

Abstract
It has now been demonstrated that the μ, δ1, δ2, and κ1 opioid receptor (OR) agonists represent the most promising group of opioids for the creation of drugs enhancing cardiac tolerance to the detrimental effects of ischemia/reperfusion (I/R). Opioids are able to prevent necrosis and apoptosis of cardiomyocytes during I/R and improve cardiac contractility in the reperfusion period. The OR agonists exert an infarct‐reducing effect with prophylactic administration and prevent reperfusion‐induced cardiomyocyte death when ischemic injury of heart has already occurred; that is, opioids can mimic preconditioning and postconditioning phenomena. Furthermore, opioids are also effective in preventing ischemia‐induced arrhythmias.
Keywords: opioids, heart, ischemia/reperfusion injury, cardioprotection, signal transduction
1. HISTORICAL BACKGROUND
The main cardiac manifestations of ischemia/reperfusion (I/R) are necrosis, apoptosis of cardiomyocytes, contractile dysfunction, and ventricular arrhythmias.1, 2, 3, 4 In the 70s it was clear that the prognosis for patients who had suffered acute myocardial infarction was highly dependent on the amount of ventricular muscle that was lost to infarction. Although it was proposed that an intervention that could reduce infarct size would save lives, there was great debate as to whether therapeutic attenuation of these negative manifestations of myocardial ischemia was even possible. This argument was settled once and for all in 1986, when three American researchers discovered the phenomenon of ischemic preconditioning (IP).5 They found that exposure to four brief periods of I/R causes the heart to become very resistant to infarction from a subsequent prolonged ischemic insult. The protective effect of IP is maintained only for 2–3 hr, making it impractical for any clinical application. However, a day later these hearts again became resistant to infarction, this time lasting about 4 days. This has been variously called “delayed preconditioning,” “late preconditioning,” or “second window of protection.”6 Still the major impediment to translating preconditioning to a clinical setting was the requirement that it has to be instituted prior to the onset of ischemia. Pretreatment is impossible, however, in the setting of acute myocardial infarction.
Although the mechanism of IP was obscure, it was strongly believed that it must target injury during ischemia and that the pretreatment requisite was absolute. It took another 17 years after the discovery of IP before it was realized that IP actually prevents reperfusion injury and treatment could be instituted right up to the time of reperfusion. In 2003, Vinten‐Johansen's group discovered “ischemic postconditioning” (IPost). It turned out that three cycles of very brief reperfusion/ischemia cycles after a prolonged ischemic insult greatly decrease the fraction of the ischemic myocardium that infarcted, often called the “infarct size‐area at risk ratio” (IS/AAR).7 This seminal discovery finally opened the door to clinical application but while IPost was theoretically possible in patients whose coronary thrombus was removed with angioplasty, it proved to be surprisingly awkward in many cases. A postconditioning drug would be a great improvement.
The mechanism of IP had been obscure until it was discovered that it resulted from protective signal transduction pathways triggered by Gi‐coupled plasma membrane receptors.8 These investigators found that adenosine was a trigger through adenosine A1 receptors but soon it was found that other Gi‐coupled receptors also participated in triggering IP's protection. In 1995, Gross's group obtained data that the infarct‐reducing effect of IP was lost after blocking opioid receptors (ORs) with naloxone9 and a year later they reported that they could precondition the heart with morphine.10 Kin et al.11 demonstrated that ORs were also involved in IPost when they showed that naloxone 5 min before reperfusion abolishes the infarct‐sparing effect of IPost.
IP or IPost with ischemia is impractical in the clinical settings. The foregoing studies generated enormous interest by physiologists and pharmacologists and provided the impetus for research aimed at finding pharmaceutical OR agonists that could mimic the phenomenon of IP and IPost. In this review, we evaluate the effect of various OR ligands on the necrosis and apoptosis of cardiomyocytes, myocardial stunning, and the incidence of ischemic and reperfusion arrhythmias. To aid the reader we have included Table I, which lists all of the OR agonists and antagonists discussed in this review.
