

Abstract
Background
Alternative sequence alignment algorithms yield different results. It is therefore useful to quantify the similarities and differences between alternative alignments of the same sequences. These measurements can identify regions of consensus that are likely to be most informative in downstream analysis. They can also highlight systematic differences between alignments that relate to differences in the alignment algorithms themselves.
Results
Here we present a simple method for aligning two alternative multiple sequence alignments to one another and assessing their similarity. Differences are categorised into merges, splits or shifts in one alignment relative to the other. A set of graphical visualisations allow for intuitive interpretation of the data.
Conclusions
AlignStat enables the easy one-off online use of MSA similarity comparisons or into R pipelines. The web-tool is available at AlignStat.Science.LaTrobe.edu.au. The R package, readme and example data are available on CRAN and GitHub.com/TS404/AlignStat.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-016-1300-6) contains supplementary material, which is available to authorized users.
Background
Multiple sequence alignments (MSAs) aim to organise a set of sequences by placing homologous residues into columns, and their accuracy affects subsequent steps in bioinformatic pipelines such as phylogenetic inference [1] and protein structure prediction [2]. However, since there is no objective function to measure true ‘biological correctness’ of an alignment, an array of alternative methods exist based on different assumpitions. These algorithms often make different alignment predictions [2], especially in MSAs with many insertions and deletions, for example in cysteine-rich proteins. Quantitative comparison and intuitive visualisation of alternative MSAs can help users make decisions as to which regions are generally agreed upon and whether any regions should be removed in further analyses. Quantitative similarity measures are also used when assessing the accuracy of alignment algorithms against benchmark MSAs, either synthetically generated [3, 4] or a curated database [5, 6], and to refine phylogenies [7].
A common method of alignment comparison is though a combination of the sum of pairs score (SPS), and total column score (CS) [8]. The sum of pairs score measures what proportion of all residue pairs within columns of one alignment are retained in a comparison alignment and the total column score measures the proportion of columns where both alignments agree completely (ie for all sequences). These methods have the benefit of including all homology information in a single score, however their interpretation can be hampered by the fact that they scale non-linearly with the degree of similarity at a site (Additional file 1: Figure S1).
Here, we use a complementary method based on a matrix of equivalency functions to allow comparable quantification of both similarity and of alternative sources of dissimilarity. Each position in the matrix corresponds to a residue of the reference MSA, with an equivalency function indicating its relationship to the corresponding residue in the comparison MSA. We present a simple set of quantitative measures and graphical visualisations for interpreting MSA comparisons. An R package generates a standardised set of comparison matrices and scores for analysis pipelines and graphing, and a user-friendly web-tool interface enables easy one-off use.
Implementation
Quantifying similarity
When alignment algorithms make different homology predictions for a set of sequences, the columns of the resulting MSAs will contain different residues. The AlignStat R package contains functions for calculating all MSA comparison statistics and creating plots quantifying differences in a manner that is equivalent for nucleotide or amino acid sequences.
Each MSA of n sequences is treated as a matrix of characters (residues plus a gap character) with the same number of rows. The two matrices are therefore defined as P (of dimensions n x p) and Q (of dimensions n x q), where each row represents an aligned sequence. Residues can occur multiple times in a sequence and so are numbered by occurrence such that each character in a row has a unique designation (Additional file 1: Figure S4). This ensures that alignment columns that contain a non-homologous occurrence of a residue are correctly distinguished. For the matrices P and Q, each column vector pair p i, q j is compared to calculate the similarity measure S ij defined in Eq. 1 (where p i is the ith column of P, and q j is the jth column of Q).
| 1 |
Where S ij is the similarity score for each column pair between P and Q, the equivalency function ε is defined in Eq. 2.
| 2 |
The similarity matrix S can be visualised using the plot_similarity_heatmap function of the AlignStat R package. Evaluating S is the most computationally expensive calculation in the AlignStat scoring method and has been implemented in C++ for maximum efficiency.
