

Abstract
Pancreatic neuroendocrine tumors (PNETs) are rare tumors, but have been increasing in incidence. Although typically thought of as indolent, more than half of patients present with metastatic disease. For many years, the only mutations commonly known in these tumors were those in the MEN1 gene. Recently, the genetics underlying PNETs have been further defined through exome sequencing. The most frequent alterations found in sporadic PNETs are in MEN1, DAXX/ATRX and a variety of genes in the mTOR pathway. Confirmation of these mutations have prompted trials with a number of drugs active in these pathways, and two were eventually approved in 2011—sunitinib and everolimus. New data additionally identify the MET and CD47 receptors as potential novel drug targets. Yet despite improvements in (PFS) with sunitinib and everolimus, further studies defining when to use these agents and factors associated with limitations in their utility are needed. As more discoveries are made in the lab that elucidate additional molecular mechanisms important in the initiation and metastasis of PNETs, continued efforts to translate these discoveries into distinct new therapies will be needed to improve patient survival.
INTRODUCTION
Pancreatic neuroendocrine tumors (PNETs) comprise only 1–2% of all pancreatic neoplasms, but have more than doubled in incidence over the past 30 years.(1–4) They are often perceived as indolent, but the majority are malignant and nearly 60% of patients will have distant metastases at the time of presentation.(5) PNETs are divided into two general categories: functional and nonfunctional, based on whether they hypersecrete hormones causing a clinical syndrome. A 2008 analysis of 1,483 PNETs in the Surveillance, Epidemiology, and End Results (SEER) database suggested that 90% of PNETs were nonfunctional. The median overall survival (OS) for all patients with PNETs was 28 months, 100 months in those with localized tumors, and this decreased to 17 months in metastatic disease. Patients with functional tumors have a median OS of 54 months, and those with non-functional tumors 26 months.(6) Survival is improved by surgery, with a median survival of 114 months in patients undergoing operation versus 35 months for those recommended to have surgery but who did not; this survival advantage also extended to patients with metastatic disease.(7–10)
PNETs are also classified by grade, which is determined either by the mitotic index or Ki-67 and an assessment of tumor morphology. High grade PNETs have > 20 mitoses/hpf or a Ki-67 index > 20%, and are often referred to as neuroendocrine carcinomas.(11) These tumors are most often poorly-differentiated, which may explain their resistance to standard PNET treatments, but can have mixed morphology (i.e. adenocarcinoma or well-differentiated components).(12) High grade, poorly-differentiated PNETs are a unique entity from their well-differentiated counterparts and are usually referred to as pancreatic neuroendocrine carcinomas (PNAC). These tumors are generally more aggressive, carry different genetic alterations, are operated upon less frequently and treated with different chemotherapeutic regimens (i.e. etoposide and cisplatin) than are PNETs. Thus for the purposes of this paper, when high grade, poorly-differentiated tumors are discussed it will be explicitly stated.
Historically, cytotoxic chemotherapy was the only other option for patients with widely metastatic PNETs. Recently, pharmaceuticals targeting cell-signaling pathways important in NET tumorigenesis have been approved by the FDA and suggest that individualized treatment with molecular therapies is not only possible, but may lead to improved survival. This review will examine the progress made deciphering the molecular mechanisms underlying PNET initiation and metastasis, and discuss how these discoveries are driving development of novel diagnostics and therapeutics.
BREAKTHROUGHS AT THE BENCH
An early discovery in the genetic underpinnings of PNETs was made in 1988, when three kindreds with multiple endocrine neoplasia type I (MEN1) were used to map the MEN1 locus to chromosome 11q13. Larsson et al. suggested that the MEN1 phenotype (parathyroid hyperplasia, pituitary adenoma, and/or PNETs) was due to a mutation in a tumor suppressor gene.(13) Chandrasekharappa et al. cloned the MEN1 gene nine years later using tumors from 15 MEN1 families, and named its protein product menin. They found 12 mutations in the gene, all likely leading to loss of function, which supported the tumor suppressor role of MEN1 (Figure 1).(14)
Figure 1. Mechanisms of genetic alteration and tumor formation.
