

Abstract
Chronic obstructive pulmonary disease (COPD) is a common, highly debilitating disease of the airways, primarily caused by smoking. Chronic inflammation and structural remodelling are key pathological features of this disease, in part caused by the aberrant function of airway smooth muscle (ASM) cells under the regulation of transforming growth factor (TGF)‐β. miRNA are short, noncoding gene transcripts involved in the negative regulation of specific target genes, through their interactions with mRNA. Previous studies have proposed that mRNA‐145 (miR‐145) may interact with SMAD3, an important downstream signalling molecule of the TGF‐β pathway. TGF‐β was used to stimulate primary human ASM cells isolated from healthy nonsmokers, healthy smokers and COPD patients. This resulted in a TGF‐β‐dependent increase in CXCL8 and IL‐6 release, most notably in the cells from COPD patients. TGF‐β stimulation increased SMAD3 expression, only in cells from COPD patients, with a concurrent increased miR‐145 expression. Regulation of miR‐145 was found to be negatively controlled by pathways involving the MAP kinases, MEK‐1/2 and p38 MAPK. Subsequent, overexpression of miR‐145 (using synthetic mimics) in ASM cells from patients with COPD suppressed IL‐6 and CXCL8 release, to levels comparable to the nonsmoker controls. Therefore, this study suggests that miR‐145 negatively regulates pro‐inflammatory cytokine release from ASM cells in COPD by targeting SMAD3.
Keywords: COPD, inflammation, microRNA
Abbreviations
ASM, airway smooth muscle
COPD, chronic obstructive pulmonary disease
CXCL8, CXC chemokine ligand 8
HSFBs, hypertrophic scar fibroblasts
IL‐6, interleukin 6
MAPKs, mitogen‐activated protein kinases
miR‐145, microRNA‐145
NF‐κB, nuclear factor‐κB
SSc, systemic sclerosis
TGF, transforming growth factor
VSMCs, vascular smooth muscle cells
WNT, wingless/integrase‐1
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory condition of the lung of high global prevalence 1 and is associated with high morbidity, mortality and socioeconomic cost 2; moreover, its contribution to deaths worldwide is predicted to increase over the course of both the current and next decade 3.
Chronic obstructive pulmonary disease is a heterogeneous condition, primarily affecting the lung, but often with significant systemic features, which can augment the morbidity of the disorder and hamper its management 2. The pulmonary component of the disease refers to a progressive and largely irreversible obstruction of airflow, due to a pathological combination of narrowing of the small airways, parenchymal destruction and structural remodelling 4. The aberrant remodelling occurs in addition, or in response, to the ongoing and worsening inflammation 5. It is associated with architectural alterations to the bronchi 6, small airways and parenchymal tissue and affects the epithelium, its underlying extracellular matrix and the surrounding smooth muscle and these changes have been correlated with disease severity 7. Although the primary cause of COPD in more economically developed countries is cigarette smoking, the majority of heavy smokers do not develop the disease 8 and indicators for determining those that will be affected remain elusive.
An increased ASM mass has been observed in both the large and small airways in COPD 6, which correlates with disease severity 7. Whether increased ASM mass in COPD is due to either hypertrophy or hyperproliferation, or a combination of both has not been definitively determined. ASM cells have also been shown to produce a number of cytokines, chemokines and growth factors in response to a variety of inflammatory stimuli, which may contribute towards the inflammatory process in COPD 9. Among these secreted cytokines are CXC chemokine ligand 8 (CXCL8) and interleukin 6 (IL‐6) both of which have been found to be elevated in the sputum of patients with COPD; even more so during exacerbations 10.
Transforming growth factor (TGF)‐β is a pleiotropic cytokine that stimulates ASM proliferation and pro‐inflammatory cytokine production 11, 12, and elevated TGF‐β secretion by pulmonary epithelial cells from patients with COPD has been observed in comparison to healthy controls 13. TGF‐β signalling is typically through the Smad‐dependent pathway 14 although other signalling pathways can also be involved, including the nuclear factor‐κB (NF‐κB) pathway, pathways involving mitogen‐activated protein kinases (MAPKs) and the Wingless/integrase‐1 (WNT) pathway 15.
In addition to the transcription of mRNA, which forms the first step of the ‘central dogma of molecular biology’, noncoding RNA are transcribed from DNA 16. miRNA bind, in a complementary manner, to the 3′ end of their target mRNA and instigate there suppression/degradation 16. Cell proliferation has been shown to be regulated by microRNA‐221 (miR‐221) in ASM cells from patients with asthma 12; in a human epithelial cell line 17; and in murine vascular smooth muscle cells (VSMCs) 18. IL‐6 and CXCL8 secretion has also been shown to be attenuated by miRNA; by miR‐221 in ASM cells from patients with asthma 12; and by miR‐146a and miR‐146b in human alveolar epithelial cells 19, 20. Our previous studies have helped to identify miR‐145 as a potential key regulator of airway smooth muscle function in COPD. Firstly, it is highly expressed in the healthy lung 21 and in healthy ASM cells specifically 22. It has also been shown to be overexpressed in the airways of patients with cystic fibrosis, and to correlate with a decrease in SMAD3 expression 23. A number of human and animal models have linked miR‐145 to mechanisms that could also contribute towards the development of COPD 24, 25. Smooth muscle cell proliferation correlated inversely with expression levels of miR‐145 in murine 26, 27, 28, leporine 29 and human 28 vasculatures. Moreover, exposure to cigarette smoke has been shown to affect expression levels of miR‐145 in the lungs of rats 30.
