

Abstract
Synaptic receptors gate the neuronal response to incoming signals, but they are not homogeneously distributed on dendrites. A spatially defined receptor distribution can preferentially amplify certain synaptic inputs, resize receptive fields of neurons, and optimize information processing within a neuronal circuit. Thus a longstanding question is how the spatial organization of synaptic receptors is achieved. Here we find that action potentials provide local signals that influence the distribution of synaptic AMPA receptors along dendrites in mouse cerebellar stellate cells. Graded dendritic depolarizations elevate CPEB3 protein at proximal dendrites where we suggest that CPEB3 binds to GluA2 mRNA suppressing GluA2 protein synthesis leading to a distance-dependent increase in synaptic GluA2 AMPARs. The activity-induced expression of CPEB3 requires increased Ca2+ and PKC activation. Our results suggest a cell-autonomous mechanism where sustained postsynaptic firing drives graded local protein synthesis thus directing the spatial organization of synaptic AMPARs.
The eTOC blurb
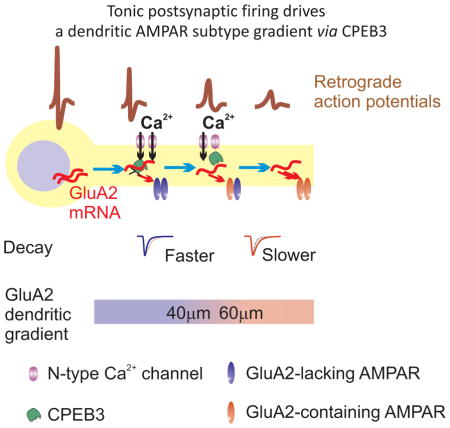
Savtchouk et al show there is a dendritic gradient in the expression of the GluA2 subunit in synaptic AMPA receptors. The gradient is maintained by tonic postsynaptic firing which controls the expression of CPEB3, a translational regulator. The postsynaptic AMPA receptor gradient optimizes information processing within a cerebellar circuit.
INTRODUCTION
Dendrites are the receptive zone for incoming signals onto a neuron, and are strategically positioned to control diverse features of synaptic activity. Synaptic receptors are a crucial determinant of the postsynaptic response, but they are not homogeneously distributed on dendrites (Gardner et al., 2001; Magee and Cook, 2000; Major et al., 2008; Nicholson et al., 2006; Pettit et al., 1997; Stricker et al., 1996; Toth and McBain, 1998). A spatially defined receptor distribution can preferentially amplify certain synaptic inputs, resize the receptive fields of neurons, and thereby optimize information processing within a neuronal circuit. This underlies the critical need to understand how the spatial organization of synapses on individual dendrites is achieved and maintained. Growing evidence supports the idea that dendrites integrate both the electrical and biochemical signals that are initiated by somatic action potentials and synaptic inputs (Hausser et al., 2000; Helmchen, 2007; Magee and Johnston, 2005). Somatic spikes can passively spread or actively travel backward in dendrites toward postsynaptic sites, and elevate intracellular Ca2+ levels by depolarizing dendritic segments. Here we have tested the hypothesis that sustained postsynaptic firing controls the pattern of synaptic glutamate receptor subunit expression and have identified the local cellular process that converts electrical signals into a spatially confined receptor distribution.
AMPA-type glutamate receptors mediate excitatory synaptic transmission in the CNS and are composed of four subunits (GluA1-4). Receptors that lack the GluA2 subunit display a number of distinct features, including a large channel conductance, rapid kinetics, and high Ca2+ permeability (Cull-Candy et al., 2006). They also exhibit a characteristic facilitation due to an activity-dependent polyamine unblock that occurs during a train of synaptic activity and which enhances the ability of excitatory postsynaptic potentials to evoke action potentials (APs) (Rozov and Burnashev, 1999; Savtchouk and Liu, 2011). GluA2 expression in neurons varies considerably with low GluA2 levels in a wide variety of neurons that display tonic activity, such as olfactory neurons, glutamatergic neurons in the lateral habenula, neostriatal cholinergic interneurons, auditory neurons in the deep cerebellar nucleus and GABAergic interneurons in several brain regions (Blakemore et al., 2006; Li et al., 2011; Liu and Cull-Candy, 2000; Maroteaux and Mameli, 2012; Samoilova et al., 1999). These Ca-permeable AMPARs play a critical role in the induction of NMDAR-independent synaptic plasticity, modulation of membrane excitability and long-range gamma oscillations (Liu and Zukin, 2007). Pyramidal neurons normally express GluA2-containing receptors, but switch to Ca-permeable, GluA2-lacking receptors after periods of hyperexcitability such as seizure or ischemia, and this leads to neuronal death (Liu et al., 2004; Noh et al., 2005). This suggests that one mechanism that could suppress GluA2 expression and promote the expression of synaptic Ca-permeable AMPARs could be sustained somatic AP firing.
Cerebellar stellate cells display spiking activity in the absence of synaptic input and somatic action potentials passively spread within the dendrites, thus elevating Ca2+ levels in proximal but not in distal dendrites (Myoga et al., 2009). Excitatory synaptic transmission onto GABAergic stellate cells is largely mediated by GluA2-lacking, Ca-permeable AMPARs, but also by some GluA2-containing receptors (Liu and Cull-Candy, 2002). The difference in the excitatory postsynaptic current (EPSC) waveforms between these two AMPAR subtypes markedly alters the ability of a synaptic response to evoke an AP (Savtchouk and Liu, 2011).
Although presynaptic activity-dependent homeostasis of postsynaptic receptor expression has been extensively studied, whether postsynaptic firing influences the subunit composition pattern of synaptic AMPARs is unknown. We propose that the change in AMPAR subunit expression along dendrites relies on a graded cellular signal, which locally influences synaptic AMPAR subunit synthesis or trafficking. A transcription-dependent regulation of synaptic AMPARs by postsynaptic firing, which has been described in cortical and CA1 pyramidal neurons (Goold and Nicoll, 2010; Ibata et al., 2008) and which is controlled by nuclear localized signals, is an unlikely mechanism for generating a dendritic AMPAR subunit gradient. In contrast CPEB3 (cytoplasmic polyadenylation element binding protein 3), an RNA binding protein that interacts with GluA2 mRNA and controls GluA2 synthesis, is a promising candidate (Huang et al., 2006; Theis et al., 2003; Vogler et al., 2009). Given that both the expression level and activity of CPEB3 can be altered by neuronal activity (Pavlopoulos et al., 2011; Wang and Huang, 2012), a cellular process involving dendritic CPEB3 and the local control of GluA2 synthesis may be sufficient to establish a synaptic GluA2 gradient along dendrites, a form of “topostatic” plasticity that homeostatically controls the spatial (or topological) distribution of AMPAR subtypes.
Here we examined the distributions of AMPAR subtypes along the dendrites of mouse cerebellar stellate cells and determined the impact of somatic AP activity on the spatial distribution of synaptic AMPARs. We find that the synaptic AMPAR subtype switches from GluA2-lacking to GluA2-containing as the distance of the synapses along the dendrites increases. This GluA2 gradient is driven by postsynaptic action potentials and requires Ca2+ entry at proximal dendrites and protein synthesis at distal dendrites. The depolarization-evoked Ca2+ rise activates PKC and elevates CPEB3 expression at proximal dendrites where we propose that CPEB3 binds to GluA2 mRNA and suppresses GluA2 protein synthesis. This leads to a distance-dependent increase in synaptic GluA2-containing AMPARs along dendrites. Our results reveal a function for postsynaptic spiking activity in spatially defining the distribution of synaptic receptors and synaptic integration, highlighting the importance of CPEB3 expression in translating dendritic depolarization into differing expression patterns of synaptic AMPAR subtypes.
RESULTS
Distance-dependent distribution of synaptic AMPAR subtypes along dendrites
We took advantage of a unique anatomic feature of the cerebellar cortex, in which dendrites of stellate cells lie within the sagittal plane and the excitatory inputs, parallel fiber axons, extend perpendicular to the dendritic plane. Thus in sagittal slices the position of a stimulating electrode evokes synaptic currents in a stellate cell at a point that corresponds to the dendritic site of parallel fiber innervation (Fig. 1A). This approach was used previously to map short-term plasticity along stellate cell dendrites (Abrahamsson et al., 2012). We examined whether synaptic AMPAR subtypes are differentially distributed along the dendrites of cerebellar stellate cells using three independent experimental approaches.
Figure 1. Distance dependent distribution of synaptic AMPAR subtypes along dendrites.

A. Left: Schematic showing that parallel fibers (vertical lines) extend orthogonally to the sagittal plane of stellate cell dendrites. The position of a stimulation electrode in a sagittal slice closely correlates with the location of a PF-stellate cell synapse. Middle: normalized I-V relationship for EPSCs evoked at each of the labeled sites. The dashed red line is the linear regression fit to the current amplitude at negative potentials (−60 to 0 mV). If EPSC amplitudes at positive potentials fall below the red line, these synaptic currents have an inwardly rectifying I-V relationship. Bottom: An example of the average current traces recorded in the soma (white circle) and evoked at several stimulation sites (colored circles) as labeled in the reconstructed image (Top right). Current amplitudes were normalized to the amplitude at −60 mV. At the distal sites the outward currents recorded at +40 mV relative to −60 mV increased. B. Left: The rectification index (RI) increases at distal synapses, indicating a greater presence of GluA2-containing AMPARs. Open circles represent the individual measurements (51 sites from 17 stellate cells and 16 mice) and the line shows the segmented average. Right: Average RI at the intermediate (40–60 μm) and distal synapses (>60 μm) is significantly larger than the RI at the proximal synapses (14–40 μm) (ANOVA, P < 0.00001). C. Left: The cumulative distribution of decay time constants for individual eEPSCs shows a significant increase in decay time at the intermediate (n=481 events) and distal synapses (n=373) compared to proximal sites (n=862) (#, P < 0.00005). Right: The decay time constant increases at distal synapses consistent with the presence of more GluA2-containing receptors (51 sites from 17 cells and 16 mice; ANOVA, P < 0.005). Bottom: The evoked EPSC amplitude at −60 mV (P < 0.05). Tukey post hoc test:*, P < 0.05; ***, P < 0.001. D. Bath application of IEM 1460 (100 μM), a GluA2-lacking AMPAR blocker, produced a greater inhibition at proximal synapses (5 sites, 1 site/cell, from 5 mice) than distal sites (6 sites, 1 site/cell from 6 mice, P = 0.005). Evoked synaptic currents were recorded before (black trace) and during the application of IEM 1460 (at 7.5–15 min, red trace) in the same stellate cells at each dendritic site. A time course of EPSC amplitude plot is shown in supplemental Figure 2A. E. Non-stationary fluctuation analysis (NSFA) of evoked EPSCs recorded at two dendritic locations. Left, example current traces of non-stationary fluctuation analysis of evoked EPSCs recorded at two dendritic locations (top, 35 μm; bottom, 45 μm). EPSCs (black lines) are superimposed to scaled average trace of 50 EPSCs (red lines). The difference between each EPSC and the scaled mean is shown as blue lines below. Middle and right, variance in the decay phase of the individual EPSCs around the mean is plotted against the averaged EPSC amplitude to produce the variance – mean current relationship. Red line is the parabolic fit from which mean single channel current was estimated (closed circles: fitted points of the data set). 50 (left) and 36 (middle) EPSCs were used for analysis. Right, the mean single channel conductance of EPSCs decreases at distal synapses (open circles present individual data; ANOVA, P < 0.0005; **, P < 0.002; ***, P < 0.0005). See also Figures S1 and S2.
