcat infection is, in this respect, comparable to an Lm-N4 infection (Figure S2A). E1 Ova (EIINFEKL) is the only known APL that supports OT-1-positive selection in vivo (Stefanski et al., 2001). The lack of a peripheral response to E1 (Figure 2F) suggests that it more closely resembles the quality of ligands (cpα1 and β-catenin) that are thought to naturally support positive selection.
A Minimum Epitope Overlap Augments the CD8+ T Cell Activation Threshold
Given the activation threshold differences in primary versus heterosubtypic infections, we sought to define the responsible mechanism. Depletion of CD4+ T cells and NK cells from Lm-WT primed mice did not restore low-affinity T cell activation (data not shown). Taking a possible role of Listeria-specific memory CD8+ T cells into consideration, we set up a system that allowed us to control the size of the antigen-specific memory population by sharing only one CD8+ T cell epitope between two consecutive infections. We constructed Listeria strains that express N4 or T4 plus the Lymphocytic choriomeningitis virus (LCMV)-derived gp33-41 epitope. We found that Lm-gp33-N4 and Lm-gp33-T4 have similar in vivo growth rates (Figure S2A) and induce similarly sized populations of endogenous gp33-specific T cells (data not shown and Figures S2B and S2C). When OT-1 T cells are primed by these Listeria strains in naive or LCMV immune hosts (Figure 3A), we observed a 2-fold reduction in OT-1 expansion to N4 and an essentially non-detectable response to T4 in LCMV immune mice (Figure 3B). This indicates that pre-existing antigen-specific memory CD8+ T cells effectively raise the T cell activation threshold and that only one epitope (or two if one also considers the H-2Kb-restricted gp34 epitope) (Hudrisier et al., 1997) is sufficient to alter the T cell response.
Figure 3.

Memory CD8+ T Cells Raise the Activation Threshold in Secondary Infections Independently of the Inflammatory Milieu
(A and B) Control (–) or LCMV-immune mice received 4 weeks after the infection 104 OT-1 and Listeria co-expressing the LCMV gp33 peptide and N4 Ova (Lm-gp33-N4) or gp33 and T4 Ova (Lm-gp33-T4). Note that only the gp33 and gp34 epitopes are shared between the consecutive infections. Frequency of blood OT-1 T cells among total CD8+ T cells was measured 6 days later. Numbers indicate the OT-1 frequency fold change between naive and immune hosts.
(C and D) 3 × 105 naive CFSE-labeled OT-1 cells were transferred at different time points into ongoing primary Lm-N4 or secondary Lm-WT and Lm-N4 infections in order to assess antigen presentation kinetics. CFSE dilution was measured 80 hr after the transfer.
(E and F) Naive and LCMV-immune mice were infected with 2,000 or 2 × 105 CFU Lm-gp33-T4. Another LCMV-immune group was co-infected with 2 × 105 CFU Lm-gp33-T4 plus 2,000 CFU Lm-WT. Splenic Listeria load was determined 4 days later (E), and the OT-1 frequency among total blood CD8+ T cells was determined 6 days later (F).
(G) Naive (–) or LCMV-immune (LCMV) mice received 104 OT-1 and 106 CFU ActA-deficient Listeria (ActA−gp33-N4 or ActA−gp33-T4). The OT-1 frequency among total blood CD8+ T cells was determined 6 days later. Numbers indicate the OT-1 ratio in naive and immune hosts.