We hypothesized that increased IL‐6 and CXCL8 release from the ASM cells of COPD patients is mediated by the TGF‐β–induced expression of miR‐145. We examined the effects of TGF‐β upon ASM IL‐6 and CXCL8 release from patients with COPD, and in healthy nonsmokers and healthy smokers. We then examined the regulation of miR‐145 with specific kinase inhibitors. Finally, we examined the effects of modulating the expression levels of miR‐145 in these cells on cytokine release and on the phosphorylation of SMAD3. miR‐145 controls the excessive cytokine release observed in ASM cells from patients with COPD, by reducing SMAD3 phosphorylation.
Materials and methods
Primary human ASM cell culture
Primary human ASM cells were previously dissected from the lungs of healthy nonsmokers, healthy smokers and patients with COPD; disease and smoking status were defined according to guidelines produced by the American Thoracic Society 31. Healthy smokers had a smoking history of at least 10 pack years. There were significant differences between FEV1 in litres, FEV1 percent predicted, and FEV1/FVC ratio between smokers and patients with COPD compared with nonsmokers but matched for age and smoking history (Table 1).
Table 1.
Patient characteristics
| Nonsmokers | Smokers | COPD | |
|---|---|---|---|
| n | 9 | 9 | 9 |
| Age (years) | 66.4 ± 12.72 | 59.2 ± 7.6 | 65.4 ± 6.6 |
| Sex (♂ – ♀) | 7 – 2 | 4 – 5 | 5 – 5 |
| Pack years smoking | N/A | 29.25 ± 3.3 | 38.32 ± 26.92 |
| FEV1 (L) | 4.02 ± 0.48 | 3.12 ± 0.78 | 1.76 ± 0.45 |
| FEV1 (% Predicted) | 104.23 ± 7.28 | 101.5 ± 4.51 | 77 ± 21.97 |
| FEV1/FVC (%) | 78.89 ± 5.98 | 77.57 ± 3.32 | 38.88 ± 15.75 |
| PC20 (mg·mL−1) | > 16 | > 16 | Too severe |
FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; PC20, provocative concentration of methacholine causing a 20% fall in FEV1. Data shown as mean ± SEM.
ASM cells were cultured and plated as previously described 11, 12, 22, 32. ASM cells were plated onto 96‐well plates for the measurement of cytokine release, and six well plates for RNA and protein extraction. Confluent cells were growth‐arrested by FCS deprivation for 24 h in Dulbecco's Modified Eagle's Medium supplemented with sodium pyruvate (1 mm), l‐glutamine (2 mm), nonessential amino acids (1 : 100), penicillin (100 U·mL−1)/streptomycin (100 mg·mL−1), amphotericin B (1.5 mg·mL−1) and BSA (0.1%). Passages 3–4 from nine different donors were used. Cells were stimulated in triplicate ± TGF‐β at the indicated concentrations.
Alternatively, ASM cells were cultured for 1 h in the presence or absence of the indicated concentrations of TPCA‐1 (an IKK‐2 inhibitor), PD098059 (a MEK‐1/2 inhibitor), SP600125 (a JNK‐1/2 inhibitor) and SB 203580 (a p38 MAP kinase inhibitor) and then stimulated with 1 ng·mL−1 of TGF‐β for 24 h. All inhibitors were obtained from Calbiochem. The supernatants were removed, and IL‐6 and CXCL8 levels were determined by DuoSet ELISA (R&D Systems, Abingdon, UK).
miRNA and mRNA Expression
The human (hsa)‐miR‐145 and SMAD3 expression levels were measured as previously described 11, 12, 22.
Transfection with miR‐145 mimics and controls
ASM cells were transfected as previously described 11, 12. A mimic for miR‐145 and controls were obtained from Ambion/Applied Biosystems, Ltd. (Paisley, UK). Transfected cells were plated into 96‐well or 6‐well plates, and left to adhere overnight before being serum starved for 6 h before stimulation with 1 ng·mL−1 TGF‐β for the indicated times.
Western blotting
Proteins were measured as previously described 12, 32, 33. Antibodies against human phospho‐S423‐S425‐Smad3 and total Smad3 were purchased from AbCam (Cambridge, UK).
Data analysis
Data were analysed using graphpad prism, version 5.03 (GraphPad Software, San Diego, CA). Data were not normally distributed (as assessed by the Kolmogorov–Smirnov test), and therefore groups were compared using the Dunn nonparametric test. All data are expressed as means ± SEMs. Significance was defined as a P value of less than 0.05.
Results
The effect of TGF‐β stimulation on CXCL8 and IL‐6 release and SMAD3 and miR‐145 expression by ASM cells after 24 h
ASM cells were stimulated with 2.5% FCS and TGF‐β at the indicated concentrations (0.001–10 ng·mL−1) for 24 h. TGF‐β induced a concentration‐dependent increase in CXCL8 and IL‐6 release from ASM cells which plateaued at 1 ng·mL−1 in the nonsmokers (P < 0.05), smokers (P < 0.01) and COPD (P < 0.001) cells (Fig. 1A,B). A significant increase in CXCL8 release was observed in the COPD ASM cells compared to the nonsmokers (P < 0.01) when the ASM cells were stimulated with 1 ng·mL−1 of TGF‐β (Fig. 1A). Furthermore, there was significant increase in IL‐6 release between the nonsmokers and smokers (P < 0.01), nonsmokers and COPDs (P < 0.001), and the smoker and COPD ASM cells (P < 0.05) (Fig. 1B).
Figure 1.
