

Significance
Approximately 25% of the human proteome is integral membrane proteins, comprising most drug targets; these include ion channels, which change shape to open and close. There are some general descriptions on the role of surrounding lipids in such shape change, but few specifics. We have previously shown that a channel (TRPV4) uses hydrogen bonding between specific residues on the TRP helix and S4–S5 linker elbow as a latch enforcing closure. Here, by using mutations, Xenopus oocyte electrophysiology, budding-yeast phenotypes, and molecular simulations, we discovered that the side chain of one of the partner amino acids binds lipids at the inner polar–nonpolar interface and that this binding counteracts the hydrogen bonds, thus helping to unlatch and open the channel.
Keywords: TRP channels, TRP domain, gating, opening mechanism, lipids
Abstract
We have some generalized physical understanding of how ion channels interact with surrounding lipids but few detailed descriptions on how interactions of particular amino acids with contacting lipids may regulate gating. Here we discovered a structure-specific interaction between an amino acid and inner-leaflet lipid that governs the gating transformations of TRPV4 (transient receptor potential vanilloid type 4). Many cation channels use a S4–S5 linker to transmit stimuli to the gate. At the start of TRPV4’s linker helix is leucine 596. A hydrogen bond between the indole of W733 of the TRP helix and the backbone oxygen of L596 secures the helix/linker contact, which acts as a latch maintaining channel closure. The modeled side chain of L596 interacts with the inner lipid leaflet near the polar–nonpolar interface in our model—an interaction that we explored by mutagenesis. We examined the outward currents of TRPV4-expressing Xenopus oocyte upon depolarizations as well as phenotypes of expressing yeast cells. Making this residue less hydrophobic (L596A/G/W/Q/K) reduces open probability [Po; loss-of-function (LOF)], likely due to altered interactions at the polar–nonpolar interface. L596I raises Po [gain-of-function (GOF)], apparently by placing its methyl group further inward and receiving stronger water repulsion. Molecular dynamics simulations showed that the distance between the levels of α-carbons of H-bonded residues L596 and W733 is shortened in the LOFs and lengthened in the GOFs, strengthening or weakening the linker/TRP helix latch, respectively. These results highlight that L596 lipid attraction counteracts the latch bond in a tug-of-war to tune the Po of TRPV4.
Integral membrane proteins comprise about a quarter of the human proteome (1), and include sensors such as G protein-coupled receptors and ion channels, which are major drug targets. Conformational changes crucial to their functions—for example, channel gating—entail intimate interactions with nearby lipids. We mostly have generalized descriptions of protein–bilayer energetics and mechanics during gating, because knowledge of specific interactions is just emerging (2–4). Take the case of the most well-studied ion channel family: the voltage-gated K+ channels (Kv). Surrounding lipids cocrystallized with Kv approximate a bilayer structure (5), and as estimated, in the budget of free-energy change during Kv gating the portion associated with the deformation of the surrounding lipid bilayer is comparable to the voltage-dependent portion (6). Though Kv is best known for its voltage sensitivity, it is in fact also highly sensitive to the bilayer stretch; its open probability can be increased by 50% by a small membrane-tension increase of only ∼1.6 mN/m (7, 8).
The present study is not primarily about the physics of mechanosensitivity, but the basic question of whether and how any localized protein–lipid interactions affect the key chemical bonds that control gating. One such bond was found in TRPV4 (transient receptor potential vanilloid type 4). In a homology model of TRPV4 based on the cryo-EM structure of TRPV1 (9, 10), L596 at the start of the S4–S5 linker helix forms a bond with W733 near the middle of the TRP helix. Not seen in Kv, this amphipathic TRP helix trails S6 and lies at the interface level of bilayer’s inner leaflet but not in contact with it. Note that this hydrogen bond is between a proton in the W733’s indole ring and the backbone oxygen, not the side chain, of L596. L596P, which twists the backbone, is among the over 50 mutant alleles causing blockages of bone development in human (11). Like other such human skeletal dysplasia mutations, L596P is a gain-of-function (GOF) allele, which increases open probability (Po) and causes pathologies presumably due to leakage (12). W733R mutants, selected by forward genetics based on toxicity in budding yeast (13), as well as systematically engineered W733X mutants, also increase Po, suggesting that the TRP helix contact with S4–S5 linker is secured by the L596–W733 H-bond and is used as a latch to keep the channel closed (14).
The S4–S5 linker is best known for its role in the electromechanical coupling between the periphery and the pore to control the S6 gate of Kv (15), and has been recently implicated in transduction of allosteric signals from lipids and ligands in TRPV1 channel (16). In our TRPV4 model, this S4–S5 linker begins at a sharp elbow connecting it to S4 (Fig. 1C). This elbow comprises four residues (G595, L596, K597, L598) exposed to lipids in a bay between the peripheral domains of neighboring subunits (Fig. 1A, arrow). Though the latch bond is between the invariant tryptophan W733 and the carbonyl oxygen of L596, L596’s isobutyl side chain faces outward and contacts the polar–nonpolar interface of the inner-leaflet lipids (Fig. 1C) (14). A hydrophobic residue (L or F) at the beginning of the linker is highly conserved in the TRPV family (14). We examined the role of this contact by analyzing the effects of amino acid substitutions at position 596 on TRPV4 gating. These effects are best explained by a balance between the side chain–membrane interaction and the bond between TRP helix and S4–S5 linker elbow, as if in a tug-of-war (Fig. 1D). Molecular dynamics (MD) simulations supported this model, showing that the contact between S4–S5 linker and TRP helix, represented by the distance between the levels of α-carbons of the latch-bond partners, varies systematically with the side chain–lipid interaction.
Fig. 1.

