

Abstract
Background
Mycobacteria, along with exospore forming Streptomyces, belong to the phylum actinobacteria. Mycobacteria are generally believed to be non-differentiating. Recently however, we showed that the mycobacterial model organism M. smegmatis is capable of forming different types of morphologically distinct resting cells. When subjected to starvation conditions, cells of M. smegmatis exit from the canonical cell division cycle, segregate and compact their chromosomes, and become septated and multi-nucleoided. Under zero nutrient conditions the differentiation process terminates at this stage with the formation of Large Resting Cells (LARCs). In the presence of traces of carbon sources this multi-nucleoided cell stage completes cell division and separates into Small Resting Cells (SMRCs). Here, we carried out RNA-seq profiling of SMRC and LARC development to characterize the transcriptional program underlying these starvation-induced differentiation processes.
Results
Changes among the top modulated genes demonstrated that SMRCs and LARCs undergo similar transcriptional changes. The formation of multi-nucleoided cells (i.e. LARCs and the LARC-like intermediates observed during SMRC formation) was accompanied by upregulation of septum formation functions FtsZ, FtsW, and PbpB, as well as the DNA translocase FtsK. The observed compaction of chromosomes was accompanied by an increase of the transcript level of the DNA binding protein Hlp, an orthologue of the Streptomyces spore-specific chromosome condensation protein HupS. Both SMRC and LARC development were accompanied by similar temporal expression patterns of candidate regulators, including the transcription factors WhiB2, WhiB3, and WhiB4, which are orthologues of the Streptomyces sporulation regulators WhiB, WhiD and WblA, respectively.
Conclusions
Transcriptional analyses of the development of mycobacterial resting cell types suggest that these bacteria harbor a novel differentiation program and identify a series of potential regulators. This provides the basis for the genetic dissection of this actinobacterial differentiation process.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3190-4) contains supplementary material, which is available to authorized users.
Keywords: SMRC, LARC, Actinobacteria, Sporulation, Evolution
Background
Mycobacteria distribute broadly across environments, including fresh water, municipal water systems and soil [1], where they encounter nutrient limitation frequently. This group of actinobacteria is known for its extraordinary capacity to survive for years under starvation conditions [2]. A laboratory starvation culture model based on saline with zero nutrients was established by Loebel and his colleagues in the 1930s [3, 4]. Upon starvation in this model, the bacilli terminate growth and enter a non-replicating state without any apparent morphological changes, but with reduced metabolism. Furthermore, the non-replicating bacteria display dramatic phenotypic drug resistance [5–7]. Recently we showed for Mycobacterium smegmatis that starvation in saline containing traces of a carbon source, as opposed to shock starvation in zero nutrient saline, triggers the development of mono-nucleoided Small Resting Cells, SMRCs. Formation of SMRCs occurred via a septated multi-nucleoided intermediate. Shock starvation in nutrient-free saline resulted as expected in apparently unaltered log-phase-sized resting cells we termed Large Resting Cells, LARCs. Surprisingly, fluorescence microscopic analyses revealed that LARCs remodeled their interior to form septated multi-nucleoided cells similar to the cell intermediate observed during SMRC development. In contrast to growing bacilli, both SMRCs and LARCs had condensed nucleoids, reduced metabolism and increased stress and antibiotic tolerance. In the first weeks of starvation, SMRCs and LARCs showed similar survival. SMRCs however, displayed an increased long-term survival when starvation was extended to 6 months, which correlated with their ability to retain intracellular lipid bodies as energy storage. Based on the morphological similarity between LARCs and the septated multi-nucleoided, LARC-like cell intermediates observed during SMRC development, we hypothesized that mycobacteria (i) undergo a previously unknown differentiation into SMRCs and LARCs; (ii) SMRCs develop through a LARC-like stage; and (iii) formation of SMRCs and LARCs should demonstrate distinct transcriptional profiles compared with non-starved mycobacteria [8]. Here, we used RNA-seq to profile the transcriptomes of M. smegmatis during SMRC and LARC development.
Results
Experimental setup for RNA-seq analyses
Exponentially growing M. smegmatis in rich 7H9 broth (0 h sample, baseline) was transferred to either phosphate buffered saline (PBS) only, or PBS containing traces of Tween80, an oleic acid ester that is used by mycobacteria as carbon source [9]. RNA samples were collected for transcriptional profiling at the following time points to capture the transcriptomes of the critical developmental stages as illustrated in Fig. 1 [8]: after 1 h and 3 h of starvation to capture immediate responses to nutrient limitation; and after 24 h when LARC formation in PBS cultures and SMRC formation in PBS-Tween80 cultures are largely completed. Fourteen-day-old starved cultures were sampled to determine long-term gene expression profiles. Genes were considered statistically significant differentially expressed when their transcript level was at least 2-fold up- or down-regulated compared to the 0 h baseline (P-value < 0.001). A list of all significantly differentially expressed genes for each time point under PBS- or PBS-Tween80-starvation conditions can be found in Additional file 1: Table S1.
