

Abstract
Neovascular age-related macular degeneration (AMD) is a leading cause of central visual acuity loss in a growing segment of the population, those over the age of 60 years. Treatment has improved over the last decade, with the availability of agents that inhibit the bioactivity of vascular endothelial growth factor (VEGF), but it is still limited, because of tachyphylaxis and potential risk and toxicity of anti-VEGF agents. The authors have sought to understand the mechanisms of choroidal endothelial cell (CEC) activation and transmigration of the retinal pigment epithelium (RPE) and of RPE barrier dysfunction, events preceding vision-threatening neovascular AMD. The authors developed physiologically relevant human RPE and CEC coculture and transmigration models that have been important in helping to understand causes of events in human neovascular AMD. The authors can control for interactions between these cells and can separately assess activation of signaling pathways in each cell type relevant during CEC transmigration. Using these models, it was found that VEGF, particularly the cell-associated VEGF splice variant VEGF189, accounts for about 40% of CEC transmigration across the RPE. This percentage is in the range of similar reports following clinical inhibition of VEGF in neovascular AMD. RPE VEGF189 working through CEC VEGF receptor 2 activates the small guanosine triphosphatase (GTPase) of the Rho family, Rac1, in CECs, which in turn facilitates CEC transmigration. Conversely, inhibition of Rac1 activity prevents CEC transmigration. Once activated, Rac1 aggregates with subunits of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, resulting in the generation of reactive oxygen species. Activated NADPH oxidase increases choroidal neovascularization in animal models of laser-induced injury. Rac1 is also downstream of the eotaxin-CCR3 pathway, another pathway important in human neovascular AMD. Studies also suggest that active Ras-related protein 1 (Rap1), another small GTPase, in RPE can strengthen the RPE barrier integrity and can resist CEC transmigration of the RPE, suggesting Rap1 activation may be another potential target for preventing neovascular AMD.
Keywords: choroidal endothelial cell (CEC), Rac1/Rap1 GTPase, retinal pigment epithelium (RPE), vascular endothelial growth factor (VEGF), NADPH oxidase
Introduction
Neovascular age-related macular degeneration (AMD) is a leading cause of central vision loss worldwide in a growing segment of the population, those over the age of 60 years. In the last decade, major strides have been made in the treatment of neo-vascular AMD. Whereas previous management with laser or photodynamic therapy, at best stabilized or improved visual acuity in less than 10% of cases, new agents that target the bioactivity of vascular endothelial growth factor (VEGF) have been reported to significantly improve visual acuity in 40% of cases.1
However, with longer-term follow-up in clinical trials, the effect of anti-VEGF agents has not been sustained2 at the level reported earlier. In part, this may reflect the need for repeated monthly injections and a relaxation in the stringency of maintaining this treatment frequency. However, there are also reports in the cancer literature that other pathways may become involved and “compensate” for the inhibition of pathologic angiogenesis.3–6 In addition, there are safety concerns from repeated intraocular injections7 and in inhibiting VEGF bioactivity on the retina, because VEGF is also a neuroprotective agent.8 For these reasons, other methods to treat neovascular AMD are being sought. The authors’ lab has taken a different approach in the treatment of neovascular AMD. The authors seek to understand the causes for the most severe vision loss that occurs in neovascular AMD – namely, the development of neovascular complexes in the neurosensory retina. Their goal is not to inhibit angiogenesis, but rather to contain or redirect aberrant angiogenesis.
Discussion
Sensory retina choroidal neovascularization
Neovascularization in the sensory retina can occur from endothelial cells (ECs) that originate from the choroid (choroidal endothelial cells [CECs]) or arise from the retinal blood vessels (retinal endothelial cells [RECs]), which make up one part of the blood–retinal barrier. In either situation, ECs are activated first to migrate to a region in the sensory retina normally devoid of blood vessels. In order to do this, several steps are needed besides activation of ECs. These steps include (1) breakdown of the blood–retinal barrier; (2) degradation of extracellular matrix and adhesion of ECs to this; (3) EC activation and migration toward a chemotactic gradient; and (4) EC proliferation.9–16 When CECs migrate into the human sensory retina17 and proliferate, the lesions are called choroidal neovascularization (CNV) (Figure 1). Less commonly, the RECs can migrate into the deeper retina and proliferate; this is called retinal angiomatous proliferation (Figure 2).18–20 In either case, the new vessels can leak fluid into and beneath the sensory retina. Based on improved visual acuity and reduced intraretinal fluid noted on optical coherence tomography (OCT) and images from fluorescein angiography (FA) following anti-VEGF treatment, it is surmised that the leakiness of the new vessel lesions is, in part, the cause of reduced visual acuity.1
Figure 1.
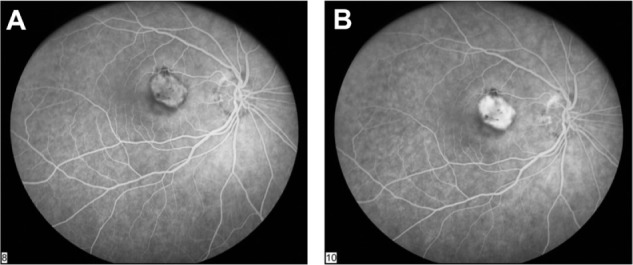
Fluorescein angiogram in the early phase (A) and in the late phase (B) showing increased fluorescence above the retinal pigment epithelium (RPE), demonstrating choroidal neovascularization in the sensory retina above the RPE.
