

Summary
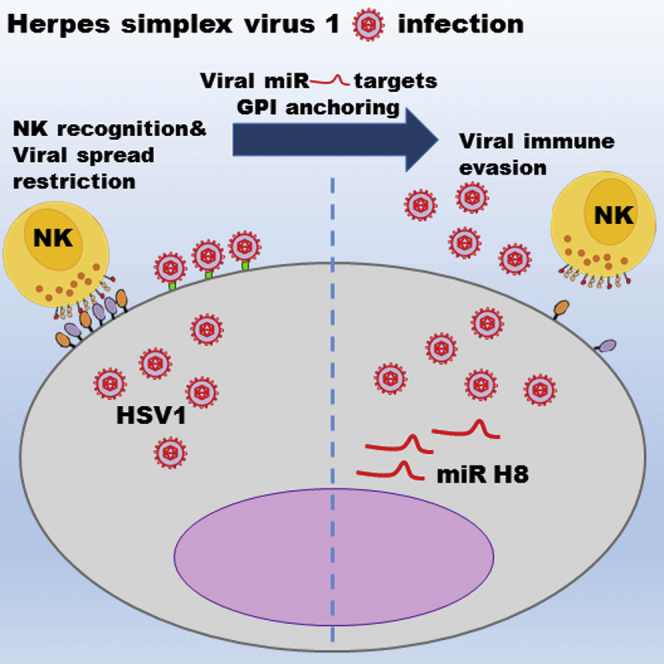
Herpes simplex virus 1 (HSV1) is a ubiquitous human pathogen that utilizes variable mechanisms to evade immune surveillance. The glycosylphosphatidylinositol (GPI) anchoring pathway is a multistep process in which a myriad of different proteins are covalently attached to a GPI moiety to be presented on the cell surface. Among the different GPI-anchored proteins there are many with immunological importance. We present evidence that the HSV1-encoded miR H8 directly targets PIGT, a member of the protein complex that covalently attaches proteins to GPI in the final step of GPI anchoring. This results in a membrane down-modulation of several different immune-related, GPI-anchored proteins, including ligands for natural killer-activating receptors and the prominent viral restriction factor tetherin. Thus, we suggest that by utilizing just one of dozens of miRNAs encoded by HSV1, the virus can counteract the host immune response at several key points.
Keywords: HSV1, microRNA, NK cells, GPI anchoring, immune evasion, tetherin
Graphical Abstract
Highlights
-
•
HSV1 miR H8 targets PIGT of the GPI anchoring pathway
-
•
Expression of the anti-viral protein tetherin is reduced and viral spread enhanced
-
•
Expression of GPI-anchored activating NK cell ligands is reduced
-
•
Recognition and elimination by NK cells decrease
Herpes simplex virus 1 is a ubiquitous human pathogen and the cause of several ailments. Enk et al. found that the HSV1-encoded miR H8 targets the GPI anchoring pathway, reducing expression of several immune-modulating proteins, thus enhancing viral spread and enabling evasion of natural killer cell elimination.
Introduction
Herpes simplex virus 1 (HSV1) is an α-herpesvirus and the etiological cause of several diseases. HSV1 establishes a life-long latent infection in the sensory nerve ganglia of the nerves innervating the primary site of infection (Arvin et al., 2007). Humans and HSV1 have co-evolved for millennia. The host immune response toward HSV1 entails both adaptive responses and innate responses, which includes both cell-intrinsic and cell-extrinsic responses (Arvin et al., 2007). The cell-intrinsic response involves cell-autonomous sensing of viral infection and subsequent production of type I interferons that induce interferon-stimulated genes, including the potent antiviral protein tetherin (Levy et al., 2011). Tetherin acts as a tether of budding viruses, adhering them to the infected cell surface and limiting viral spread (Neil, 2013). The cell-extrinsic innate immune response involves recruitment of cellular immune responders, including natural killer (NK) cells.
NK cells are innate lymphocytes that kill tumor and virally infected cells (Vivier et al., 2011) and are considered members of the innate lymphoid cell (ILC) family, classified under the ILC1 family (Artis and Spits, 2015). The NK response to target cells is dictated by a balance of signals delivered from activating and inhibitory receptors. The inhibitory NK cell receptors recognize mainly major histocompatibility complex (MHC) class I proteins (Koch et al., 2013), whereas the activating receptors recognize tumor-, pathogen-, and stress-induced ligands and self-ligands (Long et al., 2013). A principal activating NK receptor is NKG2D, which recognizes several stress-induced ligands: MHC class I polypeptide-related sequence A and B (MICA and MICB) and the UL16 binding protein 1-6 (ULBP1-6) (Lanier, 2015), variably expressed by tumor cells and upregulated by cellular stresses, like those that occur during viral infections (Iannello and Raulet, 2013). Additionally, NK cells express several other activating receptors, such as 2B4, which recognizes CD48, a cognate ligand/receptor expressed by other immune cells (Kim et al., 2014b). Other activating NK receptors, such as the natural cytotoxicity receptors (NCRs) NKp30, NKp44, and NKp46, bind mostly viral hemagglutinins and unknown cellular ligands (Koch et al., 2013).