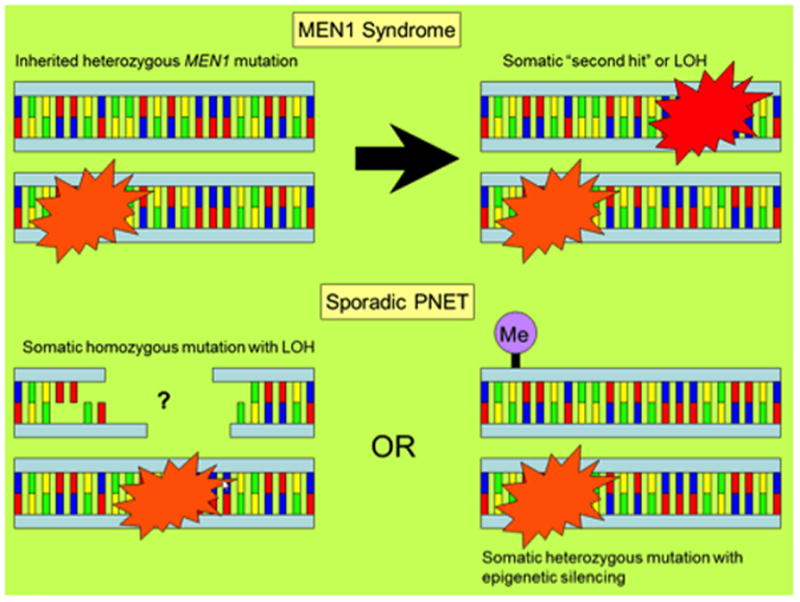
Mutations in both MEN1 and DAXX/ATRX are proposed to follow a two-hit model.(15) In multiple endocrine neoplasia type 1, an inherited heterozygous MEN1 mutation is then accompanied by a somatic second mutation in MEN1 or loss of heterozygosity (LOH), leading to tumor formation. In sporadic PNETs, alterations in MEN1 and DAXX/ATRX are frequently found to involve both copies, and tumor formation likely results from a heterozygous somatic mutation in the setting of LOH/deletion, or from a somatic heterozygous mutation with epigenetic silencing of the remaining allele.(15)
Sporadic PNETs were a genetically mysterious entity until 2011, when Jiao et al. reported frequent alterations in DAXX (death-domain associated protein), ATRX (alpha thalassemia/mental retardation syndrome X-linked), MEN1, and mTOR (mammalian target of rapamycin) pathway genes.(15) In this study, exome sequencing was performed on ten well-differentiated, sporadic, nonfunctional PNETs. They went on to evaluate 58 other PNETs for specific genes using Sanger sequencing. The mutation rates found were 44% for MEN1, 43% for DAXX/ATRX, and 15% for genes in the mTOR pathway (15%; previously described by Perren et al.(16) and Missiaglia et al.).(17) Interested in the genetic differences between PNETs and pancreatic ductal adenocarcinomas (PDAC), they compared their exome sequencing results to work by Jones et al. and saw that not only did PNETs seem to have 60% fewer genes mutated than did PDACs, but the mutations important in each tumor type were quite different. There were not any PDACs (n=114) with mutated MEN1 or DAXX/ATRX, and none of the PNETs (n=68) had mutations in KRAS, CDKN2A, TGFBR1, SMAD3 or SMAD4.(18) Jiao et al. correlated subgroups of PNETs with specific genetic mutations to clinical outcomes and found that patients with metastatic disease and mutations in both MEN1 and DAXX/ATRX had survived at least 10 years with their disease, whereas over 60% of patients without these mutations died within five years of their diagnosis, suggesting that these mutations confer more favorable PNET biology.(15) In contrast, Marinoni et al. reported worse survival in patients with DAXX/ATRX mutations.(19)
Poorly differentiated tumors behave more aggressively, and their genetics reflect this. In 2012, Yachida et al. compared protein expression in well-differentiated (n=11) versus poorly-differentiated (n=19) PNETs to explain at the cellular level what oncologists had long known—that poorly-differentiated PNETs are a distinct entity from well-differentiated PNETs, and portend a much worse prognosis. This work revealed that poorly-differentiated PNETs maintained DAXX and ATRX expression, whereas 45% of well-differentiated tumors lost expression of these proteins, supporting the earlier finding by Jiao et al. that loss of DAXX/ATRX was associated with improved tumor biology.(15) Diminished or absent expression of the tumor suppressor genes p53 and Rb were almost universally present in the poorly-differentiated tumors, whereas disruption of these pathways was rare in the well-differentiated neoplasms. They also found the majority of poorly-differentiated PNETs overexpressed Bcl-2, an inhibitor of apoptosis.(20)
Genetic alterations implicated in PNET tumorigenesis affect a variety of cellular functions and involve two major (intersecting) pathways (Figure 2).(21, 22) MEN1 encodes the protein product menin(14, 23) and participates in regulation of cell proliferation, apoptosis, and genome stability.(24–26) DAXX and ATRX form a heterodimer that function in histone recruitment and incorporation and maintenance of telomeres.(27) They likely play a role in chromatin remodeling and genomic stability, but their exact functions are unknown.(19)
Figure 2. Role of MEN1 and DAXX/ATRX mutations in PNETS.

The functions of Menin and DAXX/ATRX are incompletely understood. Menin acts as a tumor suppressor for endocrine tumors, but is required for development of some leukemias through its interaction with mixed-lineage leukemia (MLL) fusion proteins.(70) Menin appears to interact with numerous proteins and transcription factors, such as SMADs (TGFβ family), cell cycle regulators (71)(such as p27, shown to be important in SBNET tumorigenesis (72, 73)) as well as proteins involved in DNA damage-dependent cell cycle arrest or DNA repair.(24, 25) It may also act as a protein scaffold to up- and down-regulate transcription and inhibit signaling through interactions with the Map-kinase (MAPK), phosphatidylinositol-3-kinase/Akt (PI3K/Akt), and transforming growth factor beta (TGFβ) pathways, among others. Menin localizes to telomeres during cell division and has a role in regulation of histone methylation.(70) Mutations in DAXX and ATRX are found in sporadic PNETs and have been reported to correlate with both better and worse clinical outcomes compared to tumors without these mutations.(15, 19) DAXX and ATRX are directly-interacting proteins that function in histone recruitment and incorporation and maintenance of telomeres.(27) As with Menin, they likely play a role in chromatin remodeling and genomic stability, but their exact functions remain to be defined.(19)
Aberrations in the phosphatidylinositol-3-kinase (PI3K)/Akt/mTOR pathway also contribute to PNET development. PNETs are vascular tumors that express numerous growth factors (Figure 3). Aberrant expression of the receptors of these factors, in particular the receptors for VEGF, IGF-1 and FGF, activate the PI3K and RAS signaling pathways. Growth factors binding to these receptors activates PI3K, which then activates the protein kinase Akt, an inhibitor of apoptosis and a crucial driver of malignancy and chemoresistance.(28) Focal adhesion kinase (FAK) has recently been implicated in PNET proliferation via its activation of Akt. In vitro studies showed downregulation of Akt activity and decreased PNET growth when FAK is specifically inhibited.(29) The downstream effector in this pathway is mTOR. Overexpression is associated with metastasis and proliferation in PNETs.(28)
Figure 3. Targetable pathways in pancreatic neuroendocrine tumors (PNETs).