Figure 2.
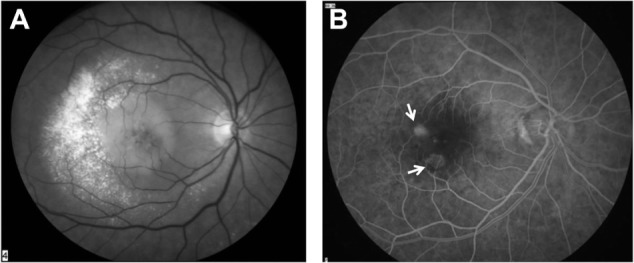
Red-free image (A) and fluorescein angiogram (B) of angiomatous lesions from the retina that have developed in the deep layers of the retina above the retinal angiomatous proliferation (indicated by arrows).
In some cases, CECs are activated to migrate and proliferate but remain within Bruch’s membrane and beneath the retinal pigment epithelium (RPE), which makes up part of the blood–retinal barrier. In these situations of sub-RPE, “occult” CNV (Figure 3), visual acuity can remain stable. However, when visual acuity declines, more than 50% of the time it is secondary to the migration of CECs across the RPE into the sensory retina.21 Therefore, CEC transmigration of the RPE is a critical step in the most common form of vision-threatening neovascular AMD. Based on these observations, the authors have focused on understanding events that are critical to this step: the impairment of the RPE blood–retinal barrier and the activation and migration of CECs.
Figure 3.
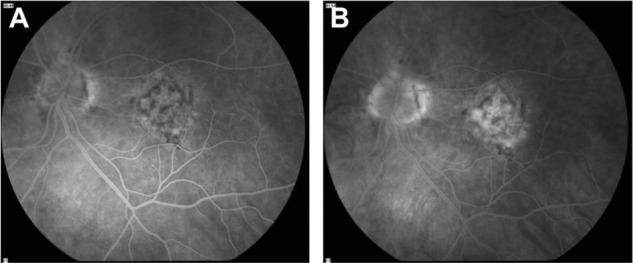
Fluorescein angiogram in the early phase (A) and in the late phase (B) showing mottled fluorescence, with a few spots of late hyperfluorescence that demonstrate where “occult” choroidal neovascularization has migrated into the sensory retina.
Limitations and importance of relevant models
It is almost impossible to safely study mechanisms of CEC transmigration in humans in vivo; therefore, the authors use in vitro methods, animal models, and information from human tissues in their approach. ECs make up the vasculature of organs throughout the body. Differences between micro-vascular and macrovascular ECs and CECs may be inherent to the cell type or to the microenvironments in which they reside. There may also be differences among species, so it may not necessarily be appropriate to generalize to all types of ECs as to signaling pathways involved in one type. The authors use human cells in their studies. They and others have found that RPE-CEC interactions are important in steps in neovascular AMD. For example, one group reported that 16 genes involved in angiogenesis were found upregulated in ECs grown in coculture with RPE compared with those grown in solo culture.22
As in any biologic process, cell culture is essential to work out mechanisms, but it is insufficient to generalize to the in vivo situation; therefore, animal models are useful. A major limitation in AMD is that many models are in rodents, which lack maculae. Models that expose animals to environmental stresses23,24 or that are transgenic are not generalizable to other pathways involved in the complexity of AMD. Some models require long time durations to develop features of AMD25 and may not be practical or economically feasible. The laser-induced CNV model represents a reproducible model of human neovascular AMD based on shared molecular mechanisms and the observation that laser injury in humans can cause CNV.26
Finally, implementation of a treatment in humans requires clinical trial testing for efficacy. However, mechanisms can rarely be worked out completely or safely in human studies. Large genetic studies and systems biology approaches have been very helpful in providing relevant associations between human AMD and genetic variants, especially when taken in context with environmental stresses and in conjunction with basic studies to determine mechanisms involved in pathologic steps of CNV.
Human heterotypic coculture and transmigration models
The authors’ lab created and uses physiologically relevant human heterotypic coculture and transmigration models (Figure 4).27 The porous insert of the transwell permits “contact” between the extracellular matrices/cell processes of the RPE and CECs in the coculture assay, analogous to in vivo situations in which CECs have already migrated through Bruch’s membrane but not yet into the sensory retina, such as in occult CNV. RPE and CECs are separately harvested and biochemical methods are used to measure activation of signaling pathways triggered in each cell type. The role of an identified pathway in CEC transmigration is then tested using specific inhibitors, genetic mutations or RNA interference (RNAi) introduced into either cell type in transmigration assays. In transmigration assays, wider pores permit vitally labeled CECs to migrate across the RPE, be distinguished from the RPE and be counted. The authors obtain CECs from human donor eyes and assure EC phenotype with EC markers. Several types of human RPE are used.27 ARPE-19 cells are easily transfected, are inexpensive and develop transepithelial electrical resistance (TER) of ∼35°ohms.cm2. The authors use these to model RPE with compromised barrier integrity. Primary adult or fetal RPE that can develop tight barrier properties (TERs > 100°ohms.cm2) is also used to corroborate findings in ARPE, because ARPE cells lack certain claudins, such as claudin 19, which is present in human RPE in vivo.28 Data obtained from coculture and transmigration assays relate to pathways important in human neovascular AMD.29–32 The authors integrate results from coculture and transmigration assays with data obtained from in vivo models and from human tissues. A three-pronged approach is taken, consisting of (1) coculture and transmigration models to determine mechanisms; (2) transgenic animals following laser injury to determine a functional in vivo correlate to the mechanism; and (3) human tissue samples to determine potential relevance to human AMD.