Detailed match scoring for comparable MSA columns
For each column in P we find its “match” in Q by finding the index j at which S ij is maximized. The match between columns, P i and Q j is then categorised leading to the dissimilarity matrix, D (of dimensions n x p x 5) based on the functions defined in Eq. 3 and Eq. 4. This matrix categorises five types of outcome when the reference and comparison alignments are compared. It is called the dissimilarity matrix because four of the five alternatives correspond to various types of mismatch.
| 3 |
Where ε k (a,b) is the kth equivalency function as defined in Eq. 4.
| 4 |
Where the five ε k (a,b) are equivalency functions (see supplementary information for formal definitions) with the following meanings. The first equivalency (ε 1) is a ‘match’, in which the two characters are identical and not gaps. The second equivalency (ε 2) is a ‘conserved gap’, when the both characters are gaps. A ‘merge’ is when P contains a gap, but Q contains any other character (ε 3). Similarly, a ‘split’ is when Q contains a gap, but P contains any other character (ε 4). Finally, a ‘shift’ is when two characters are not identical and neither are gaps (ε 5). The D matrix is visualised using the plot_dissimilarity_matrix function of the AlignStat R package.
Summary statistics
The column averages of D are used to describe the sources of dissimilarity between the reference and comparison alignments at each alignment position and each equivalency, k. This leads to the results matrix R (of dimensions 5 x p) defined by Eq. 5.
| 5 |
Where R is the results matrix, each row of which is used to summarise a source of dissimilarity from the D matrix.
The match row of the R matrix (R 1i) is visualised using the plot_similarity_summary function of the AlignStat R package. The merge, split and shift rows of the R matrix (R 3i, R 4i and R 5i) are referred to collectively as dissimilarities in AlignStat. They are visualised using the plot_dissimilarity_summary function.
A single, overall similarity score describes the weighted average similarity of the two MSAs, as defined in Eq. 6. The treatment of gaps in MSAs is complex [9, 10]. In this case, the most instructive measure is to exclude conserved gaps, to prevent results being skewed by the “similarity” of conserved gaps in low occupancy columns. Therefore, the overall score is the sum of the match characters as a proportion of characters that are not conserved gaps. A more stringent column score can also be calculated as the proportion of all columns that have a perfectly identical between the MSAs. A full worked example of the mathematical implementation is available in Additional file 1.
| 6 |
Released versions of the R package are available through the comprehensive R archive network (CRAN) and active development versions are available on github (GitHub.com/TS404/AlignStat). In order to allow AlignStat to scale to large MSAs and provide an acceptable run time the core calculation of equivalency functions and scoring statistics was implemented in C++ using the Rcpp framework [11]. A simple web interface to the AlignStat R package is implemented by the Shiny framework and is available at AlignStat.Science.LaTrobe.edu.au. The source code for the user interface is available at Github.com/iracooke/AlignStatShiny.
Results and discussion
R package and example
The AlignStat R package contains a compare_alignments function to calculate the similarity and dissimilarity matrices, and a set of plotting functions to graphically visualise the results. The main compare_alignments function reads input alignments (fasta, clustal, msf, or phylip formats) and outputs a Pairwise Alignment Comparison (PAC) object that contains the matrices and summary information. The example here is a reference MSA of cis-defensin sequences (short, divergent, cysteine-rich proteins [12]) aligned with the CysBar method, which is optimised for highly divergent cysteine-rich proteins [13], compared to an alignment by ClustalΩ [14] (Fig. 1).
Fig. 1.