Structural elements and the tug-of-war model of TRPV4 gating. A snapshot of the refined TRPV4 homology model (14), viewed from the filter (A) or gate (B) side (cytoplasmic domains not shown). Arrow marks the TRP helix/S4–S5 linker bond. There are about four lipids (tan spheres, phosphorus atoms) per bay in the inner leaflet. (C) In the TRPV4 subunit, S4 (green) is connected to the S4–S5 linker helix (blue) at a sharp elbow contacting the TRP helix (purple). See interactions with lipids in Fig. 5. (D) The expected effect of mutations based on the hydrophobicity of the introduced residue vs. the neighborhood. Mutations at L596 less hydrophobic than leucine are likely to move toward polar regions of the membrane, thus tightening the latch S4–S5 linker/TRP helix contact (low-Po LOF phenotype) (Left), whereas the more hydrophobic isoleucine would be pushed stronger into bilayer core, loosening the latch (GOF phenotype) (Right) and allowing lateral tension (γ) to open the channel. A similar unlatching effect could be caused by the pull on the TRP helix toward the polar region by charged substitutions at W733 (e.g., W733R).
Results
L596A, W, G, Q, or K Reduces, Whereas L596I Increases, the TRPV4 Current.
TRPV4 passes a nonspecific cation current that rectifies outwardly. At 3–4 d after the injection of 5 ng WT TRPV4 cRNA into Xenopus oocytes, ∼5 μA steady-state currents can be registered upon a step depolarization from −60 to 60 mV with a two-electrode voltage clamp (14) (Fig. 2A; n > 50). Oocytes with the same injection and incubation of mutant cRNAs were examined. L596A, W, G, Q, and K cRNAs each produced little or no steady-state currents above background (Fig. 2 F–J; n > 5 each). This lack of current was due to very low Po on depolarization, and not a lack of protein expression. Adding the strong TRPV4-specific agonist GSK 1016790A (GSK) to the bath activated huge currents, often beyond the capacity of our recording system (Fig. 2K and Fig. S1; n > 3 each). We refer to these channels with low Po’s as loss-of function (LOF) mutant channels. L596V or L596F cRNA produced currents not significantly different from those of the WT (Fig. 2 D and E; n > 15 each). Surprisingly, cRNA of L596I, with only an isomerized isobutyl side chain, produced very large currents and appeared to be unusually toxic to oocytes. We lowered the amount of injected cRNA to 1 ng and registered large L596I currents (Fig. 2C; n > 50), comparable to those of WT at 5 ng injection. The size of L596I current is reminiscent of those of W733X and L596P, which apparently weaken the W–L hydrogen bond (14). W733Q currents (at 0.1 ng cRNA; n > 5) are shown in Fig. 2B for comparison. We call channels with increased Po GOF mutant channels.
Fig. 2.

Representative traces of whole-oocyte currents. Currents were recorded with a two-electrode voltage clamp from whole oocytes expressing (A) WT TRPV4, (B) GOF mutant W733Q, (C) GOF mutant L596I, and (D and E) the conservative mutants L596V and L596F. (F–J) Currents from oocytes expressing LOF TRPV4 mutant L596A, L596W, L596G, L596Q, and L596K. (K) L596A currents after the application of 1 μM GSK (note current scale change). All oocytes were injected with 5 ng cRNA, except those marked.
Fig. S1.

Representative traces of L596W (A), L596Q (B), L596G (C), and L596K (D) currents after the application of 1 μM GSK (note current scale change). All oocytes were injected with 5 ng cRNA.
Activation and Inactivation.
Prominent activation of the WT TRPV4 is evident at 60 mV with outward current peaking at ∼70 ms followed by current decline due to inactivation (13) (Fig. 3A, black arrow). At 40 mV, the feeble activation is immediately followed by inactivation (τ ∼95 ms; Fig. 3A, red arrow). Though there were few discernable currents from the LOF mutants in our usual procedures (Fig. 2 F–J), measurable currents could often be seen after injecting increased amount of cRNA (10–25 ng). For example, L596A trajectory of the outward current at 60 mV resembled that of the WT at 40 mV (Fig. 3B, red arrow; inactivation τ ∼83 ms, n = 8). Even at 100 mV, WT-like activation was not observed. Massive injection of cRNA, however, could often compromise the health of the oocytes, causing larger leak current and complicating interpretation. We have therefore contrived a L596A E797K double mutant harboring a second mutation in the cytoplasmic domain that boosts the basal Po (17). Though the E797K channel had current activation and inactivation kinetics typical of GOF mutants (17), the double mutant (standard 5-ng injection) again only showed the decaying outward current even upon strong depolarizations (Fig. 3C, red arrow; two-phased inactivation τ1 ∼9 ms, τ2 ∼94 ms, n = 10). These findings indicate that the LOF channels could not be fully activated, and strongly favor the impermeable states (closed or inactivated). The kinetics of the outward currents from conservative mutant L596F resembled those of the WT. Stepping to 60 mV, the WT (Fig. 3D; n = 11) and L596F channel (Fig. 3E; n = 4) activated with τ1 ∼6 ms, τ2 ∼25 ms and inactivated in two phases (τ1 ∼450 ms, τ2 ∼1.6 s). The activation of the GOF L596I (at 1 ng injection; Fig. 3F) was similar to the WT, but its inactivation was dominated by the slow process (τ ∼1.9 s, n = 4). Lack of inactivation has been observed previously in TRPV4 with W733X’s GOF mutations, which break the latch bond (14).
Fig. 3.

Kinetics of WT and mutant TRPV4 currents. (A) Strong activation is evident in the WT (5-ng injection) at 60 mV (black arrow, n > 50), not at 40 mV (red arrow, n = 7). (B) Currents from the L596A mutant were made detectable after massive cRNA injection (15 ng). Such currents had kinetics that resembled those of the WT upon weak activation (red arrow). (C) The currents of the L596A E797K double mutant, with standard 5-ng cRNA injection, also showed weak activation (red arrow). E797K is used here as a background GOF mutation to boost basal Po. At a slower time base, the WT (D) and the conserved mutant L596F current (E) showed similar activation and inactivation kinetics, reaching steady state in 5 s (F). The GOF mutant L596I (1-ng injection) showed a slow decay even beyond 5 s.
Response to GSK or Cell Swelling.