The mitogen-activated protein kinase (MAPK) and phosphatidyl-inositol-3-kinase/mammalian target of rapamycin (PI3K/mTOR) pathways are dysregulated in sporadic and familial neuroendocrine tumors and may be targeted for treatment. Patients with multiple endocrine neoplasia type 1 (mutation in menin protein encoded by MEN1), von Hippel-Lindau syndrome (VHL), Neurofibromatosis-1 (NF1) and Tuberous Sclerosis (TSC1/2) develop neuroendocrine tumors. In non-familial PNETs, somatic mutations occur in MEN1 (44%), DAXX/ATRX (43%), and the mammalian target of rapamycin (mTOR) pathway (15%), including mutations in the phosphatidyl inositol-3-kinase (PI3K) p110 subunit PIK3CA, phosphatase and tensin homologue (PTEN), and tuberous sclerosis-2 (TSC2).(15) In both pathways, binding of ligands by receptor tyrosine kinases (RTKs) including epidermal growth factor receptors (EGFR), vascular endothelial growth factor receptors (VEGFR), platelet derived growth factor receptors (PDGFR), insulin-like growth factor 1 receptor (IGF-1R), fibroblast growth factor receptors (FGFR), transforming growth factor receptors alpha (TGFαR) and beta (TGFβR), and Met, leads to pathway activation and ultimately to transcription of genes promoting angiogenesis, growth, proliferation, cell-survival, and metastasis.(72, 74–76) VEGFR blockade by the antibody bevacizumab blocks angiogenesis and slows tumor growth in a mouse model.(77) The multi-kinase inhibitor sunitinib blocks VEGFR1-3, PDGFR-alpha and –beta, and c-kit (not shown), and was associated in a phase-III trial with significant improvement in progression-free survival (PFS, 11.4 months vs. 5.5 months with placebo, p<0.001).(54) Everolimus acts to inhibit the mTOR complex-1 (mTORC1) and is approved for treatment of metastatic PNETS.(34) VHL protein acts to stabilize hypoxia-inducible factors (HIF) to promote angiogenesis. Finally, new data suggests that PNETS overexpress MET receptors, which receive high levels of paracrine stimulation by production of their ligand, hepatocyte growth factor (HGF), by the tumor stroma, activating the MAPK and PI3K pathways. Additionally, they may evade immune surveillance by expression of CD47, blockade of which decreased tumor growth in a mouse model.(63)
DAXX and ATRX form a heterocomplex that bind to histone H3 preferentially at centromeres and telomeres and mediate chromatin remodeling (Figure 2). Mutations lead to alternate telomere lengthening(30) and aberrant chromatin architecture, which have been linked both to PNETs and development of glioblastoma multiforme.(31)
In PNETs, two commonly used cell lines for preclinical studies are BON-1 and QGP1. These have been used to study chemotherapeutic agents that target the mTOR(32) and PI3K pathways.(33) Clinical trials of the same chemotherapeutic agents demonstrated less dramatic response rates than in the in vitro studies on these cell lines.(34) In 2015, exome sequencing of BON-1 and QCP1 identified nonsynonymous single nucleotide variants at rates of 4.3 per Mb and an absence of mutations in ATRX/DAXX, and MEN1,(35) suggesting that these cell lines have genetic signatures different from those of in vivo PNETs.
TRANSLATIONAL DIAGNOSTICS
Diverse molecular investigations of PNETs have contributed to identifying potential diagnostic markers or treatment targets. NETs metastasize early, and the location of the primary tumor is unknown in approximately 15% cases.(36) The disease is usually diagnosed from a biopsy of a liver metastasis, and in these situations the primary is often discovered in the pancreas or small bowel at the time of surgical exploration. Identification of the primary site early in the workup can help guide treatment and patient selection for surgery. Our own group developed a qPCR-based gene expression classifier (GEC) using RNA from SBNETs, PNETs, and matched normal pancreas or small bowel tissue.(37) The final iteration of the GEC used four genes (BRS3, OXTR, SCTR and OPRK1) to differentiate SBNETs from PNETs using tissue obtained from liver metastases (the most common biopsy site when the primary tumor site is unknown). In a validation set of 136 liver metastases, the classifier identified 97% of SBNETs and 87% of PNETs correctly.(38) This GEC was later compared to an immunohistochemistry (IHC) algorithm created to differentiate SBNET and PNET metastases. The IHC algorithm uses CDX2 (an intestinal marker), PAX6, and Islet-1 (both pancreatic markers) as “first tier” stains. If the expression pattern is indeterminate or fails, a set of “second tier” stains (PR, PDX1, NESP55 and PrAP) is performed to achieve an IHC-based accuracy of 89%, with the GEC reserved for difficult and indeterminate cases.(39, 40)
Others have studied gene expression to identify the primary tumor site from a metastasis of an unknown primary. A 92-gene RT-PCR assay (Cancer TYPE ID, bioTheranostics, Inc., San Diego, CA, USA) uses paraffin-embedded tissue from biopsies to categorize an unknown primary into one of 28 different tumor types or 50 subtypes. This classifier had 87% sensitivity and greater than 98% specificity in a group of 790 tumors of diverse types. Seventy-five of these tumors were NETs and 99% were identified as such.(41) The classifier was then tailored to NETs only, using the expression of ELAVL4, CADPS, RGS17 and KCNJ11 as the principal discriminators between NET primary sites. Ultimately, the NET-specific GEC identified 95% of the primary sites correctly.(41) This test reports primary tumor sites as being GI carcinoid, islet cell carcinoma, lung carcinoid, merkel cell carcinoma, and small/large cell lung carcinoma.