Table I.
OR Active Drugs
| ARD‐353 | Nonpeptide δ1 and δ2 OR agonist (does not cross the BBB) |
| BNTX | δ1 OR antagonist |
| Bremazocine | κ2 OR agonist |
| BRL 52537 | κ OR agonist |
| Buprenorphine | μ and κ OR agonist |
| BW373U86 | δ OR agonist |
| Carfentanil | μ OR agonist |
| CTOP | μ OR antagonist |
| DADLE | δ OR agonist |
| Dalargin | μ and δ OR agonist |
| DALDA | μ and κ OR agonist |
| DAMGO | μ OR agonist |
| Deltorphin D | δ2 OR agonist |
| Deltorphin II | δ2 OR agonist |
| Dermorphin H | μ OR agonist |
| [Dmt(1)] DALDA | Di‐methyl tyrosine version of DALDA‐ potent μ and К OR agonist |
| DPDPE | δ1 OR agonist |
| Dynorphin | κ OR agonist |
| Eribis peptide 94 | μ and δ OR agonist |
| Fentanyl | μ OR agonist |
| FIT | δ OR agonist |
| FK 33‐824 | Selective μ OR agonist, synthetic analogue of Met‐enkephalin |
| GNTI | κ OR antagonist |
| GR‐89696 | κ2 OR agonist |
| ICI 199,441 | κ OR agonist |
| ICI 204,448 | κ OR agonist |
| MEAP | Met‐enkephalin‐Arg‐Phe ‐ μ, δ and κ OR agonist |
| Meptazinol | μ‐OR agonist and antagonist |
| Met‐enkephalin | μ and δ OR agonist |
| Methadone | μ OR agonist |
| Morphine | Nonselective OR agonist |
| Mr 2266 | κ OR antagonist |
| MrZ 2593 | Peripheral nonselective OR antagonist (does not cross BBB at 1 mg/kg) |
| Naloxone methiodide | Peripheral nonselective OR antagonist (does not cross BBB) |
| Naloxone | Nonselective OR antagonist |
| Naltrexone | Nonselective OR antagonist |
| Naltriben | δ2 OR antagonist |
| Natrindole | Highly selective δ OR‐selective antagonist |
| Nociceptin | ORL1 agonist |
| Nor‐binaltorphimine | κ OR antagonist |
| PD 129290 | κ OR agonist |
| (+)‐pentazocine | Preferential σ‐OR agonist |
| (−)‐pentazocine | κ‐OR agonist |
| Quadazocine | κ2 OR antagonist |
| Remifentanil | Nonselective OR agonist |
| SNC‐121 | Non‐peptide δ OR agonist |
| SNC‐80 | δ OR agonist |
| Sufentanil | μ OR agonist |
| TAN‐67 | δ1 OR agonist |
| Tramadol | Nonselective agonist and antagonist of ORs |
| U50,488 | κ1 OR agonist (does not cross the BBB) |
2. LOCALIZATION OF OPIOID RECEPTORS INVOLVED IN REGULATION OF HEART FUNCTION
A. Opioid Receptors in the Central Nervous System
It is well known that all discovered mammalian opioid peptides have been isolated from brain where they are most abundant12 and it is not surprising that the brain and spinal cord have a high density of ORs.13, 14, 15, 16, 17, 18, 19, 20, 21 μ OR was discovered in the spinal cord, in the periaqueductal gray matter, nucleus accumbens, amygdala and in several thalamic nuclei.15, 19 Transcripts of μ OR were found in the prefrontal cortex, nucleus accumbens, caudate putamen, and thalamus.20 δ OR was identified in spinal cord,16 caudate putamen, nucleus accumbens, and olfactory tubercle.18 Transcripts of δ OR were detected in the prefrontal cortex, nucleus accumbens, and caudate putamen.20 The κ OR was found in spinal cord.16 Transcripts for this receptor were identified in nucleus accumbens, caudate putamen, preoptic area, and hypothalamus.20 κ OR was also found in the prefrontal cortex, nucleus accumbens, hypothalamus, amygdala, ventral tegmental area, dorsal raphe nucleus, and locus coeruleus.21 The ORL1 receptor or nociceptin/orphanin FQ (N/OFQ) opioid peptide receptor (NOPr) was found in several rat brain areas, including the cerebral cortex, thalamus, subforn ical organ, habenula, hypothalamus, central gray, dorsal raphe, locus coeruleus hippocampus, amygdala, caudate nucleus, putamen, medial thalamic nuclei, and the dorsal horn of the spinal cord.14, 17