The data are representative for two (A–F) or one (G) independent experiments with n = 3–5 per group. Statistical analysis are unpaired Student’s t tests with ∗p < 0.1, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Changes in Inflammation or Duration of Antigen Presentation Are Not Responsible for Raising the Activation Threshold
We considered that rapid Listeria clearance in immune hosts might shorten the duration of antigen-presentation and lower the magnitude of concomitant inflammation (Zehn et al., 2014, Prlic et al., 2006). To address this point, we transferred CFSE-labeled OT-1 T cells into naive or Lm-WT-primed mice and challenged them with Lm-N4. We observed similar CFSE dilution in naive and immune hosts excluding major differences in antigen presentation kinetics in the two hosts (Figures 3C and 3D). Also anti-CD40-mediated DC maturation or administration of recombinant IFNα to increase inflammation left the T4 response unchanged (data not shown). Next, we established comparable levels of Listeria induced inflammation and tissue destruction in LCMV immune and naive mice by co-injecting Lm-WT and Lm-gp33-T4 into LCMV immune mice. Here, only Lm-gp33-T4 are recognized by memory T cells, whereas the Lm-WT infection propagates normally. This resulted in similar absolute Listeria titers in LCMV immune and naive mice, but the response to T4 remained unchanged in immune mice (Figure 3E). To control for an impact of the different Listeria challenge doses in naive or immune mice (Figures 1, 3A, and 3B), we used ActA-deficient Listeria. These strains cannot spread from cell to cell and are rapidly cleared by the host, allowing C57BL/6 mice to be challenged with a high Listeria dose. This creates a setup in which antigen levels are primarily dependent on the amount of the initial bacterial challenge dose, but not the in vivo expansion, of injected bacteria. We observed a lack of low-affinity T cell expansion in immune mice challenged with ActA-deficient Lm (Figure 3G). This excludes a principle shortage in antigen presentation and a lower inflammatory response as causes of the observed effect.
Transferred T Cells Are Less Effective in Raising the Activation Threshold than Endogenous T Cells
To determine whether the presence of memory T cells is sufficient to raise the activation threshold, we transferred gp33-specific memory T cells into new C57BL/6 hosts along with CD45.1/2 congenic OT-1 T cells. Contrasting our prior observation, the transferred memory P14 T cells reduced, but did not completely suppress, the OT-1 response to Lm-gp33-T4. In an Lm-gp33-N4 infection, the same P14 dose caused only a minor impact on the OT-1 response (Figure 4A). Similar outcomes were observed when P14 memory T cells were transferred 28 or 80 days after the LCMV infection (Figure S3A). We considered the possibility that the number of transferred P14 T cells was too low to suppress the T4 response effectively and therefore increased the number of transferred P14 to 106 cells. Again, the T4 response was incompletely blocked, but surprisingly, this P14 dose already caused a strong decline in the high-affinity OT-1 response (Figure 4B). These data suggest that transferring memory T cells is not sufficient to recapitulate the selective and complete exclusion of low-affinity T cells, as observed when endogenous memory T cells respond to a pathogen expressing a shared antigen (Figure 1).
Figure 4.

Endogenous Memory CD8+ T Cells Effectively Raise the Activation Threshold
(A and B) All mice received 104 OT-1 T cells and some 3 × 105 (A) or 106 (B) memory P14 T cells. The OT-1 frequency among total CD8+ T cells was determined 7 days after infecting the mice with Lm-gp33-N4 or Lm-gp33-T4. Data are representative for two independent experiments with n = 5 mice per group.
(C–E) CD45.1 P14 memory T cells were obtained from mice infected 30–90 days earlier with LCMV and transferred into naive mice. Shown are memory P14 T cell populations before and after the transfer. Bars represent individual mice, and the relative distribution of the indicated P14 subsets. The plots on the right show the frequency of KLRG1+ (C) and CD27+CD43+ subpopulations (D). Dots represent individual mice.
(E) Mice with endogenous memory cells were i.v. injected with fluorescently labeled anti-CD8 antibody and sacrificed 3 min. later. CD127/KLRG1 and CD27/CD43 expression status on antibody-positive and -negative cells are shown as indicated in (D). Data are representative for two to four independent experiments with n = 3–5 mice per group. Statistical analyses are unpaired Student’s t tests with ∗p < 0.1, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
To exclude a sole OT-1-dependent phenomenon, we reversed the strategy and used the recently described gp33C6 low-affinity APL for P14 T cells (Utzschneider et al., 2016). We transferred OT-1 memory and naive P14 T cells into naive B6 mice and infected them with Listeria expressing Ova and wild-type (Lm-gp33-N4) or the low-affinity APL gp33C6 (Lm-gp33C6-N4). Again, the memory OT-1 had a minor impact on the P14 response to high-affinity ligands and a much stronger impact on the lower-affinity gp33C6 response (Figure S3B). These data also show that a weak or strong response of de novo primed naive OT-1 or P14 T cells does not impact the response of memory P14 (Figure S3A) or OT-1 T cells (Figure S3B).
