

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder that leads to a progressive muscle wasting and paralysis. The pathological phenotypes are featured by severe motor neuron death and glial activation in the lumbar spinal cord. Proposed ALS pathogenic mechanisms include glutamate cytotoxicity, inflammatory pathway, oxidative stress, and protein aggregation. However, the exact mechanisms of ALS pathogenesis are not fully understood yet. Recently, a growing body of evidence provides a novel insight on the importance of glial cells in relation to the motor neuronal damage via the non-cell autonomous pathway. Accordingly, the aim of the current paper is to overview the role of astrocytes and microglia in the pathogenesis of ALS and to better understand the disease mechanism of ALS.
Keywords: amyotrophic lateral sclerosis, astrocyte, microglia, motor neuron, non-cell autonomous toxicity
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder with a prevalence of 2~3 per 100,000 people and is generally fatal within a few years of disease onset. Affected motor neurons in the brain stem, spinal cord, and motor cortex undergo significant loss, and it eventually causes progressive muscle wasting and paralysis in ALS patients. ALS was initially reported by Dr. Jean-Martin Charcot, a French neurologist, in 1869 [1]. Since Charcot's initial reporting, ALS received international attention when Lou Gehrig, a baseball player of the New York Yankees (Bronx, NY, USA), retired from baseball after being diagnosed with ALS in 1939. For this reason ALS has also been referred as 'Lou Gehrig's disease'. Interestingly, Gulf War veterans have a significantly increased risk (above two fold) of developing ALS [2]. Evidence has shown that the incidence of ALS has risen in recent years and it is reasonable to expect that it will continue to rise in the future. Most cases of ALS occur sporadically, but about 5~10% of ALS cases are familial ALS (FALS). In FALS, more than 90 mutations are found in superoxide dismutase 1 (SOD1) gene [3,4,5,6]. In addition, other mutations in FUS/TLS and TDP-43 genes have been known in ALS. Recently, a hexanucleotide repeat expansion of the C9orf72 gene has been identified as the most common cause of FALS discovered to date [7,8,9,10,11,12,13,14,15]. Given that mutations of the important cellular antioxidant enzyme SOD1 are a cause of FALS, it has well been proposed that oxidative stress plays a key role in the disease pathogenesis. Indeed oxidative damage and gliogenesis in both postmortem human FALS and sporadic ALS (SALS) tissue and in transgenic (mutant SOD1 (G93A)) ALS animal models have been documented [16,17]. Abnormal regulation of glutamate-dependent excitatory signal has also been identified in ALS suggesting that excessive synaptic glutamate and oxidative stress trigger motor neuronal damage. Moreover, altered calcium homeostasis, mitochondrial dysfunction, protein aggregation, cytoskeletal disruption, apoptosis, and inflammation are associated with motor neuronal damage and cell death [5,18]. Current medical care for both FALS and SALS focuses on symptom management. Supportive care can help control symptoms and make ALS more manageable for patients and their families, but this care does not significantly improve the disease progression. Even, to date, there are no effective drug therapies that slow the relentless progression of ALS [19,20,21]. In this regard, the better understanding of pathogenic mechanism of ALS may enhance the possibility for ameliorating the disease onset and progression. In this review, we focus on how non-neuronal cells are associated with the pathogenesis of ALS.
WHAT IS NON-CELL AUTONOMOUS TOXICITY?
In the past when scientists had focused on the study of neuronal function and activity, the events related to neuronal damage and cell death were only investigated from a narrow viewpoint. This view was based on the notion that neurons are damaged due to the dysfunction and deregulation by themselves (so called cell autonomous pathway), and this damage was not related to the dysfunction of any other cell types. As time went by, the view and knowledge of scientists on the mechanisms of neuronal damage have more evolved and advanced. Importantly, a growing body of evidence have proven that non-neuronal cells such as astrocytes, microglia, and oligodendrocytes directly contribute to the motor neuronal damage and cell death (so called non-cell autonomous pathway) in ALS including other neurodegenerative diseases. Indeed, the disease onset and progression is modulated via non-cell autonomous pathway in transgenic ALS [mutant SOD1 (G93A)] mice [18]. The mutant SOD1 expression within motor neurons initiates a damage process and drives the disease onset. In parallel, activation of astrocytes and microglia by mutant SOD1 markedly exacerbates the disease progression while motor neuronal mutant SOD1 has little influence on the progression of ALS. Thus, the paradigm of the non-cell autonomous toxicity has been determined and proven in several experimental conditions of ALS [22,23].