Another application of gene expression profiling in NETs is the WREN Assay, developed by Modlin and colleagues. Their model uses RNA isolated from peripheral blood samples and a qPCR panel of 51 genes to detect whether a patient has evidence of a NET. Overall, the model had a sensitivity of 85–98% and specificity of 93–97% for NET diagnosis. Subgroup analyses showed the test had 79–88% sensitivity and 94% specificity to discriminate a PNET from other NET subtypes. This test may have utility in follow up as an alternative to the standard plasma CgA assay.(42)
TRANSLATIONAL THERAPEUTICS
New insights into the range of underlying molecular alterations in endocrine cancers such as pheochromocytomas, paragangliomas,(43) thyroid cancers,(44) and PNETs have suggested that personalized therapeutics based on individual genetics might lead to better outcomes. Advancements in treatment beyond cytotoxic chemotherapy have been made, and recently approved drugs target the biological pathways underlying tumorigenesis and metastasis, enhancing specificity. Pursuing these alternative treatment pathways in PNETs is crucial, as the low proliferative rate of most PNETs makes them poorly responsive to traditional chemotherapy regimens.
Angiogenesis Inhibitors
Inhibition of angiogenesis has been validated as a useful treatment approach for some cancers and the FDA has approved angiogenesis inhibitors for metastatic colorectal cancer,(45) renal cell carcinoma,(46) hepatocellular carcinoma,(47) and in 2011, PNETs. Multiple molecular factors contribute to tumor vessel development,(48) and vascular endothelial growth factor (VEGF) represents one of the more important and best characterized (Figure 3). This receptor initiates signaling leading to blood vessel proliferation, and can be targeted directly with anti-VEGF antibodies, have its actions blocked by inhibiting its intracellular receptor, or have its production halted.(49) PNETs are highly vascular tumors and this enhanced vascularity correlates with more aggressive behavior and is likey the reason these tumors respond to these drugs.(50–52)
Sunitinib (Sutent, Pfizer) is a multitarget tyrosine kinase inhibitor that acts against the receptors for VEGF, platelet-derived growth factor (PDGF), KIT and FLT3, which are transmembrane proteins important for tumor proliferation and angiogenesis.(53) This drug was approved for use in PNETs in 2011 after a multinational, randomized phase III trial demonstrated improved outcomes in patients receiving sunitinib versus placebo. One hundred seventy-one patients were enrolled in the study, 86 of whom were randomized to the study drug, although by the end of the study 69% of the patients randomized to the placebo arm had crossed over. The median progression-free survival (PFS) of patients receiving sunitinib was 11.4 months, compared to 5.5 months in the placebo arm. Despite improved PFS, the objective response rate was rather low (9.3%), though two patients did achieve a radiologic complete response (CR). One patient randomized to sunitinib died of drug-related cardiac failure, but the majority of adverse events were grade 1 or 2 in severity.(54)
Bevacizumab (Avastin, Roche) is a monoclonal antibody that inhibits VEGF directly and has not yet been approved by the FDA for use in PNETs. This drug has been added to multiple treatment regimens with variable results. An early study randomized patients already on octreotide for metastatic NETs to additional treatment with bevacizumab (n=22) or pegylated IFN-α2b (n=22). Patients continued their pre-enrollment doses of octreotide and initially received the assigned study drug. After 18 weeks of monotherapy or upon progression, the other study drug was added to their regimen. During monotherapy treatment with bevacizumab, only one patient progressed and the addition of IFN did not halt that tumor’s progress. In all, 18% of patients in the bevacizumab arm achieved a confirmed partial radiologic response (PR), while most (77%) maintained disease stability (SD). The PFS rate at week 18 was 95% in the bevacizumab arm and 68% in the IFN arm (p=.02). This difference narrowed once treatment crossover occurred.(55)
In 2012, a group of 34 patients with either metastatic PNETs (n=15) or small bowel neuroendocrine tumors (SBNETs; n=19) treated with a combination of bevacizumab and the oral alkylating agent temozolomide were reported. Five (33%) PNET patients had a PR to treatment, with one patient obtaining a near-CR. The median PFS of PNET patients was 14.3 months (compared to 7.3 months in SBNET patients) and the OS survival in this group was 41.7 months.(56) The efficacy of temozolomide as a monotherapy has only been investigated retrospectively in a heterogeneous group of 36 NETs, 12 of which were PNETs. In that report, 1 of 12 (8%) PNETs achieved a PR and 67% maintained SD.(57) Although comparison between the two studies is difficult given their differences, the higher PR seen in the combination study suggests that the two drugs could be synergistic.
In a study combining bevacizumab with 5-fluorouracil (5FU) and streptozocin (STZ), 34 patients with metastatic PNETs were treated until progression or for a maximum of two years. Most patients had the doses of one of the three drugs adjusted during the study due to toxicity, but no patient progressed on the regimen and the median PFS was 23.7 months. Nineteen of 34 patients (56%) had a PR and 44% had SD.(58) A recent retrospective study of only STZ and 5FU in 133 patients with metastatic PNETs (100 of which were evaluable radiologically) found that 3% of patients achieved a CR, 25% achieved a PR and 8% progressed. Median PFS was 23 months.(59) Comparison of these two studies suggests that the addition of bevacizumab may exacerbate toxicity without improving survival.
Sunitinib and bevacizumab may increase a patient’s PFS, but they have not been shown to make a substantial impact on OS. Used as a single agent, the objective response rate to sunitinib is less than 10%. It is being investigated as a part of multi-drug regimens, but these data have not yet been published. The reasons for this poor response are unknown, but a recent study in metastatic renal cell carcinoma suggests that as many as 25% of patients treated with the drug have intrinsic resistance to it, and multivariate analysis correlated resistance with neutrophilia, poor performance, elevated lactate dehydrogenase levels and multiple metastatic sites (including the liver).(60) Bevacizumab has been used in combination regimens without significantly increasing the PFS or OS in patients with metastatic disease. As with sunitinib, it continues to be investigated as a part of multi-drug regimens, though its inclusion should be carefully scrutinized as its toxicity may counterbalance its efficacy.