We also examined the phenotype of P14 T cells before (endogenous situation) and after the transfer and noticed a larger population of KLRG1+ T cells among endogenous compared to transferred memory T cells and a higher proportion of CD43—CD27— cells (Figures 4C and 4D). Both populations are thought to have immediate effector function (Olson et al., 2013). Moreover, Listeria monocytogenes enters the spleen via the bloodstream and in a dendritic cell dependent fashion (Neuenhahn et al., 2006). At this stage, they will likely be in contact with effector/memory T cells that are positioned in the spleen in close proximity to the bloodstream. By injecting fluorescently labeled anti-CD8 antibody into mice, we marked the fraction of P14 T cells that are in close contact with blood (Galkina et al., 2005). Interestingly, but in line with an earlier publication (Olson et al., 2013), we saw that the KLRG1+ or CD43—CD27— T cells (Figure 4E), which show a low engraftment efficacy in our memory transfers, were enriched among the P14 cells with access to the blood stream. Therefore, we favor the conclusion that a form of tissue-resident T cell population is more effective than transferred T cells in raising the activation threshold in heterosubtypic infections.
Discussion
We show a clear difference in the TCR stimulation threshold between primary and secondary infections. Only very high-affinity ligands activate T cells in immune mice, whereas ligands such as A2 and Y3, whose EC50 for inducing a half-maximum IFNγ response differed only 2- to 4-fold from the wild-type N4 peptide (Figure 2A), trigger a much weaker response in immune compared to naive mice. This is in contrast to the situation in naive mice in which even ligands with 10,000-fold lower EC50 activate OT-1 T cells (Figure 2A). Our observations help to understand T cell responses in heterosubtypic re-infections caused for instance by different influenza strains or dengue virus serotypes. Serial infections with the distantly related Lymphocytic choriomeningitis virus and Pichinde virus were shown to induce a narrowed pathogen-specific T cell repertoire because of antigens that are shared between these infections (Cornberg et al., 2006, Welsh et al., 2010). Our data indicate that this exclusion is caused by a memory T-cell-mediated elevation of the stimulation threshold and that even minimum cross-reactivity is sufficient to cause this effect.
The exclusion of low-affinity T cells by previously established memory T cells has consequences for designing vaccine strategies that rely on a prime-boost regimen and intend to elicit a broad T cell repertoire (such as HIV vaccine trials). Several examples underline that optimum protection against pathogens is mediated by highly diverse T cell populations. This ensures cross-reactivity to pathogens expressing mutated epitopes, and it enables individuals to better handle a heterosubtypic infection with a related but non-identical pathogen. It also prevents the selection of escape variants (Price et al., 2004, Meyer-Olson et al., 2004, Cornberg et al., 2006). T cell receptor diversity in the population of antigen-specific T cells is largely facilitated by expanding T cells, which suboptimally respond to antigen (low-affinity T cells) (Zehn et al., 2009). The focusing of the T cell repertoire as we observed it may be beneficial for the ongoing secondary infection, but the narrow repertoire may severely diminish the ability to subsequently cross-react to related antigens.
Similarly, tumor immune responses rely on recruiting T cells of intermediate or low affinity given that high-affinity T cells are often eliminated by tolerance mechanisms (Enouz et al., 2012). Our data imply that vaccination strategies, which involve repetitive injections, will perform suboptimally if the vaccine vector contains previously experienced epitopes. In addition, pre-existing immunity to vaccine vectors were shown to strongly diminish the efficacy of inducing T cell immunity. Specifically, adenovirus-seropositive vaccine recipients had lower HIV-specific responses to an adenovirus 5-vectored HIV vaccine in comparison to adenovirus-seronegative recipients (Frahm et al., 2012). In line with this previous study, our data show that a minimal epitope overlap is sufficient to impact not only the quantity but also the breadth of the recruited T cell repertoire. In contrast, memory T cells do not seem to impact the clonal-like expansion of antigen-specific NK cells (Johnson et al., 2016).
