

Significance
Immune checkpoint therapies have garnered significant attention due to their ability to induce dramatic clinical responses in patients with various solid tumor malignancies, including prostate cancer. However, these therapeutic agents often elicit immune-related adverse events (irAEs) that may result in substantial morbidity. Early intervention can markedly reduce the severity of the irAEs, but biomarkers that allow for their early detection and guide their management are lacking. Based on peripheral blood samples collected longitudinally in two prostate cancer clinical trials, we propose CD8 T-cell clonal expansion within the systemic circulation as a potential correlative biomarker of immune-related adverse events that occur with ipilimumab therapy.
Keywords: ipilimumab, T cells, CD8, toxicities, prostate cancer
Abstract
Immune checkpoint therapies, such as ipilimumab, induce dramatic antitumor responses in a subset of patients with advanced malignancies, but they may also induce inflammatory responses and toxicities termed immune-related adverse events (irAEs). These irAEs are often low grade and manageable, but severe irAEs may lead to prolonged hospitalizations or fatalities. Early intervention is necessary to minimize morbidities that occur with severe irAEs. However, correlative biomarkers are currently lacking. In a phase II clinical trial that treated 27 patients with metastatic prostate cancer, we aimed to test the safety and efficacy of androgen deprivation therapy plus ipilimumab. In this study, we observed grade 3 toxicities in >40% of treated patients, which led to early closure of the study. Because ipilimumab enhances T-cell responses, we hypothesized that increased clonal T-cell responses in the systemic circulation may contribute to irAEs. Sequencing of the T-cell receptor β-chains in purified T cells revealed clonal expansion of CD8 T cells, which occurred in blood samples collected before the onset of grade 2–3 irAEs. These initial results suggested that expansion of ≥55 CD8 T-cell clones preceded the development of severe irAEs. We further evaluated available blood samples from a second trial and determined that patients who experienced grade 2–3 irAEs also had expansion of ≥55 CD8 T-cell clones in blood samples collected before the onset of irAEs. We propose that CD8 T-cell clonal expansion may be a correlative biomarker to enable close monitoring and early intervention for patients receiving ipilimumab.
Cancer immunotherapies that target the T-cell immune checkpoints, such as CTLA-4, PD-1, and PD-L1, promote tumor-specific T-cell responses. Ipilimumab, nivolumab, and pembrolizumab are immune checkpoint therapies that induce durable antitumor responses and improve survival in melanoma (1–4), nonsmall cell lung cancer (NSLC) (5, 6), and renal cell carcinoma (RCC) (7). Clinical trials with ipilimumab in men with castration-resistant prostate cancer (CRPC) have reported limited efficacy to date (1, 2, 8–12). We hypothesized that adding ipilimumab shortly after the start of androgen deprivation therapy (ADT) in men with metastatic prostate cancer, before the disease becomes castration resistant, would enhance the efficacy of the treatment. To address this hypothesis, we conducted a phase II clinical trial (ClinicalTrials.gov, #NCT01377389) in patients with metastatic castration-naïve/-sensitive prostate cancer, who received ipilimumab plus ADT for a finite duration. The clinical trial closed early due to greater than 40% grade 3 toxicities, which was a prespecified safety end point.
Immune checkpoint therapies are known to induce inflammatory tissue damage or immune-related adverse events (irAEs), including inflammation of skin (rash, dermatitis), liver (transaminitis), endocrine axis (hypophysitis, thyroiditis), or gastrointestinal tract (diarrhea, colitis) (13). In a phase III study, ∼60% of ipilimumab-treated men with CRPC experienced irAEs, of which about 25% were grade 3–4 toxicities (12), which are frequencies similar to those observed with ipilimumab monotherapy in advanced melanoma patients (1, 2, 14). Moreover, although recent studies suggest that the combination of immune checkpoint inhibitors significantly increases the rate of therapeutic benefit, the proportion of patients experiencing grade 3–4 irAEs virtually doubles when ipilimumab is combined with nivolumab (14).
Severe irAEs can be life threatening and may lead to prolonged hospitalization and morbidities. Although most of the severe irAEs can be reversed with high-dose steroids and/or other immunosuppressive therapies, they often require protracted courses of immunosuppression, which frequently results in additional complications and significant morbidities (13). Early recognition of irAEs, before their worsening to grade 3–4 severity, allows for prompt initiation of immunosuppressive treatments, which in turn curtails the toxicity more effectively and thus requires a shorter and less intense course of immunosuppression. Therefore, biomarkers that correlate with severe irAEs in patients receiving immune checkpoint therapies would greatly improve clinical management and patient outcomes.
To identify such markers, we examined longitudinally collected peripheral blood samples of patients accrued to our phase II study of finite ADT plus ipilimumab. Based on published data that clonal expansion of T cells in tumor tissues, which are likely tumor reactive, may predict for clinical benefit with immune checkpoint therapy (15), we hypothesized that clonal expansion of T cells in the systemic circulation, which are likely to be reactive against self-antigens, may correlate with irAEs. Therefore, we purified CD4 and CD8 T cells from the collected peripheral blood mononuclear cells (PBMCs) and performed next-generation sequencing of complementarity-determining region 3 (CDR3) regions in rearranged T-cell receptor (TCR) β-chains to retrospectively evaluate T-cell clonal expansion. Our data revealed that clonal expansion of CD8 T cells in blood samples that were collected before the start of irAEs correlated with the development of subsequent grade 2–3 irAEs. Our data highlight the possibility of using CD8 T-cell clonal expansion as a correlative biomarker to closely monitor and intervene early to minimize severe toxicities that occur with ipilimumab therapy.