Most of opioid peptides do not penetrate the blood–brain barrier (BBB) so their effects, when administered intravenously, are associated with activation of peripheral ORs.22, 23, 24 However, the nonpeptide OR agonists can enter the brain and activate ORs in autonomic centers regulating the functional state of the heart. It has been shown that perfusion of the fourth cerebral ventricle with the selective peptide μ OR agonist FK 33–824 induces bradycardia in the conscious dogs.25 In anesthetized dogs, [D‐Met2,Pro5]enkephalinamide perfusion through the cerebroventricular system elicited bradycardia, which was accompanied by an increase in the vagal discharge rate.26 It has been shown that intracisternal administration of opioid peptides also evoked bradycardia in unanaesthetized dogs.27 This effect was abolished by pretreatment with atropine. It has also been found that intracerebroventricular administration of the selective μ OR agonist DAMGO or the selective δ OR agonist DPDPE increased plasma catecholamine levels and blood pressure (BP) in conscious rats.28 However, DAMGO appeared to be a more potent regulator of the catecholamine level than DPDPE. At a dose of 5 nM and higher, DAMGO induced bradycardia mediated by vagal activation. The authors concluded that brain ORs regulating autonomic outflow, cardiovascular and respiratory function are mainly of the μ type, although a δ opioid system may also contribute to sympathoadrenal and respiratory effects of opioids. Thus, presented data indicate that ORs are present in the brain regions responsible for the regulation of function of the cardiovascular system and the stress response to strong stimuli.
B. Opioid Receptors in the Heart
All three OR (μ, δ, κ) transcripts were also detected in several peripheral tissues including the intestine, adrenal, kidney, and lung.29 In the stomach, δ OR and κ OR but not μ OR transcripts were found.29 mRNAs for opioid precursors were detected in adrenocortical cells.30 It has been established that μ and κ OR agonists can regulate cortisol and aldosterone secretion from the adrenocortical cells.30 The δ OR was found in a PC12 cell line derived from a pheochromocytoma of the rat adrenal medulla.31 Changes in function of these organs by activation of their ORs may indirectly affect the heart's function.
The first article reporting the existence of ORs in the myocardium was published in 1981.32 In 1988, the existence of δ OR in the myocardium was demonstrated using a radioligand binding assay.33 The next year, δ and κ ORs were also found in rat cardiac sarcolemma using this method.34 Later other investigators35 confirmed the existence of κ1 OR in the myocardium.36 Opioid‐binding sites in the myocardium were also confirmed in other studies.37, 38 In 1996, transcripts of δ and κ ORs were found in the heart.29 These data were later confirmed by Weil et al.39 None of these studies detected the μ OR in cardiomyocytes. However, in 1995, the μ3 subtype of this receptor was detected in the coronary microvascular's endothelial cells.40 This group of researchers also established that endothelial cells express a δ2 OR subtype.41 Vascular smooth muscle cells also appeared to express δ OR.42 Dumont and Lemaire were able to detect and characterize a high‐affinity [3H]nociceptin binding site in the membrane preparations of rat heart.43 Kim et al. confirmed the existence of the ORL1 receptor in cardiac myocytes.44 Thus, the view was formed that cardiac myocytes express δ OR, κ OR, and ORL1 receptor but not μ OR. This opinion was changed in 2005 when Head et al. found μ OR on the sarcolemma of cardiomyocytes using immunofluorescence microscopy.45 μ‐OR mRNA was also identified in the human right atrium. However, the amount of this receptor's mRNA in cardiomyocytes was significantly lower than the ORL1 mRNA content.46 Later, μ, δ, and κ ORs were detected immunohistochemically in human heart.47 The researchers found that μ and δ ORs are located mainly in cardiomyocytes as well as on sparse individual nerve fibers. Likewise, κ OR was identified predominantly in cardiomyocytes. This receptor was also found on intrinsic cardiac adrenergic (ICA) cells. It has been established that the δ OR is colocalized with the sensory neuron marker calcitonin gene‐related peptide (CGRP).47 Previously, similar data were obtained by Mousa et al. They identified μ OR and κ OR mRNA, as well as other OR proteins on cardiac parasympathetic, sympathetic, and sensory neurons.48 δ ORs were detected in the cholinergic neurons, small intensely fluorescent catecholaminergic cells, afferent nerve terminals, and atrial cardiomyocytes.49