Our data suggest that populations of memory T cells, which are difficult to transfer, are more effective than transferrable memory populations in raising the stimulation threshold. These cells may impair the translocation of antigen-loaded DCs from the marginal zone into the T cell zone, and this barrier function may lead to altered antigen presentation and consequently a higher stimulation threshold. This is in line with a recent report that highlights the presence of a tissue-resident T cell population in the spleen (Schenkel et al., 2014). Nonetheless, it remains difficult to precisely distinguish between qualitative and quantitative causes for the observed differences between transferred and endogenous memory T cells. What supports the notion of a qualitative difference is that the transfer of a large number of memory P14 T cells, which already diminished a high-affinity response, did not resemble the effect seen with endogenous memory T cells.
Experimental Procedures
Mice
C57BL/6 mice were obtained from Charles River, and OT-1, Rip-mOva, and CD45.1 congenic C57BL/6 mice were obtained from Jackson Laboratories. P14 mice were provided by Annette Oxenius (ETH Zürich). Low-affinity OT-3 TCR transgenic mice were described previously (Enouz et al., 2012). Mice were bred in specific pathogen-free (SPF) facilities and infected in SPF or conventional facilities at the University of Lausanne. Mice that were at least 6 weeks old were used for experiments in compliance with the University of Lausanne institutional regulations, and the experiments were approved by the veterinarian authorities of the Swiss Canton Vaud.
Purification, Labeling, and Adoptive Transfer of Cells
Single-cell suspensions were obtained by mashing spleens through a 100 μm nylon cell strainer (BD Falcon). Red blood cells were lysed with hypotonic ammonium-chloride-potassium (ACK) lysis buffer. If cells were harvested less than 5 days after the infection, then the spleens were before mashing digested with 150 μg/mL DNase I (Roche) and 200 μg/mL Liberase TL (Roche) for 40 min at 37°C. The mouse CD8+ T cell enrichment kit (Miltenyi Biotech) was used for CD8 T cell enrichment. Memory P14 cells were isolated by using fluorescein isothiocyanate (FITC), phycoerythrin (PE), or biotin-conjugated CD45.1 antibodies followed by anti-FITC, anti-PE, or anti-biotin MicroBeads (Miltenyi Biotech) according to the manufacturer’s instructions. Cells were labeled in serum-free medium with 5 μM CFSE at 37°C for 10 min.
Infections
Mice were infected intraperitoneally (i.p.) with 2 × 105 plaque-forming units (PFU) of Lymphocytic choriomeningitis virus (LCMV strain 53b, Armstrong) grown and titrated on Vero cells (Utzschneider et al., 2013). Vesicular stomatitis virus expressing SIINFEKL (N4) (Kim et al., 1998) was grown and titrated on BHK cells. Mice were infected intravenously ( i.v.) with 2 × 106 PFU. Recombinant Listeria monocytogenes strains expressing ovalbumin that contain Kb/Ova-derived APL were previously described (Zehn et al., 2010). Listeria expressing the LCMV derived gp33–41 epitope were generated according to similar protocols. The previously described pPL2 vector (Lauer et al., 2002), which contains native Ovalbumin (AA134–387) or Ovalbumin with the SIITFEKL variant (Zehn et al., 2009), were digested with SapI. Oligonucleotides encoding KAVYNFATC (gp33) or KAVYNCATC (gp33C6) plus an alanine on both sides were ligated into the SapI restriction site. The constructs were inserted into wild-type or ActA-deficient 10403s Listeria by E. coli conjugation as previously described (Lauer et al., 2002). Listeria were grown in brain heart infusion broth (Oxoid, Thermo Fisher) to mid-log phase. Then, bacterial numbers were determined by measuring the OD at 600 nm, and diluted stocks were i.v. injected in PBS. Naive mice received 1,000–3,000 colony-forming units (CFU), mice previously infected with Listeria received 5 × 104 CFU, mice previously infected with LCMV received 2 ×105 CFU, and, for infection with ActA-Listeria, 106 CFU were injected. Bacterial loads were determined by lysing spleens in PBS supplemented with 0.1% Tergitol NP-40 (Sigma-Aldrich). Serial dilutions were spread out on brain-heart infusion plates containing Streptomycin (200 μg/mL), and colonies were enumerated.