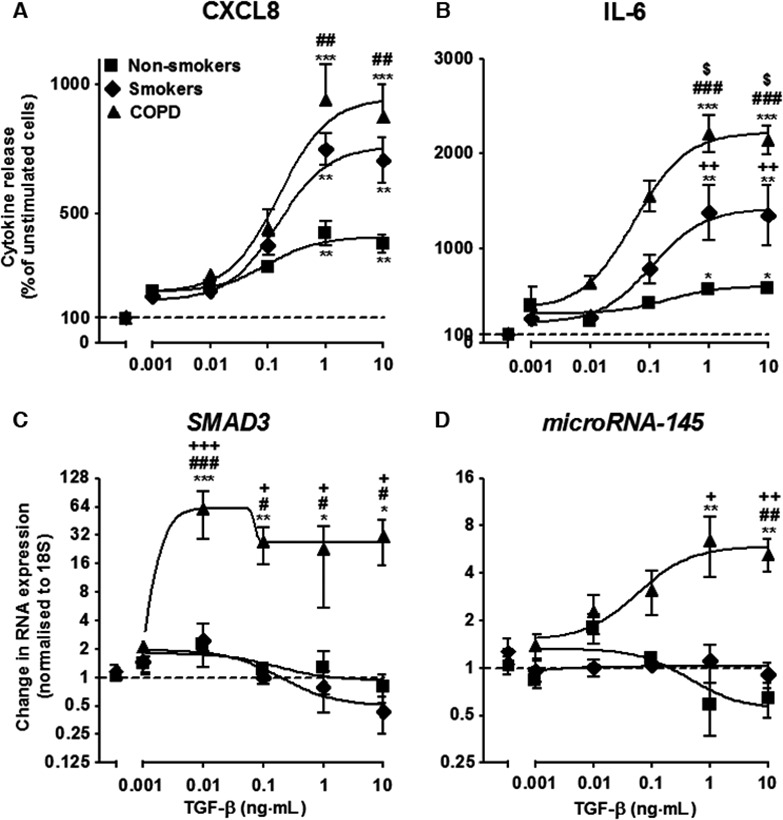
Effect of increasing concentrations of transforming growth factor–β (TGF‐β) on airway smooth muscle (ASM) CXCL8 (A) and IL‐6 release (B), SMAD3 (C) and miR‐145 (D) expression from the ASM cells of non‐smokers, smokers and patients with COPD at 24 h. Points represent the means ± SEMs from nine ASM donors in each group. */$/# P < 0.05; **/$$/## P < 0.01; ***/###/$$$ P < 0.001. Asterisks indicate comparison with no TGF‐β control. Hash signs indicate COPD vs. nonsmoker ASM cells. Dollar signs indicate COPD vs. smoker ASM cells. Plus signs indicate smoker vs. nonsmoker ASM cells.
TGF‐β (0.01 ng·mL−1) induced an increase in SMAD3 expression in ASM cells from COPD patients ~ 60‐fold higher than baseline (P < 0.001; Fig. 1C). Relatively little change to SMAD3 expression was seen in the nonsmokers and smokers compared to unstimulated cells. miR‐145 expression in ASM cells from COPD patients exhibited a concentration dependent increase which plateaued at 1 ng·mL−1 (P < 0.01) (Fig. 1D). A significant increase in expression was observed in the COPD ASM cells compared to the nonsmokers and smokers (both P < 0.01) (Fig. 1D).
The effects of specific kinase inhibitors on CXCL8 and IL‐6 release by ASM cells stimulated with FCS and TGF‐β after 24 h
In previous studies, we and others have demonstrated that cytokines can induce activation of IKK2/NF‐κB and the MAP kinases, ERK‐1/2, JNK‐1/2 and p38 MAP kinase in ASM cells and that these are inhibited in the presence of the selective pharmacological inhibitors of these 34, 35, 36, 37, 38, 39, 40, 41, 42, 43. We therefore used the biological active concentrations of these inhibitors to examine the role of the NF‐κB and MAP kinases pathways during miR‐145 expression.
Following 1 h pre‐treatment with inhibitors, ASM cells were stimulated with TGF‐β (1 ng·mL−1) and the generation of IL‐6 (Fig. 2A,D,G,J), CXCL8 (Fig. 2B,E,H,K) and miR‐145 (Fig. 2C,F,I,L) were determined at 24 h. Exposure to TPCA‐1 completely inhibited production of IL‐6 and CXCL8 in the non‐smokers at 10 μm, and a significant reduction was observed in the COPD ASM cells (both P < 0.05) (Fig. 2A,B). No effect was observed upon miR‐145 expression (Fig. 2C). The MEK‐1/2 inhibitor (10 μm) also attenuated IL‐6 and CXCL8 production (both P < 0.05) (Fig. 2D,E). Interestingly, a significant increase in miR‐145 expression was observed in the COPD ASM cells (P < 0.05) (Fig. 2F). Inhibition of the JNK‐1/2 kinase demonstrated no effect upon either cytokine release or miR‐145 expression (Fig. 2G,H,I). In contrast, inhibition of the p38 MAP kinase had differential actions upon cytokine and miR‐145 production. Blocking p38 MAP kinase inhibited CXCL8 but not IL‐6 in both the nonsmoker and COPD ASM cells (Fig. 2J,K), and a significant increase in miR‐145 expression was observed in the COPD ASM cells (Fig. 2L). Overall, pharmacological studies indicate that TGF‐β‐ induced miR‐145 expression is regulated via an MEK‐1/2‐ and p38‐dependent pathway.
Figure 2.