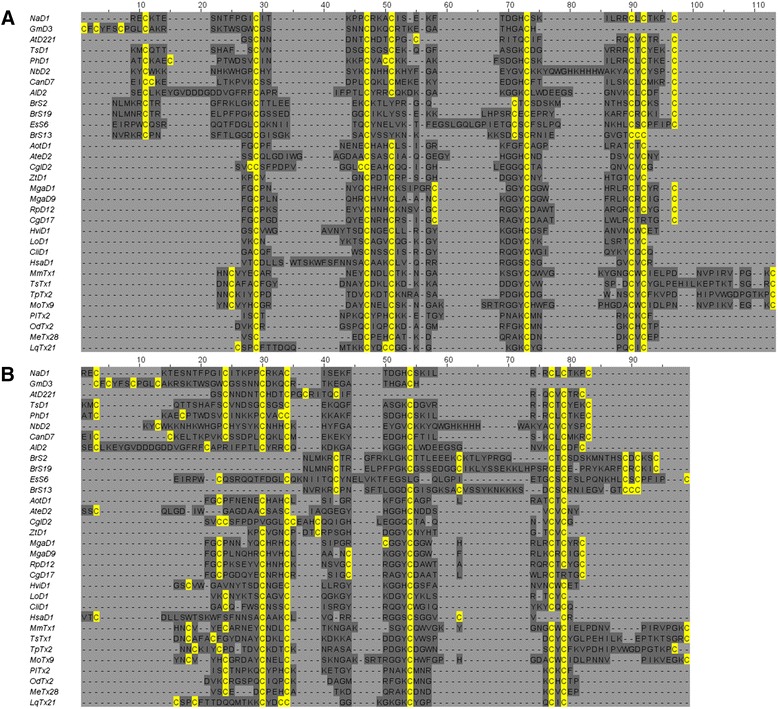
Example alignments of 32 defensin sequences coloured with cysteines in yellow, gaps in light grey, and all other residues in dark grey. a Reference alignment generated by CysBar method. b comparison alignment generated by ClustalΩ
The plot_similarity_heatmap function generates a heatmap of the similarity matrix S (Fig. 2a), analogous to a dot-plot graph used to summarise pairwise sequence alignments [15]. Similarity between each column of the two MSAs is shown such that dark diagonal lines indicate regions of high consensus, with regions of potential conflict as parallel grey lines.
Fig. 2.
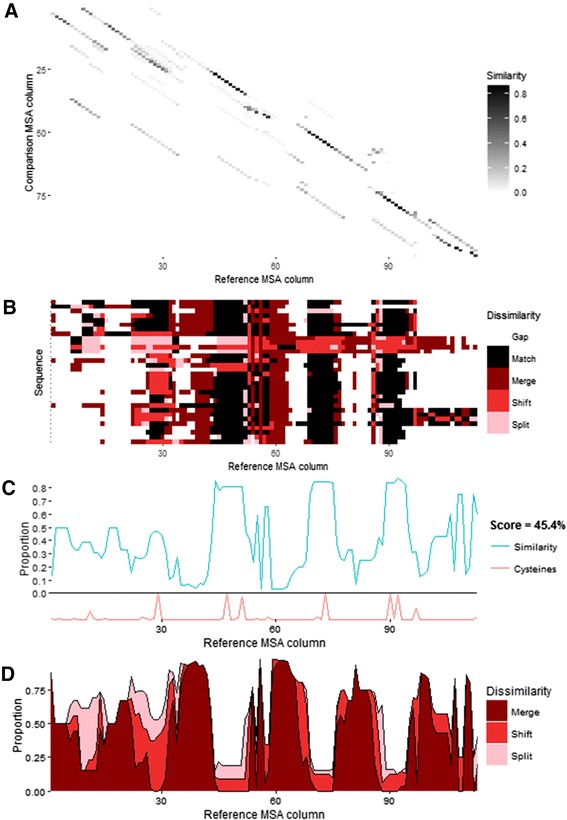
Plots of the similarity (S), difference (D) and results (R) matrices generated by compare_alignments of defensin protein MSAs (reference = CysBar alignment, comparison = ClustalΩ alignment). a Similarity matrix visualised by the plot_similarity_heatmap function. b Dissimilarity matrix visualised by the plot_dissimilarity_matrix function. c Matches in results matrix visualised by the plot_similarity_summary function. d Merges, splits and shifts in results matrix visualised by the plot_dissimilarity_proportions function
A discrete character heatmap of the dissimilarity matrix D is generated by the plot_dissimilarity_matrix function (Fig. 2b). The reference MSA is arranged on the x-axis with sequences arranged on the y-axis. For each character of the reference alignment, the heatmap colour reports whether it is a match, merge, split, shift, or conserved gap. This indicates how sequence regions (columns) or sequence sets (rows) differ between the MSAs.