MicroRNAs (miRNAs or miRs) are short non-coding RNAs that bind target mRNAs and repress their translation (Guo et al., 2010). Generally, the effect of miRNAs on target protein expression is relatively mild (Baek et al., 2008). Notably, viruses, especially of the herpesvirus family, encode for miRNAs that can modulate host gene expression (Skalsky and Cullen, 2010). Indeed, we have previously demonstrated viral miRNA-based immune-evasive mechanisms (Stern-Ginossar et al., 2007, Nachmani et al., 2010, Nachmani et al., 2009, Bauman et al., 2011). HSV1 encodes 27 known miRNAs that are variably expressed during the viral lifecycle (Cui et al., 2006, Umbach et al., 2009, Kozomara and Griffiths-Jones, 2014, Du et al., 2015). The functions of the HSV1 miRNAs are largely unknown, and it is unknown whether they affect the counter-HSV1 immune response.
Most cell surface proteins are membrane-integral; however, some are covalently attached to glycosylphosphatidylinositol (GPI) lipid anchors. The GPI anchoring pathway is a multistep process that involves more than 25 proteins and in which the GPI-transamidase (GPIT) complex covalently attaches the substrate pro-protein C terminus to the GPI moiety (Kinoshita, 2014). GPIT consists of five known proteins: PIGK, PIGS, PIGT, PIGU, and GPAA1, of which PIGK is widely accepted as the catalytic subunit (Kinoshita, 2014). Importantly, PIGT and PIGK share a disulfide bond, and PIGT is thought to be involved in GPIT complex stabilization (Kinoshita, 2014). Additionally, according to the predicted 3D model, PIGT forms a β-propeller gate that regulates access into the catalytic site of GPIT (Eisenhaber et al., 2003). Here we demonstrate that HSV1 miR H8 targets PIGT to avoid NK cell attack and the effects of tetherin.
Results
HSV1 miR H8 Downmodulates the Expression of Several Activating NK Ligands
Previously, we described viral miRNA-based mechanisms utilized by herpesviruses to downregulate the expression of MICB, an NKG2D ligand, to evade NK cell recognition and elimination (Stern-Ginossar et al., 2007, Nachmani et al., 2009, Nachmani et al., 2010). Here we investigated whether some of the 27 HSV1-encoded miRNAs might target NKG2D ligands. For this, we transduced the B cell line BJAB, which endogenously expresses five NKG2D ligands—MICA, MICB, and ULBP1–3—as well as many other known and unknown activating NK receptor ligands with lentiviral vectors encoding for artificial hairpins that are processed into mature HSV1 miRNAs, as described previously (Stern-Ginossar et al., 2007). We found that 19 of the HSV1-encoded miRNAs tested had no effect on the expression of the NKG2D stress-induced ligands (Figure S1). However, miR H8 did significantly reduce the expression level of two NKG2D ligands, ULBP2 and ULBP3, and the expression of the 2B4 ligand CD48 (Figure 1A, quantified in Figure 1B). Notably, the miR H8 effect was very pronounced (Figure 1A), unlike the expected effects (Baek et al., 2008, Bauman et al., 2011, Stern-Ginossar et al., 2007, Nachmani et al., 2009, Nachmani et al., 2010). We verified expression of miR H8 in the transduced cells (Figure 1C).
Figure 1.

HSV1 miR H8 Reduces NKG2D and 2B4 Ligand Expression
(A) MiR H8-expressing BJAB cells were stained for the indicated ligands. In all histograms, the black histograms represent the staining of miR H8/or the precursor miR H8, the empty gray histograms represent staining of control SINGFP-expressing cells, and the full gray histograms represent staining of the SINGFP cells with secondary antibody only. The background staining of cells expressing miR H8/pre-miR H8 was similar to that of SINGFP and is not shown.
(B) The fold change in mean fluorescent intensity (MFI) compared with control miRNA-expressing cells was calculated and analyzed using a single sample t test. ULBBP2, n = 5; ULBP3, n = 6; CD48, n = 6. Error bars represent SEM. ∗∗∗p > 0.005.
(C) qRT-PCR for the expression of miR H8. Mean values and SEM error bars are derived from quadruplicates.
(D) BJAB-miR H8/control vector cells were stained with the indicated fusion proteins.
(E) FACS staining of ULBP2, ULBP3, and CD48 in cells expressing pre-miR H8. Staining for all figure parts was repeated at least three times.