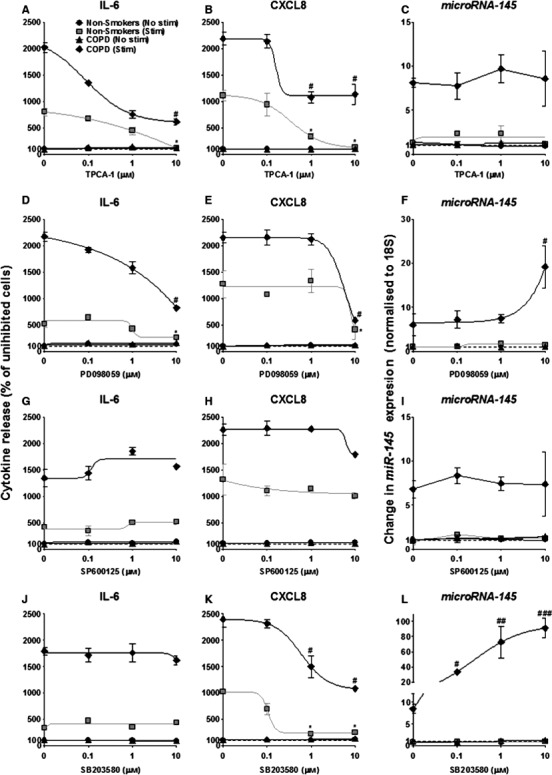
Effect of inhibitors of IKK2 and MAP kinases upon TGF‐β‐induced IL‐6 and CXCL8 release, and miR‐145 expression from the ASM cells of nonsmokers and patients with COPD at 24 h. ASM cells were pretreated for 60 min with the indicated concentrations of the inhibitors of IKK‐2 (TPCA‐1), MEK‐1/2 (PD098059), JNK‐1/2 (SP600125) and p38 MAP kinase (SB203580). Following exposure to vehicle control or TGF‐β (1 ng·mL−1) for 24 h, the release of IL‐6 and CXCL8 was determined by ELISA. miR‐145 expression was measured by RT‐PCR. Points represent the means ± SEM of nine ASM donors in each group. */# P < 0.05; ## P < 0.01; ### P < 0.001. Asterisks indicate stimulated nonsmoker comparison with no TGF‐β control. Hash signs indicate stimulated COPD comparison with no TGF‐β control.
The effect upon TGF‐β‐stimulated ASM cells of miR‐145 overexpression on CXCL8 and IL‐6 release
To clarify the role of miR‐145, we examined the effect of overexpressing miR‐145 on TGF‐β–induced CXCL8 and IL‐6 release. Transfection using Amaxa electroporation (Lonza, Slough, UK) showed that miR‐145 mimics (100 nm) inhibited CXCL8 release by approximately 47% (P < 0.01) and IL‐6 release by approximately 49% (P < 0.001), to levels comparably seen in the healthy smokers (Fig. 3A,B). Altering the endogenous levels of miR‐145 exerted no effect in either healthy or smoker ASM cells or those from patients with severe asthma. To confirm efficient transfection, we undertook parallel studies that examined the effects of a small, interfering RNA (100 nm) targeted to IL‐6. As demonstrated previously 12, 41, we showed a reduction in IL‐6 release induced by TGF‐β stimulation in ASM cells (data not shown), with no effect upon cell viability (data not shown).
Figure 3.
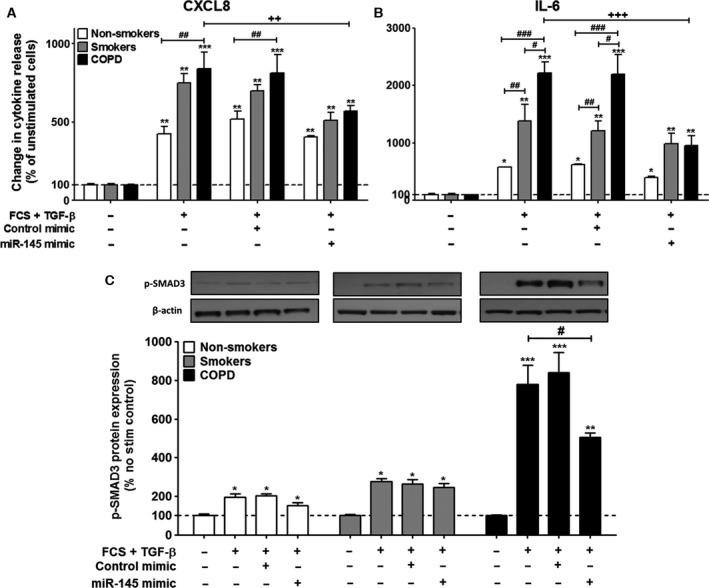
Effects of the overexpression of miR‐145 in the ASM cells of nonsmokers, smokers and patients with COPD at 24 h. ASM cells were electroporated in the presence of buffer, control mimic or miR‐145 mimic. Cells were then exposed to vehicle control or 1 ng·mL−1 TGF‐β and the release of CXCL8 (A) and IL‐6 (B) was measured by ELISA at 24 h. Furthermore, p‐SMAD3 was measure by western blotting (C). Points represent the means ± SEMs from nine ASM donors in each group. */# P < 0.05; **/##/++ P < 0.01; ***/### P < 0.001.
Effects of miR‐145 on SMAD3
We next determined whether miR‐145 could regulate SMAD3 phosphorylation. As previously demonstrated 33, TGF‐β (1 ng·mL−1) increased SMAD3 phosphorylation in the nonsmoker ASM cells at 24 h (P < 0.05) (Fig. 3C). For the first time, we demonstrate that there is a slight increase in phosphorylation in the non‐smokers, and an even greater degree of phosphorylation in the ASM cells from COPD patients induced by TGF‐β (P < 0.001) (Fig. 3C). The miR‐145 mimic (100 nm) decreased the TGF‐β‐induced phosphorylation of SMAD3 in the COPD ASM cells by approximately 50% (P < 0.05) (Fig. 3C), and demonstrated no effect upon either the nonsmoker or smoker ASM cells.