TRPs are polymodal channels able to integrate disparate chemical, thermal, mechanical, and electric stimuli. To test whether L596X mutant channels are specifically deficient in their responses to depolarization, we measured their responses to the agonist GSK and to membrane stretch upon cell swelling. At 40 mV, the steady-state currents in different GSK concentrations were recorded. As shown in Fig. S2, the dose–response curve of the LOF L596A mutant channel is clearly shifted to the right compared with the WT curve. For example, at 100 nM GSK, the WT channels were strongly activated while the L596A channels were only barely activated. The GOF L596I curve appears to be left-shifted. However, the toxic effect of L596I and the added insult of >100 nM GSK likely damaged the oocytes and resulted in smaller responses (Fig. S2). The TRPV4 gene was originally cloned based on a swelling-induced signal in expressing cells (18, 19). We tested the TRPV4 current at 60 mV during the course of oocyte swelling upon bath dilution. This current rose to a maximum over several minutes after the perfusion of a hypotonic solution in the case of the WT (12) and the conservative mutant L596F (Fig. S3 A and B). The swelling-induced current (compared with that before swelling) appeared larger in oocytes expressing the GOF mutant L596I (Fig. S3C). Swelling-induced current increase of those expressing the LOF mutant channels, if existed, was not obvious (Fig. S3 E and F); this could be because they are not sensitive to swelling or because their very low Po obscured even a several-fold increase. We therefore tested the L596A E797K double mutant using second mutation to magnify the basal Po (see above) (17). The L596A E797K double mutant was responsive to cell swelling (Fig. S3G), indicating that the LOF mutation, such as L596A, did not completely eliminate mechanosensitivity, though it responded poorly, as judged by the small proportional increase before and after swelling. The apparent stronger responses for the GOF (L596I) to ligand or swelling stimuli, the clearly feeble responses of the LOF mutant channels might indicate a central “force hub” defect and rather than deficiency in sensing a particular stimulus modality.
Fig. S2.

Steady-state currents at 60 mV induced by different concentrations of the agonist GSK. Higher GSK concentrations are clearly required to activate the current of L596A, a LOF mutant. The larger GSK effect on L596I is uncertain. Oocytes were injected with WT (2.5 ng), L596A (2.5 ng), or L596I (0.5 ng) cRNA, and tested 2∼3 d after incubation. Zero, 10, 50, 100, 400, and 1,000 nM of GSK were serially added into bath solution. Each type of cRNA was tested on three or more oocytes. Results from one representative oocyte of each are shown. Absolute current magnitude cannot be directly compared because of the difference in the amount of RNA injected and uncontrolled variations.
Fig. S3.

Response to oocyte swelling. Whole-oocyte currents tested every 20 s recorded over 15 min after the perfusion of hypotonic solution. Outward current rising upon swelling were observed in oocytes expressing the WT (A) or conservative mutant L596F (B). (C) The GOF mutant L596I TPV4 responded more strongly; the swelling-induced current (compared with that before swelling) was significantly larger than in WT in about one-third of the records (a typical trace is presented here), whereas it was more similar to WT in the rest. Little or no response was registered with the LOF mutant L596A (E) and K (F), much like the uninjected oocyte (D). Oocyte expressing TRPV4 with the L596A E797K double mutation (G) showed clear response to swelling. Typical of n ≥ 3.
Yeast Phenotypes.
TRPV4 has been functionally expressed in the budding yeast Saccharomyces cerevisiae in the membranes of an internal Ca2+ store (20). Ca2+ leakage through excessive TRPV4 activity from this store can stop yeast growth, a phenotype that has been used to select for severe GOF mutants such as W733R (13). The backbone-twisting L596P, a human mutation, also hampers yeast growth (14). Placed behind the inducible Gal4 promoter, plasmids with the GOF W733R or L596I mutant permitted growth in a noninducing medium (glucose) but stopped growth in an inducing medium (galactose; Fig. 4A, second and third rows). Neither WT nor LOF channels hamper growth when expressed (Fig. 4A). Delivered with a second selectable plasmid, GSK inhibited the growth of cells expressing the WT TRPV4 or those with conservative mutations L596F and L596V (behind a constitutive GPD promoter; Fig. 4B, columns 2–4). The growth of cells expressing LOF mutations L596A or L596Q, however, was permitted by the delivered GSK, proving that these channels rarely opened even when heterologously expressed in vivo (Fig. 4B, columns 5 and 6, middle row); they nonetheless inhibited growth at high GSK concentration, showing that they were expressed and functional, albeit with lower Po’s (bottom row). Cells expressing the L596I GOF TRPV4 inhibited growth even without GSK (Fig. 4B, rightmost column).
Fig. 4.

Growth phenotypes of budding yeast transformed with plasmids bearing GOF or LOF mutant TRPV4. (A) Starting with ∼5,000 colony-forming units, serial 10-fold dilutions were dropped on plates. Placed behind an inducible promoter, plasmids with the GOF mutants W733R and L596I (second and third row) inhibited yeast growth on the inducing galactose medium (Right) where the channels were expressed, but not in the noninducing glucose medium (Left). Those with WT, conservative mutants (L596F, V) or LOF mutants (L596A, K) did not inhibit growth. Typical of three independent tests. (B) GSK, infiltrated into the cytoplasm during transformation, stopped the plate growth of yeast constitutively expressing plasmid with WT TRPV4 or those with the conservative mutations (L596F, L596V). Those bearing TRPV4 with LOF mutations (L596A, L596Q) grew normally at low GSK concentration, but grew slower at a higher concentration. The GOF L596I hampered growth even without GSK (only small colonies can be observed on the plates). Marked are the GSK concentrations used during transformation, not those in the cytoplasm. Typical of four independent tests.
Molecular Dynamics Simulations.