Identification of the D4-Altered Peptide Ligand
A large pool of SIINFEKL-derived synthetic altered peptide ligands were tested for H-2Kb stabilization in RMA-S cells and recognition by OT-1 T cells as previously described (Zehn et al., 2009). The D4 peptide shows similar surface stabilization as the N4 peptide, but it is a weak agonist for OT-1 T cells (Figure 2A).
T Cell Expansion and Functional Tests
OT-1 cells were stimulated in RPMI (10% FCS, 100 IU/mL Penicillin, 100 IU/mL Streptomycin, 5 μM 2-ME, 5 mM HEPES; Invitrogen) with anti-CD3/CD28-coated beads (Dynabeads, Invitrogen) and cultured with 50 U/mL human IL-2 (Chiron) in 7% CO2. 6 days after activation, 2 × 105 OT-1 cells and 1 ×105 RMA cells were mixed in 96-well plates, and titrated concentrations of SIINFEKL or APL peptides (EMC microcollections) were added. After 30 min, 7 μM Brefeldin A (Sigma-Aldrich) was added, and cultures were incubated for another 3.5 hr.
Surface and Intracellular Staining and Flow Cytometry
Up to 4 × 106 cells were plated in 96-well plates in PBS supplemented with 2% FCS and 0.01% azide. Unspecific antibody binding was blocked with 2.4G2 (BioxCell). Cells were surface stained for 20 min at 4°C with fluorescently labeled anti-CD8 (53–6.7), CD127 (A7R34), KLRG1 (2F1), CD27 (LG7.F9), CD43 glyco (1B11), CD4 (GK1.5), CD45.1 (A20), and CD45.2 (104) antibodies and fixed in PBS supplemented with 1% formaldehyde, 2% glucose, and 0.03% azide. For intracellular cytokine staining, cells were fixed in PBS 2% formaldehyde and permeabilized in PBS with 0.25% Saponin and 0.25% BSA (Perm buffer). Cells were stained in Perm buffer for IFNγ (XMG1.2), TNFα (MP6-XT22), or Granzyme B (GB12). Fluorescently labeled MHC-I-gp33 multimers were used for detecting gp33-specific T cells (TC-Metrix). For in vivo staining experiments, we injected mice i.v. with fluorescently labeled anti-CD8 antibodies. Mice were sacrificed 3 min later, and harvested organs were processed as indicated above.
Data Analysis and Statistics
Flow cytometry data were obtained on a BD FACS LSRII machine and analyzed using FlowJo software (Tree Star). GraphPad Prism was used for graphic representation of data, calculation of EC50 values, and statistical calculations. Non-paired t tests (two-tailed) were used. p values ≤ 0.05 were considered significant (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001), and p values > 0.05 were considered non-significant (ns).
Author Contributions
S.G.O., M.P., and D.Z. designed the study. S.G.O. performed experiments with support by L.H.-E.-D., St.S., and S.E. Experiments added to the revised manuscript were performed by V.C. Data were analyzed by S.G.O. and V.C. The manuscript was written by S.G.O.and D.Z. Critical advice was provided by M.P.
Acknowledgments
We thank Lexus Johnson and Joe Sun for sharing their complementary findings prior to publication. This work was supported by grants from the Swiss Vaccine Research Institute, the Acteria Foundation, the Swiss National Science Foundation (CRSII3-141789, PP00P3_144883, and PP00P3_135442/1), the European Union (ERC starting grant 337043-ProtecTC), and the NIH (DP2DE023321-0 to M.P.). S.G.O. is now an employee of Pfizer, but all work was carried out previously in the laboratory of D.Z.
