

 This work is
licensed under a
This work is
licensed under a Abstract
Objective
Primary hyperparathyroidism is usually characterized by a monoclonal parathyroid tumor secreting excess parathyroid hormone (PTH). The main regulator of PTH secretion is calcium and the calcium–PTH set point is shifted in parathyroid tumor cells. We sought to investigate the relationship between tumor PTH and PTH mRNA expression and clinical presentation as well as the regulatory factors including phosphate, vitamin D, and fibroblast growth factor 23.
Design
A total of 154 parathyroid tumors were analyzed by PTH immunohistochemistry and chromogenic in situ hybridization of PTH mRNA. A subset of samples (n = 34) was analyzed using quantitative real-time PCR.
Results
Low tumor PTH mRNA level was significantly associated with low tumor PTH immunoreactivity (P = 0.026), but the two did not correlate with regard to histological distribution within individual tumors. Tumors displaying reduced PTH mRNA levels as compared with normal rim were significantly larger (P = 0.013) and showed higher expression of the calcium-sensing receptor (CASR) (P = 0.046). Weaker tumor PTH mRNA level was significantly associated with higher concentration of circulating 25-hydroxyvitamin D (P = 0.005). No significant correlation was seen between PTH immunoreactivity and patient biochemistry. Tumor weight was strongly associated with circulatory concentrations of calcium and PTH.
Conclusions
No areas with apparently higher PTH expression were identified, perhaps suggesting that hyper functioning parathyroid tumor subclones should be rare. Circulating 25-hydroxyvitamin D levels may influence tumor PTH expression in vivo. If PTH immunoreactivity reflects the tumor calcium–PTH set point, our data imply that the main determinant of disease severity should be tumor weight.
Introduction
Primary hyperparathyroidism (pHPT) is caused by parathyroid tumors, commonly a parathyroid adenoma (85–90%), or multiglandular disease/primary hyperplasia (10–15%), and rarely a parathyroid carcinoma (<1%) (1). The tumors secrete excess levels of parathyroid hormone (PTH) into the bloodstream, resulting in hypercalcemia. While disease-related mortality is low, multiple studies report increased morbidity (2, 3, 4).
The mechanisms underlying parathyroid tumor formation in the parathyroid glands are incompletely understood. Most parathyroid tumors are believed to be of monoclonal origin (5). Parathyroid adenomas frequently harbor mutations in, or loss of heterozygosity (LOH) of the multiple endocrine neoplasia type 1 (MEN1) gene; and parathyroid carcinomas often show mutations in or LOH of the cell division cycle 73 (CDC73) gene (6, 7, 8). Overall, whole-exome sequencing studies have found few somatic alterations, with just a limited number of reoccurring genes in the parathyroid adenomas (9, 10, 11).
The chief regulator of PTH secretion is serum calcium, mediated by the G-protein-coupled, membrane-bound calcium-sensing receptor (CASR) (12). The calcium–PTH set point is defined as the serum calcium level which corresponds to 50% of the maximum PTH secretion. Calcium sensitivity is reduced in parathyroid tumor cells, i.e. the calcium–PTH set point is rightward shifted, allowing for persistent PTH secretion despite relative hypercalcemia (13). Other systems also regulate the calcium–PTH set point, e.g. inhibitory feedback by the vitamin D receptor (VDR) and fibroblast growth factor 23 (FGF23)/Klotho-axes (14, 15, 16). Additional effects mediated by estrogens and progesterone have been described previously (17, 18).
Parathyroid cells synthesize PTH as a pre-pro hormone and store it in intracellular vesicles after protein cleavage. Decreased extracellular levels of serum calcium lead to lowering of intracellular calcium, resulting in PTH vesicle exocytosis (12). Regulation of PTH gene expression is mediated by serum calcium, 1-25-dihydroxyvitamin D (1, 25(OH)2D3) and phosphate levels (13, 15). Severe 25(OH)D3 deficiency may result in secondary HPT, and after prolonged deficiency patients may present with parathyroid tumors (tertiary hyperparathyroidism). Patients with pHPT frequently suffer from 25(OH)D3 deficiency, and some studies report an association with disease severity (19, 20, 21).
These regulatory systems control parathyroid physiology acutely by altering PTH secretion, intermediately by PTH gene transcription, and in the long-term by parathyroid cell proliferation. Multiple studies suggest that parathyroid tumor proliferation and/or formation are related to aberrations in these regulatory systems in pHPT.
We aimed to investigate the potential relationship between parathyroid tumor PTH secretion, PTH gene expression, and patient clinical parameters in a large, well-characterized cohort of parathyroid tumors. Tumor PTH content was measured by immunohistochemistry (IHC), and PTH gene expression by chromogenic in situ hybridization (CISH).