Fig. 1.
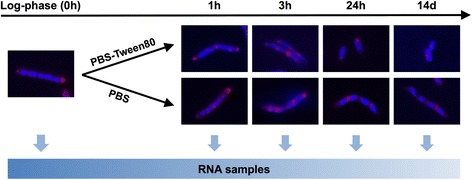
Illustration of samples collected for RNA-sequencing. Development of small and large resting cells in PBS-Tween80 and PBS was demonstrated using DAPI (blue, to visualize DNA) and FM4-64 (red, to visualize membranes) stained culture samples. As reported previously [8], 1 h and 3 h starved cultures under both shock and gentle starvation conditions formed intracellular septa; at 24 h after starvation, the formation of SMRCs and LARCs was largely completed. Samples for RNA-sequencing were collected accordingly at these critical cellular differentiation time points to determine the corresponding developmental transcriptome profiles. Log-phase samples were collected as baseline control and 14-day-old starved cultures were sampled to reflect long-term transcriptional changes. Note: although it has been suggested before that membrane and septal peptidoglycan co-localized at septal positions in dividing mycobacterial cells [41], the formation of peptidoglycan at septal positions in the starved cells remains to be determined
Global transcriptional responses to shock and gentle starvation
Overall, a relatively large fraction (~1/3) of genes showed significant changes in abundance in at least one time point during the 2-week study period (see Additional files 2: Tables S2 and Additional files 3: Table S3, Additional files 4: Figure S1 and Additional files 5: Figure S2). To examine relationships between overall transcriptional patterns among the individual time points, we used multidimensional scaling (MDS) on the 500 genes with the highest variation in expression (at any time point or growth condition in our dataset). The scree plot of variance (Fig. 2a) demonstrates that the first two dimensions of the MDS capture 77 % of the total variance in expression among these 500 genes; we therefore focused the rest of our analysis on these first two dimensions, which can be conveniently displayed in a scatter plot (Fig. 2b). All the log-phase samples were “close” to each other in this scatter plot, indicating that they were generally similar in their expression of these 500 genes. The 1 h and 3 h starved cultures (regardless of carbon source availability), in which the bacilli under both starvation conditions terminate growth and start septum formation, chromosome segregation and condensation to form septated multi-nucleoided cells (Fig. 1), are transcriptionally similar. 24 h starved cultures, when bacilli under both starvation conditions have mostly completed their microscopically observable differentiation into LARCs and SMRCs, show distinct transcriptional profiles. Consistent with the cell biological observations suggesting that differentiation is largely completed within 24 h after the onset of starvation, expression profiles between cells starved for 24 h and 14 d are reasonably similar in this visualization. There is a small shift in the transcriptional profiles from 24 h to 14 d for SMRCs, which may indicate additional developmental changes after formation of the mono-nucleoided cells which are not microscopically observable. In contrast, the 24 h and 14 d LARC samples are closely clustered; further examination of similar plots out to MDS dimension 5 (accounting for a total of >95 % of total variance) shows that these samples remained clustered as closely as the biological replicates of the 0 h samples (data not shown).
Fig. 2.
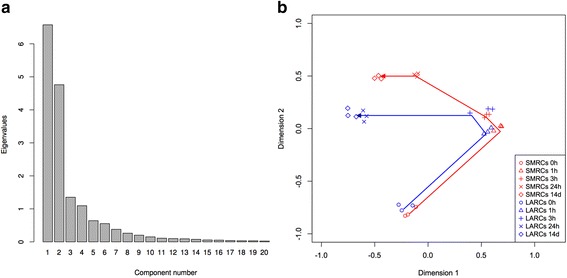
Overview of transcriptional changes. a Scree plot of eigenvalues for the dimensions extracted from a multidimensional scaling (MDS) analysis of the 500 genes with the highest variations in expression during LARC and SMRC development. Dimension 1 accounted for 45 % of the total variance, while dimension 2 accounted for 32 % of the total variance. b Projection of samples onto the first two MDS dimensions. Each sample, representing a single biological replicate of a single culture under one growth condition and one time point, is represented by a single symbol. Samples grown with trace carbon (leading to SMRCs) are colored red, while those with no carbon source (leading to LARCs) are depicted in blue. Different symbols indicate different time points as indicated in the legend at the bottom right. Points with same color and symbol represent the three biological replicates for the RNA-seq analyses. Arrows indicate the development of SMRCs and LARCs over time (0, 1, 3, 24 h and 14 d)
Taken together, the overall transcriptional profiles therefore suggest a similar genetic program in the early phase of LARC and SMRC formation, consistent with a common response to starvation and the formation of septated multi-nucleoided cells under both starvation conditions. After 24 h, when the microscopically observable differentiation into LARC and SMRC is largely completed, the respective transcriptional patterns are distinct and do not change in an appreciable way over the next 2 weeks.