Published: October 11, 2016
Footnotes
Supplemental Information contains three figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.09.072.
Supplemental Information
References
- Beura L.K., Hamilton S.E., Bi K., Schenkel J.M., Odumade O.A., Casey K.A., Thompson E.A., Fraser K.A., Rosato P.C., Filali-Mouhim A. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532:512–516. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco J.C., Boukhvalova M.S., Shirey K.A., Prince G.A., Vogel S.N. New insights for development of a safe and protective RSV vaccine. Hum. Vaccin. 2010;6:482–492. doi: 10.4161/hv.6.6.11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che J.W., Selin L.K., Welsh R.M. Evaluation of non-reciprocal heterologous immunity between unrelated viruses. Virology. 2015;482:89–97. doi: 10.1016/j.virol.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu T., Tyznik A.J., Roepke S., Berkley A.M., Woodward-Davis A., Pattacini L., Bevan M.J., Zehn D., Prlic M. Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep. 2013;3:701–708. doi: 10.1016/j.celrep.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornberg M., Chen A.T., Wilkinson L.A., Brehm M.A., Kim S.K., Calcagno C., Ghersi D., Puzone R., Celada F., Welsh R.M., Selin L.K. Narrowed TCR repertoire and viral escape as a consequence of heterologous immunity. J. Clin. Invest. 2006;116:1443–1456. doi: 10.1172/JCI27804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti D., Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu. Rev. Immunol. 2013;31:705–742. doi: 10.1146/annurev-immunol-032712-095916. [DOI] [PubMed] [Google Scholar]
- Daniels M.A., Teixeiro E., Gill J., Hausmann B., Roubaty D., Holmberg K., Werlen G., Holländer G.A., Gascoigne N.R., Palmer E. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. 2006;444:724–729. doi: 10.1038/nature05269. [DOI] [PubMed] [Google Scholar]
- Enouz S., Carrié L., Merkler D., Bevan M.J., Zehn D. Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J. Exp. Med. 2012;209:1769–1779. doi: 10.1084/jem.20120905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm N., DeCamp A.C., Friedrich D.P., Carter D.K., Defawe O.D., Kublin J.G., Casimiro D.R., Duerr A., Robertson M.N., Buchbinder S.P. Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J. Clin. Invest. 2012;122:359–367. doi: 10.1172/JCI60202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkina E., Thatte J., Dabak V., Williams M.B., Ley K., Braciale T.J. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J. Clin. Invest. 2005;115:3473–3483. doi: 10.1172/JCI24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist K.A., Tomlinson A.J., Kieper W.C., McGargill M.A., Hart M.C., Naylor S., Jameson S.C. Identification of a naturally occurring ligand for thymic positive selection. Immunity. 1997;6:389–399. doi: 10.1016/s1074-7613(00)80282-4. [DOI] [PubMed] [Google Scholar]
- Hudrisier D., Oldstone M.B., Gairin J.E. The signal sequence of lymphocytic choriomeningitis virus contains an immunodominant cytotoxic T cell epitope that is restricted by both H-2D(b) and H-2K(b) molecules. Virology. 1997;234:62–73. doi: 10.1006/viro.1997.8627. [DOI] [PubMed] [Google Scholar]
- Johnson L.R., Weizman O.E., Rapp M., Way S.S., Sun J.C. Epitope-specific vaccination limits clonal expansion of heterologous naïve T cells during viral challenge. Cel Rep. 2016;17:636–644. doi: 10.1016/j.celrep.2016.09.019. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.K., Reed D.S., Olson S., Schnell M.J., Rose J.K., Morton P.A., Lefrançois L. Generation of mucosal cytotoxic T cells against soluble protein by tissue-specific environmental and costimulatory signals. Proc. Natl. Acad. Sci. USA. 1998;95:10814–10819. doi: 10.1073/pnas.95.18.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P., Chow M.Y., Loessner M.J., Portnoy D.A., Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Olson D., Shoukry N.H., Brady K.W., Kim H., Olson D.P., Hartman K., Shintani A.K., Walker C.M., Kalams S.A. Limited T cell receptor diversity of HCV-specific T cell responses is associated with CTL escape. J. Exp. Med. 2004;200:307–319. doi: 10.1084/jem.20040638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J.P., Klasse P.J., Dolan M.J., Ahuja S.K. AIDS/HIV. A STEP into darkness or light? Science. 2008;320:753–755. doi: 10.1126/science.1154258. [DOI] [PubMed] [Google Scholar]