Thus, all four types of ORs (μ, δ, κ, and ORL1) have been found in cardiomyocytes. δ OR, κ OR, and ORL1 receptor appeared to have the highest density in cardiomyocytes. μ and δ ORs are present in the endothelial cells and vascular smooth muscle cells express δ OR. ORs have been detected on the sensory nerve terminal, on ICA cells and are probably present in the sympathetic and parasympathetic terminals in the heart. It is safe to assume that activation of any of these receptors may potentially affect the functional state of the heart.
ICA cells were identified in rodent and human heart by Huang et al.50 In 2007, they discovered localization of δ OR immunoreactivity in ICA cells in human and rat hearts.51 They demonstrated that the selective δ1 OR agonist DPDPE enhanced epinephrine51 and CGRP release52 from ICA cells in denervated rat heart and these effects were abolished by the β‐adrenergic and CGRP receptor inhibitors. The authors suggested that the cardiotropic effects of δ OR agonists are mediated through β2‐AR/CGRP signaling.
C. Opioid Receptors Modulate Neural Control of the Heart
Opioid peptides can alter the autonomic nervous regulation of the heart function. Indeed, Kett et al. established that intravenous administration of H‐Tyr‐D‐Arg‐Phe‐Lys‐NH2 (DALDA), a selective peptide μ OR agonist that does not penetrate the BBB, blunted norepinephrine‐induced baroreflex bradycardia but had no effect on the sodium nitroprusside‐evoked tachycardia.53 Pretreatment with naloxone methiodide, a peripheral OR antagonist, abolished DALDA‐induced suppression of baroreflex. These data indicate that DALDA inhibits the baroreflex through peripheral OR occupancy. Later, these investigators established that the selective peptide μ OR agonist D‐Ala2,N‐Me‐Phe4,Gly5‐ol (DAMGO) suppresses baroreflex‐mediated bradycardia in the awake sheep but the selective κ OR agonist U50,488 had no such effect.54 Peripheral μ OR stimulation can suppress vagus‐mediated baroreflex and it can be assumed that these ORs are located in the nerve endings innervating the sinoatrial node.
Urthaler et al. established that selective perfusion of the sinus node with morphine in anesthetized dogs evokes bradycardia.55 Bradycardia was not altered by atropine or vagotomy and intranodal administration of morphine had no effect on the acceleration of heart rhythm produced by stellate ganglion stimulation or by selective perfusion of the sinus node with norepinephrine. The authors concluded that morphine‐evoked bradycardia was autonomic nervous system independent and a direct effect of morphine on the sinoatrial node cells. These results were confirmed by the data of Gautret and Schmitt.56 They found that an intravenous administration of ethylketocyclazocine, a preferential κ OR agonist, induced a fall in heart rate (HR) and BP in rats anaesthetized with pentobarbital. The bradycardia and the hypotension were not altered by bilateral vagotomy and atropine, but were completely eliminated by naloxone and Mr 2266, a preferential κ OR antagonist. Ethylketocyclazocine‐induced bradycardia persisted in β‐adrenoreceptor‐blocked and pithed rats.56 These results indicate that peripheral κ OR located in the heart's conduction system can affect cardiac rhythm.