Figure 4.

(A) Diagram showing physiologically relevant human heterotypic coculture and transmigration assays with magnified inserts on right. The transwell insert does not represent Bruch’s membrane, but rather a means to control for contact between heterotypic cell processes and transmigration. (B) Diagram representing “occult” choroidal neovascularization (CNV) (top) in which choroidal endothelial cells (CECs) are activated to migrate through Bruch’s membrane and make contact with the basal aspect of the retinal pigment epithelium (RPE) (modeled by coculture assay). CECs migrate through the RPE into the sensory retina to form CNV (modeled by transmigration assay).
Notes: In Figure 4 (B), the yellow dots represent soluble vascular endothelial growth factor; black areas under the RPE represent drusen, deposits seen in clinical age-related macular degeneration.
Oxidative stress in AMD
Genetic predisposition plays an important role in AMD risk, and polymorphisms in the complement pathway, lipid metabolism, vitamin D, and retinoic acid receptor are among those that have been identified.5,33–36 In addition, environmental stresses also influence AMD risk. Risk factors of human AMD include increased age, smoking, increased body mass index, and potentially light and inflammation.37,38 Several of these factors, including smoking, inflammation, and light, can increase reactive oxygen species (ROS).23 Endogenous ROS can also play a role in experimental CNV.39 The Age-Related Eye Disease Study40 found that antioxidants reduced the progression of visual acuity loss and progression of advanced AMD, further supporting the role of ROS in the pathogenesis of neovascular AMD. The outer retina is vulnerable to oxidative stress because of high choroidal blood flow and photoreceptor photooxidation. High blood flow may increase oxygen, which increases superoxide radical generation by the mitochondria. Also, photooxidation in photoreceptors has been linked to complement activation,41 and activated complement can increase the release of RPE-produced VEGF, an important pathologic angiogenic factor in AMD. Activated complement can also increase membrane attack complex formation, which can trigger signaling of apoptosis,42 when nonlethal. ROS can also trigger signaling of angiogenesis.43,44 Therefore, many of the factors associated with AMD lead to increased generation of ROS and can mediate angiogenesis or apoptosis, which play roles in advanced forms of neovascular and atrophic AMD, respectively.
The role of aging and ROS on the VEGF signaling pathway
VEGF has been found to be one of the most important angiogenic factors involved in human pathologic angiogenesis. However, it is also a neuroprotective factor8 and it is important in vascular development and homeostasis.45 Therefore, broadly inhibiting its bioactivity to reduce pathologic angiogenesis may also have unwanted effects. The parent VEGFA messenger RNA has splice variants that have different bioactivities. The number of pro-angiogenic splice variants known in humans is approximately six, and three of these are close in protein composition to the mouse variants, each being one amino acid apart. (There are also VEGFb variants that inhibit angiogenesis but these are not discussed in this paper.) To address the question of whether sensory CNV could be inhibited by a form of VEGF that did not inhibit the physiologic beneficial aspects of VEGF, the authors first determined the splice variants that were upregulated by stresses known to be important in AMD (ie, increased age, oxidative stress, and contact between RPE and CECs to model occult CNV). When human RPE cells were exposed to several concentrations of H2O2, there was a selective and greater expression of VEGF189 than with control conditions, and also compared with the other splice variants (Figure 5A). Contact caused a dramatic change in expression of cell-associated VEGF189 compared with solo or non-contacting coculture (Figure 5B). RPE from donor eyes of humans over 70 years of age had greater expression of VEGF189 than those under 40 years of age (Figure 5C).32
Figure 5.

Vascular endothelial growth factor 189 (VEGF189) messenger RNA (mRNA) increased by (A) H2O2 (***P < 0.001 versus control), (B) CEC contact (*P = 0.05; **P = 0.001 versus solo), or (C) in eyes of donors over 70 years of age compared with those from 20 to 40 years of age (***P < 0.0001 versus young). (D) Silencing of VEGF189, (E) reduced p-VEGF receptor 2 (p-VEGFR2) in choroidal endothelial cells (CECs) grown in contacting coculture compared with non-silenced retinal pigment epithelium (RPE), and (F) inhibited CEC transmigration of RPE (*P = 0.05; ***P = 0.001 versus control siRNA).
©Copyright 2011. Association for Research in Vision and Ophthalmology. Adapted from Wang H, Geisen P, Wittchen ES, et al. The role of RPE cell-associated VEGF189 in choroidal endothelial cell transmigration across the RPE. Invest Ophthalmol Vis Sci. 2011;52(1):570–578.32
Abbreviation: siRNA, small interfering RNA.