Subjects and methods
Patients and clinical samples
This study included 143 parathyroid adenomas from a prospective cohort operated at the Karolinska University Hospital, and previously described in detail (22, 23, 24). The following clinical variables were included for statistical correlations: patient serum intact PTH, ionized calcium, creatinine, phosphate, 25(OH)D3, FGF23, and bone turnover markers: i.e., alkaline phosphatase (ALP), C-terminal cross-linking peptide of type I collagen (CTX), and type 1 procollagen (P1NP). As previously described a standardized protocol for biochemical sampling was used, allowing for accurate comparison between patient and tumor characteristics (22, 23, 24). Furthermore, all tumors were managed by a limited group of specialized personnel at the Pathology Department, ensuring a minimal difference in sample handling. In 89 adenomas, adjacent normal parathyroid tissue (“normal rim”) was present and scored as a separate entity. All samples were collected with informed patient consent and approval of the local ethics committee. The parathyroid carcinomas (n = 6) and atypical adenomas (n = 5) have been collected on a world-wide basis, and has in part previously been published as part of a historical material (22, 25); reliable biochemical data were not available for these cases. All tumors were classified according to World Health Organization (WHO) 2004 criteria (26) by an experienced endocrine pathologist. Clinical data are summarized in Supplementary Table 1, see section on supplementary data given at the end of this article.
Immunohistochemistry (IHC)
Tissue sections for IHC and CISH were prepared in direct consecutive cuts. Intracellular PTH levels were assessed using standard immunohistochemistry methodology. In short, formalin-fixated, paraffin-embedded tissue was cut into 4 µm sections and mounted on SuperFrost slides (Thermo Fisher Scientific), followed by manual xylene deparaffinization and alcohol rehydration. As determined by optimization experiments, heat-induced antigen retrieval was done in a microwave oven at 95°C for 20 min using a low pH sodium citrate buffer. Biotin blocking was performed using Avidin-Biotin Kit (Vector Laboratories; Burlingame, CA, USA). Slides were incubated with mouse monoclonal anti-PTH at 1:200 dilution (NCL-PTH-488, Leica Biosystems; Wetzlar, Germany) at 4°C overnight, followed by a horseradish peroxidase-conjugated secondary horse anti-mouse antibody at 1:200 dilution (B-200, Vector Laboratories, Burlingame, CA, USA) for 45 min in room temperature. Immunoreactivity was visualized by avidin–biotin–peroxidase complex method using Vectastain Elite Kit (Vector Laboratories, Burlingame, CA, USA) and 3,3′-diaminobenzidine (DAB) as chromogen. Counterstaining was performed using hematoxylin (Htx). Anonymized samples of normal pancreas and thyroid tissue were used as negative controls, in addition to parathyroid samples processed with omission of the primary antibody.
Chromogenic in situ hybridization (CISH)
Slide preparation and deparaffination were performed as described above. PTH mRNA was visualized using a commercially available CISH technique. The protocol is extensively explained in a study by Grabinsky and coworkers (27). We applied the RNAscope 2.0 HD Reagent Kit (cat# 310035) with a probe detecting PTH mRNA (cat# 400521), all from Advanced Cell Diagnostics, with few changes to the manufacturer´s protocol. After optimization, pretreatment 2 was lengthened to 15 min and Htx counterstaining time was lengthened to 4 min. In addition, the Bluing solution was omitted; instead, Htx counterstained slides were gently washed in distilled water for 10 min. The same negative controls as for IHC were applied for CISH. In addition, in accordance with the RNAscope protocol, dapB (dihydrodipicolinate reductase, cat# 310043, Advanced Cell Diagnostics) was used as a negative control and PPIB (Cyclophilin B, included in cat# 310035) was used as a positive control.
Slide evaluation
All samples were examined by Bright-field microscopy at ×200–400 magnification. A subset of cases was digitalized with a Hamamatsu slide scanner (Hamamatsu, Shizuoka, Japan). After reviewing the intensity for probe staining (CISH) and antibody immunoreactivity (IHC), a semi-quantitative scoring system was developed based on the cytoplasmic staining intensity distribution (both ranging from +1 to +4) (Supplementary Table 2). Thirty cases were randomly selected from the cohort and two pathologists scored them individually and blindly for validation purposes (Cohen’s weighted kappa: CISH 0.813, IHC 0.793). The remaining samples were then scored by a single pathologist. Figures were created by directly exporting images of digitally captured slides, without further processing.
Quantitative real-time PCR (qRT-PCR)
A subset of cases (n = 34) with available RNA was subject to mRNA quantification using qRT-PCR. In short, synthesized cDNA was amplified in triplicates using gene expression MasterMix and TaqMan probes (probes targeting CASR (Hs01047793_m1) and VDR (Hs00172113_m1) mRNAs). Ribosomal protein large P0 (RPLP0, Hs99999902_m1) was used as endogenous control all from (Life Technologies). Water and cDNA generated with omitted reverse transcriptase were run in parallel as negative controls. Relative expression levels were calculated by the delta-delta Ct method, samples being normalized against the mean tumor expression value (arbitrarily set to 1).
Statistical analysis
We used Fisher’s exact test for comparisons between categorical variables. When comparing continuous and categorical variables, Mann–Whitney U-test or Kruskal–Wallis test was used as applicable. Spearman’s rank-order correlation was used for continuous values. All tests were performed as two-tailed, and P-values of <0.05 were considered to be statistically significant.