However, other data indicate that opioids can exhibit vagolytic effect. The nervi vagi of isolated perfused rabbit heart were electrically stimulated and morphine, a preferential μ OR agonist, met‐enkephalin, μ OR and δ OR agonist, and D‐Ala2,D‐Leu5‐enkephalin (DADLE), a preferential δ OR agonist, reduced the vagal bradycardia with IC50 values of 148, 25, and 3.2 nM, respectively. Pretreatment with naloxone abolished the vagolytic effect of all opioids. The selective δ OR antagonist ICI 174864 eliminated met‐enkephalin effect but did not antagonize morphine's action.57 These data indicate that stimulation of both μ OR and δ OR can attenuate vagus‐mediated bradycardia but stimulation of presynaptic δ OR have a more powerful vagolytic effect. Similar data were obtained by Musha et al. in the experiments on anesthetized dogs with electrical stimulation of n. vagus.58 They confirmed that presynaptic δ OR activation prevents vagal bradycardia. In pithed rats pretreated with propranolol, vagal stimulation or injection of methacholine decreased HR.59 The selective ORL1 receptor agonist nociceptin (orphanin FQ) decreased the vagal bradycardia but did not modify the methacholine‐induced decrease in HR. The selective ORL1 receptor antagonist [Phe1ψ(CH2‐NH)Gly2]‐nociceptin(1‐13)NH2 antagonized vagolytic effect of nociceptin. Authors concluded that orphanin FQ prevents vagal bradycardia acting on the presynaptic ORL1 receptor located on the vagal terminal in the heart.59 It was established that MEAP (met‐enkephalin‐Arg‐Phe) and the selective δ2 OR agonist deltorphin II suppressed vagal bradycardia when they were delivered directly into the sinoatrial node by local microdialysis.60 The authors also found that δ OR stimulation only in the sinoatrial node prevents vagal bradycardia. In the further study, they conducted a comparative analysis of the ability of δ OR agonists to suppress vagal bradycardia during administration into the sinoatrial node and found that the vagolytic effect of opioids is mediated by δ2 ORs in the sinoatrial node.61 These data were confirmed in a subsequent study by the same group.62 They later established that δ2 ORs are located on the cholinergic vagal terminals in the sinoatrial node.63
Opioids can modulate not only the vagal discharge rate but also sympathetic outflow. Ledda and Mantelli using isolated guinea‐pig atria discovered that the nonselective OR agonist etorphine inhibits the sympathetic response induced by direct electical stimulation.64 Pretreatment with naloxone abolished this effect of etorphine. However, etorphine did not affect an inotropic effect of norepinephrine. Authors concluded that etorphine stimulates presynaptic inhibitory ORs on adrenergic nerve terminals in the heart. Later they established that the inhibitory effect of the opioid peptides could be due to stimulation of presynaptic inhibitory δ and κ ORs on adrenergic nerve terminals in the heart.65, 66 Somewhat different results were obtained by Starke et al.67 In their study using selective OR agonists and antagonists they found that, under in vitro conditions, only presynaptic κ ORs but not μ ORs or δ ORs inhibit the norepinephrine release from the sympathetic nervous innervating the rabbit heart. Fuder published results of experiments where isolated guinea‐pig atria were loaded with 3H‐(–)‐norepinephrine. In these experiments, the intrinsic nerves stimulation evoked norepinephrine efflux.68 They discovered that the nonselective OR agonist etorphine, the κ OR agonists ethylketocyclazocine, dynorphin A (1‐13), and the δ OR agonist DADLE but not the preferential μ OR agonist morphine inhibit the stimulation‐induced norepinephrine efflux in a concentration‐dependent manner. The inhibitory effect of ethylketocyclazocine and etorphine was antagonized by naloxone. The authors hypothesized that activation of presynaptic κ ORs and apparently δ ORs inhibits norepinephrine release from sympathetic nerves in the heart.68 Others showed that a strong inhibition of on the sympathetic‐mediated positive inotropic effect evoked by electrical field stimulation of guinea‐pig atria can be achieved by κ OR agonists U‐50488 and U‐69593, whereas δ OR agonists, DPDPE and BW373U86, were ineffective. This effect of κ OR agonists was reversed by the selective κ OR antagonist nor‐binaltorphimine.69