Discussion
We have made several important observations regarding the behaviour of ASM cells from patients with COPD. First, we showed that TGF‐β increased both ASM IL‐6 and CXCL8 release in the COPD patients to a greater degree than those from the nonsmoker subjects. We also observed a concurrent increase in the expression of both SMAD3 and miR‐145 in the ASM cells from the COPD patients. We next investigated the mechanisms that regulate the expression of miR‐145. We showed that expression of miR‐145 is mediated, at least in part, through activation of MEK‐1/2 and p38 in ASM cells from COPD patients. Examination of the effect of these MAP kinase inhibitors upon generation of inflammatory mediators showed that IL‐6 release was mediated via IKK2 and MEK‐1/2 while CXCL8 release was mediated via IKK2, MEK‐1/2 and p38 in both the ASM cohorts, from nonsmokers and those with COPD. Finally, we found that miR‐145 regulates the enhanced IL‐6 and CXCL8 release seen in the ASM cells from patients with COPD, that could be partly through the control of SMAD3 (Summarized in Fig. 4).
Figure 4.
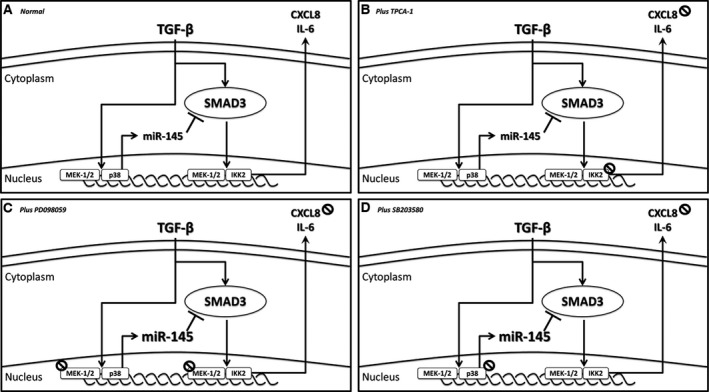
SMAD3 dependent regulation of IL‐6 & CXCL8 release by TGF‐β‐induced miR‐145 expression. In response to TGF‐β stimulation, MEK‐1/2 and p38 activation results in increased miR‐145 expression. The concurrent increase in expression and phosphorylation of SMAD3, is regulated by miR‐145 to prevent further generation and release of IL‐6 & CXCL8.
Previous observations have described the effect of inducing hyperproliferation of ASM cells with TGF‐β in both asthma 11, 12 and COPD 44. This is the first time that TGF‐β has been shown to induce both IL‐6 and CXCL8 release from primary ASM cells isolated from individuals with COPD. The regulation of smooth muscle cell phenotype has previously been shown to be correlated with expression of miR‐145. Specifically, miR‐145 has been demonstrated to regulate smooth muscle cell fate 26, 45, the contractile phenotype of VSMCs 27, 46, and acts a novel VSMC phenotypic marker in murine models 28, and prevent vein graft disease in rabbits 29. Interestingly, exposure to cigarette smoke has been shown to affect expression levels of miR‐145 in the lungs of rats 30, and has been found to be differentially expressed in lung homogenates in rats with COPD 47. Furthermore, miR‐145 (along with others) has recently been proposed to be a promising plasma based biomarker for the diagnosis of COPD 48. This is the first time that a role for miR‐145 in the ASM cells from COPD patients has been reported. We have shown that expression of miR‐145 is through activation of both MEK‐1/2 and p38. Interestingly, Hu et al. 49, have reported that activation of MEK‐1/2 suppresses miR‐145 expression in VSMCs, and Kent et al. 50, demonstrate that miR‐145 expression is inhibited through activation of the MAPK and JNK pathways in colorectal cancer. Furthermore, in cardiomyocytes, the protective activity of miR‐145 is associated with modulation of both MEK‐1/2 and JNK 51 and in gastric mucosal epithelial cell regulation of miR‐145 involves JNK 52. Similar to our results, p38 has previously been shown to be linked to miR‐145 induction in VSMCs 53 and Hong et al. 54, suggest that the p38 MAPK signalling pathway promotes miRNA biogenesis by facilitating the nuclear localization of p68. Clearly, the regulation of miR‐145 is, unsurprisingly, cell type specific.
Finally, we examined the effect of increasing miR‐145 expression in ASM cells from COPD patients upon IL‐6 and CXCL8 release. Although studies have suggested a correlation between miR‐145 expression and IL‐6 & CXCL8 release 55, 56, 57, 58, we show for the first time that increasing the expression of miR‐145 can reduce the levels of IL‐6 & CXCL8 release from the COPD ASM cells to levels comparable to that of the nonsmoker ASM cells. miR‐145 is proposed to target SMAD3 in systemic sclerosis (SSc) 59, cystic fibrosis 23, cartilage dysfunction 60, nasopharyngeal cancer 61 and in hypertrophic scar fibroblasts (HSFBs) 62, we show here that this may also be the case in ASM cells from patients with COPD.
Interestingly, miR‐145 is also likely to be important in regulating ASM cells in asthma, as it is highly expressed in the healthy lung 21 and in healthy ASM cells specifically 22, and inhibition of miR‐145 inhibits eosinophilic inflammation, mucus hypersecretion, TH2 cytokine production and airway hyperresponsiveness in house dust mite‐induced allergic mouse airways 63.