Results
Phase II Clinical Trial of Finite ADT Plus Ipilimumab in Patients with Metastatic Prostate Cancer.
To study the safety and efficacy of combining ADT plus ipilimumab in patients with metastatic castration-sensitive prostate cancer, patients received 8 mo of finite ADT [with gonadotropin-releasing hormone (GnRH) analogs such as degarelix and leuprolide], and within 1–4 wk of ADT initiation began a course of up to four doses of ipilimumab (10 mg/kg), with each dose given 4 wk apart (Fig. 1A). Our trial was designed to accrue a total of 48 patients but closed early when greater than 40% of subjects developed grade 3 drug-related toxicities, as predetermined in the safety monitoring plan. We screened 30 patients: 2 were ineligible and 1 withdrew consent before receiving ipilimumab; therefore, 27 patients were evaluable for safety and toxicity. Baseline characteristics of these 27 patients are described in Table S1. Seven patients had prior local therapy for their prostate tumor, two of which received prior ADT for 3–4 mo with radiation therapy (RT; Table S2). Fifteen of 27 patients (56%) received all four doses of ipilimumab. Twenty-four of 27 patients (89%) were evaluable for clinical responses, as 1 patient was lost to follow-up and 2 received alternative treatments as per the treating physicians’ decisions. The median follow-up time from treatment initiation was 36.2 mo [interquartile range (IQR), 23.7–44.5].
Fig. 1.

Clinical trial schema and clinical responses. (A) Clinical trial treatment [ipilimumab (IPI) plus finite ADT] and blood draw schemata. (B) Time to PSA progression from treatment initiation based on PCWG2 criteria. The y axis numbers each individual patient based on initiation of treatment with ipilimumab. Each bar represents an individual patient. The arrowhead depicts that the patient’s clinical response is ongoing. (C) Radiographic responses for patient 1. MRI of the pelvis of patient 1 at baseline and posttreatment. (a) A 2.0- × 2.4-cm infiltrating mass in the right peripheral zone of a 6.5- × 5.1- × 5.6-cm prostate. (b) A 3.4- × 1.5-cm metastasis involving the right inferior pubic ramus. (c) Resolution of the prostatic mass. (d) Resolution of the bony metastasis following treatment with ADT plus ipilimumab.
Table S1.
Baseline characteristics of patients in a phase II clinical trial
| Characteristics | Median (IQR) |
| Age, y | 62 (54–64) |
| PSA, ng/mL | 46.6 (12.9–160.6) |
| Hgb, g/dL | 14.3 (13.5–14.9) |
| Alkaline phospatase, U/L | 88 (70–117) |
| LDH, U/L | 445 (413–505) |
| Albumin, g/dL | 4.5 (4.4–4.6) |
| Gleason score | 9 (8–9) |
Table S2.
Prior local and systemic treatments in a phase II clinical trial
| Prior treatments | No. (%) |
| No localized treatments | 20 (74) |
| Localized treatments | 7 (26) |
| Radical prostatectomy (RP) | 4 (15) |
| Primary radiation therapy (RT) | 1 (4) |
| Salvage RT | 3 (11) |
| Adjuvant RT | 1 (4) |
| Brachytherapy | 1 (4) |
| Cryoablation | 1 (4) |
| Systemic treatments with RT | 2 (7) |
| ADT (3 mo) | 1 (4) |
| ADT (4 mo) | 1 (4) |
Safety and Efficacy of Finite ADT Plus Ipilimumab.
Six of 24 patients (25%) had prostate-specific antigen (PSA) progressions during ADT (<8 mo after starting treatment); therefore, these patients remained on continuous ADT. In the remaining 18 patients, following discontinuation of ADT, serum testosterone recovered to noncastrate levels (≥50 ng/dL) at a median of 87 d (IQR, 78–111). Twenty-three of the 24 patients (96%) experienced PSA progression as per Prostate Cancer Working Group 2 (PCWG2) criteria (Fig. 1B and Table 1) (16). The median time to PSA progression from ADT initiation was 10.0 mo (IQR, 5.9–13.3). Two patients (patients 1 and 4) had complete radiographic responses attributed to the combination of ADT plus ipilimumab (Fig. 1C), and patient 4 remains without PSA progression after 48.9 mo on study. Thirteen of 24 patients (54%) have developed CRPC (median time = 27.5 mo), and 8 of 24 patients (33%) have died from their disease (Table 1). Twelve of 27 patients (44%) developed grade 3 toxicities attributed to treatment. As in other trials of ipilimumab (8–10, 12, 13), the most common grade 3 irAEs were transaminitis (15%), diarrhea/colitis (11%), and hypophysitis (7%; Table S3). No grade 4 or 5 toxicities were observed in this study.
Table 1.