First, to determine AMPA subunit composition we included spermine in the patch electrode and measured synaptic currents at various potentials. Spermine and other polyamines are known to block AMPARs that do not contain GluA2 at depolarized (>0 mV) potentials, giving rise to a characteristic inwardly rectifying current-voltage (I-V) relationship (Bowie and Mayer, 1995; Kamboj et al., 1995). Synaptic currents evoked by activating parallel fiber axons with a stimulating electrode placed within 40 μm from the soma showed a reduced amplitude at +40 mV, and exhibited an inwardly rectifying I-V relationship (Fig. 1A and 1B; suppl. Fig. 1A). This suggests that synaptic currents at proximal dendrites are mediated by GluA2-lacking AMPARs. As the stimulating electrode was moved away from the soma of the stellate cell, the EPSC amplitude at positive potentials increased relative to negative potentials and the I-V relationship became more linear. The rectification index of synapses at distal dendrites (> 40 μm; 0.64 ± 0.03, n = 25) was significantly greater than that at proximal dendrites (< 40 μm; 0.45 ± 0.02; n = 26; P <0.000002; Fig. 1B). Because the change occurred at about 40 μm from the soma, the dendritic region between 40 and 60 μm was considered the intermediate dendritic zone. The change in rectification properties was due to spermine block because when a spermine-free pipette solution was used the I-V relationship of EPSCs was linear and independent of the synaptic distance (suppl. Fig. 1B). The decay time constant of synaptic currents increased with distance (Fig. 1C), consistent with greater GluA2 presence at distal sites. These results indicate that the increase in rectification index of synaptic currents along dendrites reflects an increase in GluA2 expression.
Second, we used IEM 1460, a GluA2-lacking receptor selective inhibitor to determine subunit composition of synaptic AMPARs. Because EPSCs became more linear at ~ 40 μm we compared IEM 1460 inhibitory potency at proximal (<40 μm) and distal synapses (>40 μm). At proximal dendrites the amplitude of evoked EPSCs recorded at −60 mV were markedly reduced during the application of 100 μM IEM 1460 (68 ± 3% reduction, n = 5) without an alteration in the failure rate, indicating the prevalence of GluA2-lacking receptors (Fig. 1D; suppl Fig. 2A) closer to soma. In contrast IEM 1460 blocked only 46 ± 4% of the synaptic current at intermediate/distal dendritic sites (n = 6; P < 0.002). Thus synaptic AMPA receptors change from largely GluA2-lacking at proximal, to GluA2-containing, at more distal synapses.
Third, AMPA receptors that contain GluA2 subunits have a lower single channel conductance than GluA2-lacking receptors (Bats et al., 2012). We therefore estimated the mean single channel conductance (γ) of synaptic AMPARs using peak-scaled, non-stationary fluctuation analysis (NSFA (Benke et al., 1998; Traynelis et al., 1993)). We first tested the NSFA method on computer-generated AMPA EPSCs. When simulated AMPA EPSCs were analyzed, both the conductance (5–20 pS) and the number of activated channels (100–200 channels) of the simulated EPSCs were accurately detected using NSFA (suppl. Fig. 2B). We next performed NSFA analysis on evoked EPSC recordings from slices (Fig. 1E) that displayed rapid rise and decay kinetics and which were stable throughout the recording period (mean: 32.5 ± 1.7 events; 36 synapses; suppl. Fig. 2C). The average γ of EPSCs recorded at proximal dendrites was 14.5 ± 1.0 pS (n = 18). NSFA analysis was then performed to estimate the synaptic conductance at intermediate and distal dendritic sites (> 40 μm). The γ of EPSCs decreased at 40 – 60 μm (8.7 ± 0.7 pS; n = 11; P < 0.0005) and at > 60 μm (8.6 ± 1.2 pS; n = 7; P < 0.002; Fig. 1E). There was no correlation between the mean conductance and average EPSC amplitude (suppl. Fig. 2E and F), suggesting variability in the total number of AMPARs between synapses. The smaller conductance at distal synapses was not due to dendritic filtering because prolonging action potential duration promoted the expression of inwardly rectifying AMPARs at 40–60 μm, and increased the γ of EPSCs (see below). Thus the difference in conductance is mainly due to a variation in AMPAR composition at synapses. Furthermore the single channel conductance of EPSCs is greater at synapses with a strong inward rectification and rapid decay time (suppl. Fig. 2G and H), indicating that the distance-dependent increase in EPSC rectification index is not due to an increase in transmembrane AMPAR regulatory proteins (TARP)-AMPAR interactions, which enhances single channel conductance (Bats et al., 2012). Together our results show that GluA2 expression increases with dendritic distance in stellate cells.
Ca2+ entry during action potentials determines the synaptic AMPAR phenotype along dendrites
What physiological stimulus drives the dendritic GluA2 gradient? Cerebellar stellate cells fire APs in the absence of synaptic input (Hausser and Clark, 1997). Action potentials passively spread within the dendrites of stellate cells and spike amplitude decreases at ~50 μm from the soma in P30–33 rat stellate cells with more complex dendritic structures and longer dendrites than P17–19 rat stellate cells (Myoga et al., 2009). We characterized the morphology of stellate cell dendrites from P18–23 mice. Stellate cells filled with Alexa 594 had 3.5 ± 0.4 primary dendrites with an average length of 81.2 ± 6.2 μm (n = 29 dendrites, 8 cells; suppl. Fig. 3A). Dendrites were branched with 2.4 ± 0.5 secondary branches (secondary dendrites) per primary dendrite. Similar morphological features were also found in stellate cells in DIV 18 cultures (suppl. Fig. 3B). This dendritic branching pattern matches that of stellate cells from P30–33 rats (3.8 primary dendrites with an average length of 90 μm and 2.4 secondary branches per primary dendrite (Myoga et al., 2009).
Thus P18–23 mouse stellate cells have a similar dendritic branching pattern to mature rat stellate cells and in the latter the action potential amplitude decreases in the dendrites (Myoga et al., 2009). To determine whether somatic APs also fail to elicit a Ca2+ rise at distal dendrites in mouse stellate cells, we evoked five somatic action potentials at 100 Hz and measured the amplitude of the Ca2+ transients (expressed as ΔF/F) along the dendrites. For these experiments, stellate cells were filled with Oregon Green BAPTA-1 (100 μM) and Ca2+ transients were detected using a multi-photon line scan at various dendritic locations (Fig. 2A, B). We observed the largest Ca2+ peaks at the most proximal sites (with an average ΔF/F of 35 ± 2% for distances ≤ 20 μm, n = 14); the amplitude of dendritic Ca2+ transients progressively decreased with the distance from the soma, with the steepest decrease after 40 μm (Fig. 2C). The Ca2+ signal became undetectable from baseline for distances ≥ 70 μm. This marked attenuation of the amplitude of the Ca2+ transients along the dendrites suggests that somatic APs failed to back-propagate or were unable to activate a significant number of voltage-dependent Ca2+ channels in the distal dendrites.
Figure 2. The amplitude of Ca2+ transients generated in response to somatic action potentials decreases with the distance along the dendrites of stellate cells.

A. two-dimensional projection of a two-photon 3D Z-stack for a stellate cell filled with Oregon Green BAPTA-1 (100 μM). The grey lines represent the dendritic regions where the Ca2+ transients were measured using line scans. B. Ca2+ transients (expressed as ΔF/F) generated in response to a 100 Hz-train of five somatic action potentials (lower voltage trace). The numbers on the left of the traces refer to the locations of the line scans in (A). C. Plot of the mean amplitude of the peak of the Ca2+ transients associated with the train of five back propagating APs as a function of the distance from the soma. The values of ΔF/F were binned into 10 μm intervals (20 μm for the most distal locations, ≥ 70 μm) and averaged. Open circles represent the individual measurements (63 sites from 11 stellate cells and 8 mice). D. The decrease in dendritic Ca2+ transients is spatially correlated with an increase in the rectification index of EPSCs.
The decrease in dendritic Ca2+ transients generated in response to somatic APs is spatially correlated with an increase in the rectification index of EPSCs along dendrites (Fig. 2D). We therefore tested the possibility that attenuation of APs in mouse stellate cells leads to an increase in GluA2 expression at distal dendrites. We reasoned that increasing the spike duration would allow the depolarization to propagate further along the dendrites and thereby shift the dendritic GluA2 gradient towards more distal regions. Our recent study shows that application of the potassium channel blocker TEA increases the AP width in stellate cells and enhances Ca2+ entry via voltage-gated Ca2+ channels by 40% without changing the frequency of spontaneous APs, IPSCs and EPSCs (Liu et al., 2011). We therefore incubated cerebellar slices with TEA (1 mM). Since increasing AP duration also promotes GluA2 gene expression in stellate cells (Liu et al., 2010), actinomycin D, a transcriptional inhibitor, was included during TEA incubation to inhibit gene transcription. Furthermore, to minimize any presynaptic effects of TEA, we included picrotoxin (PTX) and kynurenic acid (KYNA) to block GABAA and ionotropic glutamate receptors, respectively, in both control conditions and during TEA incubation.