(F) Sequence and predicted hybridization of HSV1 miR H8 with its target in the PIGT 3′ UTR (204–230 refers to the base number in the PIGT 3′ UTR). The miR H8 seed region is underlined (top). Shown is sequence alignment of miR H8 and the mutated 3′ UTR of PIGT (bottom).
(G) Relative luciferase activity is displayed for miR H8-expressing HeLa cells (compared with cells expressing a control HSV1 miR H15) transiently transfected with a luciferase reporter attached to a control 3′ UTR, the 3′ UTR of PIGT, or a mutated 3′ UTR of PIGT with a two-base substitution in the seed binding region of miR H8 (as depicted in F).
Shown are mean values and SEM, and error bars were derived from triplicates. The figure shows a representative experiment of four performed. ∗∗∗p > 0.005. See also Figure S1 and S2 and Tables S1 and S2.
Several other activating NK receptors, including the NCRs NKp44 and NKp46, and the short-tailed killer-cell immunoglobulin-like receptors (KIRs) bind to yet unknown ligands. To investigate whether miR H8 might affect the expression of these ligands, we utilized fusion proteins consisting of the extracellular portions of the different activating NK receptors fused to the Fc portion of the human immunoglobulin 1 (IgG1). Expression of miR H8 did not affect the expression of any of these unknown activating NK cell ligands (Figure 1D).
We also transduced BJAB cells with the full-length precursor miR of H8 (pre-H8). Using this modality, we utilized the cellular components that normally process precursor miRNAs into mature miRNAs. The results obtained using pre-H8 were practically identical to the results obtained using the miR H8 from the artificial hairpin (Figure 1E).
HSV1 miR H8 Targets PIGT of the GPI Anchoring Pathway
Next we sought to identify the direct target(s) of miR H8. For this, we utilized the RNA hybridization prediction program RNA-hybrid (Rehmsmeier et al., 2004). Initially we examined the transcripts of ULBP2, ULBP3, and CD48 for a binding site for miR H8, but no such binding sites were predicted (data not shown). Next, because ULBP2, ULBP3, and CD48 are all GPI-anchored proteins, we examined the transcripts of the proteins involved in GPI anchoring for predicted binding by miR H8. One of these, PIGT of the GPIT complex, had a predicted binding site for miR H8 (Figure 1F, top). This site was in the 3′ UTR of PIGT; it displayed full seed (bases 2–7 of the miR) complementarity and had additional supplemental binding sites in the miR 3′.
To validate this bioinformatic prediction, we utilized a dual-luciferase assay, as described previously (Nachmani et al., 2010). As can be seen in Figure 1G, when the 3′ UTR of PIGT was expressed downstream of the luciferase, normalized luciferase activity was dramatically reduced in the presence of miR H8. However, mutating 2 base pairs (bp) in the seed binding site for miR H8 (Figure 1F) reversed most of the miR H8 effect (Figure 1G). Thus, miR H8 indeed targets the 3′ UTR of PIGT at the predicted binding site.
Next, to ensure that endogenous PIGT is targeted by miR H8, we stained BJAB cells expressing miR H8 or a control miR with an anti-PIGT antibody and quantified PIGT expression using fluorescence-activated cell sorting (FACS) analysis. As can be seen in Figure 2A, quantified in Figure 2B, PIGT levels were reduced ∼40% in miR H8-expressing cells. PIGT mRNA levels remained unchanged (Figure 2C), suggesting that miR H8 induces translational inhibition. To validate that the effect of miR H8 on the expression of the activating NK ligands is indeed PIGT-dependent, we targeted PIGT directly using short hairpin RNA (shRNA). PIGT expression was reduced, as assessed by FACS staining (Figure 2D, quantified in Figure 2E) and by qRT-PCR (Figure 2F). Moreover, the expression of ULBP2, ULBP3, and CD48 was reduced to levels similar to the effect elicited of miR H8 (Figure 2G).
Figure 2.

HSV1 miR H8 Reduces the Protein Levels of PIGT
(A) Intracellular FACS staining for PIGT. Black histograms represent staining of miR H8-expressing cells, empty gray histograms represent staining of cells expressing a control miRNA, and the full gray histograms represent staining of the control miRNA with secondary antibody only. Isotype control staining and background staining of miR H8-expressing cells was similar to the control miR staining and is not shown.
(B) Fold change in MFI compared with control miRNA-expressing cells was calculate and analyzed using a single sample t test (n = 5). Error bars represent SEM.
(C) qRT-PCR for the expression of PIGT was conducted on RNA samples from control miRNA- and miR H8-expressing cells. Mean values and SEM error bars are derived from quadruplicates.
(D) Intracellular FACS staining for PIGT in cells expressing shRNA directed against PIGT (black) or a scramble shRNA (shSCR, gray). Full gray histograms represent staining of the shSCR cells with secondary antibody only. Staining was repeated five times.