Comparative transcriptional responses of selected genes
Figure 3 depicts changes of transcript levels for several individual genes over time to illustrate the largely similar kinetics in shock and gently starved cultures, allowing us to generate hypotheses regarding the regulatory program within these cells. Translation (elongation factor Tu, ribosomal protein L16) and transcription (RNA polymerase subunit alpha, DNA gyrase) functions, as well as DNA replication functions (replication initiation protein DnaA, DNA polymerase subunit beta DnaN) were immediately downregulated, consistent with the observed starvation-induced termination of growth.
Fig. 3.
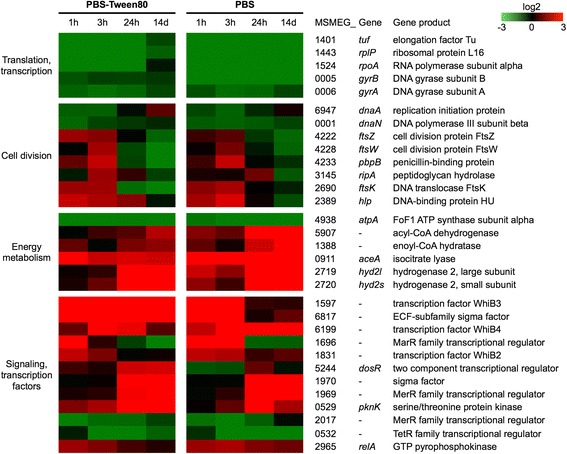
Transcript changes over time of representative genes in PBS-Tween80- and PBS-starved M. smegmatis cultures. A heat map denoting upregulated genes in red and downregulated genes in green is shown. Quantifications for the genes shown (and transcript changes of all statistically significant differentially expressed genes) are in Additional file 1: Table S1
Intriguingly, an upregulation of ftsZ, ftsW and pbpB was observed during the first 3 h in the starved non-growing cultures compared to exponentially growing cells. The corresponding proteins encoded by these three genes—Z ring protein FtsZ, cell division protein FtsW and penicillin-binding protein PpbB—have been shown to interact and form a ternary septation complex in mycobacteria which is involved in septum synthesis, especially in septal peptidoglycan biogenesis [10]. The upregulation of these septum formation functions is consistent with the observed onset of intracellular septum formation in LARC and SMRC cultures during the early hours upon starvation (Fig. 1). Concurrently the transcript level of FtsK, a DNA translocase involved in translocating any residual DNA [11], was also increased. Additionally, expression of RipA, a peptidoglycan hydrolase involved in the physical separation of daughter cells [12], was significantly upregulated during SMRC formation at 3 h.
Noteworthy is an increase in the transcript level of the histone-like DNA binding protein Hlp, associated with compaction of DNA [13, 14], mirroring the nucleoid condensation observed in LARCs and SMRCs shown in Fig. 1 [8]. Interestingly, Hlp upregulation was also observed previously when M. smegmatis shifted down to a non-replicating state induced by oxygen deprivation in nutrient rich medium [15].
Genes involved in energy metabolism were largely downregulated as illustrated by the reduction in mRNA level of ATP synthase subunits, e.g. AtpA, consistent with the observed reduced ATP level and oxygen consumption rate observed in both types of starved cultures [8]. Consistent with the observed higher utilization of fat storage bodies in LARCs when compared to SMRCs [8], upregulation of transcripts encoding β-oxidation enzymes (acyl-CoA dehydrogenase and enolyl-CoA hydratase) was more pronounced in LARCs. In line with the shift to catabolism of fatty acids, expression of isocitrate lyase AceA involved in the glyoxylate shunt [5] was increased under both starvation conditions. Interestingly, the genes encoding the subunits of the hydrogenase Hyd2, were strongly induced in both starved cultures from 24 h onwards (Fig. 3). These gene products were recently shown to be upregulated in growing carbon-limited M. smegmatis cultures and to oxidize atmospheric H2, thus providing electrons to the electron transport chain for ATP generation [16].
Differentiation into LARCs and SMRCs presumably requires changes in signaling and transcription factors. Based on the overlapping cellular differentiation pathways suggested by the microscopic analyses (Fig. 1) and the similarities in gene expression changes over time (Fig. 2b), we expected largely overlapping patterns in the expression of regulatory genes. Figure 3 (bottom), depicting the changes in transcript levels of selected strongly modulated regulatory genes, demonstrates that this is indeed the case—with some noteworthy variation between the two developmental pathways. The expression of the transcription factor WhiB3 and an ECF-subfamily sigma factor was immediately and profoundly induced (20- to 160-fold, see Additional file 1: Table S1). Whereas SMRC/PBS-Tween80 cultures maintained a high transcript level throughout (i.e. until 14 d), the two genes were downregulated from 24 h onwards in LARC/PBS cultures. In contrast, another transcription factor, WhiB4, was strongly induced throughout in LARC/PBS cultures, but more transiently upregulated in SMRC/PBS-Tween80 cultures. Other early response transcription factors were a member of the MarR family and WhiB2. These genes were only transiently upregulated during the first 1–3 h in both cultures. Late transcriptional regulators that were only strongly upregulated from 24 h onwards, i.e. after cellular differentiation was largely completed, were also identified. Examples are the dormancy survival response regulator DosR, shown previously to be involved in the survival of oxygen-depletion induced non-replicating cultures [17], another sigma factor and a MerR family regulator (Fig. 3). Another late starvation function appears to be the serine protein kinase gene pknK, suggesting a possible role of protein phosphorylation in the adaptation to starvation-induced non-replicating survival. Also shown in Fig. 3, other transcription factors, such as another MerR and a TetR family regulator were profoundly (20- to 50-fold) downregulated upon starvation. Finally, as expected, transcript abundance of the stringent response factor RelA was upregulated under both starvation conditions. Previously it was shown that the stringent response is required for starvation survival in vitro and persistence of Mycobacterium tuberculosis in animal models [18–21].