Results
PTH expression in parathyroid tumors and normal rim
All parathyroid tumors and normal rims expressed PTH protein and PTH mRNA (Table 1). PTH immunoreactivity in the adjacent normal rim was stronger than that in the corresponding tumor cells in the majority of cases (73%; 60/82), the remaining scored equal (26%; 21/82) or weaker (1%; 1/82). PTH mRNA levels were also stronger in the normal rim than those in the corresponding tumor cells in the majority of cases (76.5%; 65/85), the rest being equally strong (23.5%; 20/85). The score for staining intensity is illustrated in Fig. 1. Tumors with weak or intermediate PTH mRNA levels (+1 to +2) were significantly associated with weak tumor PTH immunoreactivity (+1 to +2) (Fisher’s exact test: P = 0.026).
Table 1.
Tumor and normal rim PTH immunoreactivity (IHC) and PTH mRNA (CISH) levels.
| Weak | Intermediate | Strong | Very strong | Tumor (T) vs normal rim (N) | |||
|---|---|---|---|---|---|---|---|
| (+1) | (+2) | (+3) | (+4) | T > N | T = N | T < N | |
| IHC | |||||||
| Adenomas | 17 | 58 | 48 | 7 | 1 | 21 | 60 |
| Atypical adenomas | 0 | 0 | 1 | 3 | n.a. | n.a. | n.a. |
| Carcinomas | 0 | 1 | 1 | 4 | n.a. | n.a. | n.a. |
| Normal rim | 0 | 14 | 46 | 22 | n.a. | n.a. | n.a. |
| CISH | |||||||
| Adenomas | 11 | 19 | 39 | 67 | 0 | 20 | 65 |
| Atypical adenomas | 0 | 0 | 2 | 2 | n.a. | n.a. | n.a. |
| Carcinomas | 0 | 2 | 1 | 1 | n.a. | n.a. | n.a. |
| Normal rim | 1 | 2 | 13 | 69 | n.a. | n.a. | n.a. |
T > N, tumor stronger than normal rim; T = N, tumor equal to normal rim; T < N, tumor weaker than normal rim; n.a., not analyzed or not applicable.
Figure 1.
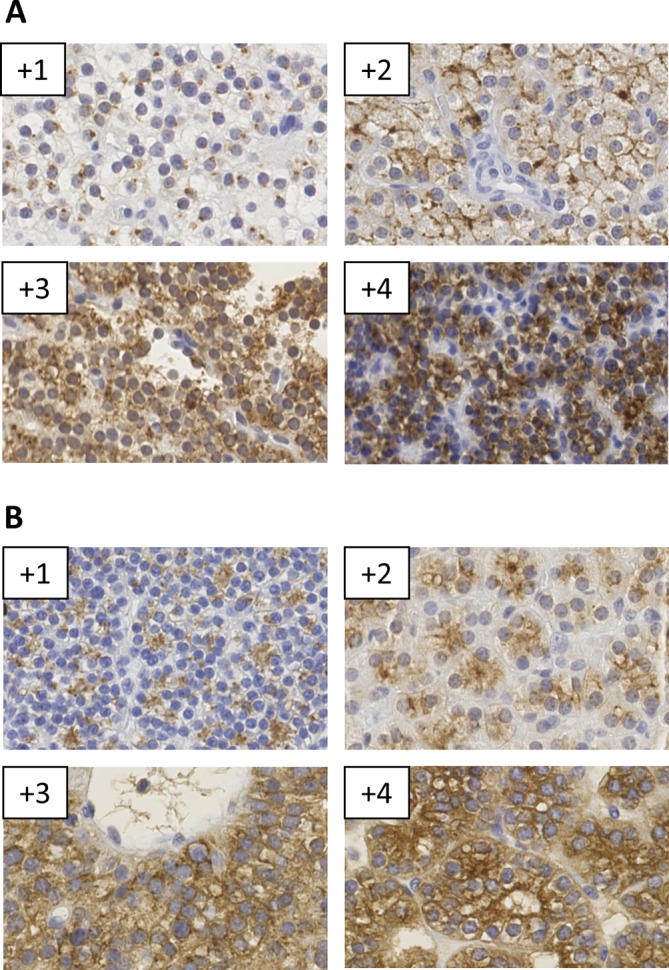
Examples of scoring intensities for (A) PTH immunohistochemistry (IHC), and (B) PTH mRNA chromogenic in situ hybridization (CISH) of parathyroid tumors.
While both PTH immunoreactivity and PTH mRNA levels frequently exhibited heterogeneous staining patterns in the tumor tissue, they too did not correlate in terms of spatial distribution within individual tumors (Fig. 2). Tumor cell areas with oxyphilic cell differentiation generally exhibited lower PTH immunoreactivity and PTH mRNA levels as compared with adjacent chief cells. However, there was no significant difference between tumors with dominant oxyphilic and chief cell differentiation (data not shown). Individual normal rims exhibited essentially uniform PTH immunoreactivity and PTH mRNA staining patterns.
Figure 2.