PI3K/Akt/mTOR Pathway Inhibitors
Downstream effectors of cell-surface receptors also represent useful targets in PNET therapy. The PI3K/Akt/mTOR pathway is critical for regulation of cell survival and proliferation, and in many tumors these pathways and members are upregulated or constitutively active. In a sample of 39 PNETs, the Akt pathway was highly active in 76% of tumors.(61) This finding serves as the basis for the use of mTOR inhibitors in PNETs. The mTOR inhibitor recently approved for use in metastatic PNETs is everolimus (Afinitor, Novartis). The RADIANT-3 study, published in 2011, randomized 410 patients with metastatic PNETs to either daily oral everolimus (n=207) or placebo (n=203). Patients treated with everolimus had a median PFS of 11 months, compared to only 4.6 months in the placebo group (p<.001). As with sunitinib, everolimus did not produce a robust objective response (only 5%), but 64% obtained some degree of tumor shrinkage. There were 12 total deaths in the everolimus arm—five related to the underlying cancer and seven due to adverse events.(34)
Everolimus has also been tested in combination with other drugs since its approval. Most recently, it was given with temozolomide in 43 patients with metastatic PNETs to determine whether the combination of the drugs could improve the objective response rate. In the 40 evaluable patients, 40% had a PR and 53% had SD—an improvement over the 5% objective response rate seen with everolimus monotherapy. The median PFS with this regimen was 15.4 months and the toxicity experienced by most patients was similar to that seen with treatment by everolimus alone. However, approximately one-third of the patients included in the study did not have progressive disease prior to enrollment, and thus response rates, especially PFS, may be overestimated.(62)
The MET receptor and CD47 have recently emerged as targets for PNET treatment. PNETs overexpress MET receptors and may receive increased MET signaling through paracrine mechanisms from the tumor stroma, leading to activation of PI3K and RAS/MEK pathways, while CD47 serves to conceal the tumor from immune surveillance.(63) Cabozantinib (Cometriq, Exelixis), a multi-receptor tyrosine kinase inhibitor approved for medullary thyroid cancer, acts preferentially on MET receptors,(64) while newly developed CD47-blocking agents may render the tumor more susceptible to macrophage and lymphocytic destruction.(63) A phase-II clinical trial of cabozantinib for advanced PNETs (ClinicalTrials.gov Identifier: NCT01466036) is currently recruiting patients.
There are no conclusive data on the use of everolimus (or any other targeted molecular agent) given in the adjuvant setting after surgical resection of the primary tumor and debulking of metastatic disease. However, there are two clinical trials actively recruiting patients to study this. One is being carried out by the Eastern Cooperative Oncology Group (Identifier: NCT02031536), where patients who have undergone an R0 or R1 resection for PNETs that have metastasized to the liver will be randomized to receive either adjuvant everolimus or placebo. The primary outcome is disease free survival, and secondary outcomes include OS and treatment toxicity. The other study (Identifier: NCT02315625) will assign patients with metastatic PNETs who have undergone an R1 or R2 resection to either adjuvant sunitinib or everolimus, based on their tumor genotype. Tumors with MEN1, PDGFR, KIT or FLT3 mutations will be treated with sunitinib, whereas tumors harboring mutations in NF1, PTEN, PI3K, AKT, mTOR or TP53 will be treated with everolimus. If a tumor has mutations in both groups the patient will be assigned to the sunitinib arm. Treatment will continue for 48 months and crossover to the other drug will occur if there is disease progression while on the study drug. The primary end point is PFS.(65) This study not only attempts to determine whether chemotherapy adds benefit after surgery, but it has the potential to offer the first “personalized” cancer treatment for patients with metastatic PNETs.
Peptide Receptor Radionuclide Therapy
Peptide receptor radionuclide therapy (PRRT) is used to treat metastatic NETs by targeting somatostatin receptor (SSTR)-expressing tumor cells. Somatostatin analogues are coupled to radioactive elements, which emit β (90Y-DOTA-Tyr3-octreotide, 90Y-DOTATOC) or a combination of β and γ-particles (177Lu-DOTA-Tyr3-octreotate, 177Lu-DOTATOC).(66) This is approved for use in Europe and is being trialed in the United States. It is most commonly used in patients not selected for surgical treatment, but some European groups are testing the modality in the neoadjuvant setting.(67) The first preliminary report from the US group performing the phase two study was promising, as a radiological response was observed in 31% of patients and 41% of patients maintained disease stability. In cases where at least 4 cycles of treatment were administered, the median PFS was 16.5 months.(68) However, only 14 of 37 patients in this study had PNETs, so although this modality should be seen as an important part of the oncologist’s treatment armamentarium as its survival impact is often coupled with a significant improvement in quality of life,(69) the results cannot be too broadly generalized.
CONCLUSIONS
Pancreatic neuroendocrine tumors are rare and potentially deadly. Data suggests that PNETs are genetically different from other NET subtypes, emphasizing the importance of developing diagnostic and therapeutic methods that specifically target the genetic alterations driving PNET initiation and metastasis. There are 93 open studies in the United States for PNETs (www.ClinicalTrials.gov) that will each add another piece to the PNET puzzle. Most studies use PFS as the primary endpoint, given the relatively long overall survival of PNET patients, but as more treatments are developed for this disease, it will be necessary to evaluate their impact on OS. Laboratories from around the world continue working to advance our understanding of the molecular mechanisms involved in these tumors, so that truly personalized medicine can one day be realized.