H2O2 was then used as a means to upregulate VEGF189 in RPE and its role was tested in the coculture and transmigration assays. Compared with RPE exposed to control, there was a significant increase in phosphorylated VEGF receptor 2 (VEGFR2), but not p-VEGFR1, in CECs grown in contact with RPE exposed previously to H2O2. CEC transmigration increased significantly when RPE cells were incubated with H2O2 (P = 0.04). Using RNAi, the authors showed that 80% silencing of the upregulated VEGF189 in H2O2-exposed RPE (Figure 5D) inhibited VEGFR2 signaling in CECs grown in contacting coculture with RPE (Figure 5E), demonstrating the importance in RPE-CEC interactions. Also using RNAi, the authors found that knockdown of the upregulated VEGF189 in RPE reduced CEC transmigration by approximately 40% (Figure 5F).32 This finding indicated that either the small remaining amount of VEGF189 resistant to knockdown was sufficient to trigger signaling in CECs or that other factors that are also partially cell associated, including VEGF165, may be acting in parallel or through another receptor. The remaining 60% of CEC transmigration may also be caused by other factors, such as eotaxin/CCR3,46 integrins, and adhesion molecules, cytokines, or growth factors that trigger signaling of pathways important in CEC migration, such as phosphoinositol 3-kinase (PI3K) or Rac1.9–16,31 Using a neutralizing antibody to all splice variants of VEGF, the authors previously reported that CEC transmigration was mediated through active CEC Rac1 and/or the PI3K pathway, but not through src kinase.31 However, when the authors compared H2O2-exposed RPE transfected with small interfering RNA (siRNA) to silence VEGF189 with control siRNA, it was found that Rac1 was activated in CECs, but phosphorylated Akt, a downstream effector of the PI3K signaling pathway, was not.32
CEC migration across the RPE overexpressing VEGF189 was virtually abolished when Rac1 activity was inhibited in CECs transfected with a green fluorescent protein (GFP)-tagged construct containing the Rac-binding domain (RBD) of Rac1 effector, plenty of SH3s (POSH) (GFP-POSH-RBD). The authors previously found the POSH effector to inhibit Rac1 activity in CECs.31 These results provide support that VEGF189 facilitates CEC migration across the RPE, and this is mediated by VEGFR2-induced activation of Rac1 in CECs.32
Since VEGFR2 signaling is important in CEC migration, the authors investigated its role in CEC chemotaxis toward a VEGF gradient by measuring the length and number of “guidance” CEC filopodia (Figure 6A). The authors found that inhibition of p-VEGFR2 using the chemical receptor tyrosine kinase inhibitor SU5416 reduced CEC filopodial length (Figure 6B) and CEC migration, but not filopodial number (Figure 6C).47 In an animal model, the effect was specific to VEGFR2 and not to integrin beta3 or VEGFR1 signaling.
Figure 6.

Choroidal endothelial cell filopodia length was reduced by vascular endothelial growth factor receptor 2 tyrosine kinase inhibitor SU5416 (A) and (B), but number was not reduced (C).
Notes: **P = 0.01; *P < 0.05.
©Copyright 2009. Elsevier. Adapted from Budd S, Byfield G, Martiniuk D, Geisen P, Hartnett ME. Reduction in endothelial tip cell filopodia corresponds to reduced intravitreous but not intraretinal vascularization in a model of ROP. Exp Eye Res. 2009;89(5):718–727.47
From these studies, it was found that environmental stresses associated with and potentially causal in AMD led to increased VEGF, especially the VEGF189 splice variant, and that signaling from RPE-generated VEGF induced CEC VEGFR2 phosphorylation resulting in increased CEC transmigration of the RPE. Also, it was found that Rac1 was activated in CECs grown in contact with RPE, and upregulated RPE-derived VEGF189 was one factor found to activate Rac1 in CECs. Activated CEC Rac1 was important in CEC transmigration.31 Furthermore, VEGF189 was not the sole cause of CEC transmigration of the RPE, in that CEC transmigration was reduced by 40% when VEGF189 was silenced32 and by about the same amount when a neutralizing antibody to VEGF was used.29 These numbers are similar to those seen in clinical trials in which 40% of patients with neovascular AMD had improvement in visual acuity1 following treatment with anti-VEGF agents. Therefore, inhibiting VEGF bioactivity does not appear sufficient to completely prevent CEC transmigration of the RPE, whereas inhibition of active Rac1 in CECs nearly abolished CEC transmigration. Developing a means to safely reduce activated Rac1 in CECs may provide a safe means to prevent CEC transmigration and CNV in the sensory retina. However, broad inhibition of Rac1 has unacceptable toxicity,48,49 so targeting specific pathways that trigger Rac1-induced CEC migration may provide a safe therapy.