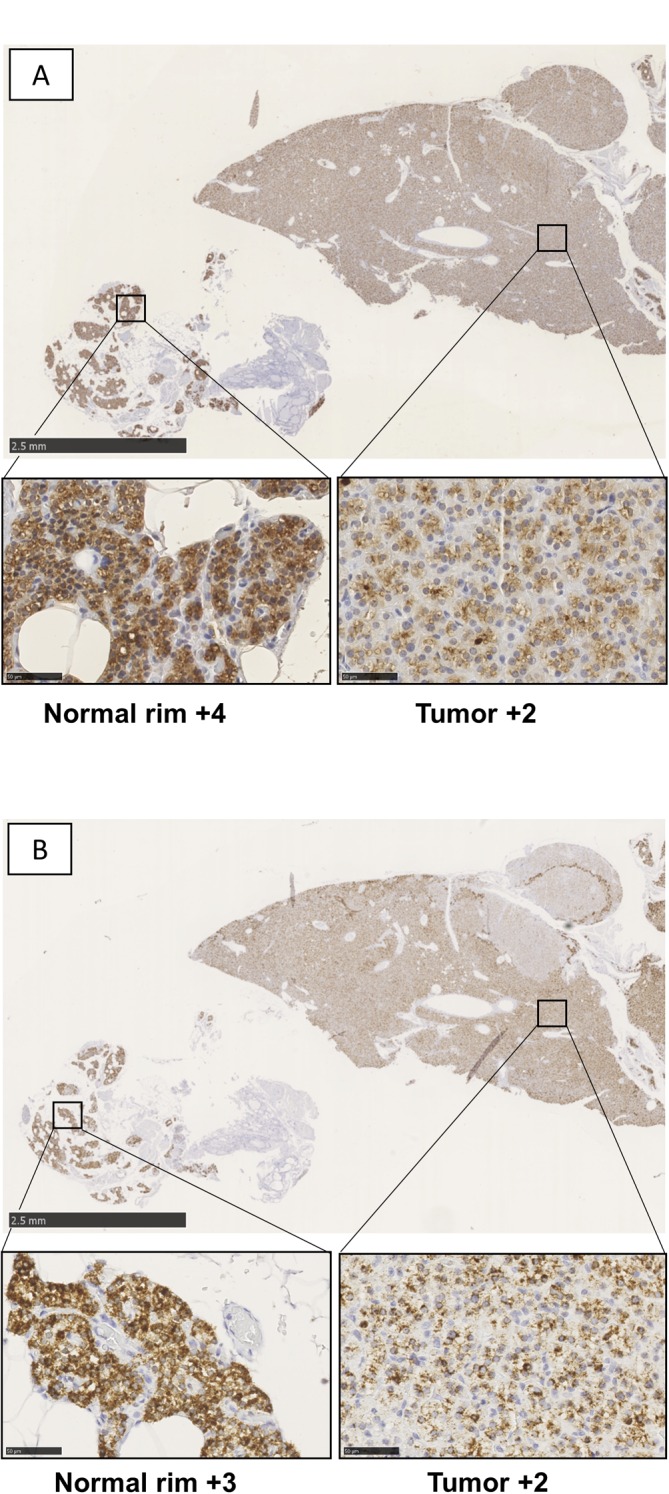
Example of parathyroid adenoma with adjacent normal rim exhibiting tumor heterogeneity and lack of histological correlation between (A) PTH immunohistochemistry (IHC, left) and (B) PTH mRNA chromogenic in situ hybridization (CISH, right).
Clinical correlations of tumor characteristics
Tumors exhibiting lower PTH mRNA levels than the corresponding normal rim were significantly larger (i.e. tumor weight) than tumors with PTH mRNA levels equivalent to their normal rim (Mann–Whitney U: P = 0.013; Supplementary Fig. 1A, see section on supplementary data given at the end of this article). To investigate this relationship, a subset of cases with available RNA was analyzed for CASR mRNA levels using qRT-PCR. Indeed, tumors with lower PTH mRNA levels than the normal rim had significantly higher levels of CASR expression (Mann–Whitney U: P = 0.046, Supplementary Fig. 1B). There was no significant correlation between tumor CASR mRNA levels and overall tumor weight (Spearman’s rank-order correlation: R = 0.299, P = 0.085).
Compared with tumors exhibiting strong or very strong PTH mRNA levels (+3 to +4), tumors with weak or intermediate (+1 to +2) expression had significantly higher patient serum 25(OH)D3 levels (Mann–Whitney U: P = 0.005, Supplementary Fig. 1C). While an interesting trend was observed, tumor PTH immunoreactivity (+1 to +4) was not significantly associated with serum circulating 25-hydroxyvitamin D levels (Kruskal–Wallis: P = 0.054). Patient serum 25(OH)D3 levels correlated significantly weakly with serum circulating PTH levels (Spearman’s rank-order correlation: R = –0.195, P = 0.026), but did not correlate with tumor weight (Spearman’s rank-order correlation: P = 0.133) or ionized calcium (Spearman’s rank-order correlation: P = 0.498). In addition, tumor Vitamin D receptor (VDR) mRNA levels, as measured by qRT-PCR, were not significantly associated with tumor PTH immunoreactivity or PTH mRNA intensity.