Incubation of cerebellar slices with actinomycin D (25 μM + PTX + KYNA) alone for 3 hrs did not alter the I-V relationship of EPSCs or the distance-dependent increase in rectification index (Fig. 3C; suppl. Fig. 4A and 4B). However following a 3 hr incubation with 1 mM TEA (+ actD + PTX + KYNA), we found that synapses located at 40–60 μm from the soma became more inwardly rectifying than controls (Fig. 3A), consistent with a decrease in GluA2 levels. The decay time constant of EPSCs was more rapid after TEA treatment relative to actinomycin D (+PTX+KYNA) control at 40–60 μm, also consistent with a local GluA2 decrease, whereas no difference was observed at synapses in proximal dendrites (<40 μm, Fig. 3B). A stronger inward rectification and more rapid decay time of EPSCs suggest an increase in GluA2-lacking receptors at 40–60 μm following TEA treatment. This hypothesis would predict a greater single channel conductance of EPSCs at intermediate dendritic sites. To test this idea we determined mean single channel conductance of EPSCs using NSFA. Indeed TEA treatment disrupted the gradient of channel conductance seen in control cells (Fig. 3D) and single channel conductance at 40–60 μm increased to 12.5 ± 1.0 pS (n = 5) from control 8.7 ± 0.7 pS (n = 11; P < 0.01). Thus prolonging the duration of APs can extend the expression of GluA2-lacking AMPARs to a more distal dendritic site than in control cells.
Figure 3. Ca2+ entry during action potentials determines synaptic AMPAR phenotype along dendrites.

A. Cerebellar slices were incubated with 1 mM tetraethylammonium (TEA) in the presence of 25 μM actinomycin D (actD), 100 μM picrotoxin (PTX) and 1 mM kynurenic acid (KYNA) for 3 hrs to increase the duration of APs and enhance Ca2+ entry (Liu et al., 2010). Left: Example of recordings at 2 locations and corresponding I-V relationship shows a decrease in outward current (at +40 mV) relative to current at −60 mV at the intermediate stimulation distance. Middle: The average RI and decay time constant of EPSCs did not increase at 40–60 μm (23 sites from 8 cells and 8 mice). Right: Increasing AP duration by TEA treatment shifted the dendritic GluA2 gradient toward more distal regions. Data from actD (+PTX+KYNA) treated and control in Fig. 1B were not different from each other (suppl Fig. 4A and 4B) and were shown as a combined control. Adjacent bins’ data points are labeled red/purple. B. The decay time constants for individual EPSCs were significantly reduced at the intermediate dendrites (40–60 μm) following TEA + ActD treatment relative to ActD control (#, P < 0.01, 203 and 248 events respectively), consistent with a decrease in synaptic GluA2. C. The rectification index and decay time constant of EPSCs increased at intermediate synapses in ActD treated control (20 sites from 8 cells and 8 animals). D. Following TEA treatment the mean single channel conductance of EPSCs at 40–60 μm was no longer reduced compared to proximal synapses. E and F. Cerebellar slices were treated with an N-type Ca2+ channel blocker, ω-conotoxin GVI-A (ω-CTX, 500 nM) for 3 hrs. I-V relationship for EPSCs evoked at the proximal sites became more linear following ω-CTX treatment (17 proximal sites and 12 distal sites from 12 cells and 9 mice). Open circles represent the individual measurements, adjacent bins’ data points are labelled blue/green. P < 0.05. See also Figure S4.
We next examined whether Ca2+ entry during APs drives the expression of GluA2-lacking AMPARs at the proximal dendrites. Ca2+ enters the dendrites of stellate cells via voltage-gated Ca2+ channels during APs (Fig. 2 and (Myoga et al., 2009)). N-type Ca2+ channels are expressed in stellate cells (Tharani et al., 1996). Following incubation of slices with 500 nM ω-conotoxin GVIA, an N type Ca2+ channel blocker for 3 hrs, the I-V relationship of EPSCs became more linear in the proximal dendrites, relative to control (Fig. 3E and 3F). Thus blocking N-type Ca2+ channels elevated synaptic GluA2 receptors in proximal dendrites and disrupted the gradient of synaptic GluA2 levels along the dendrites. These results indicate that Ca2+ entry during APs suppresses GluA2 expression or promotes GluA2-lacking receptors in proximal dendrites. Thus in contrast to a previously described presynaptic mechanism, in which elevated presynaptic activity increases synaptic GluA2 expression (Liu and Cull-Candy, 2000), postsynaptic APs suppress synaptic GluA2 levels, producing an opposite synaptic phenotype.
Synaptic activity can also increase the expression of GluA2-containing receptors at stellate cell synapses (Liu and Cull-Candy, 2000). We found that incubation with glutamate receptor and GABAA receptor blockers, kynurenic acid and picrotoxin in the presence of actinomycin D, for 3 hrs did not alter the pattern of synaptic GluA2 expression (Fig. 3C; suppl. Fig. 4A). We did not observe any difference in the paired pulse-ratio and the failure rate of EPSCs evoked at proximal and distal dendrites (Suppl. Fig. 4C), suggesting no apparent difference in glutamate release between the proximal and distal synapses. Thus synaptic activity is unlikely to drive the observed GluA2 gradient along the dendrites.
Spontaneous AP firing drives synaptic AMPAR subtype gradient via CPEB3
Thus far we found that synaptic AMPAR subtypes are differentially distributed along dendrites with GluA2-lacking receptors at proximal and GluA2-containing at distal dendrites. We also observed that inhibition of gene transcription with actinomycin D (+PTX+KYNA) did not disrupt the GluA2 gradient along dendrites (Fig. 3C and Suppl. Fig. 4A). We therefore tested the effects of a protein synthesis inhibitor, cycloheximide, on the dendritic distribution of GluA2 receptors when spermine was included in the pipette solution. Following incubation of cerebellar slices with cycloheximide for 3 hrs we observed no gradient in the rectification index of EPSCs along dendrites. Synaptic currents at a dendritic distance > 40 μm became more inwardly rectifying than controls with no alteration in the synaptic current amplitude (Fig. 4A and 4B; Suppl. Fig. 4D). Protein synthesis is thus required for the expression of GluA2 receptors in distal dendrites, and for the establishment of the GluA2 gradient along dendrites.
Figure 4. Disruption of the CPEB3-GluA2 mRNA interaction enhances synaptic GluA2 expression at the proximal dendrites in a protein synthesis-dependent manner.

A. Cerebellar slices were incubated with 100 μM cycloheximide (CHX) for 3 hrs and then synaptic currents were recorded and evoked at the stimulation sites labeled in the left panel. Spermine (100 μM) was included in the pipette solution to block synaptic GluA2-lacking receptors at positive potentials. Right: Average EPSC traces recorded at +40 and −60mV and corresponding I-V relationship. B. The increase in the RI of EPSCs at the distal synapses was abolished following CHX treatment (7 sites for proximal; 6 for intermediate; 4 sites for distal from 6 cells; 6 mice) and EPSC RI at > 40 μm was reduced compared to control (P < 0.05). C. SELEX1904 is an RNA oligomer that competes with GluA2 mRNA for binding to CPEB3 (Huang et al., 2006). If CPEB3 binding with GluA2 mRNA prevents GluA2 protein synthesis at proximal synapses, SELEX1904 should increase synaptic GluA2 expression in stellate cells. D. Left: Example of evoked EPSCs at a proximal synapse. Including SELEX1904 (10 μM) in the pipette solution increased EPSC amplitude at +40 mV and the I-V relationship of EPSCs became more linear with time, indicating an increase in synaptic GluA2 receptors. Control oligo sequence was identical to SELEX1904, but was missing several terminal nucleotides needed to stabilize the hairpin 3D conformation that disrupts CPEB3/GluA2 mRNA interaction, see Methods. Right: Anisomycin (ANS), when added to ACSF prior to and during recording, prevented the SELEX-induced increase in EPSC RI. E. RI of EPSCs remained unaltered during the first 30 minutes and then gradually increased at proximal dendrites (< 40 μm, n = 6). A control oligomer (10 μM) did not alter the amplitude (B) and RI of EPSCs (n = 5). (Two-way ANOVA, P < 0.0005 for treatment groups and for time bins; Tukey means comparison vs either control, *, P < 0.01; **: P < 0.005; or vs time bin 1, ***: P < 0.0005). F. The initial RIs are those recorded within 15 min (4 cells) and between 15–30 min (2 cells in which we were unable to find the inputs within 15 min) after obtaining the whole cell configuration. Change in RI (Δ RI) was calculated as a difference between the average RI after it reached a plateau and the initial RI (linear regression for SELEX, R2 = 0.62 and for CTL oligo, R2 = 0.11). See also Figure S4.
Protein synthesis of GluA2 subunits is regulated by an RNA binding protein, CPEB3 (Huang et al., 2006; Theis et al., 2003). We therefore tested whether CPEB3-GluA2 mRNA interactions could be responsible for the dendritic GluA2 gradient by suppressing synaptic GluA2 expression at proximal dendrites. First, does disruption of CPEB3-GluA2 mRNA interaction increase synaptic GluA2 content at proximal dendrites? Second, does AP firing regulate CPEB3 expression and thereby change AMPAR subtype?
Disruption of CPEB3-GluA2 mRNA Interaction Increases Synaptic GluA2 Expression at Proximal Synapses
To acutely disrupt a CPEB3-GluA2 mRNA interaction, we used a 49-nt oligomer, SELEX 1904, which previously was shown (Huang et al., 2006) to selectively compete with GluA2 mRNA for binding to CPEB3. We included a low concentration of SELEX 1904 in the patch electrode solution (10 μM) to disrupt the CPEB3-GluA2 mRNA interaction and monitored changes in the I-V relationship of EPSCs at proximal dendrites (<40 μm; Fig. 4C) over time. The I-V relationship of EPSCs was inwardly rectifying during the first 30 min and became more linear by 60–90 min (Fig. 4D and 4E). Synapses that showed stronger inward rectification at the beginning of recording also had a greater increase in rectification index (Fig. 4F). As a control for any non-specific effects, the inclusion of an oligomer that does not bind to CPEB3 in the pipette solution (10 μM) did not alter the rectification index of EPSCs over the time course of the recording (Fig. 4D, E, and F). If SELEX 1904 disrupts the CPEB3-GluA2 mRNA interaction and thereby increases GluA2 synthesis, the SELEX 1904-induced change should be prevented by protein synthesis inhibitors. Indeed when anisomycin was applied prior to, and throughout the duration of the recording, intracellular SELEX 1904 application failed to alter the rectification index of EPSCs (Fig. 4D and suppl. Fig. 4E). Therefore the CPEB3-GluA2 mRNA interaction in stellate cells prevents the synaptic expression of GluA2 by suppressing GluA2 protein synthesis. Increasing GluA2 expression by selective disruption of this interaction (and without interfering with any of the known mechanisms for GluA2-containing AMPAR insertion), indicates that regulation of GluA2 translation is required.