Metastatic site distribution and clinical responses in a phase II clinical trial (n = 27)
| Patient no. | Bone 1–3 lesions | Bone ≥4 lesions | Lymph node | Lung | Total no. IPI doses | Time to PSA progression (mo) | Time to CRPC (mo) | Survival (mo) |
| 1 | X | 4 | 16.1 | - | Ongoing | |||
| 2 | X | X | 4 | 5.9 | 5.9 | 31.1 | ||
| 3 | X | X | 2 | 10.6 | — | Ongoing | ||
| 4 | X | X | 4 | NR | — | Ongoing | ||
| 5 | X | X | 3 | 4.8 | 4.8 | 24.7 | ||
| 6 | X | 2 | 2.5 | 2.5 | 23.3 | |||
| 7 | X | 4 | 11.0 | 38.6 | Ongoing | |||
| 8 | X | 1 | Alternative treatment | |||||
| 10 | X | X | 2 | Lost to follow-up | ||||
| 11 | X | 3 | Alternative treatment | |||||
| 12 | X | 4 | 13.0 | — | Ongoing | |||
| 13 | X | X | 4 | 9.8 | 15.3 | Ongoing | ||
| 14 | X | 4 | 20.4 | — | Ongoing | |||
| 15 | X | X | 4 | 9.1 | 13.2 | 14.2 | ||
| 16 | X | 2 | 9.5 | 27.5 | 39.5 | |||
| 17 | X | X | 4 | 9.8 | 9.8 | Ongoing | ||
| 18 | X | 4 | 20.9 | — | Ongoing | |||
| 19 | X | 4 | 4.0 | 4.0 | 22.7 | |||
| 20 | X | 4 | 5.3 | 5.3 | 39.5 | |||
| 23 | X | 2 | 14.0 | — | Ongoing | |||
| 24 | X | 1 | 14.2 | — | Ongoing | |||
| 25 | X | 2 | 5.5 | 5.5 | Ongoing | |||
| 26 | X | 2 | 13.3 | — | Ongoing | |||
| 27 | X | X | 3 | 11.8 | 23.7 | 26.3 | ||
| 28 | X | 4 | 12.1 | — | Ongoing | |||
| 29 | X | X | X | 4 | 9.4 | 14.4 | Ongoing | |
| 30 | X | 4 | 10.0 | — | Ongoing | |||
NR, not reached.
Table S3.
Adverse events related to study drugs in a phase II clinical trial
| Adverse events | Any grade [no. of patients (%)] | Grade 3 [no. of patients (%)] | Grade 4 [no. of patients (%)] |
| Any event | 27 (100%) | 12 (44%) | — |
| Skin rash | 17 (63%) | 1 (4%) | — |
| Endocrine | |||
| Adrenal insufficiency | 1 (4%) | — | — |
| Hypophysitis | 4 (15%) | 2 (7%) | — |
| Hypothyroidism | 4 (15%) | — | — |
| Gastrointestinal | |||
| Colitis/diarrhea | 11 (41%) | 3 (11%) | — |
| Nausea | 4 (15%) | — | — |
| Hepatic | |||
| Increased ALT | 15 (56%) | 4 (15%) | — |
| Increased AST | 13 (48%) | 4 (15%) | — |
| Neurological | |||
| Confused-state | 1 (4%) | 1 (4%) | — |
| Headache | 2 (7%) | — | — |
| Sensory neuropathy | 2 (7%) | — | — |
| General | |||
| Fatigue | 9 (33%) | — | — |
| Influenza-like illness | 1 (4%) | — | — |
| Pyrexia | 4 (15%) | — | — |
| Other | |||
| Anemia | 1 (4%) | 1 (4%) | — |
| Anorexia | 1 (4%) | — | — |
| Creatinine | 4 (15%) | 1 (4%) | — |
| Edema | 4 (15%) | 1 (4%) | — |
| Hypotension | 1 (4%) | — | — |
| Leukopenia | 2 (7%) | — | — |
| Weight loss | 3 (11%) | — | — |
Evaluation for Biomarkers That May Correlate with Clinical Outcomes.
We sought to identify candidate immune biomarkers that correlate with clinical outcomes in men with metastatic castration-sensitive prostate cancer. As our group previously reported (17–19), we identified an increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker of ipilimumab therapy (Fig. S1A). To retrospectively analyze our data for potential correlative biomarkers of clinical benefit, we defined a long disease-free interval (>16 mo) before PSA progression as clinical benefit and a short disease-free interval (<8 mo) before PSA progression was defined as having no clinical benefit. We found that patients with clinical benefit were more likely to have increased frequency of CD3 T cells expressing CTLA-4 in pretreatment blood samples (Fig. S1B). We also found that men with clinical benefit had an increased ratio of CTLA-4+ CD3 T cells to PD-1+ CD3 T cells in pretreatment blood samples (Fig. S1B), suggesting that a ratio of CTLA-4+ CD3:PD-1+ CD3 T cells ≥0.9 correlates for clinical benefit from ipilimumab.
Fig. S1.

Flow cytometry-based peripheral blood candidate biomarkers. (A) CD4 T-cell ICOS expression as a pharmacodynamic marker of ipilimumab treatment. Flow cytometry analysis of ICOS expression of CD4 T cells (as a percentage of CD3CD4 T cells) before each dose of ipilimumab [IPI; baseline (pre-IPI dose 1) and following each dose] PBMCs. Each symbol represents an individual patient. The error bars represent mean and SD. (B) Baseline frequency of costimulatory markers in CD3 (Top) and CD4 (Bottom) T cells in the peripheral blood based on PSA responses. Flow cytometry analysis of baseline (pre-IPI treatment) PBMCs for expression of CTLA-4, OX-40, and PD-1 (as a percentage of CD3 and CD4 T cells) and CTLA-4:PD-1. Each symbol represents an individual patient. The error bars represent mean and SD.