In conclusion, miR‐145 is vital in controlling the increased inflammatory response of human ASM cells in patients with COPD. This finding may open a new avenue in COPD therapeutics by targeting of miRNA‐145 and diagnosis by its detection.
Author contributions
MP and LL were responsible for preparation of the manuscript; LL, KS, IP and BT conducted in‐vitro experiments; KD, AH and KFC provided the primary ASM cells; MP designed the study.
Acknowledgements
This work was supported by a fellowship from Imperial College to MMP and a grant from The Wellcome Trust (085935) to KFC. It was also supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton NHS Foundation Trust and Imperial College London. KFC is a Senior Investigator of NIHR, UK. MMP and KFC are members of Interuniversity Attraction Poles Program‐Belgian State‐Belgian Science Policy‐ project P7/30.
Edited by Tamas Dalmay
References
- 1. Buist AS, McBurnie MA, Vollmer WM, Gillespie S, Burney P, Mannino DM, Menezes AM, Sullivan SD, Lee TA, Weiss KB et al (2007) International variation in the prevalence of COPD (the BOLD Study): a population‐based prevalence study. Lancet 370, 741–750. [DOI] [PubMed] [Google Scholar]
- 2. Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M et al (2013) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 187, 347–365. [DOI] [PubMed] [Google Scholar]
- 3. Mathers CD and Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3, e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barnes PJ (2014) Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med 35, 71–86. [DOI] [PubMed] [Google Scholar]
- 5. Jeffery PK (2004) Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 1, 176–183. [DOI] [PubMed] [Google Scholar]
- 6. Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, Ousova O, Vernejoux JM, Marthan R, Tunon‐de‐Lara JM et al (2007) Bronchial smooth muscle remodeling involves calcium‐dependent enhanced mitochondrial biogenesis in asthma. J Exp Med 204, 3173–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO et al (2004) The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350, 2645–2653. [DOI] [PubMed] [Google Scholar]
- 8. Fletcher C and Peto R (1977) The natural history of chronic airflow obstruction. Br Med J 1, 1645–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prakash YS (2013) Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am J Physiol Lung Cell Mol Physiol 305, L912–L933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhowmik A, Seemungal TA, Sapsford RJ and Wedzicha JA (2000) Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax 55, 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perry MM, Durham AL, Austin PJ, Adcock IM and Chung KF (2015) BET bromodomains regulate transforming growth factor‐beta‐induced proliferation and cytokine release in asthmatic airway smooth muscle. J Biol Chem 290, 9111–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perry MM, Baker JE, Gibeon DS, Adcock IM and Chung KF (2013) Airway smooth muscle hyperproliferation is regulated by microRNA‐221 in severe asthma. Am J Respir Cell Mol Biol 50, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, Okada Y, Yamasawa F, Nakahara K and Umeda A (2001) Increased expression of transforming growth factor‐beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med 163, 1476–1483. [DOI] [PubMed] [Google Scholar]
- 14. Yeganeh B, Mukherjee S, Moir LM, Kumawat K, Kashani HH, Bagchi RA, Baarsma HA, Gosens R and Ghavami S (2013) Novel non‐canonical TGF‐beta signaling networks: emerging roles in airway smooth muscle phenotype and function. Pulm Pharmacol Ther 26, 50–63. [DOI] [PubMed] [Google Scholar]
- 15. Massague J, Seoane J and Wotton D (2005) Smad transcription factors. Genes Dev 19, 2783–2810. [DOI] [PubMed] [Google Scholar]
- 16. Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang C, Zhang J, Zhang A, Wang Y, Han L, You Y, Pu P and Kang C (2010) PUMA is a novel target of miR‐221/222 in human epithelial cancers. Int J Oncol 37, 1621–1626. [DOI] [PubMed] [Google Scholar]
- 18. Liu X, Cheng Y, Zhang S, Lin Y, Yang J and Zhang C (2009) A necessary role of miR‐221 and miR‐222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res 104, 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner‐Svensson HM and Lindsay MA (2008) Rapid changes in microRNA‐146a expression negatively regulate the IL‐1beta‐induced inflammatory response in human lung alveolar epithelial cells. J Immunol 180, 5689–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perry MM, Williams AE, Tsitsiou E, Larner‐Svensson HM and Lindsay MA (2009) Divergent intracellular pathways regulate interleukin‐1beta‐induced miR‐146a and miR‐146b expression and chemokine release in human alveolar epithelial cells. FEBS Lett 583, 3349–3355. [DOI] [PubMed] [Google Scholar]
- 21. Williams AE, Larner‐Svensson H, Perry MM, Campbell GA, Herrick SE, Adcock IM, Erjefalt JS, Chung KF and Lindsay MA (2009) MicroRNA expression profiling in mild asthmatic human airways and effect of corticosteroid therapy. PLoS One 4, e5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perry M, Tsitsiou E, Austin P, Lindsay M, Gibeon D, Adcock I and Chung K (2014) Role of non‐coding RNAs in maintaining primary airway smooth muscle cells. Respir Res 15, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Megiorni F, Cialfi S, Cimino G, De Biase RV, Dominici C, Quattrucci S and Pizzuti A (2013) Elevated levels of miR‐145 correlate with SMAD3 down‐regulation in cystic fibrosis patients. J Cyst Fibros 12, 797–802. [DOI] [PubMed] [Google Scholar]