Blocking Postsynaptic Activity with Tetrodotoxin Increases Synaptic GluA2 AMPARs and Reduces CPEB3 Expression
Our results suggest that an AP-driven Ca2+ influx reduces the proximal synaptic GluA2 content (Figure 3), and this requires CPEB3-mRNA binding (Figure 4). Failure of spontaneous AP propagation to distal dendrites elevated synaptic GluA2 expression (Fig. 3). We hypothesized that postsynaptic spiking activity in stellate cells shifts AMPAR-GluA2 composition by promoting local CPEB3 expression. To determine the effect of APs on CPEB3 expression, we therefore tested whether inhibition of somatic AP firing would increase synaptic GluA2 content and reduce the CPEB3 level in the soma of stellate cells.
We directly examined whether somatic APs controlled the synaptic AMPAR phenotype by using tetrodotoxin (TTX). TTX inhibits spiking activity not only in postsynaptic stellate cells, but also silences presynaptic neurons. This causes a reduction of neurotransmitter release, which may influence postsynaptic AMPAR phenotype in stellate cells (Liu and Cull-Candy, 2000). We therefore added GABAA and ionotropic glutamate receptor blockers, picrotoxin (PTX) and kynurenic acid (KYNA), in both control conditions and during TTX incubation for 3 hrs. TTX and KYNA were washed out prior to recordings. While evoked EPSCs were used as a direct readout of synaptic properties at known distances along the dendrites in the results shown so far (Figs 1–4), spontaneous EPSCs (sEPSCs, Fig. 5) provided a measure of the intrinsic activity of all synapses onto stellate cells. Spontaneous EPSCs enable a comparison of the average EPSCs (“average” synapse on the cell) recorded under different experimental conditions and were therefore used to investigate the molecular mechanisms by which somatic APs control synaptic AMPAR subtype expression.
Figure 5. TTX treatment enhances the protein synthesis-dependent expression of GluA2-containing AMPARs at synapses and reduces the level of CPEB3 expression in stellate cells.

A. Slices were pretreated with 1 mM kynurenic acid (KYNA) and 100 μM picrotoxin (PTX) without (control) or with 0.5 μM tetrodotoxin (TTX) for 3 hrs. Right: Slices were treated with TTX in the presence of a protein synthesis inhibitor (100 μM cycloheximide, CHX) for 3 hrs. Average current traces recorded at +40 and −60 mV and the I-V relationship of AMPAR-mediated spontaneous EPSCs in stellate cells. TTX treatment increased rectification of EPSCs, which was prevented by cycloheximide. B. Summary data. Left: RI of individual cells (open circles) and average value (filled circle). Middle and right: EPSC amplitudes at +40 and −60 mV (cont, n = 8; TTX, n = 10; TTX + CHX, n = 6; CHX, n = 4;). ANOVA, P < 0.0001; Tukey post-hoc, *, P < 0.05; **, P < 0.005; ***, P < 0.0005. C. Cerebellar cultures were prepared from GAD-65 mice, in which stellate cells expressed GFP and GFP negative neurons were mainly granule cells. TTX treatment for 3 hrs reduced CPEB3-ir in GFP positive cells. Left, CPEB3-ir. Inserts show GFP. Right, comparison of somatic CPEB3-ir in TTX treated vs control stellate cells (GFP+) and granule cells (GFP−, bottom) (GFP+, >120 cells, P < 0.00001; GFP−, 67 control and 100 TTX-treated cells from 3 cultures). D. Effects of TTX treatment on CPEB3-ir levels in GFP+ cells and GFP− cells. Bottom: Prolonged incubation with TTX for 24 hrs did not further reduce the CPEB3 levels in GFP+ cells (3 cultures). ANOVA, P < 0.002; post-hoc test, **, P < 0.005; Scale bar: 12 μm. See also Figures S5, S6 and S7.
sEPSCs in stellate cells displayed an inwardly rectifying I-V relationship, consistent with the presence of GluA2-lacking AMPARs. Incubation with PTX and KYNA did not alter the rectification index (RI) of sEPSCs (0.32 ± 0.04, n = 8, vs without PTX and KYNA, 0.34 ± 0.05 ((Liu et al., 2010); Fig. 5A) or current amplitude (−40.8 ± 2.2 vs without PTX and KYNA, −47.0 ± 3.2 pA). However following TTX treatment, the sEPSC amplitude increased at +40 mV (at this potential synaptic currents are mediated mainly by GluA2-containing receptors) from 8.3 ± 0.4 to 14.4 ± 0.9 pA (P < 0.00005; control, n = 8; TTX, n = 10), but remained unaltered at −60 mV (Fig. 5B). Consequently, the I-V relationship of sEPSCs became more linear and the rectification index increased to 0.70 ± 0.06 (P < 0.0001, Fig. 5B). Furthermore the decay time of sEPSCs was prolonged after TTX treatment (suppl. Fig. 6A). These results indicate that TTX treatment increased the synaptic expression of GluA2-containing receptors.
In addition to inclusion of GluA2 subunits, the interaction of TARP with GluA2-lacking receptors can also produce a moderate increase in rectification index (Soto et al., 2007). Since CNQX acts as an AMPAR partial agonist in the presence of TARP, a CNQX-evoked inward current is diagnostic for the association of TARP with AMPARs (Menuz et al., 2007). We therefore tested whether the TTX-induced change involved an increased interaction with TARP by examining the CNQX-induced AMPAR-mediated increase of the holding current in stellate cells. Bath application of CNQX and cyclothiazide (to reduce AMPAR desensitization) elicited an additional inward current (44.2 ± 17.3 pA; n = 4), which was not enhanced after the TTX treatment (48.9 ± 11.0 pA; n = 5, Suppl. Fig. 6B), but was blocked by AMPAR antagonist GYKI. Thus, the increase in EPSC rectification index is unlikely to result from an increase in a TARP-AMPAR association.
This result together with the two opposing effects on the dendritic GluA2 gradient by TEA and an N-type Ca2+ channel blocker (Fig. 3), suggest that postsynaptic APs and the associated Ca influx are critical for controlling synaptic GluA2 expression and establishing a distance-dependent distribution of synaptic AMPAR subtypes along dendrites.
We have shown that inhibition of protein synthesis reduced the expression of synaptic GluA2 in distal dendrites (Figure 4B), suggesting that an increased expression of GluA2 at distal synapses requires protein synthesis. We hypothesized that attenuation of AP back propagation may enhance local GluA2 synthesis and thereby synaptic GluA2 expression at distal synapses. This model predicts that suppressing somatic APs would enhance the expression of synaptic GluA2 expression by promoting protein synthesis. Cerebellar slices were incubated with TTX (PTX + KYNA) in the presence of cycloheximide (100 μM), a protein synthesis inhibitor. After this treatment, synaptic currents had an inwardly rectifying I-V relationship (RI, 0.37 ± 0.04, n = 6; vs TTX, P < 0.005; Fig. 5A) in contrast to the usual TTX-induced linear relationship. Cycloheximide prevented the TTX-induced increase in EPSC amplitude at +40 mV (11.3 ± 0.6 pA; P < 0.05; Fig. 5B). A second protein synthesis inhibitor, anisomycin (40 μM), also prevented the increase in rectification index (0.33 ± 0.04, n = 5; P < 0.0005) that was induced by TTX treatment (suppl. Fig. 6C). Incubation with anisomycin and cycloheximide alone did not alter the EPSC rectification index (Fig. 5B; suppl. Fig. 5C and 5D). These results indicate that silencing AP firing increases synaptic GluA2-containing receptors via a protein synthesis-dependent process.
We next determined the effect of spontaneous APs on CPEB3 expression in stellate cells. Cerebellar cultures were prepared from GAD65::GFP knock-in mice (expressing intracellular GFP under the GAD67 promoter), in which the GFP positive cells are stellate cells and GFP negative neurons are mainly granule cells. Stellate cells in culture also exhibited tonic firing with a spontaneous AP firing frequency (Suppl. Fig. 6A) indistinguishable from that observed in slices (Liu et al., 2010). Cerebellar cultures were incubated with TTX in the presence of PTX and KYNA to block inhibitory and excitatory transmission for 3 hrs. This treatment increased the rectification index of spontaneous EPSCs and elevated the current amplitude at +40 mV (suppl. Fig. 6B) and thus stellate cells in both cultures and slices are regulated by APs. We therefore determined whether TTX treatment regulates CPEB3 expression in cultured stellate cells. Incubation with PTX and KYNA did not alter the CPEB3-ir in stellate cells (suppl Fig. 7B). Following TTX treatment, the level of CPEB3-ir in the soma of GFP positive neurons decreased by 27.9 ± 3.3% relative to controls that were incubated in PTX and KYNA (P < 0.01; Fig. 5C). In contrast GFP signals remained unaltered (suppl. Fig. 7F). The CPEB3 expression in stellate cells decreased rapidly within 3 hrs of TTX application, but prolonged incubation with TTX for 24 hrs did not further reduce CPEB3 levels (Fig. 5D). Therefore blocking AP firing reduced overall expression of CPEB3 in stellate cells. Because Ca2+ is known to activate elongation factor 2 (eEF2) kinase, a Ca2+/calmodulin-dependent protein kinase, which phosphorylates eEF2 and thereby suppresses protein synthesis (Sutton et al., 2007) we examined the level of phosphorylated eEF2, the inactive form of eEF2. We found that TTX treatment did not alter the levels of somatic peEF2-ir in stellate cells (suppl. Fig. 7C) while incubation with NH125, an eEF2 kinase inhibitor, as a positive control reduced peEF2 levels.
In contrast to stellate cells, cerebellar granule cells do not fire APs without synaptic input and express GluA2-containing receptors. If spontaneous AP spiking activity in stellate cells is indeed responsible for an elevated CPEB3 expression, blocking AP firing should not alter CPEB3 expression in granule cells. As predicted, CPEB3-ir in GFP-negative cells was indeed unaltered by TTX incubation (Fig. 5C and 5D and suppl. Fig. 7D). Together these results support the idea that postsynaptic activity drives the expression of CPEB3. Our results are consistent with a model in which somatic APs elevate CPEB3 at proximal dendrites and attenuation of retrograde APs lowers the CPEB3 expression at distal dendrites, and thereby controls the GluA2 gradient.