We next chose to retrospectively evaluate whether clonal expansion of T cells correlates with clinical benefit and toxicities. Therefore, we purified CD4 and CD8 T cells from longitudinal blood samples of 20 patients (Fig. S2A and Table 2) and performed next-generation sequencing to determine T-cell receptor clonality (20). Clonality is a metric ranging from 0 to 1 that describes the shape of the clone frequency distribution; values approaching 1 indicate an increasingly asymmetric distribution of relative abundances and are indicative of T-cell activation and concomitant expansion of a mono- or oligo-clonal population of T cells. CD4 and CD8 T-cell clonality in pretreatment blood samples did not correlate with clinical benefit (Fig. S2B) or toxicity outcomes (Fig. S2C). To determine whether CD4 and CD8 T-cell clonal expansion in posttreatment blood samples correlate with clinical outcomes, we quantified the number of unique CD4 and CD8 T-cell clones that expanded within each patient’s repertoire after each dose of ipilimumab (Fig. 2A). Neither CD4 nor CD8 T-cell clonal expansion within the systemic circulation was significantly different between patients who were identified as having clinical benefit and those who did not (Fig. S2D).
Fig. S2.

Enrichment of CD4 and CD8 T cells from PBMCs and immunological biomarkers. (A) (Top) CD4 and CD8 T-cell expression in PBMCs based on flow cytometric analysis. Following purification of CD4 T cells (negative selection) and CD8 T cells (positive selection) from PBMCs, flow cytometric analysis was used to determine the purity of the CD4 (Bottom Right) and CD8 (Bottom Left) T-cell populations. In this patient, the CD4 population is enriched from 43.7% to 98.3% purity, and the CD8 population is enriched from 37.7% to 95.9%. (B) Baseline CD4 and CD8 T-cell clonality from pre-IPI (baseline) treatment samples were used to determine T-cell clonality and to make comparisons between patients who experienced PSA progression greater than 16 mo (clinical benefit) vs. less than 8 mo (no clinical benefit). (C) Baseline CD4 and CD8 T-cell clonality from pre-IPI (baseline) treatment samples was used to determine T-cell clonality and to make comparisons between patients who experienced grade 0–1 irAEs vs. grade 2–3 irAEs. (D) Comparison between patients who experienced PSA progression greater than 16 mo (clinical benefit) vs. less than 8 mo (no clinical benefit). The number of expanded T-cell clones was determined from post-IPI dose 1 vs. baseline samples.
Table 2.
irAEs in a subset of patients (N=20) in the Phase II clinical trial
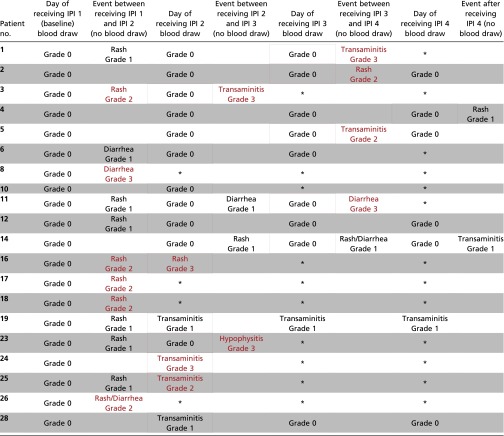 |
Represents samples that were unavailable.
Red rectangles indicate blood draw used for predicting Grade 2-3 irAEs; Blue rectangles indicate blood draw used for predicting Grade 0-1 irAEs.
Fig. 2.

Evaluation of immunological biomarkers that correlate with irAEs. (A) Scatter plots of CD4 and CD8 TCR clone frequencies within sorted T-cell populations from post-IPI dose 1 or 2 samples vs. pre-IPI (baseline) treatment samples for patients with no toxicity at the time of sample collection (patients 17 and 28), a patient experiencing grade 3 transaminitis (patient 3), and a patient experiencing grade 3 diarrhea (patient 11). (B) Comparison between patients who experienced grade 0–1 irAEs vs. grade 2–3 irAEs. The number of expanded of T-cell clones was determined in samples collected just before a grade 2–3 irAE relative to pre-IPI treatment samples vs. patients with grade 0–1 irAEs in both clinical trials. For patients with a grade 0 toxicity, we used post-IPI dose 1 samples to identify expanded clones.
To determine whether CD4 and CD8 T-cell clonal expansion correlated with irAEs, we analyzed available samples from 16 evaluable patients. CD4 T-cell clonal expansion did not significantly differ between patients experiencing grade 2–3 irAEs vs. patients experiencing grade 0–1 irAEs. However, we found that CD8 T-cell clonal expansion was significantly greater in patients developing grade 2–3 irAEs vs. grade 0–1 irAEs (Fig. 2B). Importantly, the CD8 clonal expansion was noted in blood samples obtained before the development of the grade 2–3 immune-related toxicities, and the median time between the blood draw and the onset of toxicity was 13 d (IQR, 2–24).
Circulating CD8 T-Cell Clonal Expansion Precedes the Development of Grade 2–3 irAEs.