Effects of the substitutions at the position 596, as shown by oocyte electrophysiology and yeast phenotypes above, strongly correlate with the hydrophobicity of the introduced residue, for example, by hydrophobicity scales proposed in refs. 21–23. The scales, however, are only a qualitative guide, as in reality hydrophobicity also depends on microenvironment. To seek an understanding at atomic scale, we used MD simulations of TRPV4 channel model in explicit all-atom membrane/water medium to see the neighborhood of residue 596. We have previously erected a full-length homology model of a closed TRPV4, refined through MD simulation in 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) (14). Here we explored four TRPV4 mutants based on the refined model: L596I (GOF), L596A (LOF), L596K (LOF), and W733R (GOF) (14). The simulations were not intended to reproduce the whole gating transition, which is likely much slower, but rather to visualize the environment of the “latch” residues and to reveal the initial structural changes caused by mutations underlying the phenotypes. We performed simulations and symmetry-driven structure refinement of the mutants in the same conditions as WT and compared the changes near the substituted residues. The refinement length of the WT was extended to match the mutants. The refinements have revealed that besides bonding to L596, the side chain of W733 H-bonds to the backbone oxygen of R594. It agrees with the cryo-EM structure of another TRP homolog, TRPV2 (24) (Discussion). Fig. 5A shows that the side chain of L596 in WT reaches the hydrophobic tails of POPC, but the beginning of the side chain is at the level of glycerol oxygens and contacts the H-bonded water network (Table S1). In the direction normal to the membrane, the distance between the levels of α-carbons of L596 and W733, the two latch-bond residues, was found to be 5.9 Å (Table S2). Fig. 5B shows that in the L596I GOF substitution, the I596’s side chain is located somewhat closer toward the bilayer core (green arrow). Compared with the leucine structure, the branching methyl in isoleucine is one carbon closer to membrane interface. It agrees with 20% higher contacts with water (Table S1), likely causing stronger hydrophobic push toward the core. Indeed, interactions with the polar lipid groups decreases, and the ratio of polar–nonpolar lipid contacts decreases by 23%; it concurs with isoleucine being more hydrophobic and suggests development of the drive toward the core, thus favoring disruption of TRP helix/S4–S5 linker elbow contact. The distance between the α-carbon levels slightly lengthened to 6.0 Å. The helix and the elbow moved away from the pore axis by 0.3 Å, consistent with facilitated gating. Simulations of W733R (Fig. 5C), known to be a GOF by greatly weakening the latch bond (14), show significant changes in the environment of the mutated residue. The number of waters contacting more polar arginine grows by 20%. The distance between the α-carbon levels drops to 5.6 Å; however, the electrostatic association of the 733 side chain with the backbone of L596 and R594 is disrupted, a hallmark of GOF W733X mutants. The L596 side chain acquires a less polar environment (30% lower polar–nonpolar ratio). In the case of R733, the change was caused by attraction of the arginine side chain to the oxygens of the lipid phosphates and water. In contrast to L596I, the side chains of the LOF L596A and L596K were found to be farther from the membrane core (Fig. 5 D and E, red arrows), the α-carbon distances decreased to 5.0 and 5.2 Å, respectively, and both TRP helix and S4–S5 linker elbow shifted closer to the pore axis by ∼0.3 Å. The L596A mutant has much smaller side chain, depleting polar (by 49%) and nonpolar (by 78%) contacts with lipids (137% higher polar–nonpolar ratio). The contacts with water grew by 17%; it complies with alanine being less hydrophobic and indicates loss of the attractive van der Waals interactions with the core, thus stabilizing the helix/elbow contact. The last mutant, L596K, shows very different change in contacts—positively charged lysine strongly engages with the lipid polar groups, especially the phosphates; because they are positioned closer to the bilayer surface, it drives K596 away from the core, thus strengthening the helix/elbow contact, matching the LOF phenotype of the mutant.
Fig. 5.

Changes in protein–lipid interactions caused by mutations at W733/L596 latch. (A) A snapshot from molecular dynamics simulation of WT channel showing the latch bond between the indole of W733 and the backbone oxygen of L596 as well as the location of L596’s isobutyl side chain contacting the lipid head group (oxygens marked red) and waters (aqua). Four presented homology models (B–E) were created by introducing mutation to the refined model of the WT channel, followed by refinement through MD simulation and symmetry-driven annealing in the explicit medium. The arrows indicate the direction of the residue displacement comparing to WT (A). Green arrows represent GOF mutants and where displacement favors widening the gap between S4–S5 linker and TRP helix due to (B) increased push toward membrane core (L596I) or (C) pull on TRP helix toward water and negative charges of lipids (W733R). Red arrows correspond to displacement in LOF mutants tightening S4–S5 linker/TRP helix contact caused by (D) decreased hydrophobic push from water (L596A) or (E) pull on S4–S5 linker toward lipids head groups (L596K).
Table S1.
Contacts of side chains of the residues 596 and 733 with lipids, water, and key protein residues observed in MD simulations of the WT channel and four mutants
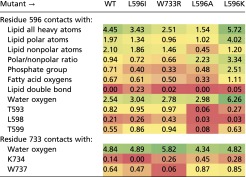 |
The contacts are quantified as the total for all nonhydrogen atoms. The data were collected over the last 0.2 ns of the second refinement cycle and averaged over two independent simulation sets. The table is color coded from the lowest (red), through medium (yellow), to the highest (green) frequency of contacts.
Table S2.
Spatial positions for the backbone of TRP helix, S4 linker elbow, and the residues of interest, 596 and 733, as observed in structures for WT channel and the mutants refined in MD simulations
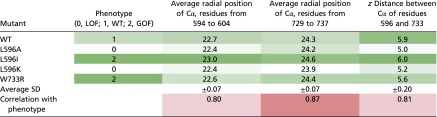 |
The positions are quantified for α-carbons of these regions, time-averaged over the last 0.2 ns of the last symmetry annealing, average of the two independent simulation sets. See SI Methods for details. Table cells with distances are color-coded from the lowest (white) to the highest (green) values within each column.
We should note that the changes in the helix/elbow forces are not confined only to a mutated side chain. As the side chain changes, its contacts with the neighboring protein groups change too. GOF mutants L596I and W733R show increased contacts of the side chain 596 with T593, L598, and T599; that shields the latter from interactions with lipids and water. In contrast, LOF L596A and L596K lower the interactions with these residues by up to 92%, thus exposing them to forces from the medium. These changes likely contribute to the push/pull balance at the latch.
Discussion
Our oocyte electrophysiological findings show that substitutions of L596 with residues that are polar or charged favor the impermeable states (closed or inactivated) and thereby greatly reduce open probability (Po). Even those that are just less hydrophobic, such as L596A, clearly reduce Po (Figs. 2 and 3). However, L596I increases Po and weakens inactivation (Figs. 2 and 3), resembling W733X or L596P mutations that weaken or break the latch bond (however, by the measures of several phenotypes, L596I is not as strong a GOF mutation as W733X) (14).