We therefore tested a prediction of this model which is that the CPEB3 level at proximal dendrites should be higher than at distal sites and should be reduced by inhibition of Ca2+ channel activity. We first determined whether the level of CPEB3 expression was lower in distal dendrites than in proximal dendrites. Cultured stellate cells from GAD67::GFP mice were immunostained for CPEB3, and the local dendritic intensity of CPEB3-ir was normalized to that of the GFP signal (i.e., the CPEB3-ir/GFP ratio). We found that the normalized CPEB3-ir was high in proximal dendrites and decreased by 20 ± 7% at 40–60 μm and 28 ± 8 % at >60 μm (Fig. 6A–C; repeated measures ANOVA, P < 0.005, n = 19 dendrites). The magnitude of the reduction in CPEB3-ir at distal dendrites (where AP propagation is attenuated) fits well with our prediction since the somatic CPEB3-ir was also reduced by 20–30 % when APs were blocked with TTX (Fig. 5C, 7D). This CPEB3-ir gradient along stellate cell processes was abolished by inhibition of N type Ca2+ channels with ω-conotoxin GVIA for 3 hrs (Fig. 6C, middle) while the GFP levels in dendrites remained unaffected by this drug (Fig. 6C, right). Following TEA (+actD+PTX+KYNA) treatment, CPEB3-ir no longer declined in intermediate dendrites, while blocking synaptic transmission and gene transcription (actD+PTX+KYNA) had no effect on the CPEB3-ir gradient (Fig. 6D). Thus the CPEB3 expression level decreases with distance along dendrites and elevating or reducing Ca2+ entry via voltage-gated Ca2+ channels can disrupt the CPEB3 gradient. In contrast to CPEB3, the level of peEF2-ir did not change along the dendrites (ΔpeEF2-ir/GFP relative to proximal dendrites: 3 ± 11% at 40–60 μm and 8 ± 10 % at >60 μm; n = 8). Thus elevated CPEB3 rather than suppression of protein synthesis by peEF2 leads to a reduction of synaptic GluA2 content at proximal dendrites. Together these data suggest that local changes in CPEB3 distribution may play an important role in establishing or maintaining a GluA2-AMPAR gradient
Figure 6. Immunohistochemical gradients of CPEB3 and GluA2 proteins in stellate cell dendrites.

A. Examples of dendritic CPEB3-ir in GFP+ neurons in cerebellar cultures. CPEB3/GFP ratio was calculated by dividing the corresponding signals in each ROI (see Methods for details). Individual channels (GFP-ir, CPEB3-ir, color-coded ratio) are diagonally offset for illustration purposes. Left: Control CPEB3 staining showed a distance-dependent decrease towards the distal dendritic regions (3 hrs incubation in PTX+KYNA). Right: the CPEB3-ir decrease was attenuated by incubating cells in 500 nM ω-conotoxin GVIA (ω-CTX) prior to immunostaining for CPEB3 (3 hrs incubation in ω-CTX+PTX+KYNA). B. The ratio of CPEB3-ir/GFP fluorescence intensity was used to quantify the CPEB3 expression at various segments along the dendrites and was normalized to the value at 5–14 μm from the soma for each dendritic process. Right: group data, represented as a color gradient of an “average” schematic dendrite. C. CPEB3-ir decreased at more distal dendrites (>60 μm) in control (n = 19 from 7 cultures, one-way ANOVA, P < 0.05, Tukey post-hoc, 14–40 vs >60 μm, P<0.05), which was abolished following ω-CTX treatment (n = 14 from 6 cultures). *, P < 0.05; ***, P < 0.005. Right: dendritic GFP fluorescence did not change after ω-CTX treatment. D. Effects of TEA treatment on the CPEB3-ir along dendrites. Cultures were treated with ActD, (+KYNA and PTX) as control or with the addition of 1 mM TEA for 3 hrs. Left: CPEB3-ir decreases at intermediate (40–60 μm) and distal (>60 μm) dendrites relative to proximal dendrites (14–40 μm) in control (n = 13; repeated measures ANOVA, P < 0.002; Tukey post hoc test: *, P < 0.05; **, P < 0.005). However following TEA treatment CPEB3-ir did not decrease at intermediate sites (n = 11; RM-ANOVA, P = 0.055). Right: changes in CPEB3-ir at 40–60 μm relative to 14–40 μm of individual dendritic processes. One sample Wilcoxon signed rank test; †, P < 0.05. E. Left: Distal dendrites of stellate cells show an increase in GluA2-ir staining. Top: a representative example of surface GluA2 staining in GAD65::eGFP stellate cell. Bottom: the dendritic GluA2 puncta were color-coded for total GluA2-ir fluorescence (area × average intensity), corresponding to the total number of GluA2 molecules present at the synapse. Right: Artificially constructed image showing predicted distribution of GluA2 puncta along an “average” dendrite, modelled on an increase in 30% puncta size and 20% intensity (suppl. Fig. 8C). This example illustrates that it is difficult to visually observe the resulting 60% GluA2 increase at distal synapses “by eye,” yet it is amenable to computer-assisted detection (see Methods for explanations). F. Left, total GluA2-ir fluorescence along a dendritic process increased with the distance from the soma (11 processes from 5 cultures). Middle, Total GluA2-ir of individual processes for proximal vs distal puncta (paired Wilcoxon signed rank test, P < 0.005). Right, both RI in slices (see Fig. 1) and GluA2-ir showed similar increase with the dendritic distance. See also Figure S8.
Figure 7. Spontaneous action potentials regulate the expression of CPEB3 via activation of PKC.

A. Quantification of PKC-ir. Left, confocal images of a GABAergic interneuron (GFP). Dashed lines outline each compartment. Right, line scans showing the levels of PKC-ir, GFP and RedDot2 (RD2 for nuclear staining) across the soma. B. Translocation of PKC to the cytoplasm after TTX treatment. Top: PKC-ir in GFP neurons treated with TTX or PKC modulators. Bottom: distribution of PKC-ir in each compartment. C. Left, mean membrane and cytoplasmic PKC-ir levels. Right, the PKC memb/PKC cyto ratio. TTX treatment reduced membrane staining and increased cytoplasmic CPEB3-ir, thereby lowering the PKC memb/PKC cyto ratio. These changes were reversed by co-application of PMA and TTX. Chelerythrine (CHE) or bisindolylmaleimide (BIS), PKC inhibitors, also reduced cytoplasmic translocation of PKC and the PKC memb/PKC cyto ratio. D. Effect of the same treatments on CPEB3-ir level (> 3 cultures). Scale bars: 5 (A, B). C (ANOVA, P < 0.002), D (repeated measures ANOVA, P < 0.02), post-hoc test, *, P < 0.05; **, P < 0.01 ***, P < 0.005. See also Figure S7.
We next used an antibody that binds to the extracellular domain of GluA2 to examine the distribution of surface GluA2 immunoreactivity (ir) along the dendrites of cultured stellate cells. Cerebellar neurons were co-immunostained for GluA2 and MAP2 without membrane permeabilization. MAP2, an intracellular protein, was used to detect any permeabilization of the dendrites of GFP-expressing stellate cells and these dendrites were excluded from further analysis of surface GluA2 expression. Many GluA2-ir puncta were located opposite vGlut puncta (suppl. Fig. 8A), suggesting that these AMPARs are likely to be activated by presynaptic glutamatergic inputs. We found a higher level of GluA2-ir (total puncta fluoresce = size × fluorescence intensity) at distal dendrites than at proximal dendrites (~50% increase, Fig. 6E and F) but a lower puncta density at >60 μm (Suppl. Fig. 8C). This spatial change is closely correlated with the increase in rectification index of EPSCs along dendrites of stellate cells observed in slices (Fig. 6F and Suppl Fig. 8B). Thus GluA2 expression also increases with dendritic distance in cultured stellate cells.
Action potentials regulate CPEB3 expression via a PKC-dependent pathway
Blocking AP firing in stellate cells by TTX and attenuation of dendritic AP back propagation reduces Ca2+ entry that occurs through voltage-gated Ca2+ channels. Because repetitive Ca2+ spikes can activate protein kinase C and PKC detects the pattern of action potential firing in sensory neurons (Wan et al., 2012), we tested the idea that Ca2+ entry during APs enhances CPEB3 expression in stellate cells by activating PKC.
We first examined whether blocking AP firing in stellate cells suppressed PKC activity. Once PKC is activated, it is translocated to the plasma membrane (Codazzi et al., 2001). We thus determined the intensity of membrane staining of PKC (“active”) relative to PKC-ir in the cytoplasm (Fig. 7A). As a positive control we treated the cultures with PMA, a PKC activator for 30 min and found that GFP-positive neurons showed clear membrane staining with little cytoplasmic PKC-ir (Fig. 7B). We quantified the distribution of PKC using two methods: first, using line scans across the somata of the GFP-positive neurons to monitor the level of PKC-ir; and second, by quantifying total PKC-ir in the membrane, cytoplasm and nucleus (Fig. 7A).
In control stellate cells incubated with PTX and KYNA, we observed a prominent membrane PKC-ir and cytoplasmic PKC-ir (41 ± 2 vs 37 ± 0.5% of total PKC-ir, 33 cells; Fig. 7B, 7C). In contrast, after TTX treatment the level of cytoplasmic PKC-ir increased to 49 ± 3 % (P < 0.02), while the membrane PKC-ir diminished (26 ± 3 %; 33 cells, P < 0.05). The PKC membrane:PKC cytoplasm ratio decreased from 1.95 ± 0.3 in control to 1.02 ± 0.2 in TTX treated cells (P < 0.01; Fig. 7C). When PKC was activated with PMA (0.1 μM) at the end of the TTX treatment, we found that PMA completely abolished the TTX-induced change in PKC-ir (Fig. 7B–C). To rule out any sampling bias we calculated the thickness of the membrane staining and found that this was the same in all experimental conditions (suppl. Fig. 7E). Thus the reduction in PKC membrane translocation following TTX treatment indicates a decrease in PKC activity.
We next determined whether reducing PKC activity suppressed CPEB3 expression in the soma of stellate cells. Following a 3 hr incubation of cerebellar cultures with the PKC inhibitor, chelerythrine (1 μM), we found that the level of somatic CPEB3-ir in GFP-positive neurons decreased by 20 ± 4 % (4 cultures; RM ANOVA, P < 0.02; Fig. 7D and suppl. Fig. 7C). A second PKC inhibitor, bisindolylmaleimide I (0.3 μM), also reduced CPEB3-ir by 18 ± 2 % (P < 0.02), indicating that PKC activity can regulate CPEB3 expression. If silencing postsynaptic neuronal activity with TTX reduces CPEB3 expression via preventing PKC activation, the presence of a PKC inhibitor during the TTX treatment should not cause a further reduction in CPEB3-ir. Indeed the reduction in CPEB3-ir following co-incubation of cultures with TTX and chelerythrine was indistinguishable from that after TTX or chelerythrine treatment alone (Fig. 7D, suppl. fig. 7E). This change (27 ± 2%) was significantly lower than the predicted value if TTX and the PKC inhibitor reduced CPEB3 through two independent (and thus additive) pathways (37 ± 2 %; P < 0.05). Therefore TTX treatment occludes the suppression of CPEB3 levels by a PKC inhibitor, suggesting that the PKC signaling pathway mediates the postsynaptic activity-dependent regulation of CPEB3 expression. This implies that activation of PKC should prevent the TTX-induced decrease in CPEB3. Indeed in the presence of the PKC activator PMA, TTX failed to reduce CPEB3 expression in stellate cells (Fig. 7D, suppl. Fig. 7G). In contrast to CPEB3 expression, TTX treatment and enhanced or suppressed PKC activity did not alter the GFP signal (suppl. Fig. 7F). Together these results indicate that blocking somatic APs with TTX reduces the activity of PKC in cerebellar stellate cells, leading to a decrease in CPEB3 expression.