- Neuenhahn M., Kerksiek K.M., Nauerth M., Suhre M.H., Schiemann M., Gebhardt F.E., Stemberger C., Panthel K., Schröder S., Chakraborty T. CD8alpha+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity. 2006;25:619–630. doi: 10.1016/j.immuni.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Olson J.A., McDonald-Hyman C., Jameson S.C., Hamilton S.E. Effector-like CD8+ T cells in the memory population mediate potent protective immunity. Immunity. 2013;38:1250–1260. doi: 10.1016/j.immuni.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price D.A., West S.M., Betts M.R., Ruff L.E., Brenchley J.M., Ambrozak D.R., Edghill-Smith Y., Kuroda M.J., Bogdan D., Kunstman K. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection. Immunity. 2004;21:793–803. doi: 10.1016/j.immuni.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Prlic M., Hernandez-Hoyos G., Bevan M.J. Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J. Exp. Med. 2006;203:2135–2143. doi: 10.1084/jem.20060928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman A.L. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat. Rev. Immunol. 2011;11:532–543. doi: 10.1038/nri3014. [DOI] [PubMed] [Google Scholar]
- Santori F.R., Kieper W.C., Brown S.M., Lu Y., Neubert T.A., Johnson K.L., Naylor S., Vukmanović S., Hogquist K.A., Jameson S.C. Rare, structurally homologous self-peptides promote thymocyte positive selection. Immunity. 2002;17:131–142. doi: 10.1016/s1074-7613(02)00361-8. [DOI] [PubMed] [Google Scholar]
- Schenkel J.M., Fraser K.A., Masopust D. Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J. Immunol. 2014;192:2961–2964. doi: 10.4049/jimmunol.1400003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanski H.E., Mayerova D., Jameson S.C., Hogquist K.A. A low affinity TCR ligand restores positive selection of CD8+ T cells in vivo. J. Immunol. 2001;166:6602–6607. doi: 10.4049/jimmunol.166.11.6602. [DOI] [PubMed] [Google Scholar]
- Sun J.C., Beilke J.N., Lanier L.L. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider D.T., Legat A., Fuertes Marraco S.A., Carrié L., Luescher I., Speiser D.E., Zehn D. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat. Immunol. 2013;14:603–610. doi: 10.1038/ni.2606. [DOI] [PubMed] [Google Scholar]
- Utzschneider D.T., Alfei F., Roelli P., Barras D., Chennupati V., Darbre S., Delorenzi M., Pinschewer D.D., Zehn D. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J. Exp. Med. 2016;213:1819–1834. doi: 10.1084/jem.20150598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh R.M., Che J.W., Brehm M.A., Selin L.K. Heterologous immunity between viruses. Immunol. Rev. 2010;235:244–266. doi: 10.1111/j.0105-2896.2010.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachi P.P., Ampudia J., Zal T., Gascoigne N.R. Altered peptide ligands induce delayed CD8-T cell receptor interaction--a role for CD8 in distinguishing antigen quality. Immunity. 2006;25:203–211. doi: 10.1016/j.immuni.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Zehn D., Lee S.Y., Bevan M.J. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehn D., Turner M.J., Lefrançois L., Bevan M.J. Lack of original antigenic sin in recall CD8(+) T cell responses. J. Immunol. 2010;184:6320–6326. doi: 10.4049/jimmunol.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehn D., Roepke S., Weakly K., Bevan M.J., Prlic M. Inflammation and TCR signal strength determine the breadth of the T cell response in a bim-dependent manner. J. Immunol. 2014;192:200–205. doi: 10.4049/jimmunol.1302289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Bevan M.J. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.