Acknowledgments
Grant Support
This work was supported by NIH grants P50 CA174521-01 (to J.R. Howe) and T32 CA148062-01 (to J.E. Maxwell and S.K. Sherman).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Franko J, Feng W, Yip L, Genovese E, Moser AJ. Non-functional neuroendocrine carcinoma of the pancreas: incidence, tumor biology, and outcomes in 2,158 patients. J Gastrointest Surg. 2010;14:541–8. doi: 10.1007/s11605-009-1115-0. [DOI] [PubMed] [Google Scholar]
- 2.Schimmack S, Svejda B, Lawrence B, Kidd M, Modlin IM. The diversity and commonalities of gastroenteropancreatic neuroendocrine tumors. Langenbecks Arch Surg. 2011;396:273–98. doi: 10.1007/s00423-011-0739-1. [DOI] [PubMed] [Google Scholar]
- 3.Kuo JH, Lee JA, Chabot JA. Nonfunctional pancreatic neuroendocrine tumors. Surg Clin North Am. 2014;94:689–708. doi: 10.1016/j.suc.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Fraenkel M, Kim MK, Faggiano A, Valk GD. Epidemiology of gastroenteropancreatic neuroendocrine tumours. Best Pract Res Clin Gastroenterol. 2012;26:691–703. doi: 10.1016/j.bpg.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Roland CL, Bian A, Mansour JC, Yopp AC, Balch GC, Sharma R, et al. Survival impact of malignant pancreatic neuroendocrine and islet cell neoplasm phenotypes. J Surg Oncol. 2012;105:595–600. doi: 10.1002/jso.22118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19:1727–33. doi: 10.1093/annonc/mdn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill JS, McPhee JT, McDade TP, Zhou Z, Sullivan ME, Whalen GF, et al. Pancreatic neuroendocrine tumors: the impact of surgical resection on survival. Cancer. 2009;115:741–51. doi: 10.1002/cncr.24065. [DOI] [PubMed] [Google Scholar]
- 8.Chamberlain RS, Canes D, Brown KT, Saltz L, Jarnagin W, Fong Y, et al. Hepatic neuroendocrine metastases: does intervention alter outcomes? J Am Coll Surg. 2000;190:432–45. doi: 10.1016/s1072-7515(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 9.Sarmiento JM, Heywood G, Rubin J, Ilstrup DM, Nagorney DM, Que FG. Surgical treatment of neuroendocrine metastases to the liver. J Am Coll Surg. 2003;197:29–37. doi: 10.1016/S1072-7515(03)00230-8. [DOI] [PubMed] [Google Scholar]
- 10.Mayo SC, de Jong MC, Pulitano C, Clary BM, Reddy SK, Gamblin TC, et al. Surgical management of hepatic neuroendocrine tumor metastasis: results from an international multi-institutional analysis. Ann Surg Oncol. 2010;17:3129–36. doi: 10.1245/s10434-010-1154-5. [DOI] [PubMed] [Google Scholar]
- 11.Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO classification of tumours of the digestive system. 4. Lyon (France): WHO Press; 2010. [Google Scholar]
- 12.Tang LH, Basturk O, Sue JJ, Klimstra DS. A practical approach to the classification of WHO grade 3 (G3) well-differentiated neuroendocrine tumor (WD-NET) and poorly differentiated neuroendocrine carcinoma (PD-NEC) of the pancreas. Am J Surg Pathol. 2016;40:1192–202. doi: 10.1097/PAS.0000000000000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988;332:85–7. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- 14.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi S, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–7. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 15.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perren A, Komminoth P, Saremaslani P, Matter C, Feurer S, Lees JA, et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol. 2000;157:1097–103. doi: 10.1016/S0002-9440(10)64624-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245–55. doi: 10.1200/JCO.2008.21.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marinoni I, Kurrer AS, Vassella E, Dettmer M, Rudolph T, Banz V, et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. 2014;146:453–60. e5. doi: 10.1053/j.gastro.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Yachida S, Vakiani E, White CM, Zhong Y, Saunders T, Morgan R, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36:173–84. doi: 10.1097/PAS.0b013e3182417d36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi C, Klimstra DS. Pancreatic neuroendocrine tumors: pathologic and molecular characteristics. Semin Diagn Pathol. 2014;31:498–511. doi: 10.1053/j.semdp.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Francois R, Iyer R, Seshadri M, Zajac-Kaye M, Hochwald SN. Current understanding of the molecular biology of pancreatic neuroendocrine tumors. J Natl Cancer Inst. 2013;105:1005–17. doi: 10.1093/jnci/djt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemmens I, Merregaert J, Van de Ven WJ, Kas K, Zhang CX, Giraud S, et al. Construction of a 1.2-Mb sequence-ready contig of chromosome 11q13 encompassing the multiple endocrine neoplasia type 1 (MEN1) gene. Genomics. 1997;44:94–100. doi: 10.1006/geno.1997.4872. [DOI] [PubMed] [Google Scholar]
- 24.Poisson A, Zablewska B, Gaudray P. Menin interacting proteins as clues toward the understanding of multiple endocrine neoplasia type 1. Cancer Lett. 2003;189:1–10. doi: 10.1016/s0304-3835(02)00509-8. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol. 2007;265–266:34–41. doi: 10.1016/j.mce.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agarwal SK, Kennedy PA, Scacheri PC, Novotny EA, Hickman AB, Cerrato A, et al. Menin molecular interactions: insights into normal functions and tumorigenesis. Horm Metab Res. 2005;37:369–74. doi: 10.1055/s-2005-870139. [DOI] [PubMed] [Google Scholar]