HOW DO ASTROCYTES MIND MOTOR NEURONS?
A major pathological feature of ALS is the generation and migration of new cells, specifically astrocytes, within and around damaged regions of the spinal cord [24]. Astrocytes respond to cellular stresses by proliferating and adopting a reactive phenotype characterized by the development of long and thick processes with an increased content of glial fibrillary acidic protein (GFAP). Interestingly, a similar increase in GFAP immunoreactivity was found when cultured primary spinal cord astrocytes were exposed to oxidative stress, suggesting that such morphological changes may be triggered by stress signals [24]. It seems likely that epigenetic alterations induced by mutant SOD1 (mtSOD1) and other pathological stresses are involved in the transformation of astrocytes to a neurotoxic reactive phenotype. In this scenario, non-cell autonomous cell death of motor neurons in ALS could result from either a loss of normal astrocytic support and/or the secretion of neurotoxic cytokines. Several studies have proven this idea as following: co-culture of astrocytes expressing mtSOD1 (G93A) or exposure to conditioned medium derived from astrocytes expressing mtSOD1 (G93A) damages both primary motor neurons and embryonic stem cell-derived motor neurons [25,26]. Previous studies have suggested that cytokines and other toxic factors released from SOD1(G93A) astrocytes may trigger motor neuronal damage [27,28,29,30]. For example, in vitro studies by Ferraiuolo et al. (2011) show that SOD1(G93A) astrocytes are toxic to normal motor neurons by reducing metabolic support from lactate release and activating pro-nerve growth factor-p75 receptor signaling pathway [27]. Interestingly, SOD1 (G93A) astrocytes specifically express NLRP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasome complexed with the NLR protein NLRP3, the adaptor ASC and pro-caspase 1, indicating that astrocytes mediate the neuroinflammation in ALS [28]. Moreover, transforming growth factor-β1 (TGF-β1) is increased in SOD1(G93A) astrocytes, and astrocyte-specific overexpression of TGF-β1 in SOD1(G93A) mice accelerates disease progression in a non-cell-autonomous manner [29]. On the other hand, the elevation of Bid, a BCL-2 family protein, in SOD1(G93A) astrocytes suggests that Bid activation may contribute to astrocyte activation and motor neuronal damage in ALS [30]. In this study, Bid is necessary for activating nuclear factor-κB in astrocytes to mediate pro-inflammatory stimuli, which represents that Bid is not directly toxic to motor neuron but indirectly modulates the astrocyte-dependent non-cell autonomous toxicity. Together, it has been successfully proven that astrocytic cytokines and toxin could determine disease progression and are critical to the pathogenesis of ALS.
Excitatory amino acid transporter-2 (EAAT2) is known as a typical glial glutamate transporter that uptakes neurotransmitters glutamate and aspartate from the synaptic cleft [31]. It is believed that EAAT2 uptakes more than 90% of glutamate into glia. In normal condition, astrocytes uptake glutamate and turn it into glutamine, and nourish motor neurons by supplying them as energy source. However, when astrocytes become reactive, the expression of EAAT2 gene is decreased and subsequently an excess amount of extracellular synaptic glutamate may lead to excitocytotoxicity in motor neurons in the spinal cord of ALS. Indeed, as the dysfunction of EAAT2 is implicated in ALS, the level of EAAT2 is reduced in the motor cortex and spinal cord of ALS patients [32]. Moreover, the decrease of EAAT2 activity impairs motor neuron survival in mouse models of ALS [33]. Otherwise, not only does chemical induction of EAAT2 activity improve motor neuron survival in an in vitro model of chronic excitotoxicity but it also extends the survival of transgenic ALS mice [34,35]. When EAAT2 transgenic mice is crossed with mutant SOD1 (G93A) mice, it shows a significant delay in motor symptom such as grip strength decline but not in the onset of paralysis [36]. Interestingly, Foran et al., (2011) reports that sumoylated carboxy-terminal fragment of EAAT2 (CTE-SUMO1) is accumulated in the nucleus of astrocytes in the spinal cord of SOD1(G93A) mice [37]. The expression of CTE-SUMO1 in spinal cord astrocytes produces extrinsic toxicity by inducing caspase-3 activation and impairs axonal growth of motor neurons in a co-culture system. This study provides an unconventional role of EAAT2 in that EAAT2 participates in motor neuron degeneration through the direct cytotoxic effect of its truncated peptide but not through the activity of glutamate transporter. All together, growing evidence supports that regulation of EAAT2 activity accounts for motor neuronal survival and death in ALS via a non-cell autonomous pathway.