The similarity of the MSAs is summarised as a line graph by the plot_similarity_summary function (Fig. 2c). The average column match is shown for each reference MSA column, normalised to the proportion of characters that are not gaps. Cysteine proportion can also optionally be reported, since the alignment accuracy of cysteine-rich proteins often correlates with key cysteine motifs. Likewise, a stacked area plot summarising the sources of dissimilarity is generated by the plot_dissimilarity_summary function (Fig. 2d). It presents the average merge, split and shift occurrence for each reference MSA column, also normalised to proportion of characters that are not gaps.
When a ‘true’ reference alignment is known (either simulated, or manually curated) the overall similarity statistics can be used to compare which alternative alignment methods most accurately recreate the reference MSA, and the columnwise similarity statistics indicate the causes of any discrepancies. In this case, higher scores indicate a higher recapitulation by the comparison alignment of the homologous residues in the reference alignment. When the ‘true’ alignent is unknown, as is often the case for real datasets, then the similarity statistics quantify consensus and uncertainty between the alignments. In this case, columns with higher scores indicate agreement of which residues are agreed upon as homologous between the two MSAs. Low scores indicate significant discrepancies, which may occur due to repeat regions, insertions and deletions, or low conservation.
For the defensin example, the highest similarity between the MSAs clusters around the conserved cysteine columns. However, misalignment of non-homologous cysteines and frequent merger of low occupancy inter-cysteine regions by ClustalΩ lead to a similarity score of 45.5 %. The splitting of cysteine columns in the defensin alignment by ClustalΩ indicates which loop insertions and deletions prevent the algorithm from finding true structurally homologous cysteines. In this case, cysteines were split from one column to be merged in with non-homologous cysteines. Similarly, cysteines at the N-terminal end of the proteins are are erroneously split, losing information on their homology. Additionally, an entire set of four sequences was clearly translocated to the right, misaligning all cysteines and inter-cysteine regions. These differences in predicted homology significantly affect any phylogenetics or structure homology modelling using the alignment. By comparison, a ClustalΩ alignment of conserved S1 proteases differs only by minor translocations (similarity score of 81 %) compared to the curated benchmark BALI alignment [5] (Additional file 1: Figures S2 and S3). This reflects far higher reproduction of the curated S1 protease reference alignment by ClustalΩ, particularly in the strucurally conserved protein core regions.
Online web-tool
A webserver at AlignStat.Science.LaTrobe.edu.au performs the AlignStat method and outputs the set of graphs generated by the R script plot functions described above. The matrices and output graphs can then be downloaded. Example data is also provided to perform a test run. The server is capable of performing the method on MSAs of up to 1000 sequences, each with 1000 alignment columns. Additionally, both online and offline versions of AlignStat can compute and visualise sum of pairs analyses of alignments (Additional file 1: Figure S3). This online web-tool implementation allows for easy use of the method without needing to be familiar with the R programming.
Conclusions
The online and offline AlignStat tools allow the quantitative comparison and graphical interpretation of alternative MSAs of a set of sequences. Summarising similarity and dissimilarity aids interpretation of alternative MSAs. In particular understanding the differences between two MSAs can demonstrate significantly different homology predictions for important residues. These measures therefore complement and extend existing offline sum of pairs tools such as SuiteMSA and MQAT [16, 17]. The R package function can be placed into analysis pipelines, and the online web-tool provides a user-friendly graphical interface.
Availability and requirements
Project name: AlignStat
Project home page: AlignStat.Science.LaTrobe.edu.au
Repository: GitHub.com/TS404/AlignStat
Operating system(s): Platform independent
Programming language: R
Other requirements: R 3.1 or higher
License: Academic Free License 3.0
Acknowledgements
The authors thank H. Lockwood and A. Buchanan for their critical reading of the mathematics.
Funding
TS was supported by the Australian Research Council. IC was supported by the Victorian Life Sciences Computation Initiative, a collaboration between Melbourne, Monash and La Trobe Universities and an initiative of the Victorian Government, Australia. The funding bodies had no further roles.
Authors’ contributions
TS conceived the method and wrote the R script. IC implemented the online webtool. Both authors read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent to publish
There was no use of human participants, data or tissues.
Ethics and consent to participate
There was no use of human or animal participants, data or tissues.