- 24. Perry MM, Adcock IM and Chung KF (2015) Role of microRNAs in allergic asthma: present and future. Curr Opin Allergy Clin Immunol 15, 156–162. [DOI] [PubMed] [Google Scholar]
- 25. Brook PO, Perry MM, Adcock IM and Durham AL (2015) Epigenome‐modifying tools in asthma. Epigenomics 7, 1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN and Srivastava D (2009) miR‐145 and miR‐143 regulate smooth muscle cell fate and plasticity. Nature 460, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L and Braun T (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119, 2634–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES and Zhang C (2009) MicroRNA‐145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res 105, 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohnaka M, Marui A, Yamahara K, Minakata K, Yamazaki K, Kumagai M, Masumoto H, Tanaka S, Ikeda T and Sakata R (2014) Effect of microRNA‐145 to prevent vein graft disease in rabbits by regulation of smooth muscle cell phenotype. J Thorac Cardiovasc Surg 148, 676–682. [DOI] [PubMed] [Google Scholar]
- 30. Izzotti A, Calin GA, Arrigo P, Steele VE, Croce CM and De FS (2009) Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J 23, 806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. American Thoracic Society. Am J Respir Crit Care Med 152, S77–S121. [PubMed] [Google Scholar]
- 32. Perry MM, Hui CK, Whiteman M, Wood ME, Adcock I, Kirkham P, Michaeloudes C and Chung KF (2011) Hydrogen sulfide inhibits proliferation and release of IL‐8 from human airway smooth muscle cells. Am J Respir Cell Mol Biol 45, 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michaeloudes C, Sukkar MB, Khorasani NM, Bhavsar PK and Chung KF (2010) TGF‐beta regulates Nox4, MnSOD and catalase expression and IL‐6 release in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 300, L295–L304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wuyts WA, Vanaudenaerde BM, Dupont LJ, Demedts MG and Verleden GM (2003) Involvement of p38 MAPK, JNK, p42/p44 ERK and NF‐kappaB in IL‐1beta‐induced chemokine release in human airway smooth muscle cells. Respir Med 97, 811–817. [DOI] [PubMed] [Google Scholar]
- 35. Oltmanns U, Issa R, Sukkar MB, John M and Chung KF (2003) Role of c‐jun N‐terminal kinase in the induced release of GM‐CSF, RANTES and IL‐8 from human airway smooth muscle cells. Br J Pharmacol 139, 1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang CC, Lin WN, Lee CW, Lin CC, Luo SF, Wang JS and Yang CM (2005) Involvement of p42/p44 MAPK, p38 MAPK, JNK, and NF‐kappaB in IL‐1beta‐induced VCAM‐1 expression in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 288, L227–L237. [DOI] [PubMed] [Google Scholar]
- 37. Laporte JD, Moore PE, Lahiri T, Schwartzman IN, Panettieri RA Jr and Shore SA (2000) p38 MAP kinase regulates IL‐1 beta responses in cultured airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 279, L932–L941. [DOI] [PubMed] [Google Scholar]
- 38. Laporte JD, Moore PE, Abraham JH, Maksym GN, Fabry B, Panettieri RA Jr and Shore SA (1999) Role of ERK MAP kinases in responses of cultured human airway smooth muscle cells to IL‐1beta. Am J Physiol 277, L943–L951. [DOI] [PubMed] [Google Scholar]
- 39. Hedges JC, Singer CA and Gerthoffer WT (2000) Mitogen‐activated protein kinases regulate cytokine gene expression in human airway myocytes. Am J Respir Cell Mol Biol 23, 86–94. [DOI] [PubMed] [Google Scholar]
- 40. Hallsworth MP, Moir LM, Lai D and Hirst SJ (2001) Inhibitors of mitogen‐activated protein kinases differentially regulate eosinophil‐activating cytokine release from human airway smooth muscle. Am J Respir Crit Care Med 164, 688–697. [DOI] [PubMed] [Google Scholar]
- 41. Larner‐Svensson HM, Williams AE, Tsitsiou E, Perry MM, Jiang X, Chung KF and Lindsay MA (2010) Pharmacological studies of the mechanism and function of interleukin‐1beta‐induced miRNA‐146a expression in primary human airway smooth muscle. Respir Res 11, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Issa R, Xie S, Lee KY, Stanbridge RD, Bhavsar P, Sukkar MB and Chung KF (2006) GRO‐alpha regulation in airway smooth muscle by IL‐1beta and TNF‐alpha: role of NF‐kappaB and MAP kinases. Am J Physiol Lung Cell Mol Physiol 291, L66–L74. [DOI] [PubMed] [Google Scholar]
- 43. Shan L, Redhu NS, Saleh A, Halayko AJ, Chakir J and Gounni AS (2010) Thymic stromal lymphopoietin receptor‐mediated IL‐6 and CC/CXC chemokines expression in human airway smooth muscle cells: role of MAPKs (ERK1/2, p38, and JNK) and STAT3 pathways. J Immunol 184, 7134–7143. [DOI] [PubMed] [Google Scholar]