DISCUSSION
Synaptic AMPARs are critical in determining the postsynaptic response, but are not uniformly distributed on dendrites. A greater number of AMPARs are present at distal compared to proximal dendrites to compensate for dendritic filtering in hippocampal pyramidal neurons (Magee and Cook, 2000; Nicholson et al., 2006; Pettit et al., 1997; Stricker et al., 1996). Synaptic AMPARs of distinct subunit composition are also differentially distributed on spatially segregated dendrites or in an input specific manner in auditory and hippocampal neurons (Gardner et al., 2001; Toth and McBain, 1998). Such a gradient of synaptic AMPAR subtypes is predicted to afford the synapse with not only different postsynaptic responses but also distinct forms of synaptic plasticity in a distance-dependent manner, enhancing the computational power of individual neurons. However the mechanism that controls synaptic strength/properties based on dendritic distance from the soma is not known. Somatic APs actively propagate or passively spread along dendrites, and both dendritic depolarization and Ca2+ entry often attenuate with distance (Gasparini and Migliore, 2015; Hausser et al., 2000). This may supply a local dendritic signal to synapses that influences the distribution of synaptic receptors. Here we provide experimental evidence supporting the idea that dendrites not only integrate synaptic signals, but also control the spatial distribution of synaptic AMPAR subtype in retrograde. We identified a CPEB3 expression gradient, which is locally regulated by electrical signals and directly controls GluA2 protein synthesis, and is responsible for the distance-dependent differential expression of synaptic AMPAR subtypes. Our finding that a depolarization-evoked Ca2+ rise activates PKC and thus elevates the CPEB3 levels, provides a molecular link between electrical signaling and CPEB3 expression. We conclude that sustained AP activity in postsynaptic neurons elevates CPEB3 levels, suppresses GluA2 translation and promotes GluA2-lacking AMPAR expression at proximal dendrites, establishing a GluA2 gradient along dendrites (suppl. Fig. 8D).
A GluA2 gradient can give rise to a distance-dependent postsynaptic response to presynaptic input. We have previously shown that incorporation of GluA2 subunits into an AMPAR prolongs the decay time constant of EPSCs and enhances the probability that synaptic inputs evoke APs (Savtchouk and Liu, 2011). Thus synapses at distal dendrites have a higher rate of EPSP-AP coupling. However GluA2-lacking AMPARs exhibit paired pulse potentiation due to the release of polyamine block by the second stimulus, and therefore proximal synapses would respond more effectively to a train of stimuli, such as those evoked by sensory stimulation (suppl. Fig. 8E)(Abrahamsson et al., 2012; Chadderton et al., 2004; Savtchouk and Liu, 2011). Furthermore since GluA2-lacking AMPARs are highly Ca2+ permeable and can trigger anti-Hebbian synaptic plasticity, synapses at proximal (but not at distal) dendritic locations can undergo a switch in AMPAR phenotype to GluA2-containing receptors, leading to a reduction in synaptic response (Savtchouk and Liu, 2011). Activation of Ca permeable AMPARs can also lead to a lasting reduction in glutamate release at parallel fiber to stellate cell synapses (Soler-Llavina and Sabatini, 2006). Thus a distance-dependent distribution of synaptic AMPAR subtypes would enable multiple forms of synaptic plasticity to occur depending on the location of the synapse and so provide a mechanism for bidirectional synaptic plasticity along dendrites. Indeed AP back propagation can influence synaptic plasticity in cortical neurons in a distance-dependent manner (Sjostrom and Hausser, 2006).
Although presynaptic activity is known to regulate synaptic AMPARs, whether postsynaptic APs influence the composition of synaptic AMPARs remains poorly understood. Spontaneous spiking activity has long been recognized as a common feature of many neurons including olfactory neurons, glutamatergic neurons in the lateral habenula, neostriatal cholinergic interneurons, auditory neurons in DCN and GABAergic interneurons in several brain regions (Bennett and Wilson, 1999; Hausser and Clark, 1997; Kowski et al., 2009). While such activity has been implicated in interneuron migration and the establishment of connectivity in the olfactory sensory map (De Marco Garcia et al., 2011; Yu et al., 2004), the function of tonic activity in an established circuit is not well understood. Previous studies have revealed that many of these tonically active neurons also have synaptic GluA2-lacking receptors (Blakemore et al., 2006; Li et al., 2011; Liu and Cull-Candy, 2000; Samoilova et al., 1999). Our results show that loss of tonic activity enhances synaptic GluA2 expression, and provides evidence supporting the idea that intrinsic activity drives the expression of GluA2-lacking AMPARs. In contrast to spontaneously active neurons, cortical and CA1 pyramidal neurons express a high level of GluA2 mRNA, and postsynaptic activity can suppress GluA2 expression via a transcription-dependent mechanism (Goold and Nicoll, 2010; Ibata et al., 2008). Emerging evidence indicates that experience, such as learning or seizure episodes, can alter intrinsic membrane excitability (Breton and Stuart, 2009; Makara et al., 2009) and convert neurons that are normally silent in the absence of synaptic inputs to ones that display tonic activity (Matthews et al., 2008; Shah et al., 2004). Such a sustained enhancement in the intrinsic activity of postsynaptic neurons may promote the expression of synaptic GluA2-lacking receptors in depolarized dendrites, and thereby alter the activity of a neuronal network. Indeed learning and sensory experiences have been shown to increase GluA2-lacking receptors in the amygdala and sensory cortices (Clem and Barth, 2006; Clem and Huganir, 2010; Goel et al., 2006). Thus our results provide evidence for a role for postsynaptic activity in synaptic homeostasis, which gives rise to a characteristic distance-dependent spatial distribution of synaptic AMPAR subtypes.
We also suggest the local cellular process that converts electrical signals into a spatially confined receptor distribution. CPEB3 protein binds to GluA2 mRNA and regulates GluA2 translation, and is critical to learning and memory, as mutations in the CPEB3 gene are associated with memory loss (Huang et al., 2006; Theis et al., 2003; Vogler et al., 2009). Thus an activity-dependent regulation of CPEB3 expression is likely to influence memory formation. We have demonstrated that disruption of the CPEB3-GluA2 mRNA interaction rapidly alters functional synaptic AMPAR phenotype from GluA2-lacking to GluA2-containing receptors at proximal dendrites, providing direct evidence that the CPEB3-GluA2 interaction suppresses synaptic GluA2 receptors. Intriguingly we found a CPEB3-ir gradient along dendrites, which appears to closely mimic the GluA2 gradient we report in these cells. However we are currently limited to using a single, semi-quantitative technique (immunocytochemistry) to visualize CPEB3 protein and therefore further confirmation of the gradient-like distribution of CPEB3 needs to be performed once improved tools are available (e.g. with fluorescently tagged endogenous CPEB3 in transgenic mice). We also found that both the CPEB3 and the synaptic GluA2 gradient are disrupted by Ca2+ channel blockers. Thus it is likely that CPEB3 in dendrites controls the local synthesis of GluA2 and the content of synaptic GluA2. Our finding that membrane depolarization elevates CPEB3 levels suggests somatic APs act as a centrifugal signal that controls the dendritic synthesis and expression of synaptic receptors. The dendritic GluA2-ir that was located near the soma (<14 μm) appeared to be higher than in the proximal region (14–40 μm) and this is consistent with a greater RI of the EPSCs in this dendritic region (sup. Fig. 1A). The expression of somatic receptors is controlled by a different regulatory protein (s) than synaptic AMPARs (Bats et al., 2012), and may undergo a distinct form of activity-dependent regulation. Consistent with the idea of an activity-dependent increase in CPEB3, seizure activity increases the expression of CPEB3 mRNA in hippocampal pyramidal cells that normally express low levels of CPEB3(Theis et al., 2003). This however contrasts with an NMDAR-triggered down-regulation of CPEB3 levels via calpain-dependent protein degradation (Wang and Huang, 2012), suggesting that somatic APs and synaptic activity may produce two opposing effects on CPEB3 levels. A recent study showed that spatial learning promotes mono-ubiquitination of CPEB3 by Neutralized1 and enhances the expression of GluA2 and 1. (Pavlopoulos et al., 2011). Thus neuronal activity alters synaptic AMPAR subtype and number by two distinct mechanisms, namely regulation of the expression and the activity of CPEB3, respectively.
We found that postsynaptic action potentials activate PKC, and tonic PKC activity is critical for elevating and maintaining the CPEB3 levels that suppress GluA2 expression in stellate cells. This is a novel cell-autonomous plasticity mechanism by which the basal activity of a postsynaptic cell controls the expression of its own GluA2 receptors. Such a mechanism does not rely on synaptic input or changes in gene transcription, and thus fundamentally differs from the regulation of synaptic AMPAR phenotype by presynaptic activity and neuromodulators in response to external stimuli. However, the interplay between the cell-autonomous and input-dependent types of plasticity is yet to be explored. For example, we have previously shown that noradrenaline release during acute stress enhances GluA2 gene transcription and thus synaptic GluA2 expression in stellate cells (Liu et al., 2010). Overwhelming evidence indicates that glutamate secretion from presynaptic terminals can alter postsynaptic AMPARs via PICK1-dependent recycling of GluA2 and protein synthesis-dependent pathways. The latter commonly involves regulation of activity-regulated cytoskeleton-associated proteins and CaMKII by fragile X mental retardation protein, which alters receptor trafficking (Bear et al., 2004; Gardner et al., 2005; Kelly et al., 2009; Liu and Cull-Candy, 2005; Luscher and Huber, 2010; Sun and June Liu, 2007; Sutton and Schuman, 2006). In contrast, postsynaptic firing directly regulates CPEB3 levels and GluA2 translation, a mechanism that is distinct from the regulation of receptor trafficking and transcription. The possibility that somatic APs may also regulate AMPAR trafficking remains to be tested. While presynaptic activity promotes GluA2 expression, somatic action potentials reduce the synaptic GluA2 content. Thus these two processes are regulated by two distinct physiological signals, produce two opposite synaptic phenotypes and utilize two different mechanisms. Suppression of GluA2 protein synthesis by somatic action potentials may provide a mechanism to reverse the synaptic activity-induced increase in GluA2 levels.