Neither PTH immunoreactivity nor PTH mRNA levels were significantly associated with patient serum FGF23, PTH, ionized calcium, phosphate levels or bone turnover markers.
Tumor weight significantly correlated with serum ionized calcium concentration (Spearman’s rank-order correlation: R = 0.254, P = 0.004) and PTH expression (Spearman’s rank-order correlation: R = 0.432, P < 0.001) and was inversely correlated with serum phosphate concentration (Spearman’s rank-order correlation: R = –0.268, P = 0.002).
Parathyroid carcinomas and atypical adenomas had significantly stronger tumor PTH immunoreactivity
All atypical adenomas (n = 4) and carcinomas (n = 6) investigated expressed PTH and PTH mRNA. While the number of cases was limited in this study, the PTH immunoreactivity in atypical adenomas (Fisher’s exact test: P = 0.002) and carcinomas (Fisher’s exact test: P = 0.003) was more frequently scored as stronger in comparison with parathyroid adenomas.
Discussion
We observed that a majority of parathyroid adenomas exhibited weaker PTH immunoreactivity and PTH mRNA intensity compared with corresponding normal parathyroid rim. The attenuated PTH immunoreactivity plausibly reflects a decreased amount of PTH vesicles within the tumor, and may be explained by decreased PTH mRNA expression, alterations in PTH protein stability and/or reflect the rightward shift in the parathyroid tumor cells’ calcium–PTH set point as a result of increased PTH secretion. While most tumors exhibited some degree of staining heterogeneity, we speculate that this spatial variance is a reflection of biological variation over time in individual cells. This would to some degree explain the lack of spatial correlation between tumor PTH mRNA and protein levels. Indeed, tumors with lower PTH mRNA levels, as measured by CISH, were significantly associated with lower PTH immunoreactivity, suggesting that reduced PTH expression may partly explain the decreased tumor PTH levels in these cases. We found that a majority of parathyroid tumors had generally lower levels of PTH mRNA than the corresponding normal rim. Previously, studies have found both lower and higher PTH mRNA expression in parathyroid tumors as compared with normal parathyroids (28, 29, 30, 31, 32). Similar to other studies, we did not observe a relationship between PTH IHC or PTH CISH and serum calcium and serum PTH levels (33, 34). However, a strong correlation was observed between tumor weight and serum PTH and serum calcium levels. If the intensity of tumor PTH immunoreactivity is a reflection of the rightward shift in the calcium–PTH set point, our data indicate that it is the sheer tumor mass, rather than the pathological regulation of PTH, that determines the severity of the disease. While these relationships have been previously presented, this study is the first to integrate both PTH IHC and CISH with patients’ clinical chemistry in a well-characterized large cohort of parathyroid tumors.
Recently, a study by Shi and colleagues has demonstrated that a significant number of parathyroid tumors are of polyclonal origin, with heterogeneous endocrine properties (35). We were unable to distinguish specific areas within individual tumors where PTH immunoreactivity and the in situ signal correlated. Our findings are in line with Shi and colleagues, which also reported a lack of correlation between calcium-mediated suppressibility of PTH secretion and disease severity. Taken together, these data argue against a specific entity where a hyperfunctioning tumor subclone is responsible for driving the hyperparathyroidism.
Previous studies have reported downregulation of VDR and CASR mRNA levels in parathyroid tumors as compared with the normal rim. The level of downregulation was unrelated to patient serum calcium, PTH or 25(OH)D3 levels. Carling and coworkers reported a correlation between tumor VDR levels and patient serum calcium, but neither this study nor Varshney and coworkers could confirm these findings (32, 36, 37, 38). In our study samples, tumors whose PTH mRNA levels were lower than adjacent normal rim were significantly larger than tumors with equivalent levels. These cases also had significantly higher levels of tumor CASR expression. In theory, downregulation of CASR levels would allow for a continuously reduced feedback, which would result in both tumor growth and reduced calcium-mediated suppression of PTH gene expression. Interestingly, tumor CASR protein levels have previously been coupled to the degree of shift in the calcium–PTH set point, but not to tumor size (13). It is tempting to speculate that this subgroup of tumors has an etiology that is independent of changes in the calcium–PTH set point. Speculatively, a genetic event resulting in parathyroid proliferation while leaving PTH-regulatory systems intact – in this case exemplified by intact CASR expression – could account for a phenotype with relatively larger tumor size and lower PTH expression per tumor cell. Unfortunately, the lack of a functional cellular model for pHPT currently limits the possibility of functionally testing this hypothesis. Future genetic studies may be able to cluster and correlate genetic aberrations to the level of tumor PTH secretion and clinical phenotype. We observed that tumors with lower PTH mRNA levels as measured by CISH had significantly higher levels of circulating 25(OH)D3. While the difference was seemingly small (Supplementary Figure 1C) and circulating 25(OH)D3 levels did not significantly correlate with PTH immunoreactivity (P = 0.054), a mechanism for this relationship has been previously described in in vitro settings (39, 40). Some, but not all, studies have reported various correlations between patient vitamin D levels and disease severity (PTH levels, tumor weight, bone turnover). The rationale behind these discrepancies may be due to differences in the study populations, including the degree of vitamin D insufficiency. Specifically, the impact of vitamin D insufficiency may be limited in primary hyperparathyroidism diagnosed at an early stage (18, 19, 20, 21, 32, 40, 41). Indeed, our semi-quantitative immunohistochemistry may be too crude to accurately quantify the tumor PTH levels for weaker statistical relationships. While the value of vitamin D supplementation in pHPT is currently debated, it has been reported to lower serum PTH levels (40, 41). Since our cohort was built up by consecutively collected parathyroid adenomas, it should be the representative of the mild pHPT currently presented in Western populations. Patient circulating serum 25-hydroxyvitamin D levels exhibited a weak correlation with serum PTH level, but were not associated with tumor weight or bone turnover markers (data not shown). While observational, our study suggests that circulating 25(OH)D3 levels may influence tumor PTH expression in a clinical setting, but further studies are required to assess the possible gains of vitamin D supplementation in pHPT.