On average, valine, phenylalanine, and leucine have a very similar hydrophobicity (21–23). Phenylalanine replaces leucine in other channels of the TRPV subfamily, explaining the near-normal channel behavior of the L596V and L596F mutants. The Po and kinetic variations are consistent with the phenotypes in transgenic yeast, an entirely different in vivo system (Fig. 4). These findings indicate that the hydrophobicity of residue 596 is a key determinant of TRPV4 gating.
Structural Interpretations.
Interpretations of L596X phenotypes can be based on the drive on side chain 596 toward more hydrophobic or hydrophilic environments. However, such interpretations require knowledge of the nearby atomic groups and feasibility for the residue to experience change in the external force with mutations. Hence, the MD simulations were performed (Fig. 5). The interaction between L596 side chain and its environment should create a hydrophobic force, normal to the membrane plane, driving the side chain toward the membrane core, counteracting the latch bond between W733’s indole and backbone oxygens of L596 and R594 (Fig. 5A). L596I mutation brings the branching methyl group one carbon closer to the polar region, so water would push I596 stronger toward the membrane core. This push weakens the latch between the S4–S5 linker and TRP-helix as reflected in slight increase in the α-carbon distance of the bonding partners and outward displacement of TRP helix and S4–S5 linker elbow (Fig. 5B and Table S2). Destabilization of the latch contact can be achieved from the opposite side, by mutating the other partner, W733, to arginine (Fig. 5C), which disrupts bonding to the elbow backbone and replaces it with the pull of the TRP-helix toward water, away from the membrane core. In contrast, when applied to the opposite side of the helix–elbow contact, the weakened pull to the core inhibits TRPV4. The LOF L596A mutation significantly decreases the number of contacts, which strongly diminishes the hydrophobic force from water toward the membrane core as well as the attractive van der Waals interactions with hydrocarbon tails, as evident by the decrease in α-carbon distance strengthening S4–S5 linker/TRP helix association (Fig. 5D). The LOF mutation L596K has its positively charged side chain pulled to interact with negative charges of the oxygens of POPC’s phosphate group, also resulted in the decrease in α-carbon distance (Fig. 5E). Both LOF mutations result in the helix and elbow displacement closer to the pore axis—opposite the expected gating direction. Recall that the TRP helix is directly bound to S6, and the elbow between S4 and the S4–S5 linker likely controls the S6 gate through S5/S6 helices coupling (Fig. 1 B–D). Thus, gating depends on the relative position of the S4–S5 linker and the TRP helix, and this position depends on a balance between the strength of the latch bond and the side chain 596-to-lipid interaction in the direction normal to the membrane. In this tug-of-war, cartooned in Fig. 1D, disrupting the latch is likely to facilitate the expansion of the TRP helix and the S4-to-linker elbow coupled to the gate in the lateral direction under the action of membrane tension; this is supported by the strong correlation of the outward displacement of both TRP helix and the S4-to-linker with GOF phenotype and their shift back toward the pore axis in LOF mutants as is observed in simulations (Table S2). A possible reason for this facilitation of expansion by the contact disruption is that these two domains are likely to move laterally in somewhat different directions on channel opening, because they are positioned at an angle to each other, are connected to different hinge points to other protein domains, and are receiving tension from different membrane regions. MD simulations indicate that, in the closed state, the TRP helix does not contact the lipids in the bay (Figs. 1A and 5A), but might receive tension from its C terminus protruding to the membrane. Unlike the TRP domain, the S4-to-linker elbow has direct contact with the lipids in the bay (Figs. 1A and 5A).
The S4-to-Linker Elbow.
At this elbow, besides the side chain of L596, the backbone oxygen of G595, and the backbone NH of K597 and L598 (but not their side chains) also contact lipids in the bay. These contacts, as parts of a team in the tug-of-war, should also contribute to the force counteracting the latch bonds of W733 with L596 and R594. It is beyond the scope of this paper to examine all these contacts. As an example, we tested K597. K597E is clearly a LOF mutation causing a very low Po (Fig. S4). In MD simulation, our structural model has the side chain of K597 facing water and its backbone hydrogen binds to the phosphate oxygens of lipids to receive tension from lipids. K596E mutation likely causes the formation of E597–K730 salt bridge, which would add an extra tie between the TRP helix and S4–S5 linker and could turn the residue 596 around so that the backbone would lose contact with the phosphate oxygens of the lipid and restrain TRP helix in place, restricting movement under tension.
Fig. S4.

Representative traces of whole-cell currents from the oocytes expressing K597E. The currents were recorded before (A) and after (B) addition of 1 mM GSK. Oocytes were injected with 5 ng cRNA, and tested after 3 or 4 d incubation. Voltage protocol was as in Fig. 2. n > 5.
TRP Channels Are Polymodal.
Here, depolarization (Figs. 2 and 3), an agonist (Fig. 2 and Fig. S2), and cell swelling (Fig. S3) are all shown to open WT TRPV4. Responses to all three stimuli are clearly weaker in the LOFs, although their being stronger in L596I (GOF) appears to be somewhat attenuated by the toxicity. In the complex environment, for the live yeast cells (presumably experiencing multiple stimuli), both the LOF and the GOF phenotypes of the TRPV4 mutants are clearly observed (Fig. 4). A parsimonious interpretation is therefore not because the sensitivities of all three sensors or receptors are coincidentally changed, but because the strength of the central latch bond systematically varies with the mutations. These results therefore indicate that all three stimuli funnel through the S4–S5 linker and/or TRP helix to mechanically open the S6 gate.
Model.