Taken together, the largely similar transcript level changes in PBS- and PBS-Tween80-starved cultures are consistent with the observed similarities in cellular differentiation events and physiological changes of M. smegmatis under both starvation conditions [8]. The dynamic transcript level changes of regulator genes suggest the presence of a starvation-induced genetic differentiation program.
Discussion
Upon starvation, M. smegmatis is capable of forming at least two morphologically distinct types of resting cells. Starvation in PBS containing traces of a carbon source triggers the development of mono-nucleoided Small Resting Cells, SMRCs. Starvation in PBS with zero carbon source results in the formation of Large Resting Cells, LARCs, which show internal remodeling to septated multi-nucleoided cells similar to the intermediates observed during SMRC development. Based on these similarities we had previously proposed a model in which nutrient starvation with or without traces of a carbon source triggers a differentiation program that results in the formation of septated multi-nucleoided cells [8]. Under zero nutrient conditions the differentiation process terminates at this stage with the formation of LARCs. In the presence of traces of carbon sources this septated multi-nucleoided cell stage completes cell division and separates into SMRCs (Fig. 4). We have now shown by transcriptional profiling that morphological changes during SMRC and LARC development are generally mirrored by large-scale changes in gene transcription. In particular, early stages of starvation with or without trace carbon result in similar morphological and genetic changes in M. smegmatis, while the later stages (24 h and beyond) that are morphologically distinct are also transcriptionally distinct. The formation of LARCs and the septated multi-nucleoided LARC-like intermediates observed during SMRC formation is accompanied by the upregulation of the septum formation functions FtsZ, FtsW, and PbpB, as well as the DNA translocase FtsK. Compaction of chromosomes is accompanied by an increase of the transcript level of the histone binding protein Hlp. Both SMRC and LARC development is accompanied by similar temporal expression patterns of candidate transcriptional regulators. Taken together, the correlated morphological and transcriptional changes strongly suggest that mycobacteria harbor a previously unrecognized differentiation program activated by starvation. We have further identified several potential regulators that may provide further insight into resting cell development.
Fig. 4.
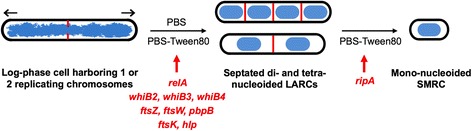
Nutrient-starvation induced differentiation in M. smegmatis. Model depicting starvation induced differentiation of log-phase cells into LARCs and SMRCs. Under zero-nutrient starvation (PBS), the development terminates at the septated, multi-nucleoided LARC stage. In the presence of traces of a carbon source (PBS-Tween80), LARCs undergo cell division and separate into SMRCs. Representative genes upregulated early upon starvation are indicated (as shown in Fig. 3). Blue: DNA, red: septa, black: cell envelope. Arrows indicate polar growth of log-phase cells
Transcriptome profiles of nutrient-limited growing M. smegmatis in continuous cultures have been studied previously [22, 23]. As reported in those studies [22], we also observed marked adaptations in energy metabolism, including upregulation of glyoxylate shunt pathway (e.g. isocitrate lyase AceA) and activation of alternative hydrogenases (e.g. hydrogenase Hyd2) for energy generation. However, consistent with the fact that the carbon-limited continuous cultures were growing, DNA replication functions (e.g. replication initiation protein DnaA, DNA polymerase subunit DnaN) which were immediately downregulated in our non-growing cultures, showed no significant transcriptional changes in the previously reported nutrient-limited growing cells [22]. Moreover, key genes and potential regulators such as ftsZ, ftsW, pbpB, ftsK and whiB3 that were upregulated dramatically under our starvation conditions, showed no differential expression in continuous, carbon-limited growing cultures. Overall, our data are generally consistent with previously reported transcriptome data for growing nutrient-limited mycobacterial cultures. Additionally, using a batch culture starvation system generating non-growing bacteria, we identified a novel transcriptional differentiation program that governs the recently uncovered starvation-induced morphological differentiation processes, i.e. SMRC and LARC formation.