Our results show that changes in synaptic AMPA receptor subtypes occur within a few hours following disruption of GluA2 mRNA-CPEB3 interactions and pharmacological inhibition of protein synthesis. This process is slower than the synaptic activity-induced plasticity via AMPAR trafficking, but faster than the overall AMPAR turnover and endocytosis rate, which ranges from hours to days (Cohen et al., 2013; Ehlers, 2000). Since several AMPAR pools with different turnover rates contribute to the overall AMPAR maintenance, one possibility is that dendritic GluA2 synthesis represents a pool that is regulated by postsynaptic APs and enables a more rapid cycling rate of GluA2 at dendritic sites (Cohen et al., 2013). A gradient of CPEB3 proteins along dendrites enables both the spatial and subunit specificity of dendritic AMPA receptor distribution although the overall turnover and endocytosis rate are not different among AMPAR subunits (Cohen et al., 2013; Ehlers, 2000).
Dendritic gradients have emerged as a common feature of neurons in the CNS. In addition to synaptic receptors, the density of voltage-gated ion channels also changes with dendritic distance (Hoffman et al., 1997; Kole et al., 2006; Magee, 1998; Norenberg et al., 2010; Sjostrom and Hausser, 2006). Our experiments indicate the existence of a cell-autonomous mechanism in which retrograde action potentials control dendritic CPEB3 levels and thereby synaptic AMPAR subunit expression. These results highlight the importance of local synthesis of synaptic receptors as a way to establish a spatially graded expression pattern. We conclude that postsynaptic AP firing sets up the dendritic gradient of synaptic AMPAR subtypes and serves as a powerful means to control excitatory drive and the response to incoming synaptic signals. A dendritic GluA2 gradient would preferentially amplify proximal synaptic inputs and enable these synapses to undergo anti-Hebbian synaptic plasticity to reduce synaptic drive. Such a spatially defined synaptic response and plasticity at the parallel fiber-stellate cell synapse may enhance the ability of cerebellar circuitry to process sensory information and experience-dependent learning. Given the importance of activity-dependent change in AMPAR phenotype, for example in learning and in the neuronal death that is associated with various neurological disorders (reviewed in (Liu and Zukin, 2007)), a postsynaptic action potential driven synaptic plasticity would present a novel mechanism for a modification of AMPAR subtypes in dendrites.
Experimental Procedures
Complete details of experimental conditions, methods, and analysis are provided in Supplemental Experimental Procedures.
Slice preparation and incubation
C57/BL6 mice (postnatal day 18–23) were decapitated in accordance with the animal welfare guidelines of LSU Health Sciences Center and Penn State University. Cerebellar slices were prepared as described previously (Liu et al., 2010; Savtchouk and Liu, 2011). Following decapitation, cerebellar slices (250–300 μm) were obtained with a Leica VT1200 vibrating microslicer. Cerebellar dissection and slicing were performed in an ice-cold slicing solution (mM: 125 NaCl, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 25 glucose, saturated with 95% O2-5% CO2, pH 7.4). Slices were maintained in external artificial cerebrospinal fluid (ACSF, in mM: 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 25 glucose) at room temperature. In several experiments cerebellar slices were incubated in ACSF containing 1 mM kynurenic acid and 0.1 mM picrotoxin (as control) or with the addition of drugs for at least 3 hours before recording.
Electrophysiology
Whole cell patch clamp recordings were obtained using an Axoclamp 700A or 700B amplifier (Axon Instruments). Stellate cells located in the outer two thirds of the molecular layer were visually identified under DIC using a 60x upright water immersion objective and by the presence of action potentials in the cell attached configuration and spontaneous synaptic currents in the whole cell configuration as described previously (Liu and Cull-Candy, 2000). Whole-cell recordings were performed using 5–7 MΩ borosilicate glass pipettes, and the cell series resistance was monitored throughout the experiment. The recording was terminated if the series resistance changed by more than 20–30%. All recordings were performed at room temperature.
Evoked EPSCs
Voltage clamp recordings were performed in sagittal slices using a cesium-based internal solution (in mM: 135 CsCl, 10 EGTA-Cs, 10 HEPES, 4 ATP-Na, 4 MgCl2, 5 TEA, and 1 QX314, pH adjusted to 7.3). To determine the subunit composition of synaptic AMPA receptors, 100 μM spermine was included in the internal solution unless otherwise indicated. Spermine and endogenous polyamines block GluA2-lacking (but not GluA2-containing) AMPARs at positive potentials. Therefore, synaptic GluA2 content at a synapse was assessed by comparing the synaptic conductance at positive and negative potentials from the I-V relationship. Synaptic AMPAR currents were evoked by placing a monopolar glass stimulating electrode filled with ACSF at various distances from the soma of a patched cell and applying brief (~400 μs) voltage pulses (5–25 V) in the presence of 200 μM PTX and 10 μM R-CPP at 0.3 Hz to block GABA and NMDA receptors, respectively. The stimulation strength was adjusted such that 50% of stimuli evoked EPSCs to minimize stimulation of multiple synapses.
To achieve a local, targeted activation of the parallel fibers we used a stimulation pipette with a small opening (5–8 MΩ), ejecting minimal currents to achieve threshold stimulation (50% failure rate).
Dendritic distance measurements
In a few pilot experiments we included 20 μM Alexa Fluor 488 in the pipette solution to visualize the dendritic processes and more accurately measure the synaptic distance. However Alexa 488, when included in the pipette solution, alters the rectification index of EPSCs (suppl. Fig. 1C), and therefore was not used to reveal dendritic morphology in electrophysiology experiments. We therefore estimated the dendritic length by the Cartesian distance between recording and stimulating electrodes.
CPEB3 oligonucleotide (Aptamer) experiments
To examine whether CPEB3 was involved in the synaptic GluA2 gradient, we used a synthetic RNA oligonucleotide sequence previously shown to disrupt the interaction of GluA2 mRNA and CPEB3 (Huang et al, 2006).
Ca2+ imaging
For Ca2+ imaging experiments (Gasparini, 2011), whole-cell patch-clamp somatic recordings were performed using a Dagan BVC-700 amplifier in the active ‘bridge’ mode. Trains of 5 somatic action potentials at 100 Hz were elicited by the injection of brief current steps (300–600 pA, 2 ms-duration each). Patch pipettes were filled with a solution containing (in mM): K-methylsulphonate 130, HEPES 10, NaCl 4, Mg2ATP 4, Tris2GTP 0.3, phosphocreatine 14, and Oregon Green BAPTA-1 (OGB-1) hexapotassium salt 0.1; the resistance of the electrodes was 4–6 MΩ. OGB-1 was excited using a Chameleon Ultra laser (Coherent, Santa Clara, CA) emitting ultra-fast, pulsed light at 920 nm; the emitted fluorescence was detected using an Ultima scanner (Prairie Technologies, Madison, WI) mounted over an Olympus (Center Valley, PA) BX61WI microscope.
Primary cerebellar cell culture
Cerebellar cell cultures were prepared using P7 wildtype and mutant mouse pups expressing eGFP under the GAD65 (GAD65::GFP) or GAD67 promoter (GAD67::GFP; from the Jackson Lab). Cells were maintained for 18–27 days in vitro at 37°C in 5% CO2. Stellate cells were visually identified by green fluorescent protein expression.
Immunocytochemistry
Cultured cerebellar neurons were washed in PBS and fixed in 4% paraformaldehyde in PBS for 20 minutes. A full list of antibodies, staining conditions, and image acquisition and analysis is provided in the Supplemental Experimental Procedures.
Statistics
All values are expressed as mean ± S.E.M. Statistical significance was assessed using ANOVA, repeated measures ANOVA, or Student’s t tests (paired, unpaired two-tailed or one sample) as appropriate if a data set passed the Shapiro-Wilk normality test. Otherwise, Mann-Whitney test and Wilcoxon signed rank test were used. The Kolmogorov–Smirnov test was used for comparison of cumulative distribution plots. Summary of statistical analysis are shown in suppl. Table 1.
Supplementary Material
Highlights.
Synaptic GluA2-containing AMPA receptors increase along dendrites of stellate cells
Postsynaptic action potentials and graded Ca2+ entry drive the GluA2 gradient.
Action potential firing activates PKC and elevates CPEB3 expression.
Elevated CPEB3 levels at proximal dendrites suppresses GluA2 protein synthesis.