Based on our findings, we envision the physiological gating of WT TRPV4 to be as follows: At rest, the tug-of-war at L596 holds the S4 linker elbow and the TRP helix in a balance that favors closure. Certain stimuli (e.g., depolarization) act on the linker, while other stimuli (e.g., Ca2+-calmodulin) act on the TRP helix or on both of them (e.g., membrane stretch). These actions establish new balances in favor of opening. Topology and sequence conservation indicate important functions of the S1–S4 peripheral domain and the S4–S5 linker in TRPV channels, likely similar to those in Kv (7, 8). The TRP helix is followed by cytoplasmic domains used in Ca2+-calmodulin regulation (17); it also has bonds with the pre-S1 helix and the preceding domains at the N-terminal end, and is therefore capable of receiving cytoplasmic stimuli. The TRP helix–S4 linker elbow contact might unlatch in response to the motion of the ankyrin repeats that have been implicated in gating (10) and inactivation (24). Thus, the tug-of-war between the elbow and the TRP helix can be considered the “force hub” to integrate various stimuli, explaining polymodality. The TRP helix is not found in Kv, Cav, or Nav, and may have evolved for this integration and as a safeguard to enforce closure at rest. The tug-of-war mechanism may be general to many TRP channels; this holds true in the recently published TRPV1 structure in membrane nanodiscs (16), as well as cryo-EM TRPV2 structure (24) (Fig. S5). The TRPV2 elbow, however, is one residue longer. As a result, instead of a H-bond to the backbone of F517 (homolog of L596), the tryptophan on TRP helix interacts with the backbone oxygens of Q518 and R515, structurally close to the two H-bonds to L596 and R594 oxygens in our refined TRPV4 models (Fig. S5).The tryptophan is invariant among all TRPV family members, and a TRPV3 W692G substitution here is a GOF allele causing Olmsted syndrome, a human skin disorder (25). The typical TRP-box motif is not present in TRPA family; however, TRPA1 structures have revealed a TRP-like helix in an analogous position, a proposed convergent point of allosteric chemonociception signals (26). Instead of a tryptophan/leucine couple, there is an arginine/glutamate pair capable of forming a salt bridge between the side chains.
Fig. S5.

Comparison of the TRPV4 original and refined models with the cryoelectron microscopic structure of TRPV2. An overall architecture of the channel in general and of TRP helix contact with S4–S5 linker elbow is similar between the model of TRPV4 and TRPV2 structure. A distinct feature of TRPV2 is a one-residue longer loop between the S4 helix and S4–S5 linker. In TRPV4 the residue K597 is part of S4–S5 linker helix, whereas the residue in the homologous position of TRPV2 (Q518) is not part of the helix and adopts conformation similar to L596. TRPV2 residue F517 (homologous to L596) is in the position intermediate between L596 and G596 of TRPV4. The backbone oxygen of F517 is facing away from the tryptophan and cannot H-bond to it (similar to the backbone in TRPV1 structure with capsaicin). However, the backbone oxygen of Q518 of TRPV2 is in a position similar to the oxygen of L596 of TRPV4 and can potentially serve a similar structural function. Unlike the TRPV4 linker, which has only L596 side chain contacting the lipid bilayer, the linker in TRPV2 has two side chains, F517 and L518, exposed to lipids. This region in TRPV2 might be more tolerant to the point mutations comparing to L596 in TRPV4. The position of tryptophan on the TRP helix is similar to the one in TRPV4: in TRPV2, W658 (homolog of W733) is positioned under the S4–S5 linker elbow. The side chain NH capable of H-bonding is located between the backbone oxygens of Q518 (distance 3.65 Å) and R515 (3.08 Å); note that it is closer to R515 and it agrees with our simulation results on refined models of WT and mutant channels where W733 side chains have switched to R694.
Nonetheless, the elbow-TRP domain tug-of-war is not likely the only focus where the forces from lipid work directly, given that the inner part of S5 helix is exposed to lipids in the bay, along with the entire periphery of S1–S4 domain. Protruding into the membrane is also the C-terminal extension of the TRP helix, which can transmit the lipid forces to loosen the latch contact and facilitate opening. Not all interactions between lipids and amino acid side chains are as pivotal as that of L596, however. For example, I529 of S2 in the peripheral domain is in contact with lipids, but I529A has no discernable effects on gating (Fig. S6). A comprehensive “force roadmap” of the channel response to tension requires further exploration at the bench and in silico.
Fig. S6.

Exemplar traces of whole-cell currents from the oocyte expressing I529A. Oocytes were injected with 5 ng I529A cRNA and tested 3 or 4 d after incubation. Voltage protocol was as in Fig. 2. n > 5.
Prospective.
In contrast to a general physical description of forces and energies associated with channel–lipid interaction during gating, we revealed and simulated, in atomic detail, a key focal chemical interaction between amino acids and lipids. Identifying the cruxes of specific interaction between amino acids and lipids should help fundamental understanding of other membrane proteins, be they channels, receptors, or enzymes.
Methods
Xenopus oocytes were injected with in vitro-synthesized TRPV4 cRNAs and recorded with a TEVC (two-electrode voltage clamp) after 3–4 d (14, 27). Plasmid-borne TRPV4s were expressed and growth-phenotyped in yeast (14, 28). We modeled TRPV4 in a POPC bilayer (14) using Modeler and VMD (29, 30). MD simulations were done with NAMD (31) in an all-atom medium with CHARMM36 (32, 33), force field, TIP3P water model (34), and PME electrostatics (35) with a 12-Å cutoff as an NPT ensemble in a periodic cell at 303.15 °K. We refined structures with symmetry-driven simulated annealing (36). Results were analyzed and visualized using VMD (30). See SI Methods for details.
SI Methods
Mutagenesis, cRNA Synthesis, and Oocyte Treatment.
Mutations were introduced in pGH19-TRPV4 using QuikChange lightning site-directed mutagenesis kit (Agilent Technologies). The entire cDNA of WT and mutants TRPV4 alleles were verified by sequencing. Synthesis and injection of cRNA were described previously (14, 27). The oocytes 3∼4 d after incubation were used for electrophysiological recording.
Electrophysiological Techniques.
Two-electrode voltage-clamp recording and instrument were as described in previous studies (13, 27). Pipettes were filled with 3 M KCl. The base bath solution contained (in mM) 66 KMeSO4, 100 sorbitol, 1.8 Ba(MeSO4)2, and 5 K+ Hepes, pH 7.2 (all from Sigma). GSK1016790A (100 nM; Sigma) was directly added into bath solution. Oocyte swelling was induced by perfusing the bath with a hypotonic solution with 100 mM sorbitol subtraction (27, 28). All recordings were performed at room temperature.
Yeast Expression of TRPV4 and Its Mutant Alleles.