In comparison to the astrocytic phenotype in ALS, different astrocytic behaviors in relation to the excitotoxicity may be derived due to either the different damage region of CNS (brain versus spinal cord) or the different stress stimuli (bolus excitotoxicity versus chronic oxidative stress). For instance, GFAP-positive astrocytes appear extensively around the damage sites 7 days after injection of N-ethyl-D-aspartic acid (NMDA) while EAAT2- and GFAP-positive astrocytes disappear in a kainic acid (KA)-injected cortical region of the brain [38]. This study shows that two excitotoxic injury models exhibit quite different pattern of astrocyte behaviors such as astrogliogenesis versus astrocyte loss that are distinguished from the pathology of ALS. Accordingly, it will be challenging to pursue how the difference of region or stress stimuli concerts and affects astrocyte behaviors in future studies.
HOW ARE ASTROCYTES ADAPTED TO ENVIRONMENTAL STRESSES AND WHAT ARE THE SURVIVAL MECHANISMS OF ASTROCYTES UNDER ALS CONDITION?
Our group has previously addressed this question using primary astrocytes from the spinal cord of wild type (WT) and ALS transgenic [mutant SOD1 (G93A)] mice. Our study shows that astrocyte survival is correlated with the elevation of Ets-2 transcription factor and with Bcl-xL expression [39]. The transcriptional activation of Bcl-xL by Ets-2 compensates oxidative stress by preventing astrocytes from apoptotic or necrotic cell death during the pathogenesis of ALS. Because we observed that motor neurons do not induce Bcl-xL in response to oxidative stress, we suggest that molecular mechanisms of Ets-2-mediated and Bcl-xL-dependent survival pathways may vary among different cell types [39]. Then why are motor neurons of ALS not rescued by the surviving astrocytes? We propose a plausible mechanism that the Ets-2 and Bcl-xL pathway improves astrocyte survival but it occurs too late to prevent earlier motor neuronal damage, or perhaps survived reactive astrocytes release toxic molecules to propagate motor neuron damage (Fig. 1). However, whether this might be expected to occur at an earlier stage, before astrocyte activation is reached its threshold, remains to be further investigated.
Fig. 1. Astrocytes are associated with non-cell autonomous motor neuronal damage in ALS. For example, cellular stresses elevate Bcl-xl gene expression in astrocytes, and the increase level of mitochondrial Bcl-xl prevents oxidative damage in astrocytes under ALS condition. Meanwhile, the increased iNOS/NOS2 expression and the decreased EAAT2/GLT-1/SLC1A2 expression in astrocytes lead to increased NO release and decreased glutamate uptake in the synaptic cleft of spinal cord. Consequently, elevation of glutamate and NO triggers motor neuronal damage and cell death via non-cell autonomous pathway.

Oxidative stress due to the mutation of SOD1 is highly implicated in the pathogenesis of ALS. Not only does superoxide anion (O2-) lead to cellular damage including oxidation of DNA and protein and lipid peroxidation but nitric oxide (NO) is also thought to play a key pathogenic role in ALS [40]. Motor neurons are particularly vulnerable to oxidative stress in ALS which is a phenomena attributed to a low level of antioxidant enzymes and a high content of easily oxidized substrates [5,24,40]. NO is synthesized by NO synthases (NOSs) from arginine, which is a rate-limiting factor for NO production. We have reported that neuronal NOS (nNOS)-positive motor neurons are depleted while inducible NOS (iNOS)-positive reactive astrocytes are increased in ALS transgenic [mutant SOD1 (G93A)] mice [41]. The expression of iNOS/NOS2 was correlated with the increases of astrocyte activation and NO levels while nNOS/NOS1 expression was decreased in ALS transgenic [mutant SOD1 (G93A)] mice. The high levels of NO interact with superoxide and form highly toxic peroxynitrite. Consistent with findings previously reported by Przedborski and colleagues, increased levels of NO may further exacerbate oxidative stress and trigger motor neuron death [40,41,42]. As similar to ALS transgenic mice, accumulation of 8-hydroxy-2-deoxyguanosine, a marker of oxidative DNA damage, and elevated levels of peroxinitration damage (production of nitrotyrosine residues by covalent interactions of NO) have also been found in human ALS [43,44,45,46]. These data support a prominent role of oxidative stress derived from reactive astrocytes during the pathogenesis of ALS (Fig. 1).