(E) Fold change in MFI calculated for several experiments as in (D) (n = 5). Error bars represent SEM.
(F) qRT-PCR for the expression of PIGT in shPIGT- and sh-SCR-expressing cells as in (C).
(G) Cells expressing shPIGT and shSCR stained for the expression of the activating NK ligands displayed. Black histograms represent staining of shPIGT-expressing cells, empty gray histograms represent staining of cells expressing shSCR, and the full gray histograms represent staining of shSCR with secondary antibody only. The background staining of shPIGT-expressing cells was similar to the shSCR staining and is not shown (for D and G).
All experiments were performed at least three times. ∗p > 0.05. See also Figure S1 and Table S1.
HSV1 miR H8 Reduces NK-Dependent Killing
The functionality of the reductions observed in the levels of ULBP2, ULBP3, and CD48 was tested using NK cytotoxicity assays. First we co-incubated our miR H8 and control cells with the NK cell line YTS-ECO, known to kill target cells exclusively via 2B4-CD48 interactions (Chuang et al., 2000). Cells expressing miR H8 were killed significantly less than control miRNA-expressing cells (Figure 3A). Moreover, when miR H8 and control cells were incubated with primary bulk NK cells, the same effect was observed. This reduction in NK cell killing was also seen when the target cells expressed shPIGT but not shScramble (Figure 3B). Furthermore, when miR H8-expressing cells were rescued using exogenous PIGT, the miR H8 effect was reversed (Figure 3C).
Figure 3.

HSV1 miR H8 Inhibits NK Killing
(A) HSV1 miR H8- and control miRNA-expressing BJAB cells labeled with 35S methionine were co-cultured for 5 hr with the NK cell line YTS-ECO at the effector-to-target (E:T) ratios indicated.
(B) Primary bulk NK cells were incubated for 5 hr with BJAB cells expressing miR H8, a control miR, shPIGT, or a control shScramble labeled using 35S methionine as above.
(C) BJAB cells expressing miR H8 were co-transduced to overexpress exogenous PIGT (PIGT OE) or an empty vector (ds-empty). These cells and control miR-expressing cells were used in NK killing assays as in (B).
Representative experiments were repeated twice for (A) and (C) and four times for (B). Target killing is displayed as a percentage of the total. Shown are mean values ± SEM. ∗p > 0.05, ∗∗p > 0.01, ∗∗∗p > 0.005.
HSV1 miR H8-Mediated Reduction in PIGT Affect Tetherin Expression
Next we tested whether miR H8’s effect on PIGT might affect the expression of the GPI anchored protein tetherin (also known as BST2/CD317). For this, we stained our HSV1 miR H8- and control miRNA-expressing cells with an anti-tetherin antibody and observed that expression of miR H8 significantly reduced the expression levels of tetherin (Figure 4A; quantified in Figure 4B). Importantly, pre-H8 caused the same marked reduction in tetherin expression (Figure 4C), as did shPIGT (Figure 4D). Next, to examine the functionality of the decrease in tetherin expression, we infected miR H8- and control empty vector-expressing cells with HSV1-RFP. 72 hr after infection, the supernatants were used to titrate the viral load in Vero cells. As expected, miR H8-expressing cells with low tetherin expression were significantly more susceptible to HSV1 infection and produced more viral progeny than control vector-expressing cells. Indeed, shPIGT elicited the same effect and so did shTetherin, but to a stronger degree (Figure 4E). Additionally, reconstitution of PIGT in miR H8-expressing cells (H8-PIGT) reversed this effect compared with miR H8-expressing cells transfected with an empty vector (H8-ds) (Figure 4F). In this setting, the cells expressing shTetherin were also the cells that displayed the highest release of virus, and the controls, expressing a control miR with the empty vector (Control miR-ds) and an shScramble, displayed the lowest viral release. When relative viral release was measured (explained in Experimental Procedures), cells expressing miR H8 showed a higher proportion of viral release, as did the shTetherin- and shPIGT-expressing cells compared with their appropriate controls (Figure 4G). Finally, to validate that the miR H8 effect on HSV1 spread is mediated via PIGT and tetherin, we expressed tetherin exogenously in A549 cells and transduced these cells with miR H8. As can be seen in Figure 4H, miR H8 also enhanced viral release in this setting. Thus, the effect of HSV1 miR H8 on PIGT expression and the downstream effects on GPI-anchored proteins facilitate viral immune evasion at several different checkpoints.
Figure 4.

HSV1 miR H8 Expression Reduces Tetherin Surface Presentation, Enhancing Viral Spread
(A, C, and D) BJAB cells expressing miR H8 (A), pre-miR H8 (C), or shPIGT (D) stained for tetherin (black histograms). Empty gray histograms represent control miRNA-expressing (A and C) or scramble shRNA-expressing (C) cells. The full gray histogram represents staining with secondary antibody only.