The cDNA fragment of WT or mutant TRPV4 was PCR amplified using high-fidelity PfuUltra polymerase (Stratagene) and integrated into the inducible Ura-selectable yeast expression plasmid p416GAL. After sequence confirmation, the resulting plasmids or the empty vector was introduced into yvc1Δ yeast strain (BY4742) (MATα, his3Δ,1 leu2Δ0, lys2Δ0, ura3Δ0, tok1::KanMX4, yvc1::HIS3) according to the high-efficiency yeast transformation protocols (37). Growth phenotype of yeast cells harboring plasmids with TRPV4 or mutants alleles was investigated using spot assay. Starting at 1,000 cells/µL, 5 µL of culture in serial 10-fold dilutions were spotted on a solid expressive medium [2% (wt/vol) galactose and 1% raffinose] or a repressive medium [2% (wt/vol) glucose], and incubated at 30 °C for 2–3 d (13, 14).
Delivery of GSK into Yeast Cytoplasm.
The BY4742 yvc1Δ yeast strain transformed with p416GPD plasmid (20) carrying WT or mutant TRPV4 was subjected to a second round of LiCl-based transformation (37) with an empty Leu-selectable plasmid pEVP11/Aeq (20). Zero, 50, or 400 μM of GSK1016790A (Sigma) was added directly in the transformation solution. Transformants were selected on CMD (-Ura -Leu) solid medium and incubated at 30 °C for 2–3 d.
Homology Modeling.
Previously published refined homology model of TRPV4 in POPC bilayer (14) was used as a template for the models of the mutant channels. Mutations representing GOF (L596I, W733R) and LOF (L596A, L596K) phenotypes were introduced using Modeller (29) and PSFGEN plug-in to VMD (30). All of the spatial rearrangements, structure visualization, and analysis for this work were performed using VMD v1.9.1 (30).
Molecular Dynamic Simulations.
Methods of simulation and refinement of the WT channel have been reported previously (14). With these mutants we have performed two cycles of computational refinement, each cycle consisting of 5 ns unrestrained MD simulation and 1 ns symmetry-driven simulated annealing. The WT channel was refined for an additional two cycles to match the total simulation length of the mutants. To increase sampling of the lipid microenvironment, we have also assembled and run an independent copy of the whole set of simulations (including the refinement of the WT channel and all of the mutants) with the same starting channel model but repositioned into a new location at the preequilibrated lipid bilayer. All of the simulations were run following the same protocol. The final analysis results represent the average of these two independent sets of simulations. Although these simulations are not intended to address the complete transition to the open conformation or long-term changes in the closed state caused by mutations, they do allow us to see the expected neighborhood of residue 596 and the trends in protein–lipid interactions as well as the tightness of S45/TRP helix contact in response to the introduced mutations, which reveals physical determinants of W733-L596 latch stability. For comparison, the thermal dynamics of lipids in our system simulated at 303.15 °K allows a lipid group (e.g., choline residue of POPC or a hydrophobic tail) to explore conformational space comparable in size with the group itself on a nanosecond time scale, which allows the membrane to adjust to the introduced mutation over the simulation timescale. Moreover, on the test case of potassium channels, this combination of unrestrained simulation and annealing was shown to allow a considerable refinement of the homology model already in the first round (36), the feature we leverage to see the initial change in protein structure in the vicinity of mutation.
All MD simulations were performed in an explicit medium (all-atom protein, lipids, water, and ions) using the NAMD package (31). All simulations were performed in the NPT ensemble with CHARMM36 (32, 33) force field and TIP3P water model (34). The acidic and basic residues were set in their default protonation state at the neutral pH, as suggested by estimations using PROPKA (38). The N and C termini were set dissociated. The models were surrounded by a 1,007-molecule POPC bilayer membrane (membrane patch inherited from the refined WT structure simulation; the size of the patch allows sufficient separation between the mirror images of the whole channel with periodic boundary conditions to avoid direct contacts). The assembled system was hydrated with ∼141,000 waters. Two Ca2+ ions were present in the selectivity filter at the starting positions derived from WT simulation. To maintain the electroneutrality of the system, 393 K+ and 397 Cl− ions were present (up to the equivalent of 150 mM salt concentration) in L596I and L596A simulations and 391 K+ and 399 Cl− ions in L596K and W733R simulations (to ensure electroneutrality). Langevin dynamics (39, 40) was used to maintain constant pressure (1 atm) and constant temperature (303.15 °K) in a flexible simulation cell. Periodic boundary conditions were maintained and the particle mesh Ewald method (35) with a real space cutoff distance of 12 Å and a grid width of 1.00 Å. Energy minimization of the initial mutant models was performed using the conjugate gradient for 1,000 steps. Next, the systems were simulated completely unrestrained for 5 ns. To ensure that the Ca2+ ions will not leave the filter, we used Tcl scripting in NAMD to apply soft boundary conditions for ions that would prevent them from leaving the 12 Å vicinity of the center of mass of the selectivity filter. The returning force was set as a harmonic string with stiffness of 1 kcal⋅mol⋅A for the distance measured from ion center to the sphere boundary. However, we did not apply any force while ions were within this sphere. The boundary conditions were present through all of the restrained and unrestrained simulations. However, the ions were found to reside at the starting binding points and did not attempt to leave or approach the boundary; therefore, no forces were applied to ions through the most of the simulation time.
To quantify the tightness of the contact between the S4–S5 linker and the TRP helix, we measured the distance between the α-carbons of the residues W733 and L596 that form the latch H-bond between the NH group of the tryptophan side chain and backbone oxygen of the leucine. Using the direct distance between the H-bonding atoms has certain caveats—a considerable fraction of time W733 side chain switches the hydrogen bond at a slight angle to the backbone oxygen of the nearby residue R594. That connection appears to have a comparable strength and would also stabilize the S4–S5 linker connection to the TRP helix. As such, we chose the distance between the levels of α-carbons of the residues at positions 733 and 596 in the direction normal to the membrane as it does not noticeably change with the switch of the H-bond between the two alternative positions but increases when the residues are pulled apart by introduced mutations. Beside the direct H-bonding, there are also weaker but numerous interactions between the atoms of the TRP helix and S4–S5 linker elbow, as well as their interactions with the medium. To characterize the positions of these groups, we considered an averaged position of the α-carbons for two groups of atoms: TRP helix residues 729–737 and S4 linker elbow 594–604. For WT channel and all of the mutants, the coordinates for the α-carbons were sampled with a 1-ps step over the last 0.2 ns of the last refinement cycle. The data were averaged between the two independent simulation sets. The results are presented in Table S2.