- 27.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075–80. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolin EM. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013;335:1–8. doi: 10.1016/j.canlet.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 29.Francois RA, Maeng K, Nawab A, Kaye FJ, Hochwald SN, Zajac-Kaye M. Targeting focal adhesion kinase and resistance to mTOR inhibition in pancreatic neuroendocrine tumors. J Natl Cancer Inst. 2015:107. doi: 10.1093/jnci/djv123. pii: djv123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 32.Zitzmann K, Ruden J, Brand S, Goke B, Lichtl J, Spottl G, et al. Compensatory activation of Akt in response to mTOR and Raf inhibitors - a rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010;295:100–9. doi: 10.1016/j.canlet.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 33.Pitt SC, Chen H, Kunnimalaiyaan M. Inhibition of phosphatidylinositol 3-kinase/Akt signaling suppresses tumor cell proliferation and neuroendocrine marker expression in GI carcinoid tumors. Ann Surg Oncol. 2009;16:2936–42. doi: 10.1245/s10434-009-0591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao JC, Shah MH, Ito T, Lombard Bohas C, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. NEJM. 2011;364:514–23. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boora GK, Kanwar R, Kulkarni AA, Pleticha J, Ames M, Schroth G, et al. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet. 2015;208:374–81. doi: 10.1016/j.cancergen.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Wang SC, Parekh JR, Zuraek MB, Venook AP, Bergsland EK, Warren RS, et al. Identification of unknown primary tumors in patients with neuroendocrine liver metastases. Arch Surg. 2010;145:276–80. doi: 10.1001/archsurg.2010.10. [DOI] [PubMed] [Google Scholar]
- 37.Carr JC, Boese EA, Spanheimer PM, Dahdaleh FS, Martin M, Calva D, et al. Differentiation of small bowel and pancreatic neuroendocrine tumors by gene-expression profiling. Surgery. 2012;152:998–1007. doi: 10.1016/j.surg.2012.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherman SK, Maxwell JE, Carr JC, Wang D, Bellizzi AM, Sue O’Dorisio M, et al. Gene expression accurately distinguishes liver metastases of small bowel and pancreas neuroendocrine tumors. Clin Exp Metastasis. 2014;31:935–44. doi: 10.1007/s10585-014-9681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bellizzi AM. Assigning site of origin in metastatic neuroendocrine neoplasms: a clinically significant application of diagnostic immunohistochemistry. Adv Anat Pathol. 2013;20:285–314. doi: 10.1097/PAP.0b013e3182a2dc67. [DOI] [PubMed] [Google Scholar]
- 40.Maxwell JE, Sherman SK, Stashek KM, O’Dorisio TM, Bellizzi AM, Howe JR. A practical method to determine the site of unknown primary in metastatic neuroendocrine tumors. Surgery. 2014;156:1359–65. doi: 10.1016/j.surg.2014.08.008. discussion 65–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerr SE, Schnabel CA, Sullivan PS, Zhang Y, Huang VJ, Erlander MG, et al. A 92-gene cancer classifier predicts the site of origin for neuroendocrine tumors. Mod Pathol. 2014;27:44–54. doi: 10.1038/modpathol.2013.105. [DOI] [PubMed] [Google Scholar]
- 42.Modlin IM, Drozdov I, Kidd M. The identification of gut neuroendocrine tumor disease by multiple synchronous transcript analysis in blood. PLoS One. 2013;8:e63364. doi: 10.1371/journal.pone.0063364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jochmanova I, Pacak K. Pheochromocytoma: the first metabolic endocrine cancer. Clin Cancer Res. 2016;22 doi: 10.1158/1078-0432.CCR-16-0606. xxxx–xxxx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raue F, Frank-Raue K. Thyroid cancer: risk-stratified management and personalized care. Clin Cancer Res. 2016;22 doi: 10.1158/1078-0432.CCR-16-0484. xxxx–xxxx. [DOI] [PubMed] [Google Scholar]
- 45.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 46.Rini BI, Halabi S, Taylor J, Small EJ, Schilsky RL. Cancer and Leukemia Group B 90206: a randomized phase III trial of interferon-a or interferon-a plus anti-vascular endothelial growth factor antibody (bevacizumab) in metastatic renal cell carcinoma. Clin Cancer Res. 2004;10:2584–6. doi: 10.1158/1078-0432.ccr-03-0605. [DOI] [PubMed] [Google Scholar]
- 47.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 48.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 49.Capurso G, Fazio N, Festa S, Panzuto F, De Braud F, Delle Fave G. Molecular target therapy for gastroenteropancreatic endocrine tumours: biological rationale and clinical perspectives. Crit Rev Oncol Hematol. 2009;72:110–24. doi: 10.1016/j.critrevonc.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 50.Terris B, Scoazec JY, Rubbia L, Bregeaud L, Pepper MS, Ruszniewski P, et al. Expression of vascular endothelial growth factor in digestive neuroendocrine tumors. Histopathology. 1998;32:133–8. doi: 10.1046/j.1365-2559.1998.00321.x. [DOI] [PubMed] [Google Scholar]
- 51.Rubbia-Brandt L, Terris B, Giostra E, Dousset B, Morel P, Pepper MS. Lymphatic vessel density and vascular endothelial growth factor-C expression correlate with malignant behavior in human pancreatic endocrine tumors. Clin Cancer Res. 2004;10:6919–29. doi: 10.1158/1078-0432.CCR-04-0397. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Jia Z, Li Q, Wang L, Rashid A, Zhu Z, et al. Elevated expression of vascular endothelial growth factor correlates with increased angiogenesis and decreased progression-free survival among patients with low-grade neuroendocrine tumors. Cancer. 2007;109:1478–86. doi: 10.1002/cncr.22554. [DOI] [PubMed] [Google Scholar]
- 53.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–37. [PubMed] [Google Scholar]
- 54.Raymond E, Dahan L, Raoul J, Bang Y, Borbath I, Lombard Bohas C, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–13. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 55.Yao JC, Phan A, Hoff PM, Chen HX, Charnsangavej C, Yeung SC, et al. Targeting vascular endothelial growth factor in advanced carcinoid tumor: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol. 2008;26:1316–23. doi: 10.1200/JCO.2007.13.6374. [DOI] [PubMed] [Google Scholar]