Rac1 in ROS generation and CNV
Besides activating CECs to migrate, Rac1 is also a subunit required for NADPH oxidase activation and ROS generation. Therefore, NADPH oxidase activation and ROS-triggered signaling were studied, and it was found that hypoxia triggered NADPH oxidase activation in cultured ECs and induced angiogenesis in a model that fluctuated oxygen levels and led to an angiopathic retinopathy in vivo.50 Retinal VEGF expression was increased in the model,51 and both VEGF and NADPH oxidase appeared to trigger the Janus kinase/signal transducers and activators of transcription pathway and, specifically, STAT3 phosphorylation, which mediated aberrant angiogenesis.52 Therefore, it was predicted that VEGF would activate NADPH oxidase in CECs. Indeed, VEGF activated Rac1 in CECs (Figure 7A), and active Rac1 then mediated NADPH oxidase-generated ROS (Figure 7B). Furthermore, the activation of Rac1 occurred upstream and independent of ROS generation, since treatment with apocynin did not significantly affect Rac1 activity (Figure 7A). In mice that were silenced for a subunit of NADPH oxidase, p47phox, or that received apocynin to inhibit NADPH oxidase activation, dihydroethidium fluorescence (indicator of ROS) in choroidal flat mounts (Figure 7C) and CNV volume following laser injury (Figure 7D) were significantly reduced.39
Figure 7.

(A) Vascular endothelial growth factor (VEGF) treatment induced Ras-related C3 botulinum toxin substrate 1 (Rac1) activation, and Rac activity was not reduced by inhibition of NADPH oxidase with apocynin; thus, Rac1 was upstream of NADPH oxidase (P < 0.05 versus control [Con], by a post hoc Bonferroni t-test). (B) VEGF-induced reactive oxygen species (ROS) detected by dichlorodihydrofluorescein (DCF) diacetate assay was inhibited in choroidal endothelial cells silenced for Rac1 (P < 0.05). (C) ROS visualized with dihydroethidium (DHE) fluorescence and (D) Laser-induced choroidal neovascularization measured in choroidal flat mounts was inhibited by apocynin in mice (*P < 0.05).
©Copyright 2010. Elsevier. Adapted from Monaghan-Benson E, Hartmann J, Vendrov AE, et al. The role of vascular endothelial growth factor-induced activation of NADPH oxidase in choroidal endothelial cells and choroidal neovascularization. Am J Pathol. 2010;177(4):2091–2102.39
Abbreviations: APO, apocynin; GTP, guanosine triphosphate; PBS, phosphate-buffered saline; RFU, relative fluorescence units; siRNA, small interfering RNA.
The authors also tested Rac1 activity downstream of other pathways important in neovascular AMD. They had collaborated with several laboratories to study the chemokine receptor CCR3 in AMD.46 CCR3 expression was increased in aged and AMD eyes compared with young donor eyes. Inhibition of the CCR3 using antibodies, chemical inhibitors, and transgenic mice reduced CEC migration and laser-induced CNV.46 In addition, CCR3 triggered activation of Rac1 in CECs.
RPE barrier properties and the role of Rac1
The evidence above showed the importance of RPE-CEC interactions in CEC transmigration across the RPE, an important step preceding the transition from occult (sub-RPE CNV) to CNV in the sensory retina. One of the properties of the RPE is the maintenance of the outer blood–retinal barrier. In homeostasis, the barriers of epithelial cells are constantly reassembling and disassembling to meet physiologic needs.53 Stresses, such as ROS, can disrupt barriers and their function.54 In human AMD, an impaired RPE barrier permits fluid, molecules, or other factors to enter the sensory retina and disrupt its structure, attract cells, and reduce visual acuity. FA and OCT detect clinical changes. It was wondered whether impaired RPE barrier integrity would permit growth factors to enter the sensory retina and act as chemoattractants for activated, migrating CECs.
The authors became interested in the potential role of Ras-related protein 1 (Rap1), a guanosine triphosphatase (GTPase) of the Ras superfamily, which is important in epithelial and endothelial junctions and motility. Like other GTPases, Rap1 is activated by guanine nucleotide exchange factors that mediate release of guanosine diphosphate and allow binding of guanosine triphosphate (GTP). Rap1 is inactivated when GTPase-activating proteins (GAPs) hydrolyze the GTP. When active, Rap1 interacts with downstream effectors and has a number of functions, including junctional assembly and integrity of epithelial cells55,56 and ECs.57 Knockout of Rap1 is lethal,58 but its two isoform-specific knockouts are viable.59–61 The activity of GTPases, rather than expression of the protein alone, is believed most important to cell function. This makes study in vivo difficult. However, pull-down assays designed to specifically detect GTP-bound Rap1 (ie, the active form) can be used to measure GTPase activity in vitro. In addition, through the use of viral constructs to alter endogenous Rap1 expression and/or activity, the effect on cell function in vitro can be determined.
The authors first tested the activity of Rap1 in human RPE grown in solo culture or in contacting coculture with CECs. Total Rap1 in cell lysates remained unchanged, but activity of Rap1 was reduced in RPE grown in contact with CECs compared with those grown in solo culture (Figure 8A). This suggested that CEC contact with RPE inhibited a pathway involving Rap1 that would normally provide a junctional strengthening function, such as that observed in human umbilical vein ECs.57 The authors used tools they developed to manipulate Rap1 activity. Rap1 activity was increased by exogenous expression of a constitutively active version of Rap1 protein (Figure 8B, left), or inhibited by expression of RapGAP57 (Figure 8B, right). The authors found that inhibition of Rap1 by expressing a RapGAP, or by adenoviral constructs containing siRNA to Rap1, reduced barrier integrity of the RPE and delayed junctional reassembly during a calcium switch assay.62 Our work provides evidence that Rap1 activity is important in the RPE’s ability to maintain junctional integrity during physiologic and perhaps pathologic stresses. If proven in vivo, there may potentially be pharmacologic methods to increase Rap1 activity and strengthen RPE barrier integrity.