IS MICROGLIAL ACTIVATION A GOOD SIGN OR A BAD SIGN TO MOTOR NEURONS?
Despite its controversy, microglia are also known to be linked to motor neuronal damage and the pathogenesis of ALS via the non-cell autonomous pathway [22,47]. Interestingly, deletion of NF-κB signaling in microglia rescues motor neurons from microglial-mediated death in vitro and extended survival in ALS mice by deregulating proinflammatory microglial activation. In contrast, selective NF-κB inhibition in ALS astrocytes was not sufficient to rescue motor neuron death [48]. In this context, the microglia-mediated damage and toxicity to motor neurons are driven through the diversity of death mechanisms. Using the mice carrying deletable mutant SOD1 transgene by the action of Cre recombinase, Yamanaka and Yamashita have shown that diminishing mutant SOD1 toxicity within microglia significantly slowed the disease progression of ALS. This finding suggests that, in part, microglia contribute to neurodegenerative process of ALS [49].
On the other hand, in order to examine whether proliferating microglia leads to motor neuron degeneration in ALS mice, Gowing et al. (2008) generated double transgenic mice with CD11b-TK(mut-30) and mutant SOD1(G93A) in which a 50% reactive microglia is specifically reduced in the lumbar spinal cord [50]. Unexpectedly, reduction of reactive microglia had no effect on the degeneration of motor neuron. This study implies that proliferating microglia-expressing mutant SOD1 (G93A) does not play a pivotal role in triggering neuronal damage in an animal model of ALS. This study raises a question regarding whether different stages of microglia are involved in different modes of action for protecting versus being involved in the damaging of motor neurons through yet unidentified mechanisms. We suggest that future studies are necessary to uncover the precise action mechanism behind the obscure role of microglia in ALS.
WHY IS IT NOT CON SISTENT TO OBSERVE THE ROLE OF MICROGLIA IN THE NEURODEGENERATIVE PROCESS OF ALS?
Is microglia activation beneficial or disadvantageous to motor neurons? Microglia function is necessary for surveilancing the condition of motor neurons and for restoring tissue injury in response to acute and reversible stress: microglia are beneficial before the threshold limit reached. However, constitutive activation of microglia by a chronic and irreversible stress such as ALS stress may transform them as a non-cell autonomous player to be toxic to motor neurons: microglia are disadvantageous after they become fully activated.