It is interesting to note that orthologues of some of the genes upregulated during the formation of mycobacterial resting cells are involved in the formation of exospores (another distinct type of resting cell), in Streptomyces. Streptomyces, like Mycobacterium, belongs to the phylum actinobacteria. The two bacterial groups separated about 1.3 billion years ago [24, 25]. In response to nutrient depletion, Streptomyces initiates growth of aerial hyphae. These multi genomic structures septate to form chains of mono-nucleoided pre-spore compartments, and then break up into individual small resting cells called exospores [26]. Two categories of genes, bld and whi, have been shown to be the key developmental regulators for the exosporulation process [27]. Interaction among Bld proteins, e.g. BldN, BldD, BldH, has been shown to be important in repressing vegetative growth and initiating the formation of aerial mycelium [27–30]; while five central whi genes—whiA, whiB, whiG, whiH and whiI—are critical for early stages of the conversion from aerial hyphae to spores [27, 31, 32]. Besides these central Whi family regulators, additional WhiB-like transcription factors including WhiD and WblA, are required for morphological differentiation in Streptomyces for the placement of septa and spore maturation [33, 34]. The majority of the sporulation-specific Streptomyces transcriptional regulators have no orthologues in mycobacterial genomes. However interestingly, mycobacterial orthologues of sporulation specific Streptomyces WhiB, WhiD and WblA—WhiB2, WhiB3, and WhiB4 [24]—were strongly upregulated during the development of SMRCs/LARCs. Apparent molecular similarities between mycobacterial resting cell formation and Streptomyces exospore formation also extend to some of the key functional sporulation genes. The mycobacterial orthologue of the Streptomyces DNA translocase FtsK, which mobilizes chromosomal DNA through the closing septum in sporulating aerial hyphae [35], was upregulated during mycobacterial resting cell formation. The mycobacterial orthologue of the Streptomyces sporulation-specific DNA-packaging protein HupS [36], Hlp, was also found to be upregulated during LARC/SMRC formation. It is interesting to note that an additional domain in HupS/Hlp, which makes the DNA binding protein unusual compared the canonical HU proteins in bacteria, can only be found in mycelial actinobacteria and Mycobacterium spp. [24]. Although SMRC formation in mycobacteria and exospore formation in Streptomyces are obviously different processes, the similarities we have observed in the cell biology (formation of septated multi-nucleoided intermediates followed by the separation of mono-nucleoided resting cells) and gene transcription during both differentiation processes may hint at an ancient conserved regulatory program that has diverged to produce the distinct resting cell types, SMRCs/LARCs and exospores in modern actinobacteria.
Conclusions
Overall, our comparative developmental transcriptome studies suggest that M. smegmatis harbors a novel transcriptional differentiation program that governs a starvation induced morphological differentiation process. The identification of a set of differentially expressed regulatory genes provides the basis for a reverse genetic approach for the functional dissection of the regulatory program underlying the development of nutrient starvation induced mycobacterial resting cells.
Methods
Bacterial strain and culture conditions
Mycobacterium smegmatis mc2155 (ATCC 700084) obtained from the American Type Culture Collection (Manassas, Virginia, USA) was cultivated in Middlebrook 7H9 broth supplemented with 0.5 % bovine albumin, 0.2 % glucose, 0.085 % NaCl, 0.5 % glycerol, 0.0003 % catalase and 0.05 % Tween80 at 37 °C with agitation. In nutrient starvation experiments, log-phase cultures with an optical density at 600 nm (OD600) of 0.5 were pelleted by centrifugation at 25 °C/3200 rpm for 10 min. The harvested cells were then washed three times with PBS-0.025 % Tween80 or PBS-0.025 % Tyloxapol (PBS, Gibco 14080-055) and diluted in the corresponding starvation media to a final OD600 of 0.10–0.15. 100 ml of this suspension was then transferred into a 1 L roller bottle (Corning, COR430195) and starved for 14 days (rolling at 2 rpm, 37 °C).
FM4-64, DAPI staining and fluorescence microscopy
For membrane and DNA staining, samples collected at different time points were first fixed in 2 % paraformaldehyde in PBS for 30 min, washed with PBS and then stained as previously described [8] with membrane dye FM4-64 (Molecular Probes, T3166) at a 1 μg/ml for 1 h, followed by staining with DAPI (Molecular Probes, D1306) at 10 μg/ml for 10 min. For visualization, dual stained samples were mounted to slides and observed under epifluorescence microscope Olympus BX60 using U-M61002 and U-MWIG filters for DAPI and FM4-64, respectively.