Figure 8.
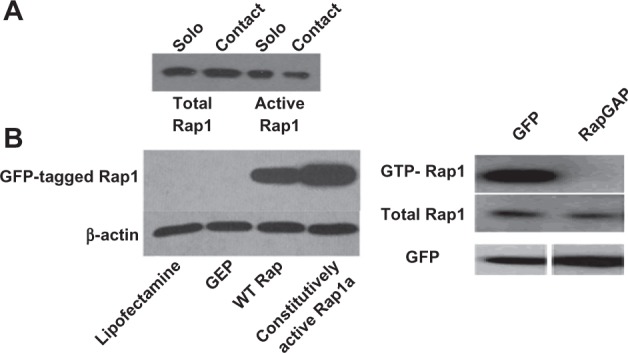
(A) Ras-related protein 1 (Rap1) activity is reduced in retinal pigment epithelium (RPE) grown in contact with choroidal endothelial cells (CECs) compared with solo RPE. (B) (Left) RPE cells transfected with a constitutively active green fluorescent protein (GFP)-tagged Rap1 show more protein in the pull-down than wild-type (WT) Rap or controls, indicating greater activity; (Right) expression of Rap GTPase-activating proteins (RapGAP), which acts by promoting guanosine triphosphate (GTP) to guanosine diphosphate hydrolysis, reduces active Rap1.
Conclusion
Neovascular AMD is a leading cause of central visual acuity loss in a growing segment of the population, those over the age of 60 years. Treatment has improved over the last decade but, because of tachyphylaxis and potential risk and toxicity of anti-VEGF agents, it is still limited. The authors have presented their study and have reviewed other studies in order to understand the mechanisms of CEC activation and transmigration of the RPE and of RPE barrier dysfunction. The heterotypic coculture and transmigration models used have proven to be relevant methods for testing pathways important in human neovascular AMD.29–32 The authors have found that the small GTPase Rac1 is important for CEC activation and transmigration and it is downstream of two signaling pathways that are important in human neovascular AMD, the VEGF-VEGFR231 and eotaxin-CCR346 pathways. Further, studies suggest that active Rap1 in RPE may play a role in strengthening RPE barrier integrity and resisting CEC transmigration of the RPE. Further study is warranted.62
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.CATT Research Group Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364(20):1897–1908. doi: 10.1056/NEJMoa1102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eghoj MS, Sorensen TL. Tachyphylaxis during treatment of exudative age-related macular degeneration with ranibizumab. Br J Ophthalmol. 2011 Jul 6; doi: 10.1136/bjo.2011.203893. Epub. [DOI] [PubMed] [Google Scholar]
- 3.Dias JR, Rodrigues EB, Maia M, Magalhaes O, Jr, Penha FM, Farah ME. Cytokines in neovascular age-related macular degeneration: fundamentals of targeted combination therapy. Br J Ophthalmol. 2011 May 5; doi: 10.1136/bjo.2010.186361. Epub. [DOI] [PubMed] [Google Scholar]
- 4.Bhutto IA, Kim SY, McLeod DS, et al. Localization of collagen XVIII and the endostatin portion of collagen XVIII in aged human control eyes and eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45(5):1544–1552. doi: 10.1167/iovs.03-0862. [DOI] [PubMed] [Google Scholar]
- 5.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102(20):7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamoreaux WJ, Fitzgerald MEC, Reiner A, Hasty KA, Charles ST. Vascular endothelial growth factor increases release of gelatinase A and decreases release of tissue inhibitor of metalloproteinases by microvascular endothelial cells in vitro. Microvasc Res. 1998;55(1):29–42. doi: 10.1006/mvre.1997.2056. [DOI] [PubMed] [Google Scholar]
- 7.Day S, Acquah K, Mruthyunjaya P, Grossman DS, Lee PP, Sloan FA. Ocular complications after anti-vascular endothelial growth factor therapy in Medicare patients with age-related macular degeneration. Am J Ophthalmol. 2011 Jun 9; doi: 10.1016/j.ajo.2011.01.053. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishijima K, Ng YS, Zhong L, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171(1):53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zarbin MA. Age-related macular degeneration: review of pathogenesis. Eur J Ophthalmol. 1998;8(4):199–206. doi: 10.1177/112067219800800401. [DOI] [PubMed] [Google Scholar]
- 10.Kamei M, Hollyfield JG. TIMP-3 in Bruch’s membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999;40(10):2367–2375. [PubMed] [Google Scholar]
- 11.Sheridan C, Williams R, Grierson I. Basement membranes and artificial substrates in cell transplantation. Graefes Arch Clin Exp Ophthalmol. 2004;242(1):68–75. doi: 10.1007/s00417-003-0800-z. [DOI] [PubMed] [Google Scholar]
- 12.Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122(4):598–614. doi: 10.1001/archopht.122.4.598. [DOI] [PubMed] [Google Scholar]
- 13.Stone EM, Braun TA, Russell SR, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351(4):346–353. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- 14.Mousa A, Lorelli W, Campochiaro PA. Role of hypoxia and extracellular matrix-integrin binding in the modulation of angiogenic growth factors secretion by retinal pigmented epithelial cells. J Cell Biochem. 1999;74(1):135–143. [PubMed] [Google Scholar]
- 15.Frank RN. Growth factors in age-related macular degeneration: pathogenic and therapeutic implications. Ophthalmic Res. 1997;29(5):341–353. doi: 10.1159/000268032. [DOI] [PubMed] [Google Scholar]
- 16.Renno RZ, Youssri AI, Michaud N, Gragoudas ES, Miller JW. Expression of pigment epithelium-derived factor in experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002;43(5):1574–1580. [PubMed] [Google Scholar]