Acknowledgments
This work was supported by National Science Foundation Grant IBN-0344559 and National Institutes of Health Grants NS58867 and MH095948 (S.Q.J.L.) and NS069714 (to S. Gasparini). We thank Drs. Kathryn L. Carzoli, Christophe J. Dubois, Matthieu Maroteaux, Yu Liu, Charles Nichols and Matthew Whim for experimental advice and helpful discussions, and the LSUHSC Neuroscience COBRE multi photon core (P30-GM103340).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahamsson T, Cathala L, Matsui K, Shigemoto R, Digregorio DA. Thin dendrites of cerebellar interneurons confer sublinear synaptic integration and a gradient of short-term plasticity. Neuron. 2012;73:1159–1172. doi: 10.1016/j.neuron.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Bats C, Soto D, Studniarczyk D, Farrant M, Cull-Candy SG. Channel properties reveal differential expression of TARPed and TARPless AMPARs in stargazer neurons. Nature neuroscience. 2012;15:853–861. doi: 10.1038/nn.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends in neurosciences. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Benke TA, Luthi A, Isaac JT, Collingridge GL. Modulation of AMPA receptor unitary conductance by synaptic activity. Nature. 1998;393:793–797. doi: 10.1038/31709. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ. Spontaneous activity of neostriatal cholinergic interneurons in vitro. The Journal of neuroscience. 1999;19:5586–5596. doi: 10.1523/JNEUROSCI.19-13-05586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore LJ, Resasco M, Mercado MA, Trombley PQ. Evidence for Ca (2+)-permeable AMPA receptors in the olfactory bulb. American journal of physiology Cell physiology. 2006;290:C925–935. doi: 10.1152/ajpcell.00392.2005. [DOI] [PubMed] [Google Scholar]
- Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Breton JD, Stuart GJ. Loss of sensory input increases the intrinsic excitability of layer 5 pyramidal neurons in rat barrel cortex. The Journal of physiology. 2009;587:5107–5119. doi: 10.1113/jphysiol.2009.180943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Clem RL, Barth A. Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron. 2006;49:663–670. doi: 10.1016/j.neuron.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codazzi F, Teruel MN, Meyer T. Control of astrocyte Ca (2+) oscillations and waves by oscillating translocation and activation of protein kinase C. Current biology: CB. 2001;11:1089–1097. doi: 10.1016/s0960-9822(01)00326-8. [DOI] [PubMed] [Google Scholar]
- Cohen LD, Zuchman R, Sorokina O, Muller A, Dieterich DC, Armstrong JD, Ziv T, Ziv NE. Metabolic turnover of synaptic proteins: kinetics, interdependencies and implications for synaptic maintenance. PloS one. 2013;8:e63191. doi: 10.1371/journal.pone.0063191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Current opinion in neurobiology. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- De Marco Garcia NV, Karayannis T, Fishell G. Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature. 2011;472:351–355. doi: 10.1038/nature09865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, Huganir RL. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron. 2005;45:903–915. doi: 10.1016/j.neuron.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Gardner SM, Trussell LO, Oertel D. Correlation of AMPA receptor subunit composition with synaptic input in the mammalian cochlear nuclei. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21:7428–7437. doi: 10.1523/JNEUROSCI.21-18-07428.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S, Migliore M. Action Potential Backpropagation. In: Jaeger D, Jung R, editors. Encyclopedia of Computational Neuroscience. New York, NY: Springer New York; 2015. pp. 133–137. [Google Scholar]
- Goel A, Jiang B, Xu LW, Song L, Kirkwood A, Lee HK. Cross-modal regulation of synaptic AMPA receptors in primary sensory cortices by visual experience. Nature neuroscience. 2006;9:1001–1003. doi: 10.1038/nn1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Hausser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signaling. Science. 2000;290:739–744. doi: 10.1126/science.290.5492.739. [DOI] [PubMed] [Google Scholar]
- Helmchen F. Biochemical compartmentalization in dendrites. In: Stuart Greg, Spruston Nelson, Häusser Michael., editors. Dendrites. 2. Oxford: Oxford University Press; 2007. [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Huang YS, Kan MC, Lin CL, Richter JD. CPEB3 and CPEB4 in neurons: analysis of RNA-binding specificity and translational control of AMPA receptor GluR2 mRNA. The EMBO journal. 2006;25:4865–4876. doi: 10.1038/sj.emboj.7601322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. The Journal of physiology. 1995;486(Pt 2):297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly L, Farrant M, Cull-Candy SG. Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nature neuroscience. 2009;12:593–601. doi: 10.1038/nn.2309. [DOI] [PubMed] [Google Scholar]
- Kole MH, Hallermann S, Stuart GJ. Single Ih channels in pyramidal neuron dendrites: properties, distribution, and impact on action potential output. The Journal of neuroscience. 2006;26:1677–1687. doi: 10.1523/JNEUROSCI.3664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowski AB, Veh RW, Weiss T. Dopaminergic activation excites rat lateral habenular neurons in vivo. Neuroscience. 2009;161:1154–1165. doi: 10.1016/j.neuroscience.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, Malinow R. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470:535–539. doi: 10.1038/nature09742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Lau L, Wei J, Zhu D, Zou S, Sun HS, Fu Y, Liu F, Lu Y. Expression of Ca (2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron. 2004;43:43–55. doi: 10.1016/j.neuron.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Cull-Candy SG. Activity-dependent change in AMPA receptor properties in cerebellar stellate cells. The Journal of neuroscience. 2002;22:3881–3889. doi: 10.1523/JNEUROSCI.22-10-03881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Cull-Candy SG. Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nature neuroscience. 2005;8:768–775. doi: 10.1038/nn1468. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends in neurosciences. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Liu Y, Formisano L, Savtchouk I, Takayasu Y, Szabo G, Zukin RS, Liu SJ. A single fear-inducing stimulus induces a transcription-dependent switch in synaptic AMPAR phenotype. Nature neuroscience. 2010;13:223–231. doi: 10.1038/nn.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Savtchouk I, Acharjee S, Liu SJ. Inhibition of Ca2+-activated large-conductance K+ channel activity alters synaptic AMPA receptor phenotype in mouse cerebellar stellate cells. Journal of neurophysiology. 2011;106:144–152. doi: 10.1152/jn.01107.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. The Journal of neuroscience. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Cook EP. Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nature neuroscience. 2000;3:895–903. doi: 10.1038/78800. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Plasticity of dendritic function. Current opinion in neurobiology. 2005;15:334–342. doi: 10.1016/j.conb.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Major G, Polsky A, Denk W, Schiller J, Tank DW. Spatiotemporally graded NMDA spike/plateau potentials in basal dendrites of neocortical pyramidal neurons. Journal of neurophysiology. 2008;99:2584–2601. doi: 10.1152/jn.00011.2008. [DOI] [PubMed] [Google Scholar]
- Makara JK, Losonczy A, Wen Q, Magee JC. Experience-dependent compartmentalized dendritic plasticity in rat hippocampal CA1 pyramidal neurons. Nature neuroscience. 2009;12:1485–1487. doi: 10.1038/nn.2428. [DOI] [PubMed] [Google Scholar]
- Maroteaux M, Mameli M. Cocaine evokes projection-specific synaptic plasticity of lateral habenula neurons. The Journal of neuroscience. 2012;32:12641–12646. doi: 10.1523/JNEUROSCI.2405-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EA, Weible AP, Shah S, Disterhoft JF. The BK-mediated fAHP is modulated by learning a hippocampus-dependent task. PNAS. 2008;105:15154–15159. doi: 10.1073/pnas.0805855105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz K, Stroud RM, Nicoll RA, Hays FA. TARP auxiliary subunits switch AMPA receptor antagonists into partial agonists. Science. 2007;318:815–817. doi: 10.1126/science.1146317. [DOI] [PubMed] [Google Scholar]
- Myoga MH, Beierlein M, Regehr WG. Somatic spikes regulate dendritic signaling in small neurons in the absence of backpropagating action potentials. The Journal of neuroscience. 2009;29:7803–7814. doi: 10.1523/JNEUROSCI.0030-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DA, Trana R, Katz Y, Kath WL, Spruston N, Geinisman Y. Distance-dependent differences in synapse number and AMPA receptor expression in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:431–442. doi: 10.1016/j.neuron.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. PNAS. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norenberg A, Hu H, Vida I, Bartos M, Jonas P. Distinct nonuniform cable properties optimize rapid and efficient activation of fast-spiking GABAergic interneurons. PNAS. 2010;107:894–899. doi: 10.1073/pnas.0910716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlopoulos E, Trifilieff P, Chevaleyre V, Fioriti L, Zairis S, Pagano A, Malleret G, Kandel ER. Neuralized1 activates CPEB3: a function for nonproteolytic ubiquitin in synaptic plasticity and memory storage. Cell. 2011;147:1369–1383. doi: 10.1016/j.cell.2011.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DL, Wang SS, Gee KR, Augustine GJ. Chemical two-photon uncaging: a novel approach to mapping glutamate receptors. Neuron. 1997;19:465–471. doi: 10.1016/s0896-6273(00)80361-x. [DOI] [PubMed] [Google Scholar]
- Samoilova MV, Buldakova SL, Vorobjev VS, Sharonova IN, Magazanik LG. The open channel blocking drug, IEM-1460, reveals functionally distinct alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors in rat brain neurons. Neuroscience. 1999;94:261–268. doi: 10.1016/s0306-4522(99)00326-7. [DOI] [PubMed] [Google Scholar]
- Savtchouk I, Liu SJ. Remodeling of synaptic AMPA receptor subtype alters the probability and pattern of action potential firing. The Journal of neuroscience. 2011;31:501–511. doi: 10.1523/JNEUROSCI.2608-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Hausser M. A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron. 2006;51:227–238. doi: 10.1016/j.neuron.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina GJ, Sabatini BL. Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nature neuroscience. 2006;9:798–806. doi: 10.1038/nn1698. [DOI] [PubMed] [Google Scholar]
- Soto D, Coombs ID, Kelly L, Farrant M, Cull-Candy SG. Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nature neuroscience. 2007;10:1260–1267. doi: 10.1038/nn1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker C, Field AC, Redman SJ. Statistical analysis of amplitude fluctuations in EPSCs evoked in rat CA1 pyramidal neurones in vitro. The Journal of physiology. 1996;490(Pt 2):419–441. doi: 10.1113/jphysiol.1996.sp021155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, June Liu S. Activation of extrasynaptic NMDA receptors induces a PKC-dependent switch in AMPA receptor subtypes in mouse cerebellar stellate cells. The Journal of physiology. 2007;583:537–553. doi: 10.1113/jphysiol.2007.136788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron. 2007;55:648–661. doi: 10.1016/j.neuron.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Tharani Y, Thurlow GA, Turner RW. Distribution of omega-Conotoxin GVIA binding sites in teleost cerebellar and electrosensory neurons. The Journal of comparative neurology. 1996;364:456–472. doi: 10.1002/(SICI)1096-9861(19960115)364:3<456::AID-CNE6>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Theis M, Si K, Kandel ER. Two previously undescribed members of the mouse CPEB family of genes and their inducible expression in the principal cell layers of the hippocampus. PNAS. 2003;100:9602–9607. doi: 10.1073/pnas.1133424100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K, McBain CJ. Afferent-specific innervation of two distinct AMPA receptor subtypes on single hippocampal interneurons. Nature neuroscience. 1998;1:572–578. doi: 10.1038/2807. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Silver RA, Cull-Candy SG. Estimated conductance of glutamate receptor channels activated during EPSCs at the cerebellar mossy fiber-granule cell synapse. Neuron. 1993;11:279–289. doi: 10.1016/0896-6273(93)90184-s. [DOI] [PubMed] [Google Scholar]
- Vogler C, Spalek K, Aerni A, Demougin P, Muller A, Huynh KD, Papassotiropoulos A, de Quervain DJ. CPEB3 is associated with human episodic memory. Frontiers in behavioral neuroscience. 2009;3:4. doi: 10.3389/neuro.08.004.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Jiang XY, Negroiu AM, Lu SG, McKay KS, Abrams TW. Protein kinase C acts as a molecular detector of firing patterns to mediate sensory gating in Aplysia. Nature neuroscience. 2012;15:1144–1152. doi: 10.1038/nn.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CF, Huang YS. Calpain 2 activated through N-methyl-D-aspartic acid receptor signaling cleaves CPEB3 and abrogates CPEB3-repressed translation in neurons. Molecular and cellular biology. 2012;32:3321–3332. doi: 10.1128/MCB.00296-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CR, Power J, Barnea G, O’Donnell S, Brown HE, Osborne J, Axel R, Gogos JA. Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron. 2004;42:553–566. doi: 10.1016/s0896-6273(04)00224-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.