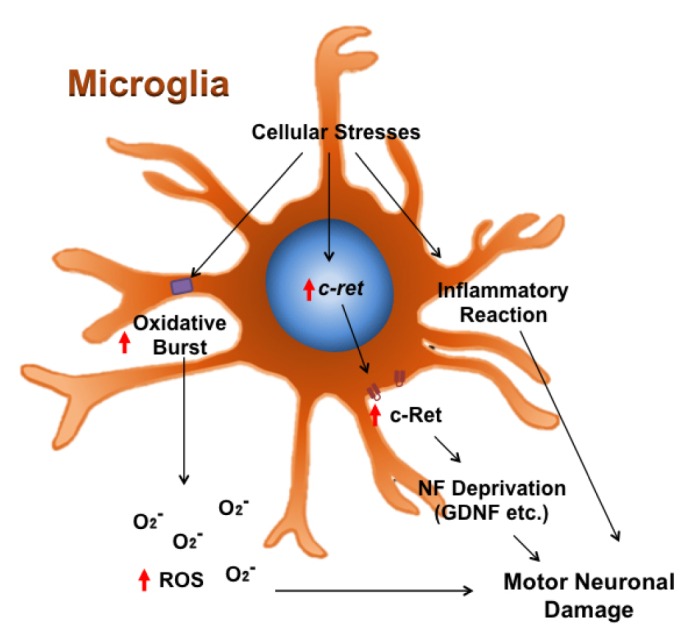
We have previously found that the expression of c-Ret is altered in motor neurons of the lumbar spinal cord in ALS transgenic [mutant SOD1 (G93A)] mice and ALS [mutant SOD1 (G85R) and (G93A)] motor neuronal cell lines [51]. c-Ret oncoprotein is a protein kinase receptor and responds to glial cell line-derived neurotrophic factor (GDNF). c-Ret-mediated signal transduction is important to maintain cellular activity and survival function. Notably, the levels of non-phosphorylated and phosphorylated c-Ret were markedly elevated in active microglia of the lumbar spinal cord of ALS mice in an age-dependent manner. Our findings suggest that ALS stress-induced expression of c-Ret in microglia may trigger non-cell autonomous toxic signals and exacerbate damage responses in motor neurons by disturbing the GDNF signaling pathway in motor neurons [51]. Our previous study does not provide a direct evidence that microglia contribute to non-cell autonomous motor neuronal damage in ALS. However, based on our findings, we suggest an indirect contribution of microglia to motor neuronal damage. For instance, the increased level of c-Ret in microglia elevates interaction with GDNF. As a result, the c-Ret and GDNF interaction promotes the survival of microglia whereas the subsequent deprivation of NFs by activated microglia in the niche of spinal cord may lead to motor neuronal damage (Fig. 2).
Fig. 2. Microglia may contribute to non-cell autonomous motor neuronal damage in ALS. Under ALS condition, cellular stresses elevate c-Ret gene expression in microglia but not in motor neurons. The increased level of c-Ret in microglia interacts with neurotrophic factors (NFs) such as glia derived neurotrophic factor (GDNF). The c-Ret and GDNF interaction in microglia improves their survival under ALS condition, whereas the deprivation of NFs in the niche of spinal cord by activated microglia may result in motor neuronal damage.
CONCLUSION
A vicious cycle of ALS stresses transforms astrocytes and microglia from Dr. Jekyll to Mr. Hyde
In the pathogenesis of ALS, non-motor neuronal cells such as astrocytes and microglia undergo a series of molecular and cellular changes in that these cells become unprofitable to motor neurons, leading to irrecoverable neurodegeneration. The mechanism of non-cell autonomous motor neuron death is closely associated with the pathophysiological change in ALS that is apparently distinguished from cell autonomous pathway.
Neuroinflammation is now identified as a key contributor to motor neuron damage in ALS [52,53,54]. Reactive astrocytes and microglia are triggers of neuroinflammation that accelerate disease progression [55,56] which is further exacerbated by ongoing neuronal injury [53]. Inflammatory cytokines released by astrocytes and microglia may facilitate glutamate excitotoxicity thereby linking neuroinflammation and excitotoxic death [18,57,58].
Taken together, previous findings suggest that the molecular and cellular adaptation between astrocytes, microglia, and motor neurons may be differently modulated by epigenetic components upon ALS stresses. In this paradigm, due to chronic oxidative stress or other irreversible mechanisms, a critical threshold limit is reached and that reactive astrocytes and microglia trigger the pathological processes that subsequently lead to a non-cell autonomous death of motor neurons in ALS. This idea suggests that future therapeutic strategy for the treatment of ALS should be aimed at specific interception of pro-oxidant and pro-death signals in a cell-type specific manner [59,60,61,62].
ACKNOWLEDGEMENTS
J.L. is an awardee of Les Turner ALS Foundation Grant. This study was supported by NIH NS52724 (H.R.) and Flagship from Korea Institute of Science and Technology (2E26200 and 2E26663 H.R.). This work was also supported by the National Research Foundation of Korea (NRF) grant (2016M3C7A1904233 H.R.) from the Korea government (MEST).