(B) Fold change in tetherin staining MFI (as in A) of miR H8 compared with control miRNA-expressing cells, calculated and analyzed using a single sample t test (n = 6).
(E) HSV1 viral titers calculated in Vero cells incubated with supernatants from BJAB cells expressing HSV1 miR H8, a control miRNA, shScramble, shPIGT, or shTetherin and infected with HSV1 for 72 hr (n = 2).
(F) HSV1 viral titers calculated in Vero cells incubated with supernatants from BJAB cells expressing HSV1 miR H8 concomitantly with exogenous PIGT/empty vector (H8-PIGT/H8-ds) and infected with HSV1. BJAB cells expressing a control miRNA and empty vector (control miR-ds) were used as a negative control, whereas BJABs expressing shTetherin were used as a positive one (shTet).
(G) Relative HSV1 release calculated as a proportion of total viral titers (supernatant and cell-associated). The abbreviations are the same as in (E) and (F).
(H) A549 cells overexpressing exogenous tetherin and HSV1 miR H8 or an empty control vector were infected with HSV1 for 72 hr. Supernatants were then used to calculate viral titers in Vero cells.
All error bars represent SEM. For (A), (C), and (D), n = 6. For (E)–(H), n = 2. ∗p > 0.05, ∗∗p > 0.01, ∗∗∗p > 0.005.
Discussion
Here we demonstrate that miR H8 significantly reduces the membrane expression of the NKG2D ligands ULBP2 and ULBP3 as well as the levels of CD48, a ligand for 2B4. We demonstrate that miR H8 targets the 3′ UTR of PIGT of the GPIT complex seed dependently. Next we show that these effects are biologically significant and reduce NK cell killing of miR H8-expressing cells. Finally, we show that the dominant cell-intrinsic antiviral protein tetherin, which is GPI-anchored, is also affected by miR H8 expression. Tetherin is a cell-intrinsic, interferon-inducible viral restriction factor (Neil, 2013). The importance of tetherin in the anti-HSV1 response is emphasized by the viral countermeasures employed to subvert its activity. Two recent papers found that HSV1 counteracts tetherin via its viral host shutoff gene as well as by its glycoprotein M (Blondeau et al., 2013, Zenner et al., 2013). However, the effect of miR H8 via PIGT on tetherin levels was prominent enough to be of biological significance in titration experiments even when these mechanisms were at play.
Interestingly, the effect of miR H8 on PIGT levels and GPI anchoring did not encompass all GPI-anchored proteins. Namely, ULBP1 (which is also GPI-anchored) levels did not change. This can be explained by preferential loading of different pro-proteins into the remaining GPIT complexes if the predicted role of PIGT as the “gatekeeper” (Eisenhaber et al., 2003) of GPIT is correct. Moreover, even the affected proteins did not completely disappear from the cell surface. Indeed, partial activity of the GPIT can be expected because it was reported previously that a complete lack of PIGT is required for complete blocking of GPI anchoring (Krawitz et al., 2013), and hypomorphic activity of PIGT resulting in partial loss of GPI-anchored protein presentation has been observed (Kvarnung et al., 2013). Thus, our results that demonstrate a decrease in PIGT levels of approximately 40% are consistent with previous findings.
Recent research characterized the expression dynamics of HSV1 miRNAs (Du et al., 2015), and miR H8 was found to be expressed exclusively during reactivation from latency. This finding is important when taken in conjunction with the results presented here because the role of miR H8 is somewhat clarified. The function of immune cells, particularly CD8+ T cells, in maintaining viral latency and inhibiting HSV1 reactivation has been well established, and the role of NK cells in controlling HSV1 infection is also well documented (Egan et al., 2013, Moraru et al., 2012). Additionally, during HSV1 reactivation, the infected cells undergo various stresses stemming from the de novo expression of viral transcripts and proteins. Taken together, these findings point to the importance of immune evasion during early HSV1 reactivation before the main viral protein-based mechanisms are fully active. For this, utilization of viral miRNAs is ideal because miRNAs are considered non-immunogenic (Boss and Renne, 2011).
We do not know why miR H8 is expressed only during reactivation; its effects could be utilized by the virus to subvert tetherin during lytic infection as well. One possible explanation could be that, during reactivation, HSV1 needs to replicate and egress from a few selected neurons to establish widespread infection, and the effects miR H8 might help the virus overcome this initial limitation. On the other hand, during most of the lytic infection, a much larger mass of cells is infected, and thus a much higher viral load is enabled simply by virtue of infected cell mass.