We sought to provide additional evidence that CD8 T-cell clonal expansion precedes the development of subsequent grade 2–3 irAEs. Therefore, we formulated a receiver operating characteristic (ROC) curve that yielded an area under the curve of 0.87 (values closer to 1 denote an excellent classifier) using 35 available blood samples collected from the 16 patients (Fig. 3A). A logistic regression model demonstrates that the number of expanded CD8 T-cell clones had a strong association with irAEs (P = 0.01) with an odds ratio (OR) of 1.02 (95% CI, 1.01, 1.04) or a 2% increase in risk of irAE for each additional expanded clone (Table S4), which equates to a 27% increase of irAE for an increase of 10 expanded clones (OR, 1.27; 95% CI, 1.06, 1.51). Taken together, we found that the expansion of 55 or more CD8 T-cell clones resulted in a sensitivity of 100% of patients who would experience grade 2–3 irAEs.
Fig. 3.

CD8 clonal expansion precedes grade 2–3 irAEs. (A) The ROC curve of expanded CD8 clones. (B) Comparison between pooled patients from two clinical trials who experienced grade 0–1 irAEs vs. grade 2–3 irAEs. The number of expanded T-cell clones was determined in samples collected just before a grade 2–3 irAE relative to pre-IPI treatment samples vs. patients with grade 0–1 irAEs in both clinical trials. For patients with a grade 0 toxicity, we used post-IPI dose 1 samples to identify expanded clones.
Table S4.
Specificity and sensitivity based on CD8 T-cell clonal expansion
| No. expanded clones or more | Model predicted probability of irAE | % True positives detected (sensitivity) | % Negatives found as positive (1 − specificity) |
| 55 | 0.10 | 100% | 42% |
| 65 | 0.13 | 89% | 38% |
| 97 | 0.24 | 78% | 19% |
| 133 | 0.42 | 67% | 12% |
| 157 | 0.56 | 56% | 4% |
Logistic regression model:
To further test this concept, we retrospectively evaluated CD8 T-cell clonal expansion in blood samples collected from a second, independent, cohort of 11 patients enrolled on a separate clinical trial whereby patients with localized prostate cancer received ADT plus ipilimumab therapy before radical prostatectomy (ClinicalTrials.gov, #NCT01194271). Nine of 11 patients (82%) in this study developed grade 2–3 irAEs. Analysis of blood samples collected before the development of these grade 2–3 toxicities revealed CD8 T-cell clonal expansion of 55 or greater in all nine of these patients (Table 3). These data indicate that CD8 T-cell clonal expansion may be a biomarker that would enable increased surveillance of patients on treatment such that patients found to have 55 or more expanded clones would be expected to have a 100% probability of developing grade 2–3 irAEs at a later time.
Table 3.
Correlation between CD8 T cell clonal expansion and irAEs in a second clinical trial
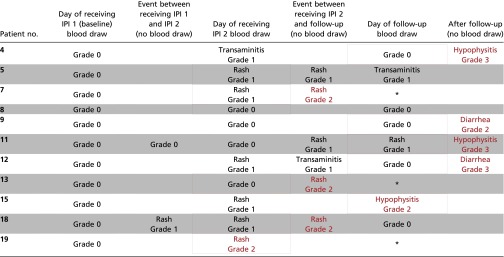 |
Represents samples that were unavailable.
Red rectangles indicate blood draw used for predicting Grade 2-3 irAEs.
Blue rectangles indicate blood draw used for predicting Grade 0-1 irAEs.
In a pooled retrospective analysis of independent cohorts of patients from our two clinical trials, consisting of a total of 27 patients, we observed a statistically significant difference in clonal expansion of CD8 T cells in peripheral blood for patients who developed grade 0–1 irAEs compared with those who developed grade 2–3 irAEs (Fig. 3B). These findings will need to be evaluated prospectively in larger cohorts of patients for confirmation and to determine feasibility for incorporation into clinical practice, which would provide a useful tool for physicians to determine when to institute close monitoring of patients receiving ipilimumab.
Discussion
Although limited by the toxicities encountered, our experience suggests that a subset of patients with prostate cancer may derive significant benefit from the addition of ipilimumab. We observed sustained responses following combination therapy with ipilimumab plus finite ADT in 4 of 24 patients and complete radiographic responses in 2 patients. Remarkably, one of these patients continues to require no additional treatment after completing finite ADT plus ipilimumab over 4 y ago. Importantly, the frequency of ICOS+CD4 T cells in peripheral blood increased following ipilimumab treatment suggesting that ipilimumab plus ADT combination therapy had similar pharmacodynamic impact as previously reported for ipilimumab monotherapy. In addition, consistent with the concept that T cells expressing CTLA-4 may represent an activated subset of cells, whereas those with low levels of PD-1 may characterize a less exhausted subpopulation, we identified baseline CTLA-4+ CD3 T cells and the ratio of CTLA-4+ CD3 T cells to PD-1+ CD3 T cells within the systemic circulation as potential correlative biomarkers of clinical benefit with ipilimumab therapy, which will need to be tested in larger cohorts of patients. Previous studies found that high expression of CTLA-4 and PD-1 on CD4 T cells in baseline peripheral blood samples as potential correlative biomarkers of clinical benefit in men with CRPC treated with ipilimumab combinations (21, 22).