References
- 1.Kumar DR, Aslinia F, Yale SH, Mazza JJ. Jean-Martin Charcot: the father of neurology. Clin Med Res. 2011;9:46–49. doi: 10.3121/cmr.2009.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt S, Allen KD, Loiacono VT, Norman B, Stanwyck CL, Nord KM, Williams CD, Kasarskis EJ, Kamel F, McGuire V, Nelson LM, Oddone EZ. Genes and environmental exposures in veterans with amyotrophic lateral sclerosis: the GENEVA study. Rationale, study design and demographic characteristics. Neuroepidemiology. 2008;30:191–204. doi: 10.1159/000126911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak–Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 4.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 5.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 6.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 7.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 8.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 10.Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ ITALSGEN Consortium. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo JM, Ule J, Hirth F, Rogelj B, Houart C, Shaw CE. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wood H. Neurodegenerative disease: C9orf72 RNA foci--a therapeutic target for ALS and FTD? Nat Rev Neurol. 2013;9:659. doi: 10.1038/nrneurol.2013.244. [DOI] [PubMed] [Google Scholar]
- 16.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 17.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 18.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Festoff BW, Suo Z, Citron BA. Prospects for the pharmacotherapy of amyotrophic lateral sclerosis : old strategies and new paradigms for the third millennium. CNS Drugs. 2003;17:699–717. doi: 10.2165/00023210-200317100-00002. [DOI] [PubMed] [Google Scholar]
- 20.Rothstein JD. Of mice and men: reconciling preclinical ALS mouse studies and human clinical trials. Ann Neurol. 2003;53:423–426. doi: 10.1002/ana.10561. [DOI] [PubMed] [Google Scholar]
- 21.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 22.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbeito LH, Pehar M, Cassina P, Vargas MR, Peluffo H, Viera L, Estévez AG, Beckman JS. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Brain Res Rev. 2004;47:263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferraiuolo L, Higginbottom A, Heath PR, Barber S, Greenald D, Kirby J, Shaw PJ. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134:2627–2641. doi: 10.1093/brain/awr193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. 2015;63:2260–2273. doi: 10.1002/glia.22891. [DOI] [PubMed] [Google Scholar]
- 29.Endo F, Komine O, Fujimori-Tonou N, Katsuno M, Jin S, Watanabe S, Sobue G, Dezawa M, Wyss-Coray T, Yamanaka K. Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015;11:592–604. doi: 10.1016/j.celrep.2015.03.053. [DOI] [PubMed] [Google Scholar]
- 30.König HG, Coughlan KS, Kinsella S, Breen BA, Prehn JH. The BCL-2 family protein Bid is critical for pro-inflammatory signaling in astrocytes. Neurobiol Dis. 2014;70:99–107. doi: 10.1016/j.nbd.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Karki P, Smith K, Johnson J, Jr, Aschner M, Lee EY. Genetic dys-regulation of astrocytic glutamate transporter EAAT2 and its implications in neurological disorders and manganese toxicity. Neurochem Res. 2015;40:380–388. doi: 10.1007/s11064-014-1391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 33.Pardo AC, Wong V, Benson LM, Dykes M, Tanaka K, Rothstein JD, Maragakis NJ. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp Neurol. 2006;201:120–130. doi: 10.1016/j.expneurol.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 34.Ganel R, Ho T, Maragakis NJ, Jackson M, Steiner JP, Rothstein JD. Selective up-regulation of the glial Na+-dependent glutamate transporter GLT1 by a neuroimmunophilin ligand results in neuroprotection. Neurobiol Dis. 2006;21:556–567. doi: 10.1016/j.nbd.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 35.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 36.Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA, Lin CL. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet. 2003;12:2519–2532. doi: 10.1093/hmg/ddg267. [DOI] [PubMed] [Google Scholar]