- 44. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, Bao W, Pavlidis S, Barnes PJ, Kanerva J et al (2015) Oxidative stress‐induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 136, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamaguchi S, Yamahara K, Homma K, Suzuki S, Fujii S, Morizane R, Monkawa T, Matsuzaki Y, Kangawa K and Itoh H (2011) The role of microRNA‐145 in human embryonic stem cell differentiation into vascular cells. Atherosclerosis 219, 468–474. [DOI] [PubMed] [Google Scholar]
- 46. Hutcheson R, Terry R, Chaplin J, Smith E, Musiyenko A, Russell JC, Lincoln T and Rocic P (2013) MicroRNA‐145 restores contractile vascular smooth muscle phenotype and coronary collateral growth in the metabolic syndrome. Arterioscler Thromb Vasc Biol 33, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li B, Zhou X, Chen L, Feng C and Li T (2014) Expression of microRNAs in lung homogenates in rats with chronic obstructive pulmonary disease. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 26, 905–909. [DOI] [PubMed] [Google Scholar]
- 48. Wang M, Huang Y, Liang Z, Liu D, Lu Y, Dai Y, Feng G and Wang C (2016) Plasma miRNAs might be promising biomarkers of chronic obstructive pulmonary disease. Clin Respir J 10, 104–111. [DOI] [PubMed] [Google Scholar]
- 49. Hu B, Song JT, Qu HY, Bi CL, Huang XZ, Liu XX and Zhang M (2014) Mechanical stretch suppresses microRNA‐145 expression by activating extracellular signal‐regulated kinase 1/2 and upregulating angiotensin‐converting enzyme to alter vascular smooth muscle cell phenotype. PLoS One 9, e96338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kent OA, Fox‐Talbot K and Halushka MK (2013) RREB1 repressed miR‐143/145 modulates KRAS signaling through downregulation of multiple targets. Oncogene 32, 2576–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li R, Yan G, Zhang Q, Jiang Y, Sun H, Hu Y, Sun J and Xu B (2013) miR‐145 inhibits isoproterenol‐induced cardiomyocyte hypertrophy by targeting the expression and localization of GATA6. FEBS Lett 587, 1754–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Luo XJ, Liu B, Dai Z, Li TB, Li NS, Zhang XJ, Yang ZC, Li YJ and Peng J (2013) Expression of apoptosis‐associated microRNAs in ethanol‐induced acute gastric mucosal injury via JNK pathway. Alcohol 47, 481–493. [DOI] [PubMed] [Google Scholar]
- 53. Blumensatt M, Wronkowitz N, Wiza C, Cramer A, Mueller H, Rabelink MJ, Hoeben RC, Eckel J, Sell H and Ouwens DM (2014) Adipocyte‐derived factors impair insulin signaling in differentiated human vascular smooth muscle cells via the upregulation of miR‐143. Biochim Biophys Acta 1842, 275–283. [DOI] [PubMed] [Google Scholar]
- 54. Hong S, Noh H, Chen H, Padia R, Pan ZK, Su SB, Jing Q, Ding HF and Huang S (2013) Signaling by p38 MAPK stimulates nuclear localization of the microprocessor component p68 for processing of selected primary microRNAs. Sci Signal 6, ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oliva EN, Cuzzola M, Aloe Spiriti MA, Poloni A, Lagana C, Rigolino C, Morabito F, Galimberti S, Ghio R, Cortelezzi A et al (2013) Biological activity of lenalidomide in myelodysplastic syndromes with del5q: results of gene expression profiling from a multicenter phase II study. Ann Hematol 92, 25–32. [DOI] [PubMed] [Google Scholar]
- 56. Yu CC, Tsai LL, Wang ML, Yu CH, Lo WL, Chang YC, Chiou GY, Chou MY and Chiou SH (2013) miR145 targets the SOX9/ADAM17 axis to inhibit tumor‐initiating cells and IL‐6‐mediated paracrine effects in head and neck cancer. Cancer Res 73, 3425–3440. [DOI] [PubMed] [Google Scholar]
- 57. Li W, Ma K, Zhang S, Zhang H, Liu J, Wang X and Li S (2015) Pulmonary microRNA expression profiling in an immature piglet model of cardiopulmonary bypass‐induced acute lung injury. Artif Organs 39, 327–335. [DOI] [PubMed] [Google Scholar]
- 58. Zhou J, Chaudhry H, Zhong Y, Ali MM, Perkins LA, Owens WB, Morales JE, McGuire FR, Zumbrun EE, Zhang J et al (2015) Dysregulation in microRNA expression in peripheral blood mononuclear cells of sepsis patients is associated with immunopathology. Cytokine 71, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu H, Li Y, Qu S, Luo H, Zhou Y, Wang Y, Zhao H, You Y, Xiao X and Zuo X (2012) MicroRNA expression abnormalities in limited cutaneous scleroderma and diffuse cutaneous scleroderma. J Clin Immunol 32, 514–522. [DOI] [PubMed] [Google Scholar]
- 60. Yang B, Kang X, Xing Y, Dou C, Kang F, Li J, Quan Y and Dong S (2014) Effect of microRNA‐145 on IL‐1beta‐induced cartilage degradation in human chondrocytes. FEBS Lett 588, 2344–2352. [DOI] [PubMed] [Google Scholar]
- 61. Huang H, Sun P, Lei Z, Li M, Wang Y, Zhang HT and Liu J (2015) miR‐145 inhibits invasion and metastasis by directly targeting Smad3 in nasopharyngeal cancer. Tumour Biol 36, 4123–4131. [DOI] [PubMed] [Google Scholar]
- 62. Zhu HY, Li C, Zheng Z, Zhou Q, Guan H, Su LL, Han JT, Zhu XX, Wang SY, Li J et al (2015) Peroxisome proliferator‐activated receptor‐gamma (PPAR‐gamma) agonist inhibits collagen synthesis in human hypertrophic scar fibroblasts by targeting Smad3 via miR‐145. Biochem Biophys Res Commun 459, 49–53. [DOI] [PubMed] [Google Scholar]
- 63. Collison A, Mattes J, Plank M and Foster PS (2011) Inhibition of house dust mite‐induced allergic airways disease by antagonism of microRNA‐145 is comparable to glucocorticoid treatment. J Allergy Clin Immunol 128, 160–167. [DOI] [PubMed] [Google Scholar]