Immune checkpoint therapies are expanding their clinical indications from melanoma to other solid tumors (5, 7) and even hematological diseases such as refractory Hodgkin’s lymphoma (23). Furthermore, combining immune checkpoint therapies, such as ipilimumab plus nivolumab, appear to increase the rate of clinical benefit. However, the frequency of toxicities is also increased with combination therapy (14). As combination therapy has such significant clinical benefit and is likely to become a broad standard in oncology, a better understanding and management of its associated toxicities is needed.
Consistent with the experiences in other tumor types, common irAEs in this and other trials included rash/pruritus, diarrhea/colitis, and transaminitis. Interestingly, immune-mediated toxicities are relatively similar independent of the tumor type and immune checkpoint being targeted. These observations suggest that irAEs develop from similar mechanisms of action, especially because immune checkpoint therapies target T-cell responses. Although most irAEs induced by immune checkpoint therapies can be reversed with high-dose steroids and/or other immunosuppressive therapies, prolonged immunosuppression can have severe consequences, including acquisition of life-threatening opportunistic infections (24, 25). Cumulative experiences show that the earlier immunosuppressive therapies are initiated, the shorter the required course will be; however, early manifestations of irAEs are often subtle, and the start of immunosuppressive therapies is frequently delayed. Therefore, a biomarker that can be used to monitor severe irAEs and allow for early intervention is critically needed.
Previously published data indicate that increased T-cell clonality in the tumor microenvironment correlates with clinical response to anti–PD-1 therapy (15). Because immune checkpoint therapies affect T-cell function, it may be that T-cell responses in the tumor tissues would be beneficial but T-cell responses in the systemic circulation may indicate responses against self-antigens, which may lead to toxicities. Our data indicate that peripheral blood CD8 T-cell clonal expansion precedes the onset of grade 2–3 irAEs. Specifically, expansion of 55 or more CD8 T-cell clones in the peripheral blood strongly correlated with subsequent development of grade 2–3 irAEs. This finding was substantiated using an independent cohort of prostate cancer patients who also received ADT plus ipilimumab, whereby the expansion of 55 or more CD8 T-cell clones occurred before the development of grade 2–3 irAEs. Although there are likely many mechanisms that contribute to irAEs, our study highlights CD8 T-cell clonal expansion as one plausible factor. Clonal expansion of CD8 T cells in the peripheral blood could become an important clinical tool to guide early intervention for immune checkpoint toxicities across tumor types, but this will need to be tested prospectively in additional cohorts of patients.
Although it remains to be defined how to optimally integrate ipilimumab into the current armamentarium of prostate cancer therapies, these data indicate that additional studies in patients with advanced metastatic prostate cancer are warranted to evaluate other immune checkpoint treatment strategies with ipilimumab, possibly with a lower dose of 3 mg/kg, as is used in patients with metastatic melanoma, instead of 10 mg/kg, as was used here. Importantly, the frequency of severe irAEs appears to be directly correlated with the dose of ipilimumab (26, 27).
Nonetheless, for the current immune checkpoint agents, irAEs have been reported for all of the approved doses to date. Cumulative data indicate that early diagnosis and intervention for irAEs are important for successful resolution of the toxicities and use of minimal doses of steroids or other immunosuppressive therapies. The clinical challenge is that the manifestations of these toxicities are often subtle and the start of immunosuppressive therapies is frequently delayed. Therefore, the possibility of using CD8 T-cell clonal expansion as a biomarker that may potentially predict for severe irAEs and allow for early intervention represents a first step forward and will require additional evaluation.
Methods
Patients.
We conducted an open-label, single-center, phase II study of ADT in combination with ipilimumab. To be eligible, patients had to be at least 18 y of age and have histologically confirmed prostate carcinoma, radiographic evidence of metastatic disease, and noncastrate disease or must have received ADT within 1 mo of starting ipilimumab. Prior hormonal therapy for nonmetastatic disease was allowed if the patient had been off ADT for 1.5 times the time that they were on it. Other eligibility criteria included adequate hematological, renal, and hepatic function and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1. The study was approved by the institutional review board of The University of Texas M.D. Anderson Cancer Center. Written informed consent for participation in the study was obtained from all participants.
Analysis of the T-Cell Receptor β-Expressing Repertoire in Peripheral Blood.
Genomic DNA from sorted cells was extracted using the Qiagen Symphony according to the manufacturer’s instructions. Samples were quantified using Dropsense96 and diluted for library preparation. Amplification and sequencing of CDR3 regions in rearranged TCR β-chains was performed using the immunoSEQ Assay (Adaptive Biotechnologies). The immunoSEQ assay combines multiplex PCR with high-throughput sequencing and a sophisticated bioinformatics pipeline for TCRβ CDR3 region analysis (28, 29). Diversity is calculated as Shannon’s Entropy by , where pi is the proportional abundance of clone i, and N is the number of unique clones detected in the sample. Clonality is defined as 1 − Pielou’s eveness metric and is calculated by . Diversity describes the richness of the sample (i.e., the number of unique sequences) and their proportional evenness, whereas clonality describes the shape of the distribution of proportional abundances and ranges from 0 to 1. Clonality values approaching one indicate an increasingly asymmetric distribution in which a few clones are present at high frequencies (28, 29).
SI Methods
Treatment and Evaluation.