The local environment of the residues 596 and 733 in the refined models was quantified as the number of contacts of the nonhydrogen atoms of the side chains of these residues and various groups of lipids, water, ions, and some key residues of the channel. The contacts were counted with the thresholds based on the position of the first minimum of the radial distribution function of the contacting atom types (an approximation of the first contacting shell). The data were collected with a 1-ps step over the last 0.2 ns of the last refinement cycle, and then averaged between the two independent simulation sets. The results are presented in Table S1.
Acknowledgments
Support for this work was provided by NIH Grant GM096088 (to J.T., S.H.L., and C.K.); the Vilas Trust of the University of Wisconsin (J.T., S.H.L., and C.K.); and the Department of Biology, University of Maryland (A.A.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1613523113/-/DCSupplemental.
References
- 1.Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10(6):1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 2.Lundbaek JA, Birn P, Girshman J, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. 1996;35(12):3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- 3.Phillips R, Ursell T, Wiggins P, Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459(7245):379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Battle AR, et al. Lipid-protein interactions: Lessons learned from stress. Biochim Biophys Acta. 2015;1848(9):1744–1756. doi: 10.1016/j.bbamem.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 5.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450(7168):376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 6.Reeves D, Ursell T, Sens P, Kondev J, Phillips R. Membrane mechanics as a probe of ion-channel gating mechanisms. Phys Rev E Stat Nonlin Soft Matter Phys. 2008;78(4 Pt 1):041901. doi: 10.1103/PhysRevE.78.041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt D, MacKinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc Natl Acad Sci USA. 2008;105(49):19276–19281. doi: 10.1073/pnas.0810187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt D, del Mármol J, MacKinnon R. Mechanistic basis for low threshold mechanosensitivity in voltage-dependent K+ channels. Proc Natl Acad Sci USA. 2012;109(26):10352–10357. doi: 10.1073/pnas.1204700109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504(7478):107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao E, Liao M, Cheng Y, Julius D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature. 2013;504(7478):113–118. doi: 10.1038/nature12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013;14(2):152–163. doi: 10.1038/embor.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loukin S, Su Z, Kung C. Increased basal activity is a key determinant in the severity of human skeletal dysplasia caused by TRPV4 mutations. PLoS One. 2011;6(5):e19533. doi: 10.1371/journal.pone.0019533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loukin S, Su Z, Zhou X, Kung C. Forward genetic analysis reveals multiple gating mechanisms of TRPV4. J Biol Chem. 2010;285(26):19884–19890. doi: 10.1074/jbc.M110.113936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teng J, Loukin SH, Anishkin A, Kung C. L596-W733 bond between the start of the S4-S5 linker and the TRP box stabilizes the closed state of TRPV4 channel. Proc Natl Acad Sci USA. 2015;112(11):3386–3391. doi: 10.1073/pnas.1502366112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science. 2005;309(5736):903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 16.Gao Y, Cao E, Julius D, Cheng Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature. 2016;534(7607):347–351. doi: 10.1038/nature17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loukin SH, Teng J, Kung C. A channelopathy mechanism revealed by direct calmodulin activation of TrpV4. Proc Natl Acad Sci USA. 2015;112(30):9400–9405. doi: 10.1073/pnas.1510602112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liedtke W, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103(3):525–535. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2(10):695–702. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 20.Loukin SH, Su Z, Kung C. Hypotonic shocks activate rat TRPV4 in yeast in the absence of polyunsaturated fatty acids. FEBS Lett. 2009;583(4):754–758. doi: 10.1016/j.febslet.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manavalan P, Ponnuswamy PK. Hydrophobic character of amino acid residues in globular proteins. Nature. 1978;275(5681):673–674. doi: 10.1038/275673a0. [DOI] [PubMed] [Google Scholar]
- 22.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 23.Sweet RM, Eisenberg D. Correlation of sequence hydrophobicities measures similarity in three-dimensional protein structure. J Mol Biol. 1983;171(4):479–488. doi: 10.1016/0022-2836(83)90041-4. [DOI] [PubMed] [Google Scholar]
- 24.Zubcevic L, et al. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat Struct Mol Biol. 2016;23(2):180–186. doi: 10.1038/nsmb.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Z, et al. Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome. Am J Hum Genet. 2012;90(3):558–564. doi: 10.1016/j.ajhg.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paulsen CE, Armache JP, Gao Y, Cheng Y, Julius D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature. 2015;520(7548):511–517. doi: 10.1038/nature14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teng J, Loukin S, Zhou X, Kung C. Yeast luminometric and Xenopus oocyte electrophysiological examinations of the molecular mechanosensitivity of TRPV4. J Vis Exp. 2013 doi: 10.3791/50816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loukin S, Zhou X, Su Z, Saimi Y, Kung C. Wild-type and brachyolmia-causing mutant TRPV4 channels respond directly to stretch force. J Biol Chem. 2010;285(35):27176–27181. doi: 10.1074/jbc.M110.143370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eswar N, et al. Comparative protein structure modeling using Modeller. Curr Protoc Protein Sci. 2006 doi: 10.1002/0471140864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 31.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKerell AD, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 33.Klauda JB, et al. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J Phys Chem B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgensen WLCJ. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 35.Darden T, York D, Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089. [Google Scholar]
- 36.Anishkin A, Milac AL, Guy HR. Symmetry-restrained molecular dynamics simulations improve homology models of potassium channels. Proteins. 2010;78(4):932–949. doi: 10.1002/prot.22618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gietz RD, Schiestl RH. Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2(1):38–41. doi: 10.1038/nprot.2007.15. [DOI] [PubMed] [Google Scholar]
- 38.Rostkowski M, Olsson MH, Søndergaard CR, Jensen JH. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct Biol. 2011;11:6. doi: 10.1186/1472-6807-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martyna GJTDJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 40.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103(11):4613. [Google Scholar]