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/EJE-15-1062.
Declaration of interest
The authors have no conflicts of interest to disclose.
Funding
This study was supported by Cancer Society in Stockholm, Swedish Cancer Society, Swedish Research Council, Stockholm County Council, and Karolinska Institutet.
Author contribution statement
FH drafted the manuscript. FH, CJ, NK, CL, I-LN, and AH reviewed the manuscript. FH and NK performed the experiments. FH, CJ, and AH performed slide scoring. FH and I-LN performed statistical analysis. NK and AH designed the study
Acknowledgments
The authors are indebted to Ms Lisa Ånfalk for excellent sample handling.
References
- 1.DeLellis RA, Mazzaglia P, Mangray S. Primary hyperparathyroidism: a current perspective. Archives of Pathology & Laboratory Medicine 2008. 132 1251–1262. [DOI] [PubMed] [Google Scholar]
- 2.Yu N, Donnan PT, Flynn RW, Murphy MJ, Smith D, Rudman A, Leese GP Increased mortality and morbidity in mild primary hyperparathyroid patients. The parathyroid epidemiology and audit research study (PEARS). Clinical Endocrinology 2010. 73 30–34. [DOI] [PubMed] [Google Scholar]
- 3.Siilin H, Rastad J, Ljunggren O, Lundgren E. Disturbances of calcium homeostasis consistent with mild primary hyperparathyroidism in premenopausal women and associated morbidity. Journal of Clinical Endocrinology and Metabolism 2008. 93 47–53. ( 10.1210/jc.2007-0600) [DOI] [PubMed] [Google Scholar]
- 4.Fraser WD. Hyperparathyroidism. The Lancet 2009. 374 145–58. ( 10.1016/S0140-6736(09)60507-9) [DOI] [PubMed] [Google Scholar]
- 5.Arnold A, Staunton CE, Kim HG, Gaz RD, Kronenberg HM. Monoclonality and abnormal parathyroid hormone genes in parathyroid adenomas. New England Journal of Medicine 1988. 318 658–662. ( 10.1056/NEJM198803173181102) [DOI] [PubMed] [Google Scholar]
- 6.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997. 276 404–407. ( 10.1126/science.276.5311.404) [DOI] [PubMed] [Google Scholar]
- 7.Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, Buisson N, De Witte K, Salandre J, Lenoir G, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European consortium on MEN1. Human Molecular Genetics 1997. 611 77–83. [DOI] [PubMed] [Google Scholar]
- 8.Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nature Genetics 2002. 32 676–680. [DOI] [PubMed] [Google Scholar]
- 9.Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP, Carling T. Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. Journal of Clinical Endocrinology and Metabolism 2012. 97 E1774–E1781. ( 10.1210/jc.2012-1743) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newey PJ, Nesbit MA, Rimmer AJ, Attar M, Head RT, Christie PT, Gorvin CM, Stechman M, Gregory L, Mihai R, et al. Whole-exome sequencing studies of nonhereditary (sporadic) parathyroid adenomas. Journal of Clinical Endocrinology and Metabolism 2012. 97 E1995–E2005. ( 10.1210/jc.2012-2303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soong CP, Arnold A. Recurrent ZFX mutations in human sporadic parathyroid adenomas. Oncoscience 2014. 1 360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coburn JW, Elangovan L, Goodman WG, Frazaõ JM. Calcium-sensing receptor and calcimimetic agents. Kidney International Supplements 1999. 73 S52–S58. ( 10.1053/ajkd.2001.22077) [DOI] [PubMed] [Google Scholar]
- 13.Cetani F, Picone A, Cerrai P, Vignali E, Borsari S, Pardi E, Viacava P, Naccarato AG, Miccoli P, Kifor O, et al. Parathyroid expression of calcium-sensing receptor protein and in vivo parathyroid hormone-Ca(2+) set-point in patients with primary hyperparathyroidism. Journal of Clinical Endocrinology and Metabolism 2000. 85 4789–4794. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. Journal of Clinical Investigations 2007. 117 4003–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lips P. Vitamin D physiology. Progress in Biophysics and Molecular Biology 2006. 92 4–8. ( 10.1016/j.pbiomolbio.2006.02.016) [DOI] [PubMed] [Google Scholar]