- 56.Chan JA, Stuart K, Earle CC, Clark JW, Bhargava P, Miksad R, et al. Prospective study of bevacizumab plus temozolomide in patients with advanced neuroendocrine tumors. J Clin Oncol. 2012;30:2963–8. doi: 10.1200/JCO.2011.40.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ekeblad S, Sundin A, Janson ET, Welin S, Granberg D, Kindmark H, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007;13:2986–91. doi: 10.1158/1078-0432.CCR-06-2053. [DOI] [PubMed] [Google Scholar]
- 58.Ducreux M, Dahan L, Smith D, O’Toole D, Lepere C, Dromain C, et al. Bevacizumab combined with 5-FU/streptozocin in patients with progressive metastatic well-differentiated pancreatic endocrine tumours (BETTER trial)--a phase II non-randomised trial. Eur J Cancer. 2014;50:3098–106. doi: 10.1016/j.ejca.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 59.Antonodimitrakis C, Sundin A, Wassberg C, Granberg D, Skogseid B, Eriksson B. Streptozocin and 5-FU for the treatment of pancreatic neuroendocrine tumors: efficacy, prognostic factors and toxicity. Neuroendocrinology. 2016;103:345–53. doi: 10.1159/000439086. [DOI] [PubMed] [Google Scholar]
- 60.Lim SH, Hwang IG, Ji JH, Oh SY, Yi JH, Lim DH, et al. Intrinsic resistance to sunitinib in patients with metastatic renal cell carcinoma. Asia Pac J Clin Oncol. 2016 Mar 31; doi: 10.1111/ajco.12465. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 61.Shah T, Hochhauser D, Frow R, Quaglia A, Dhillon AP, Caplin ME. Epidermal growth factor receptor expression and activation in neuroendocrine tumours. J Neuroendocrinol. 2006;18:355–60. doi: 10.1111/j.1365-2826.2006.01425.x. [DOI] [PubMed] [Google Scholar]
- 62.Chan JA, Blaszkowsky L, Stuart K, Zhu AX, Allen J, Wadlow R, et al. A prospective, phase 1/2 study of everolimus and temozolomide in patients with advanced pancreatic neuroendocrine tumor. Cancer. 2013;119:3212–8. doi: 10.1002/cncr.28142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krampitz GW, George BM, Willingham SB, Volkmer JP, Weiskopf K, Jahchan N, et al. Identification of tumorigenic cells and therapeutic targets in pancreatic neuroendocrine tumors. Proc Natl Acad Sci U S A. 2016;113:4464–9. doi: 10.1073/pnas.1600007113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maxwell JE, Sherman SK, O’Dorisio TM, Howe JR. Medical management of metastatic medullary thyroid cancer. Cancer. 2014;120:3287–301. doi: 10.1002/cncr.28858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Neychev V, Steinberg SM, Cottle-Delisle C, Merkel R, Nilubol N, Yao J, et al. Mutation-targeted therapy with sunitinib or everolimus in patients with advanced low-grade or intermediate-grade neuroendocrine tumours of the gastrointestinal tract and pancreas with or without cytoreductive surgery: protocol for a phase II clinical trial. BMJ Open. 2015;5:e008248. doi: 10.1136/bmjopen-2015-008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34:228–52. doi: 10.1016/j.yfrne.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 67.van Vliet EI, van Eijck CH, de Krijger RR, Nieveen van Dijkum EJ, Teunissen JJ, Kam BL, et al. Neoadjuvant treatment of nonfunctioning pancreatic neuroendocrine tumors with [177Lu-DOTA0,Tyr3]octreotate. J Nucl Med. 2015;56:1647–53. doi: 10.2967/jnumed.115.158899. [DOI] [PubMed] [Google Scholar]
- 68.Delpassand ES, Samarghandi A, Zamanian S, Wolin EM, Hamiditabar M, Espenan GD, et al. Peptide receptor radionuclide therapy with 177Lu-DOTATATE for patients with somatostatin receptor-expressing neuroendocrine tumors: the first US phase 2 experience. Pancreas. 2014;43:518–25. doi: 10.1097/MPA.0000000000000113. [DOI] [PubMed] [Google Scholar]
- 69.Brabander T, Teunissen JJ, Van Eijck CH, Franssen GJ, Feelders RA, de Herder WW, et al. Peptide receptor radionuclide therapy of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2016;30:103–14. doi: 10.1016/j.beem.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 70.Matkar S, Thiel A, Hua X. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci. 2013;38:394–402. doi: 10.1016/j.tibs.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–54. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maxwell JE, Sherman SK, Li G, Choi AB, Bellizzi AM, O’Dorisio TM, et al. Somatic alterations of CDKN1B are associated with small bowel neuroendocrine tumors. Cancer Genet. 2015 Sep 15; doi: 10.1016/j.cancergen.2015.08.003. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013;45:1483–6. doi: 10.1038/ng.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sherman SK, Howe JR. Translational research in endocrine surgery. Surg Oncol Clin N Am. 2013;22:857–84. doi: 10.1016/j.soc.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chan J, Kulke M. Targeting the mTOR signaling pathway in neuroendocrine tumors. Curr Treat Options Oncol. 2014;15:365–79. doi: 10.1007/s11864-014-0294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Djukom C, Porro LJ, Mrazek A, Townsend CM, Hellmich MR, Chao C. Dual inhibition of PI3K and mTOR signaling pathways decreases human pancreatic neuroendocrine tumor metastatic progression. Pancreas. 2014;43:88–92. doi: 10.1097/MPA.0b013e3182a44ab4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kasuya K, Nagakawa Y, Suzuki M, Tanaka H, Ohta H, Itoi T, et al. Anti-vascular endothelial growth factor antibody single therapy for pancreatic neuroendocrine carcinoma exhibits a marked tumor growth-inhibitory effect. Exp Ther Med. 2011;2:1047–52. doi: 10.3892/etm.2011.349. [DOI] [PMC free article] [PubMed] [Google Scholar]