- 17.Hartnett ME, Elsner AE. Characteristics of exudative age-related macular degeneration determined in vivo with confocal and indirect infrared imaging. Ophthalmology. 1996;103(1):58–71. doi: 10.1016/s0161-6420(96)30731-8. [DOI] [PubMed] [Google Scholar]
- 18.Hartnett ME, Weiter JJ, Staurenghi G, Elsner AE. Deep retinal vascular anomalous complexes in advanced age-related macular degeneration. Ophthalmology. 1996;103(12):2042–2053. doi: 10.1016/s0161-6420(96)30389-8. [DOI] [PubMed] [Google Scholar]
- 19.Hartnett ME, Weiter JJ, Garsd A, Jalkh AE. Classification of retinal pigment epithelial detachments associated with drusen. Graefes Arch Clin Exper Ophthalmol. 1992;230(1):11–19. doi: 10.1007/BF00166756. [DOI] [PubMed] [Google Scholar]
- 20.Yannuzzi LA, Negrao S, Iida T, et al. Retinal angiomatous proliferation in age-related macular degeneration. Retina. 2001;21(5):416–434. doi: 10.1097/00006982-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Stevens TS, Bressler NM, Maguire MG, et al. Occult choroidal neovascularization in age-related macular degeneration: a natural history study. Arch Ophthalmol. 1997;115(3):345–350. doi: 10.1001/archopht.1997.01100150347006. [DOI] [PubMed] [Google Scholar]
- 22.Dardik R, Livnat T, Nisgav Y, Weinberger D. Enhancement of angiogenic potential of endothelial cells by contact with retinal pigment epithelial cells in a model simulating pathological conditions. Invest Ophthalmol Vis Sci. 2010;51(12):6188–6195. doi: 10.1167/iovs.09-5095. [DOI] [PubMed] [Google Scholar]
- 23.Espinosa-Heidmann DG, Suner IJ, Catanuto P, Hernandez EP, Marin-Castano ME, Cousins SW. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest Ophthalmol Vis Sci. 2006;47(2):729–737. doi: 10.1167/iovs.05-0719. [DOI] [PubMed] [Google Scholar]
- 24.Wu T, Handa JT, Gottsch JD. Light-induced oxidative stress in choroidal endothelial cells in mice. Invest Ophthalmol Vis Sci. 2005;46(4):1117–1123. doi: 10.1167/iovs.04-0517. [DOI] [PubMed] [Google Scholar]
- 25.Ambati J, Anand A, Fernandez S, et al. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9(11):1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 26.Ambati J. Age-related macular degeneration and the other double helix: the Cogan Lecture. Invest Ophthalmol Vis Sci. 2011;52(5):2166–2169. doi: 10.1167/iovs.11-7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geisen P, McColm JR, King BM, Hartnett ME. Characterization of barrier properties and inducible VEGF expression of several types of retinal pigment epithelium in medium-term culture. Curr Eye Res. 2006;31(9):739–748. doi: 10.1080/02713680600837408. [DOI] [PubMed] [Google Scholar]
- 28.Peng S, Rao VS, Adelman RA, Rizzolo LJ. Claudin-19 and the barrier properties of the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2011;52(3):1392–1403. doi: 10.1167/iovs.10-5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geisen P, McColm JR, Hartnett ME. Choroidal endothelial cells transmigrate across the retinal pigment epithelium but do not proliferate in response to soluble vascular endothelial growth factor. Exp Eye Res. 2006;82(4):608–619. doi: 10.1016/j.exer.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 30.Hartnett ME, Lappas A, Darland D, McColm JR, Lovejoy S, D’Amore PA. Retinal pigment epithelium and endothelial cell interaction causes retinal pigment epithelial barrier dysfunction via a soluble VEGF-dependent mechanism. Exp Eye Res. 2003;77(5):593–599. doi: 10.1016/s0014-4835(03)00189-1. [DOI] [PubMed] [Google Scholar]
- 31.Peterson LJ, Wittchen ES, Geisen P, Burridge K, Hartnett ME. Heterotypic RPE-choroidal endothelial cell contact increases choroidal endothelial cell transmigration via PI 3-kinase and Rac1. Exp Eye Res. 2007;84(4):737–744. doi: 10.1016/j.exer.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Geisen P, Wittchen ES, et al. The role of RPE cell-associated VEGF189 in choroidal endothelial cell transmigration across the RPE. Invest Ophthalmol Vis Sci. 2011;52(1):570–578. doi: 10.1167/iovs.10-5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 35.Silveira AC, Morrison MA, Ji F, et al. Convergence of linkage, gene expression and association data demonstrates the influence of the RAR-related orphan receptor alpha (RORA) gene on neovascular AMD: a systems biology based approach. Vision Res. 2010;50(7):698–715. doi: 10.1016/j.visres.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neale BM, Fagerness J, Reynolds R, et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC) Proc Natl Acad Sci U S A. 2010;107(16):7395–7400. doi: 10.1073/pnas.0912019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch Ophthalmol. 2005;123(6):774–782. doi: 10.1001/archopht.123.6.774. [DOI] [PubMed] [Google Scholar]
- 38.Seddon JM, Willett WC, Speizer FE, Hankinson SE. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA. 1996;276(14):1141–1146. [PubMed] [Google Scholar]