RNA isolation and preparation
RNA samples from 3 biological replicates were collected for transcriptional profiling at each time point. Briefly, 10 ml of exponentially growing cultures at OD600 of 0.5 (t = 0) or 50 ml of starved cultures at 1 h, 3 h, 24 h and 14 days were harvested by centrifugation (25 °C, 3200 rpm, 10 min). The pellet was resuspended in 1 ml of Trizol (Invitrogen). Bacterial cultures in Trizol were then disrupted by bead beating (FastPrep Instrument and BLUE Kit tubes (BIO 101); two cycles of 30 s at maximum speed with cooling on ice between cycles). The samples were centrifuged for 1 min at 4 °C/13,000 rpm and the supernatant was transferred to a new tube containing 200 μl chloroform, mixed and incubated at RT for 5 min. After centrifugation at 4 °C/13,000 rpm for 10 min, RNA was extracted from the aqueous phase with isopropanol according the supplier’s manual followed by a treatment with DNase I, Amplification Grade (Invitrogen) treatment with subsequent ammonium acetate / isopropanol precipitation. Quality and quantity of total RNA were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies) and a NanoDrop 1000 spectrophotometer (Kisker). The Gram-Positive Bacteria Ribo-Zero (Epicentre) rRNA Magnetic Removal Kit was used to remove bacterial rRNA from total RNA preparations.
RNA sequencing and transcriptome analysis
RNA-seq libraries were prepared according to the TruSeq RNA Sample Preparation v2 Guide (Illumina) without fragmentation and without size selection. Using an Agilent Bioanalyzer high sensitivity DNA kit, we confirmed an average final library size approximately 350 bp rendering fragmentation obsolete. Therefore the library insert fragmentation time at 94 °C was set to 0 min. The quality of the cDNA libraries was assessed using the DNA-1000 kit (Agilent Technologies) on a 2100 Bioanalyzer and quantified with the Qubit 2.0 Fluorometer (Life Technologies). Two libraries of 15 different samples each were indexed, pooled, loaded and clustered onto individual lanes of a Rapid Run flowcell with a cBot instrument (Illumina). In total 30 different samples were analyzed on a Hi-Seq 1500 sequencer (Illumina) using a TruSeq Rapid SBS Kit and 2 × 51 cycles including 7 cycles indexing to obtain 50-bp paired-end reads.
RNA sequencing reads were mapped to the M. smegmatis mc2155 reference genome (NCBI accession: NC_008596) using BWA (v0.5.9) with default parameters [37]. Sequencing reads mapping to predicted open reading frames (ORFs) were quantified using HTSeq [38] based on the NCBI RefSeq annotation. Sequence counts for annotated ribosomal sequences and the tRNAs were filtered out prior to subsequent analysis. Differential expression analyses were performed in R (version 2.15.1) using the Bioconductor package, edgeR [39], and generalized linear modeling to account for gene expression dependencies on growth condition and on time. Differentially expressed genes were defined as genes with at least 2-fold change between samples, with P-value < 0.001 and false detection rate (FDR) value < 0.005. MDS plots were generated using the plotMDS function in R, selecting the 500 genes with the highest variation in expression (based on standard deviations calculated using all samples, including all growth conditions and time points) in the entire data set. COG enrichment analysis was performed using DAVID (v6.7) [40] with default parameters on the top 3000 significantly genes (based on P-value and FDR) among the data sets being compared.
Acknowledgements
We would like to thank Karin Hahnke for technical support of RNA sequencing.
Funding
This work was supported by the Singapore Ministry of Health’s National Medical Research Council under its TCR Flagship Grant NMRC/TCR/011-NUHS/2014 to TD and is part of the Singapore Programme of Research Investigating New Approaches to Treatment of Tuberculosis (SPRINT-TB; www.sprinttb.org) led by Nick Paton. MLW receives a research scholarship from National University of Singapore. JCSC and SLC are supported by the National Research Foundation, Prime Minister’s Office, Singapore under its NRF Research Fellowship Scheme (NRF-RF2010-10) and the Genome Institute of Singapore (GIS)/Agency for Science, Technology and Research (A*STAR).
Availability of data and materials
The dataset discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE69983 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE69983).