The abovementioned reactivation exclusivity of miR H8 expression presents a distinct problem: all experiments that aim to ascertain the effect of miR H8 in a bona fide infection will require a setting in which HSV1 is reactivated. To date, however, all existing experimental models of HSV1 reactivation involve research animals (Du et al., 2015, Ramakrishna et al., 2015, Yao et al., 2014, Kobayashi et al., 2012, Kim et al., 2014a, Roehm et al., 2011), and because miR H8 targets the human PIGT transcript exclusively (Figures S2 and S3), performing such experiments in animals would be non-informative. Thus, an HSV1 latency reactivation model in human cells is needed in combination with specific miR H8 knockout (KO) viruses or an anti-miR H8 antagomiR to determine the physiological importance of our findings. Importantly, however, the expression levels of miR H8 in our system were comparable with the levels observed during reactivation in mouse ganglions (Figure 1C in this paper and Figure 3 by Du et al., 2015), when measured using the same qRT-PCR TAQMAN probes. Thus, even though we did not demonstrate the importance of miR H8 in a bona fide HSV1 reactivation setting, our experiments using the ectopic expression of miR H8 closely resemble the settings observed during HSV1 reactivation.
In conclusion, in this paper we discover that HSV1 miR H8 targets PIGT of the GPI anchoring pathway, enabling HSV1 to vitiate the host immune response, inhibiting elimination by NK cells and curbing the cell-intrinsic capacity to limit viral spreading. Thus, by using a single microRNA to target a basic cellular pathway, HSV1 is able to subvert the host immune response at several key points. The importance of understanding HSV1 biology and life cycle has recently been reiterated as its possible roles in Alzheimer’s disease (Itzhaki, 2014) and even cancer (Yun et al., 2015) are being discovered and investigated.
Experimental Procedures
Cell Culture, Lentiviral Constructs, Production, and Transduction
Cells were kept in their common corresponding media. RNA artificial hairpins (Table S1), orthologs of pre-miRNA, were generated and cloned into lentiviral vectors. Lentiviruses were generated in 293T cells and used to transduce target cells. Transduction efficiency was assessed by GFP, and only cell populations with >90% efficiency were used for experiments. A detailed description of the primers and procedures used for cell culture and cloning is included in the Supplemental Experimental Procedures.
Cytotoxicity Assays and NK Cell Preparation
The cytotoxic activity of NK cells against various targets was assessed in 5-hr 35S release assays as described previously (Mandelboim et al., 1996). NK cells were isolated from peripheral blood using the human NK cell isolation kit and the Cell-Sep kit (STEMCELL Technologies) according to the manufacturer’s instructions. The NK purity was close to 100% as determined by FACS analysis.
Viral Titer Experiments
2 × 105 BJAB cells expressing miR H8 or an empty Sin-GFP vector were infected with HSV-1 OK14 at a multiplicity of infection (MOI) of 0.2 for 72 hr. Supernatants were collected and used to infect Vero cells, and, 48 hours post infection (hpi), viral plaques were counted. For relative viral release assays, cells were infected with HSV1 as above for 24 hr. The cell pellet was then frozen and thawed three times to release viral progeny, and the supernatant and cell pellet fractions were used for titration in Vero cells as above. HSV1 release proportions = supernatant virus/(supernatant virus + cell-associated virus). The HSV-1 OK14 was a gift from Oren Kobiler and generated by cotransfecting BamHI-digested pHSV1(17+)Lox-mRFPVP26 and purified HSV-1(17+) DNA (O. Kobiler and L.W. Enquist, personal communication). The pHSV1(17+)Lox-mRFPVP26 was a kind gift from Drs. Katinka Döhner and Beate Sodeik (Nagel et al., 2012, Nagel et al., 2014).
Flow Cytometry
For flow cytometry, cells were incubated overnight at equal densities. Cells were incubated on ice for 1 hr with the primary antibody at a concentration of 0.2 μg/well. Appropriate secondary antibodies were applied for 30 min on ice at a concentration of 0.75 μg/well. Unless otherwise noted, cells expressing HSV1 miR-H15 were used as a control for the miR H8-expressing cells. For a detailed account and a list of antibodies used, see the Supplemental Experimental Procedures.
qRT-PCR
Details of the qRT-PCR procedure and a list of primers are included in the Supplemental Experimental Procedures and Table S2.
Luciferase Assay
The generation of the firefly luciferase constructs was as described previously (Stern-Ginossar et al., 2007). For more details, please consult the Supplemental Experimental Procedures and Table S3.
Statistical Methods
For statistical significance, Student’s t test analysis was used. A statistical test was considered significant when p < 0.05.
Author Contributions
J.E. conceived and performed all experiments and wrote the paper. A.L., Y.W., R.Y., and Y.C.A. performed experiments. D.G.W. and O.M. supervised the project. O.M. secured funding and wrote the paper.