- 16.Moosgaard B, Vestergaard P, Heickendorff L, Melsen F, Christiansen P, Mosekilde L. Plasma 25-hydroxyvitamin D and not 1,25-dihydroxyvitamin D is associated with parathyroid adenoma secretion in primary hyperparathyroidism: a cross-sectional study. European Journal of Endocrinology 2006. 155 237–244. ( 10.1530/eje.1.02197) [DOI] [PubMed] [Google Scholar]
- 17.Duarte B, Hargis GK, Kukreja SC. Effects of estradiol and progesterone on parathyroid hormone secretion from human parathyroid tissue. Journal of Clinical Endocrinology and Metabolism 1988. 66 584–587. ( 10.1210/jcem-66-3-584) [DOI] [PubMed] [Google Scholar]
- 18.Haglund F, Ma R, Huss M, Sulaiman L, Lu M, Nilsson IL, Höög A, Juhlin CC, Hartman J, Larsson C. Evidence of a functional estrogen receptor in parathyroid adenomas. Journal of Clinical Endocrinology and Metabolism 2012. 97 4631–4639. ( 10.1210/jc.2012-2484) [DOI] [PubMed] [Google Scholar]
- 19.Moosgaard B, Vestergaard P, Heickendorff L, Melsen F, Christiansen P, Mosekilde L. Vitamin D status, seasonal variations, parathyroid adenoma weight and bone mineral density in primary hyperparathyroidism. Clinical Endocrinology 2005. 63 506–513. ( 10.1530/eje.1.02197) [DOI] [PubMed] [Google Scholar]
- 20.Rao DS, Honasoge M, Divine GW, Phillips ER, Lee MW, Ansari MR, Talpos GB, Parfitt AM. Effect of vitamin D nutrition on parathyroid adenoma weight: pathogenetic and clinical implications. Journal of Clinical Endocrinology and Metabolism 2000 85 1054–1058. [DOI] [PubMed] [Google Scholar]
- 21.Ozbey N, Erbil Y, Ademoglu E, Ozarmagan S, Barbaros U, Bozbora A. Correlations between vitamin D status and biochemical/clinical and pathological parameters in primary hyperparathyroidism. World Journal of Surgery 2006. 30 321–326. ( 10.1016/j.ejim.2006.04.013) [DOI] [PubMed] [Google Scholar]
- 22.Haglund F, Rosin G, Nilsson IL, Juhlin CC, Pernow Y, Norenstedt S, Dinets A, Larsson C, Hartman J, et al. Tumour nuclear oestrogen receptor beta 1 correlates inversely with parathyroid tumour weight. Endocrine Connections 2015. 4 76–85. ( 10.1530/EC-14-0109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norenstedt S, Pernow Y, Brismar K, Sääf M, Ekip A, Granath F, Zedenius J, Nilsson IL. Primary hyperparathyroidism and metabolic risk factors, impact of parathyroidectomy and vitamin D supplementation, and results of a randomized double-blinded study. European Journal of Endocrinology 2013. 6 795–804. ( 10.1530/EJE-13-0547) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norenstedt S, Pernow Y, Zedenius J, Nordenström J, Sääf M, Granath F, Nilsson IL. Vitamin D supplementation after parathyroidectomy: effects on bone mineral density – a randomized double-blind study. Journal of Bone and Mineral Research 2014. 4 960–967. ( 10.1002/jbmr.2102) [DOI] [PubMed] [Google Scholar]
- 25.Juhlin CC, Villablanca A, Sandelin K, Haglund F, Nordenström J, Forsberg L, Bränström R, Obara T, Arnold A, Larsson C, et al. Parafibromin immunoreactivity: its use as an additional diagnostic marker for parathyroid tumor classification. Endocrine-Related Cancer 2007. 14 501–512. ( 10.1677/ERC-07-0021) [DOI] [PubMed] [Google Scholar]
- 26.DeLellis RA, Lloyd RV, Heitz PU, Eng C. Tumours of endocrine organs. In World Health Organization Classification of Tumour Pathology and Genetics, Lyon, France: IARC Press, 2004. [Google Scholar]
- 27.Grabinski TM, Kneynsberg A, Manfredsson FP, Kanaan NM. A method for combining RNAscope in situ hybridization with immunohistochemistry in thick free-floating bran sections and primary neuronal cultures. PLoS One 2015. 10 e0120120 ( 10.1371/journal.pone.0120120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kendall CH, Roberts PA, Pringle JH, Lauder I. The expression of parathyroid hormone messenger RNA in normal and abnormal parathyroid tissue. Journal of Pathology 1991. 165 111–118. ( 10.1002/(ISSN)1096-9896) [DOI] [PubMed] [Google Scholar]
- 29.Weber CJ, Russell J, Costanzo MK, Karp F, Benjamin M, Hardy MA, Feind CR. Relationships of parathyroid hormone, parathyroid secretory protein, parathyroid hormone messenger RNA, parathyroid secretory protein mRNA, and replication in human parathyroid adenoma and secondary hyperplasia tissues and cultures. Surgery 1992. 112 1089–1094. [PubMed] [Google Scholar]