Abbreviations
- MSA
Multiple sequence alignment
- PAC
Pairwise Alignment Comparison
- SPS
Sum of pairs score
- CS
Total column score
- CRAN
Comprehensive R archive network
Additional file
Supplementary methods and figures. Worked example of full mathematical implementation for a 6x4 MSA. Supplementary Figures S1–S8. on comparison of scores and additional S1 protease family example. (PDF 1300 kb)
Contributor Information
Thomas Shafee, Email: T.Shafee@LaTrobe.edu.au.
Ira Cooke, Email: Ira.Cooke@jcu.edu.au.
References
- 1.Ogden H, Rosenberg MS. Multiple sequence alignment accuracy and phylogenetic inference. Syst Biol. 2006;55:314–28. doi: 10.1080/10635150500541730. [DOI] [PubMed] [Google Scholar]
- 2.Edgar RC, Batzoglou S. Multiple sequence alignment. Curr Opin Struct Biol. 2006;16:368–73. doi: 10.1016/j.sbi.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Pang A, Smith AD, Nuin P a S. Tillier ERM: SIMPROT: using an empirically determined indel distribution in simulations of protein evolution. BMC Bioinformatics. 2005;6:236. doi: 10.1186/1471-2105-6-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fletcher W, Yang Z. INDELible: a flexible simulator of biological sequence evolution. Mol Biol Evol. 2009;26:1879–88. doi: 10.1093/molbev/msp098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson JD, Koehl P, Ripp R, Poch O. BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins Struct Funct Genet. 2005;61:127–36. doi: 10.1002/prot.20527. [DOI] [PubMed] [Google Scholar]
- 6.Sauder JM, Arthur JW, Dunbrack RL. Large-scale comparison of protein sequence alignment algorithms with structure alignments. Proteins. 2000;40:6–22. doi: 10.1002/(SICI)1097-0134(20000701)40:1<6::AID-PROT30>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Penn O, Privman E, Landan G, Graur D, Pupko T. An alignment confidence score capturing robustness to guide tree uncertainty. Mol Biol Evol. 2010;27:1759–67. doi: 10.1093/molbev/msq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson JD, Plewniak F, Poch O. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 1999;27:2682–90. doi: 10.1093/nar/27.13.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simmons MP, Ochoterena H. Gaps as characters in sequence-based phylogenetic analyses. Syst Biol. 2000;49:369–81. doi: 10.1093/sysbio/49.2.369. [DOI] [PubMed] [Google Scholar]
- 10.Egan AN, Crandall K a. Incorporating gaps as phylogenetic characters across eight DNA regions: ramifications for North American Psoraleeae (Leguminosae) Mol Phylogenet Evol. 2008;46:532–46. doi: 10.1016/j.ympev.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Eddelbuettel D, François R. Rcpp : Seamless R and C ++ integration. J Stat Softw. 2011;40:1–8. [Google Scholar]
- 12.Shafee TMA, Lay FT, Hulett MD, Anderson MA. The defensins consist of two independent, convergent protein superfamilies. Mol Biol Evol. 2016;33(9):2345–356. doi:10.1093/molbev/msw106. [DOI] [PubMed]
- 13.Shafee TMA, Robinson AJ, van der Weerden N, Anderson MA. Structural homology guided alignment of cysteine rich proteins. Springerplus. 2016;5:27. doi: 10.1186/s40064-015-1609-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbs AJ, Mcintyre GA. The diagram, a method for comparing sequences. Eur J Biochem. 1970;16:1–11. doi: 10.1111/j.1432-1033.1970.tb01046.x. [DOI] [PubMed] [Google Scholar]
- 16.Anderson CL, Strope CL, Moriyama EN. SuiteMSA: visual tools for multiple sequence alignment comparison and molecular sequence simulation. BMC Bioinformatics. 2011;12:184. doi: 10.1186/1471-2105-12-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pervez MT, Babar ME, Nadeem A, Aslam N, Raza A, Aslam M, Hussain T, Qadri S, Ahmad S, Shoaib M. MQAT: An efficient quality assessment tool for large multiple sequence alignments. Life Sci J. 2013;10(SPEC. ISSUE 9):9–16. [Google Scholar]