Acknowledgments
This study was supported by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement 320473-BacNK. Further support came from the Israel Science Foundation, the GIF Foundation, the ICRF professorship grant, the Israeli Science Foundation, the Helmholtz Israel grant, and the Rosetrees Trust (all to O.M.). O.M. is a Crown Professor of Molecular Immunology.
We are grateful to Prof. Amos Panet (Hebrew University) for helpful discussions, Dr. Oren Kobiler (Tel Aviv University) for providing reagents and expertise, and Prof. Eran Bacharach (Tel Aviv University) for helpful discussions.
Published: October 18, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, three figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.09.077.
Supplemental Information
References
- Artis D., Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- Arvin A., Campadelli-Fiume G., Mocarski E., Moore P.S., Roizman B., Whitley R., Yamanishi K., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge University Press; Cambridge: 2007. [PubMed] [Google Scholar]
- Baek D., Villén J., Shin C., Camargo F.D., Gygi S.P., Bartel D.P. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman Y., Nachmani D., Vitenshtein A., Tsukerman P., Drayman N., Stern-Ginossar N., Lankry D., Gruda R., Mandelboim O. An identical miRNA of the human JC and BK polyoma viruses targets the stress-induced ligand ULBP3 to escape immune elimination. Cell Host Microbe. 2011;9:93–102. doi: 10.1016/j.chom.2011.01.008. [DOI] [PubMed] [Google Scholar]
- Blondeau C., Pelchen-Matthews A., Mlcochova P., Marsh M., Milne R.S., Towers G.J. Tetherin restricts herpes simplex virus 1 and is antagonized by glycoprotein M. J. Virol. 2013;87:13124–13133. doi: 10.1128/JVI.02250-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss I.W., Renne R. Viral miRNAs and immune evasion. Biochim. Biophys. Acta. 2011;1809:708–714. doi: 10.1016/j.bbagrm.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang S.S., Kim M.H., Johnson L.A., Albertsson P., Kitson R.P., Nannmark U., Goldfarb R.H., Mathew P.A. 2B4 stimulation of YT cells induces natural killer cell cytolytic function and invasiveness. Immunology. 2000;100:378–383. doi: 10.1046/j.1365-2567.2000.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C., Griffiths A., Li G., Silva L.M., Kramer M.F., Gaasterland T., Wang X.J., Coen D.M. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J. Virol. 2006;80:5499–5508. doi: 10.1128/JVI.00200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T., Han Z., Zhou G., Roizman B. Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc. Natl. Acad. Sci. USA. 2015;112:E49–E55. doi: 10.1073/pnas.1422657112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan K.P., Wu S., Wigdahl B., Jennings S.R. Immunological control of herpes simplex virus infections. J. Neurovirol. 2013;19:328–345. doi: 10.1007/s13365-013-0189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber B., Maurer-Stroh S., Novatchkova M., Schneider G., Eisenhaber F. Enzymes and auxiliary factors for GPI lipid anchor biosynthesis and post-translational transfer to proteins. BioEssays. 2003;25:367–385. doi: 10.1002/bies.10254. [DOI] [PubMed] [Google Scholar]
- Guo H., Ingolia N.T., Weissman J.S., Bartel D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannello A., Raulet D.H. Immune surveillance of unhealthy cells by natural killer cells. Cold Spring Harb. Symp. Quant. Biol. 2013;78:249–257. doi: 10.1101/sqb.2013.78.020255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki R.F. Herpes simplex virus type 1 and Alzheimer’s disease: increasing evidence for a major role of the virus. Front. Aging Neurosci. 2014;6:202. doi: 10.3389/fnagi.2014.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.Y., Shiflett L.A., Linderman J.A., Mohr I., Wilson A.C. Using homogeneous primary neuron cultures to study fundamental aspects of HSV-1 latency and reactivation. Methods Mol. Biol. 2014;1144:167–179. doi: 10.1007/978-1-4939-0428-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.J., Kim M., Kim H.M., Lim S.A., Kim E.O., Kim K., Song K.H., Kim J., Kumar V., Yee C. Homotypic NK cell-to-cell communication controls cytokine responsiveness of innate immune NK cells. Sci. Rep. 2014;4:7157. doi: 10.1038/srep07157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita T. Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 2014;90:130–143. doi: 10.2183/pjab.90.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M., Kim J.Y., Camarena V., Roehm P.C., Chao M.V., Wilson A.C., Mohr I. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J. Vis. Exp. 2012;62:3823. doi: 10.3791/3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J., Steinle A., Watzl C., Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013;34:182–191. doi: 10.1016/j.it.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawitz P.M., Höchsmann B., Murakami Y., Teubner B., Krüger U., Klopocki E., Neitzel H., Hoellein A., Schneider C., Parkhomchuk D. A case of paroxysmal nocturnal hemoglobinuria caused by a germline mutation and a somatic mutation in PIGT. Blood. 2013;122:1312–1315. doi: 10.1182/blood-2013-01-481499. [DOI] [PubMed] [Google Scholar]