Authors’ contributions
MLW and TD conceived the project and designed the strategy. MLW carried out all the experiments except RNA-seq. MG, HJM and SHEK carried out the RNA-seq experiments. JCSC and SC carried out the RNA-seq analyses. MLW and TD analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare having no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- COG
Clusters of orthologous groups
- LARC
Large resting cell
- MDS
Multidimensional scaling
- OD
Optical density
- PBS
Phosphate buffered saline
- SMRC
Small resting cell
Additional files
List of Mycobacterium smegmatis mc2155 genes with statistically significant differential expression (log2 fold change > 1, p < 0.001) after starvation in PBS or PBS-Tween80 at 1, 3, 24 h and 14 d. (XLSX 2064 kb)
Functional classification of gene numbers up- and down-regulated during shock starvation in PBS-Tween80. (PDF 762 kb)
Functional classification of gene numbers up- and down-regulated during shock starvation in PBS. (PDF 1796 kb)
Venn Diagrams showing overlap of significantly differentially expressed genes between SMRCs and LARCs at each time point. Upper diagrams indicate up-regulated genes and lower diagrams indicate down-regulated genes. The numbers of genes in each region of the diagrams are indicated. (PDF 29 kb)
Venn Diagrams showing overlap of significantly differentially expressed genes among the four time points (1 h, 3 h, 24 h, 14 d) during SMRC and LARC development respectively. Upper diagrams indicate up-regulated genes and lower diagrams indicate down-regulated genes. The numbers of genes in each region of the diagrams are indicated. (PDF 322 kb)
References
- 1.Goodfellow M, Magee JG. Taxonomy of mycobacteria. In: Gangadharam PRJ, Jenkins PA, editors. Mycobacteria: I Basic Aspects. US: Springer; 1997. pp. 1–71. [Google Scholar]
- 2.Nyka W. Studies on the effect of starvation on mycobacteria. Infect Immun. 1974;9:843–850. doi: 10.1128/iai.9.5.843-850.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loebel RO, Shorr E, Richardson HB. The influence of foodstuffs upon the respiratory metabolism and growth of human tubercle bacilli. J Bacteriol. 1933;26(2):139–166. doi: 10.1128/jb.26.2.139-166.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loebel RO, Shorr E, Richardson HB. The influence of adverse conditions upon the respiratory metabolism and growth of human tubercle bacilli. J Bacteriol. 1933;26(2):167–200. doi: 10.1128/jb.26.2.167-200.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gengenbacher M, Rao SP, Pethe K, Dick T. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology. 2010;156(Pt 1):81–87. doi: 10.1099/mic.0.033084-0. [DOI] [PubMed] [Google Scholar]
- 6.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002;43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 7.Xie Z, Siddiqi N, Rubin EJ. Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2005;49(11):4778–4780. doi: 10.1128/AAC.49.11.4778-4780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu M-L, Gengenbacher M, Dick T. Mild nutrient starvation triggers the development of a small-cell survival morphotype in mycobacteria. Front Microbiol. 2016;7:947. doi: 10.3389/fmicb.2016.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomioka H. Purification and characterization of the Tween-hydrolyzing esterase of Mycobacterium smegmatis. J Bacteriol. 1983;155:1249–1259. doi: 10.1128/jb.155.3.1249-1259.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Datta P, Dasgupta A, Singh AK, Mukherjee P, Kundu M, Basu J. Interaction between FtsW and penicillin-binding protein 3 (PBP3) directs PBP3 to mid-cell, controls cell septation and mediates the formation of a trimeric complex involving FtsZ, FtsW and PBP3 in mycobacteria. Mol Microbiol. 2006;62(6):1655–1673. doi: 10.1111/j.1365-2958.2006.05491.x. [DOI] [PubMed] [Google Scholar]
- 11.Sherratt DJ, Arciszewska LK, Crozat E, Graham JE, Grainge I. The Escherichia coli DNA translocase FtsK. Biochem Soc Trans. 2010;38(2):395–398. doi: 10.1042/BST0380395. [DOI] [PubMed] [Google Scholar]
- 12.Bishai W, Hett EC, Chao MC, Deng LL, Rubin EJ. A mycobacterial enzyme essential for cell division synergizes with resuscitation-promoting factor. PLoS Pathog. 2008;4(2):e1000001. doi: 10.1371/journal.ppat.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhowmick T, Ghosh S, Dixit K, Ganesan V, Ramagopal UA, Dey D, Sarma SP, Ramakumar S, Nagaraja V. Targeting Mycobacterium tuberculosis nucleoid-associated protein HU with structure-based inhibitors. Nat Commun. 2014;5:4124. doi: 10.1038/ncomms5124. [DOI] [PubMed] [Google Scholar]
- 14.Anuchin AM, Goncharenko AV, Demina GR, Mulyukin AL, Ostrovsky DN, Kaprelyants AS. The role of histone-like protein, Hlp, in Mycobacterium smegmatis dormancy. FEMS Microbiol Lett. 2010;308(2):101–107. doi: 10.1111/j.1574-6968.2010.01988.x. [DOI] [PubMed] [Google Scholar]
- 15.Lee BH, Murugasu-Oei B, Dick T. Upregulation of a histone-like protein in dormant Mycobacterium smegmatis. Mol Gen Genet. 1998;260(5):475–479. doi: 10.1007/s004380050919. [DOI] [PubMed] [Google Scholar]