- 37.Foran E, Bogush A, Goffredo M, Roncaglia P, Gustincich S, Pasinelli P, Trotti D. Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia. 2011;59:1719–1731. doi: 10.1002/glia.21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong HK, Ji KM, Min KJ, Choi I, Choi DJ, Jou I, Joe EH. Astrogliosis is a possible player in preventing delayed neuronal death. Mol Cells. 2014;37:345–355. doi: 10.14348/molcells.2014.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J, Kannagi M, Ferrante RJ, Kowall NW, Ryu H. Activation of Ets-2 by oxidative stress induces Bcl-xL expression and accounts for glial survival in amyotrophic lateral sclerosis. FASEB J. 2009;23:1739–1749. doi: 10.1096/fj.08-121046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J, Ryu H, Ferrante RJ, Morris SM, Jr, Ratan RR. Translational control of inducible nitric oxide synthase expression by arginine can explain the "arginine paradox". Proc Natl Acad Sci U S A. 2003;100:4843–4848. doi: 10.1073/pnas.0735876100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J, Ryu H, Kowall NW. Differential regulation of neuronal and inducible nitric oxide synthase (NOS) in the spinal cord of mutant SOD1 (G93A) ALS mice. Biochem Biophys Res Commun. 2009;387:202–206. doi: 10.1016/j.bbrc.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almer G, Vukosavic S, Romero N, Przedborski S. Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;72:2415–2425. doi: 10.1046/j.1471-4159.1999.0722415.x. [DOI] [PubMed] [Google Scholar]
- 43.Warita H, Hayashi T, Murakami T, Manabe Y, Abe K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Brain Res Mol Brain Res. 2001;89:147–152. doi: 10.1016/s0169-328x(01)00029-8. [DOI] [PubMed] [Google Scholar]
- 44.Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 45.Niebrój-Dobosz I, Dziewulska D, Kwieciński H. Oxidative damage to proteins in the spinal cord in amyotrophic lateral sclerosis (ALS) Folia Neuropathol. 2004;42:151–156. [PubMed] [Google Scholar]
- 46.Abe K, Pan LH, Watanabe M, Konno H, Kato T, Itoyama Y. Upregulation of protein-tyrosine nitration in the anterior horn cells of amyotrophic lateral sclerosis. Neurol Res. 1997;19:124–128. doi: 10.1080/01616412.1997.11740784. [DOI] [PubMed] [Google Scholar]
- 47.Brites D, Vaz AR. Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci. 2014;8:117. doi: 10.3389/fncel.2014.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamanaka K, Yamashita H. ALS and microglia--a player for non-cell-autonomous neuron death. Brain Nerve. 2007;59:1163–1170. [PubMed] [Google Scholar]
- 50.Gowing G, Philips T, Van Wijmeersch B, Audet JN, Dewil M, Van Den Bosch L, Billiau AD, Robberecht W, Julien JP. Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J Neurosci. 2008;28:10234–10244. doi: 10.1523/JNEUROSCI.3494-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryu H, Jeon GS, Cashman NR, Kowall NW, Lee J. Differential expression of c-Ret in motor neurons versus non-neuronal cells is linked to the pathogenesis of ALS. Lab Invest. 2011;91:342–352. doi: 10.1038/labinvest.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, West M, Williamson KS. On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal. 2006;8:2075–2087. doi: 10.1089/ars.2006.8.2075. [DOI] [PubMed] [Google Scholar]
- 53.McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- 54.Sargsyan SA, Monk PN, Shaw PJ. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia. 2005;51:241–253. doi: 10.1002/glia.20210. [DOI] [PubMed] [Google Scholar]
- 55.Henkel JS, Engelhardt JI, Siklós L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312:60–73. doi: 10.1016/s0042-6822(03)00181-8. [DOI] [PubMed] [Google Scholar]
- 57.Pickering M, Cumiskey D, O'Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- 58.Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85:2059–2070. doi: 10.1002/jnr.21325. [DOI] [PubMed] [Google Scholar]
- 59.Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH, Jr, Ferrante RJ. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- 60.Lee J, Ryu H, Kowall NW. Motor neuronal protection by L-arginine prolongs survival of mutant SOD1 (G93A) ALS mice. Biochem Biophys Res Commun. 2009;384:524–529. doi: 10.1016/j.bbrc.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jung MK, Kim KY, Lee NY, Kang YS, Hwang YJ, Kim Y, Sung JJ, McKee A, Kowall N, Lee J, Ryu H. Expression of taurine transporter (TauT) is modulated by heat shock factor 1 (HSF1) in motor neurons of ALS. Mol Neurobiol. 2013;47:699–710. doi: 10.1007/s12035-012-8371-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee J, Ryu H, Keum G, Yoon YJ, Kowall NW, Ryu H. Therapeutic targeting of epigenetic components in amyotrophic lateral sclerosis (ALS) Curr Med Chem. 2014;21:3576–3582. doi: 10.2174/0929867321666140706131825. [DOI] [PubMed] [Google Scholar]