Patients received ADT for 8 mo, in a setting in which universal recurrence was expected. Between 1 and 4 wk after initiation of ADT, ipilimumab at 10 mg/kg i.v. was added for a planned four doses given at 4-wk intervals. The primary end point was to estimate the proportion of patients treated with ipilimumab plus ADT that had PSA ≤0.2 ng/mL at 7 mo. Secondary end points included the assessment of time to testosterone recovery (≥50 ng/dL), time to progression of disease, and overall survival; the characterization of safety and drug-related adverse events; and the profiling of baseline and treatment-induced immunological changes.
The trial was originally designed in 2009 to use a Simon’s optimal two-stage design to stop the trial if ≤7 among the first 16 patients achieved a PSA ≤0.2 ng/mL at month 7. However, accrual started in 2011 and based on new data indicating that evidence of the efficacy of ipilimumab could emerge at later time points, the study was changed to a single-stage design. It was assumed that finite ADT plus ipilimumab would result in 60% of patients achieving PSA ≤0.2 ng/mL at month 7. A proportion of ≤40% would result in rejection of the combination therapy. A sample size of 48 patients (computed using nQuery Advisor 7) would have an 80% power to detect this 20% difference using a χ2 test and with a two-sided type I error rate of 0.05.
The safety monitoring plan predetermined the suspension of accrual if ≥40% of treated subjects experienced ≥grade 3 drug-related toxicities; ≥10% experienced ≥grade 3 irAEs that could not be alleviated or controlled by appropriate care within 14 d of the initiation of such therapy; or ≥2 ipilimumab-related deaths occurred, unless attributed to progression. AEs were graded according to the Common Terminology Criteria for Adverse Events, version 3.0.
Collection of PBMCs.
Whole blood was collected in cell preparation tubes (CPTs) containing sodium heparin (BD Vacutainer) and centrifuged at 800 × g for 25 min. The interface cells were harvested and washed twice with PBS with 10% (vol/vol) FCS at 500 and 450 × g for 10 min, respectively. PBMCs were then resuspended in complete RPMI 1640 with 10% (vol/vol) autologous plasma and 10% (vol/vol) DMSO for storage.
Flow Cytometry Analysis.
Multiparameter flow cytometric analysis of different cell subsets was performed in the harvested PBMCs. CD4 and CD8 T cells were stained with CD4-APCCy7 and CD8-Amcyan, respectively, and gates were set according to isotype controls and for cells to be lineage negative (CD14, CD16, CD19, CD20, CD56, CD303, and γ/δ; all antibodies were from BD Biosciences, except for CD303, Miltenyi). Antibodies used for evaluating different subsets of CD4 and CD8 T cells included ICOS PECy7 (eBioscience), PD-1-Pacific Blue (Biolegend), OX40-PE (BD Biosciences), and CTLA-4-PECy5 (BD Biosciences). Gates were set according to isotype controls. Acquisition of data were carried out on a FACS Canto II (BD Biosciences) and analyzed with the software FlowJo (Tree Star).
Isolation of CD4 and CD8 T Cells from PBMCs.
Isolation of CD4 and CD8 T cells was performed with an EasySep magnet following the manufacturer’s protocol (Stemcell Technologies). The quality of the CD4 and CD8 T-cell purifications was confirmed by flow cytometry. Sorted CD4 and CD8 T cells were sent to Adaptive Biotechnologies for further analysis.
Statistical Analyses.
Differences in candidate biomarker levels between groups of patients were compared using the Mann–Whitney test. Differences within the same patients at subsequent time points were compared with the paired Mann–Whitney test. The numbers of CD8 expanded clones were determined as described in a prior study was based on using a false discovery rate (FDR) of 0.05 for selecting each clone as expanded or not (30). An FDR of 0.05 was selected because the numbers of expanded clones started to become stable for many patients around 0.05 compared with smaller values. Larger FDR levels were not considered. An ROC curve was created from the logistic regression model. The numbers of expanded clones at different times in the same patient are assumed to behave independently. A logistic regression model was implemented to assess the role of CD8 expanded clones in identifying patients who are at risk for developing grade 2–3 irAEs. Examination of the data indicates this may be a reasonable assumption even though the small sample size of repeated measures on the same person does not allow this assumption to be fully evaluated. The results from the initial cohort of patients were used to determine whether the cutoff established would also provide similar results in the second, independent, cohort of patients. Logistic regression and ROC curves were implemented in SAS 9.3 (SAS Institute). All other analyses and figures were performed in Prism version 6.05 (GraphPad Software).
Acknowledgments
We thank Drs. Paul Corn and Nizar Tannir for helping accrue patients on this clinical trial. Statistical support was provided by the M.D. Anderson Biostatistics Resource Group. This work was also performed in part by M. D. Anderson’s institutional Immunotherapy Platform, and we thank Ashura Khan, Program Director, for assistance. Most importantly, a special thanks to all of the patients for participation in this clinical study. These studies were supported in part by Bristol-Myers-Squibb (BMS). The research work was also supported by Stand Up To Cancer-Cancer Research Institute Cancer Immunology Dream Team Translational Research Grant SU2C-AACR-DT1012 (to J.P.A., P.S., S.K.S., and J.G.) (Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research); a Prostate Cancer Foundation (PCF) challenge grant in immunology (to J.P.A. and P.S.); a PCF 2014 young investigator award (to S.K.S.); a Conquer Cancer Foundation-American Society of Clinical Oncology 2012 young investigator award (to J.G.); Cancer Prevention Research in Texas Grant RP120108 (to P.S.); NIH/National Cancer Institute (NCI) Grant R01 CA1633793 (to P.S.); NIH/NCI Grant K12 CA088084 (to J.G.); and NIH/NCI Award P30CA016672. The Genitourinary Cancers Program of the Cancer Center support grant shared resources at The University of Texas M.D. Anderson Cancer Center. P.S. and J.P.A. are members of the Parker Institute for Cancer Immunotherapy at The University of Texas M.D. Anderson Cancer Center.