- 16.Greening C, Berney M, Hards K, Cook GM, Conrad R. A soil actinobacterium scavenges atmospheric H2 using two membrane-associated, oxygen-dependent [NiFe] hydrogenases. Proc Natl Acad Sci U S A. 2014;111(11):4257–4261. doi: 10.1073/pnas.1320586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boon C, Dick T. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J Bacteriol. 2002;184(24):6760–6767. doi: 10.1128/JB.184.24.6760-6767.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahl JL, Kraus CN, Boshoff HI, Doan B, Foley K, Avarbock D, Kaplan G, Mizrahi V, Rubin H, Barry CE., 3rd The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc Natl Acad Sci U S A. 2003;100(17):10026–10031. doi: 10.1073/pnas.1631248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, Barry CE., 3rd The stringent response of Mycobacterium tuberculosis is required for long-term survival. J Bacteriol. 2000;182(17):4889–4898. doi: 10.1128/JB.182.17.4889-4898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boutte CC, Crosson S. Bacterial lifestyle shapes stringent response activation. Trends Microbiol. 2013;21(4):174–180. doi: 10.1016/j.tim.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klinkenberg LG, Lee JH, Bishai WR, Karakousis PC. The stringent response is required for full virulence of Mycobacterium tuberculosis in guinea pigs. J Infect Dis. 2010;202(9):1397–1404. doi: 10.1086/656524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berney M, Cook GM. Unique flexibility in energy metabolism allows mycobacteria to combat starvation and hypoxia. PLoS One. 2010;5(1):e8614. doi: 10.1371/journal.pone.0008614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petridis M, Benjak A, Cook GM. Defining the nitrogen regulated transcriptome of Mycobacterium smegmatis using continuous culture. BMC Genomics. 2015;16:821. doi: 10.1186/s12864-015-2051-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandra G, Chater KF. Developmental biology of Streptomyces from the perspective of 100 actinobacterial genome sequences. FEMS Microbiol Rev. 2014;38(3):345–379. doi: 10.1111/1574-6976.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Battistuzzi FU, Feijao A, Hedges SB. A genomic timescale of prokaryote evolution: insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol Biol. 2004;4:44. doi: 10.1186/1471-2148-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flardh K, Buttner MJ. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat Rev Microbiol. 2009;7(1):36–49. doi: 10.1038/nrmicro1968. [DOI] [PubMed] [Google Scholar]
- 27.McCormick JR, Flardh K. Signals and regulators that govern Streptomyces development. FEMS Microbiol Rev. 2012;36(1):206–231. doi: 10.1111/j.1574-6976.2011.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bibb MJ, Molle V, Buttner MJ. sigma(BldN), an extracytoplasmic function RNA polymerase sigma factor required for aerial mycelium formation in Streptomyces coelicolor A3(2) J Bacteriol. 2000;182(16):4606–4616. doi: 10.1128/JB.182.16.4606-4616.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elliot MA, Bibb MJ, Buttner MJ, Leskiw BK. BldD is a direct regulator of key developmental genes in Streptomyces coelicolor A3(2) Mol Microbiol. 2001;40(1):257–269. doi: 10.1046/j.1365-2958.2001.02387.x. [DOI] [PubMed] [Google Scholar]
- 30.Champness WC. New loci required for Streptomyces coelicolor morphological and physiological differentiation. J Bacteriol. 1988;170(3):1168–1174. doi: 10.1128/jb.170.3.1168-1174.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chater KF, Bruton CJ, Plaskitt KA, Buttner MJ, Mendez C, Helmann JD. The developmental fate of S. coelicolor hyphae depends upon a gene product homologous with the motility sigma factor of B. subtilis. Cell. 1989;59(1):133–143. doi: 10.1016/0092-8674(89)90876-3. [DOI] [PubMed] [Google Scholar]
- 32.Elliot MA, Buttner MJ, Nodwell JR. Multicellular development in Streptomyces. In: Whitworth DE, editor. Myxobacteria: Multicellularity and Differentiation. Herndon: ASM Press; 2008. pp. 419–438. [Google Scholar]
- 33.Fowler-Goldsworthy K, Gust B, Mouz S, Chandra G, Findlay KC, Chater KF. The actinobacteria-specific gene wblA controls major developmental transitions in Streptomyces coelicolor A3(2) Microbiology. 2011;157(Pt 5):1312–1328. doi: 10.1099/mic.0.047555-0. [DOI] [PubMed] [Google Scholar]
- 34.Molle V, Palframan WJ, Findlay KC, Buttner MJ. WhiD and WhiB, homologous proteins required for different stages of sporulation in Streptomyces coelicolor A3(2) J Bacteriol. 2000;182(5):1286–1295. doi: 10.1128/JB.182.5.1286-1295.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Yu Y, He X, Zhou X, Deng Z, Chater KF, Tao M. Role of an FtsK-like protein in genetic stability in Streptomyces coelicolor A3(2) J Bacteriol. 2007;189(6):2310–2318. doi: 10.1128/JB.01660-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salerno P, Larsson J, Bucca G, Laing E, Smith CP, Flardh K. One of the two genes encoding nucleoid-associated HU proteins in Streptomyces coelicolor is developmentally regulated and specifically involved in spore maturation. J Bacteriol. 2009;191(21):6489–6500. doi: 10.1128/JB.00709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 41.Singh B, Nitharwal RG, Ramesh M, Pettersson BM, Kirsebom LA, Dasgupta S. Asymmetric growth and division in Mycobacterium spp.: compensatory mechanisms for non-medial septa. Mol Microbiol. 2013;88(1):64–76. doi: 10.1111/mmi.12169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE69983 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE69983).