- 30.Luts L, Bergenfelz A, Alumets J, Sundler F. Parathyroid function and histology in patients with parathyroid adenoma: correlation of clinical and morphologic findings. World Journal of Surgery 1997. 21 553–563. ( 10.1007/PL00012285) [DOI] [PubMed] [Google Scholar]
- 31.Weber CJ, Russell J, Chryssochoos JT, Hagler M, McGarity WC. Parathyroid hormone content distinguishes true normal parathyroids from parathyroids of patients with primary hyperparathyroidism. World Journal of Surgery 1996. 20 1010–1014. ( 10.1007/s002689900154) [DOI] [PubMed] [Google Scholar]
- 32.Varshney S, Bhadada SK, Saikia UN, Sachdeva N, Behera A, Arya AK, Sharma S, Bhansali A, Mithal A, Rao SD. Simultaneous expression analysis of vitamin D receptor, calcium-sensing receptor, cyclin D1, and PTH in symptomatic primary hyperparathyroidism in Asian Indians. European Journal of Endocrinology 2013. 169 109–116. ( 10.1530/EJE-13-0085) [DOI] [PubMed] [Google Scholar]
- 33.Tomita T. Immunocytochemical staining patterns for parathyroid hormone and chromogranin in parathyroid hyperplasia, adenoma, and carcinoma. Endocrine Pathology 1999. 2 145–156. [DOI] [PubMed] [Google Scholar]
- 34.Weber CJ, Russell J, Chryssochoos JT, Hagler M, McGarity WC. Parathyroid hormone content distinguishes true normal parathyroids from parathyroids of patients with primary hyperparathyroidism. World Journal of Surgery 1996 20 1010–1014. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y, Hogue J, Dixit D, Koh J, Olson JA., Jr Functional and genetic studies of isolated cells from parathyroid tumors reveal the complex pathogenesis of parathyroid neoplasia. Proceedings of the National Academy of Sciences of the United States of America 2014. 111 3092–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varshney S, Bhadada SK, Sachdeva N, Arya AK, Saikia UN, Behera A, Rao SD. Methylation status of the CpG islands in vitamin D and calcium-sensing receptor gene promoters does not explain the reduced gene expressions in parathyroid adenomas. Journal of Clinical Endocrinology and Metabolism 2013. 98 e1631–e1635. ( 10.1530/EJE-13-0085) [DOI] [PubMed] [Google Scholar]
- 37.Carling T, Rastad J, Akerström G, Westin G. Vitamin D receptor (VDR) and parathyroid hormone messenger ribonucleic acid levels correspond to polymorphic VDR alleles in human parathyroid tumors. Journal of Clinical Endocrinology and Metabolism 1998. 83 2255–2259. [DOI] [PubMed] [Google Scholar]
- 38.Carling T, Rastad J, Szabó E, Westin G, Åkerström G. Reduced parathyroid vitamin D receptor messenger ribonucleic acid levels in primary and secondary hyperparathyroidism. Journal of Clinical Endocrinology and Metabolism 2000. 85 2000–2003. [DOI] [PubMed] [Google Scholar]
- 39.Naveh-Many T, Silver J. Regulation of parathyroid hormone gene expression by hypocalcemia, hypercalcemia, and vitamin D in the rat. Journal of Clinical Investigations 1990. 86 1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ritter CS, Armbrecht HJ, Slatopolsky E, Brown AJ. 25-Hydroxyvitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney International 2006. 70 654–659. [DOI] [PubMed] [Google Scholar]
- 41.Rao DS, Agarwal G, Talpos GB, Phillips ER, Bandeira F, Mishra SK, Mithal A. Role of vitamin D and calcium nutrition in disease expression and parathyroid tumor growth in primary hyperparathyroidism: a global perspective. Journal of Bone and Mineral Research 2002. 17 N75–N80. [PubMed] [Google Scholar]
- 42.Sudhaker Rao D, Han ZH, Phillips ER, Palnitkar S, Parfitt AM. Reduced vitamin D receptor expression in parathyroid adenomas: implications for pathogenesis. Clinical Endocrinology 2000. 53 373–381. [DOI] [PubMed] [Google Scholar]
- 43.Rao DS, Honasoge M, Divine GW, Phillips ER, Lee MW, Ansari MR, Talpos GB, Parfitt AM. Effect of vitamin D nutrition on parathyroid adenoma weight: pathogenetic and clinical implications. Journal of Clinical Endocrinology and Metabolism 2000. 85 1054–1058. [DOI] [PubMed] [Google Scholar]
- 44.Stein EM, Dempster DW, Udesky J, Zhou H, Bilezikian JP, Shane E, Silverberg SJ. Vitamin D deficiency influences histomorphometric features of bone in primary hyperparathyroidism. Bone 2011. 48 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]