Footnotes
Conflict of interest statement: J.P.A., consultant/advisory [self: Jounce, Kite Pharma, Neon, and Amgen; spouse: Bristol-Myers-Squibb (BMS), GSK, AstraZeneca, Amgen Constellation, Jounce, and Kite Pharma]; investment interest (self: Jounce, Kite Pharma, Neon, and Amgen; spouse: Jounce, Kite Pharma, and Evelo); owner and product development (self: patents licensed to BMS, Jounce, and Merck; spouse: patent licensed to Jounce). P.S., consultant/advisor (self: BMS, GSK, AstraZeneca, Amgen, and Constellation); investment interest (self: Jounce, Kite Pharma, and Evelo; spouse: Jounce, Kite, and Neon); investigator (self: BMS, GSK, and AstraZeneca); owner and product development (self: owns patent licensed to Jounce; spouse: patents licensed to BMS, Jounce, and Merck). C.J.L., commercial research grants [Astellas, BMS, Janssen (J&J), Medivation, Bayer, and Sanofi]; consultant/advisory board (Astellas, Sanofi, Janssen, and Bayer). E.Y., stock/ownership (Adaptive Biotechnologies); R.O.E., stock/ownership (Adaptive Biotechnologies); M.V., stock/ownership (Adaptive Biotechnologies); H.S.R., stock/ownership (Adaptive Biotechnologies); patents/royalties (Adaptive Biotechnologies); stock/ownership (Adaptive Biotechnologies); honorariums (BMS); consultant/advisory (Adaptive Biotechnologies).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1611421113/-/DCSupplemental.
References
- 1.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 3.Robert C, et al. KEYNOTE-006 investigators Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 4.Weber JS, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 5.Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brahmer J, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motzer RJ, et al. CheckMate 025 Investigators Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Small EJ, et al. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2007;13(6):1810–1815. doi: 10.1158/1078-0432.CCR-06-2318. [DOI] [PubMed] [Google Scholar]
- 9.Beer T, et al. Phase I trial of ipilimumab (IPI) alone and in combination with radiotherapy (XRT) in patients with metastatic castration resistant prostate cancer (mCRPC) J Clin Oncol. 2008;26(15S):5004. [Google Scholar]
- 10.Gerritsen W, et al. Expanded phase I combination trial of GVAX immunotherapy for prostate cancer and ipilimumab in patients with metastatic hormone-refractory prostate cancer (mHPRC) J Clin Oncol. 2008;26(15S):5146. [Google Scholar]
- 11.Lynch TJ, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30(17):2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 12.Kwon ED, et al. CA184-043 Investigators Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–712. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao J, et al. Review of immune-related adverse events in prostate cancer patients treated with ipilimumab: MD Anderson experience. Oncogene. 2015;34(43):5411–5417. doi: 10.1038/onc.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larkin J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scher HI, et al. Prostate Cancer Clinical Trials Working Group Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26(7):1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liakou CI, et al. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci USA. 2008;105(39):14987–14992. doi: 10.1073/pnas.0806075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carthon BC, et al. Preoperative CTLA-4 blockade: Tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res. 2010;16(10):2861–2871. doi: 10.1158/1078-0432.CCR-10-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng Tang D, et al. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res. 2013;1(4):229–234. doi: 10.1158/2326-6066.CIR-13-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cha E, et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med. 2014;6(238):238ra70. doi: 10.1126/scitranslmed.3008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santegoets SJ, et al. T cell profiling reveals high CD4+CTLA-4 + T cell frequency as dominant predictor for survival after prostate GVAX/ipilimumab treatment. Cancer Immunol Immunother. 2013;62(2):245–256. doi: 10.1007/s00262-012-1330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwek SS, et al. Pre-existing levels of CD4 T cells expressing PD-1 are related to overall survival in prostate cancer patients treated with ipilimumab. Cancer Immunol Res. 2015;3(9):1008–1016. doi: 10.1158/2326-6066.CIR-14-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kyi C, Hellmann MD, Wolchok JD, Chapman PB, Postow MA. Opportunistic infections in patients treated with immunotherapy for cancer. J Immunother Cancer. 2014;2:19. doi: 10.1186/2051-1426-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arriola E, et al. Immunosuppression for ipilimumab-related toxicity can cause pneumocystis pneumonia but spare antitumor immune control. OncoImmunology. 2015;4(10):e1040218. doi: 10.1080/2162402X.2015.1040218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fong L, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res. 2009;69(2):609–615. doi: 10.1158/0008-5472.CAN-08-3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolchok JD, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11(2):155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 28.Robins HS, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114(19):4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlson CS, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi: 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 30.DeWitt WS, et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol. 2015;89(8):4517–4526. doi: 10.1128/JVI.03474-14. [DOI] [PMC free article] [PubMed] [Google Scholar]