- 39.Monaghan-Benson E, Hartmann J, Vendrov AE, et al. The role of vascular endothelial growth factor-induced activation of NADPH oxidase in choroidal endothelial cells and choroidal neovascularization. Am J Pathol. 2010;177(4):2091–2102. doi: 10.2353/ajpath.2010.090878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Age-Related Eye Disease Study Research Group A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103(44):16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou J, Kim SR, Westlund BS, Sparrow JR. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009;50(3):1392–1399. doi: 10.1167/iovs.08-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9(6):731–739. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 44.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res. 2006;71(2):226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 45.Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D’Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A. 2009;106(44):18751–18756. doi: 10.1073/pnas.0905010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda A, Baffi JZ, Kleinman ME, et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460(7252):225–230. doi: 10.1038/nature08151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Budd S, Byfield G, Martiniuk D, Geisen P, Hartnett ME. Reduction in endothelial tip cell filopodia corresponds to reduced intravitreous but not intraretinal vascularization in a model of ROP. Exp Eye Res. 2009;89(5):718–727. doi: 10.1016/j.exer.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hernández E, De La Mota-Peynado A, Dharmawardhane S, Vlaar CP. Novel inhibitors of Rac1 in metastatic breast cancer. P R Health Sci J. 2010;29(4):348–356. [PubMed] [Google Scholar]
- 49.Firestein GS. ‘Rac’-ing upstream to treat rheumatoid arthritis. Arthritis Res Ther. 2010;12(1):109. doi: 10.1186/ar2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saito Y, Uppal A, Byfield G, Budd S, Hartnett ME. Activated NAD(P)H oxidase from supplemental oxygen induces neovascularization independent of VEGF in retinopathy of prematurity model. Invest Ophthalmol Vis Sci. 2008;49(4):1591–1598. doi: 10.1167/iovs.07-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Budd SJ, Thompson H, Hartnett ME. Association of retinal vascular endothelial growth factor with avascular retina in a rat model of retinopathy of prematurity. Arch Ophthalmol. 2010;128(8):1014–1021. doi: 10.1001/archophthalmol.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Byfield G, Budd S, Hartnett ME. The role of supplemental oxygen and JAK/STAT signaling in intravitreous neovascularization in a ROP rat model. Invest Ophthalmol Vis Sci. 2009;50(7):3360–3365. doi: 10.1167/iovs.08-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sallee JL, Wittchen ES, Burridge K. Regulation of cell adhesion by protein-tyrosine phosphatases: II. Cell-cell adhesion. J Biol Chem. 2006;281(24):16189–16192. doi: 10.1074/jbc.R600003200. [DOI] [PubMed] [Google Scholar]
- 54.Rao RK, Basuroy S, Rao VU, Karnaky KJ, Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. 2002;368(Pt 2):471–481. doi: 10.1042/BJ20011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Price LS, Hajdo-Milasinovic A, Zhao J, Zwartkruis FJ, Collard JG, Bos JL. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J Biol Chem. 2004;279(34):35127–35132. doi: 10.1074/jbc.M404917200. [DOI] [PubMed] [Google Scholar]
- 56.Hogan C, Serpente N, Cogram P, et al. Rap1 regulates the formation of E-cadherin-based cell-cell contacts. Mol Cell Biol. 2004;24(15):6690–6700. doi: 10.1128/MCB.24.15.6690-6700.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wittchen ES, Worthylake RA, Kelly P, Casey PJ, Quilliam LA, Burridge K. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J Biol Chem. 2005;280(12):11675–11682. doi: 10.1074/jbc.M412595200. [DOI] [PubMed] [Google Scholar]
- 58.Chrzanowska-Wodnicka M. Regulation of angiogenesis by a small GTPase Rap1. Vascul Pharmacol. 2010;53(1–2):1–10. doi: 10.1016/j.vph.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Yan J, De P, et al. Rap1a null mice have altered myeloid cell functions suggesting distinct roles for the closely related Rap1a and 1b proteins. J Immunol. 2007;179(12):8322–8331. doi: 10.4049/jimmunol.179.12.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC., 2nd Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005;115(3):680–687. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan J, Li F, Ingram DA, Quilliam LA. Rap1a is a key regulator of fibroblast growth factor 2-induced angiogenesis and together with Rap1b controls human endothelial cell functions. Mol Cell Biol. 2008;28(18):5803–5810. doi: 10.1128/MCB.00393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wittchen ES, Hartnett ME. The small GTPase Rap1 is a novel regulator of RPE cell barrier function. Invest Ophthalmol Vis Sci. 2011 doi: 10.1167/iovs.11-7295. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]