- Kvarnung M., Nilsson D., Lindstrand A., Korenke G.C., Chiang S.C., Blennow E., Bergmann M., Stödberg T., Mäkitie O., Anderlid B.M. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J. Med. Genet. 2013;50:521–528. doi: 10.1136/jmedgenet-2013-101654. [DOI] [PubMed] [Google Scholar]
- Lanier L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015;3:575–582. doi: 10.1158/2326-6066.CIR-15-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D.E., Marié I.J., Durbin J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 2011;1:476–486. doi: 10.1016/j.coviro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long E.O., Kim H.S., Liu D., Peterson M.E., Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelboim O., Reyburn H.T., Vales-Gomez M., Pazmany L., Colonna M., Borsellino G., Strominger J.L. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J. Exp. Med. 1996;184:913–922. doi: 10.1084/jem.184.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraru M., Cisneros E., Gómez-Lozano N., de Pablo R., Portero F., Cañizares M., Vaquero M., Roustán G., Millán I., López-Botet M., Vilches C. Host genetic factors in susceptibility to herpes simplex type 1 virus infection: contribution of polymorphic genes at the interface of innate and adaptive immunity. J. Immunol. 2012;188:4412–4420. doi: 10.4049/jimmunol.1103434. [DOI] [PubMed] [Google Scholar]
- Nachmani D., Stern-Ginossar N., Sarid R., Mandelboim O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe. 2009;5:376–385. doi: 10.1016/j.chom.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Nachmani D., Lankry D., Wolf D.G., Mandelboim O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010;11:806–813. doi: 10.1038/ni.1916. [DOI] [PubMed] [Google Scholar]
- Nagel C.H., Döhner K., Binz A., Bauerfeind R., Sodeik B. Improper tagging of the non-essential small capsid protein VP26 impairs nuclear capsid egress of herpes simplex virus. PLoS ONE. 2012;7:e44177. doi: 10.1371/journal.pone.0044177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel C.H., Pohlmann A., Sodeik B. Construction and characterization of bacterial artificial chromosomes (BACs) containing herpes simplex virus full-length genomes. Methods Mol. Biol. 2014;1144:43–62. doi: 10.1007/978-1-4939-0428-0_4. [DOI] [PubMed] [Google Scholar]
- Neil S.J. The antiviral activities of tetherin. Curr. Top. Microbiol. Immunol. 2013;371:67–104. doi: 10.1007/978-3-642-37765-5_3. [DOI] [PubMed] [Google Scholar]
- Ramakrishna C., Ferraioli A., Calle A., Nguyen T.K., Openshaw H., Lundberg P.S., Lomonte P., Cantin E.M. Establishment of HSV1 latency in immunodeficient mice facilitates efficient in vivo reactivation. PLoS Pathog. 2015;11:e1004730. doi: 10.1371/journal.ppat.1004730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehm P.C., Camarena V., Nayak S., Gardner J.B., Wilson A., Mohr I., Chao M.V. Cultured vestibular ganglion neurons demonstrate latent HSV1 reactivation. Laryngoscope. 2011;121:2268–2275. doi: 10.1002/lary.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalsky R.L., Cullen B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010;64:123–141. doi: 10.1146/annurev.micro.112408.134243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N., Elefant N., Zimmermann A., Wolf D.G., Saleh N., Biton M., Horwitz E., Prokocimer Z., Prichard M., Hahn G. Host immune system gene targeting by a viral miRNA. Science. 2007;317:376–381. doi: 10.1126/science.1140956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach J.L., Nagel M.A., Cohrs R.J., Gilden D.H., Cullen B.R. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 2009;83:10677–10683. doi: 10.1128/JVI.01185-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E., Raulet D.H., Moretta A., Caligiuri M.A., Zitvogel L., Lanier L.L., Yokoyama W.M., Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H.W., Ling P., Tung Y.Y., Hsu S.M., Chen S.H. In vivo reactivation of latent herpes simplex virus 1 in mice can occur in the brain before occurring in the trigeminal ganglion. J. Virol. 2014;88:11264–11270. doi: 10.1128/JVI.01616-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S.J., Jeong P., Kang H.W., Kim Y.H., Kim E.A., Yan C., Choi Y.K., Kim D., Kim J.M., Kim S.K. Urinary MicroRNAs of Prostate Cancer: Virus-Encoded hsv1-miRH18 and hsv2-miR-H9-5p Could Be Valuable Diagnostic Markers. Int. Neurourol. J. 2015;19:74–84. doi: 10.5213/inj.2015.19.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenner H.L., Mauricio R., Banting G., Crump C.M. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J. Virol. 2013;87:13115–13123. doi: 10.1128/JVI.02167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.