Similar data were obtained in the in vivo experiments on pithed animals. Thus in pithed rabbits, it was found that ethylketocyclazocine decreased BP, the endogenous plasma norepinephrine level, and the 3H‐norepinephrine release rate.70 These effects were inhibited by naloxone. Investigators concluded that ethylketocyclazocine inhibits norepinephrine release from postganglionic sympathetic neurons, apparently by stimulation of ORs at the terminal axons. Later they established that the preferential κ2 OR agonist bremazocine prevents the 3H‐norepinephrine release and BP elevation in response to electrically (2 Hz) stimulated sympathetic outflow in pithed rabbits but has no effect on the BP increase evoked by an intravenous infusion of norepinephrine.71 The inhibitory effects of bremazocine were antagonized by naloxone. These results indicate that the κ2 OR stimulation inhibits norepinephrine release and consequently lowers BP by activation of peripheral, probably prejunctional, κ OR. This function of κ OR was later confirmed in the experiments of Caffrey's group,72 while Feuerstein et al. excluded a possible role of μ OR in the regulation of sympathetic outflow in pithed rats.73 Malinowska et al. showed in the experiments on pithed rats pretreated with atropine that the postganglionic sympathetic nerves innervating the rat heart have presynaptic ORL1 receptor and its activation inhibits the sympathetic outflow.59 Thus, the data show that activation of peripheral ORs can inhibit the cardiotropic effects of parasympathetic and sympathetic nerve stimulation. However, in the conscious animals the effect of opioid peptides can be quite the opposite. In particular, it has been shown that in unanesthetized sheep and dogs, intravenous administration of enkephalins or nociceptin may cause transient rise in BP and HR associated with enhanced sympathetic outflow.74, 75, 76 These effects were associated with activation of ORs located outside the BBB in the area postrema, a BBB‐deficient small, elevated area in the lateral wall of the inferior recess of the fourth ventricle.74, 75, 76 It was established that stimulation of central μ and δ OR also can increase plasma catecholamine levels and BP.12, 28
In 1990, Giuliani et al. demonstrated that electrical stimulation of the left atria of reserpine‐pretreated guinea pigs in the presence of atropine produces a positive inotropic effect involving activation of capsaicin‐sensitive afferents.77 μ OR agonists dermorphin, DAMGO, and morphine all inhibited this effect. The authors concluded that capsaicin‐sensitive nerves in the atrium have μ OR, which inhibit transmitter release from sensory nerve terminals.77 In a similar model, these investigators found that the selective ORL1 agonist nociceptin inhibits a positive inotropic response induced by electrical field stimulation.78 However, nociceptin (the selective ORL1 agonist) did not affect the positive inotropic effect of exogenous CGRP. Therefore, the authors suggested that nociceptin inhibits CGRP release by activation of ORL1 receptors localized on the afferent nerve endings in atria.78
The adrenal medulla can also be involved in the cardiovascular effects of opioids. Gulati and Bhargava studied cardiovascular effects of intravenous administration of κ OR agonists bremazocine, tifluadom, and U‐50,488 in anesthetized rats.79 All three opioids evoked bradycardia. Bremazocine and U‐50,488 decreased BP. The hemodynamic effects of the opioids were blocked by bilateral adrenal demedullation. The peripherally acting OR antagonist naltrexone methylbromide blocked the cardiovascular effects of U‐50,488. Based on these results, the investigators suggest that cardiovascular effects of κ OR agonists are mediated through the adrenal medulla and peripheral κ OR stimulation.79 The mechanism of this effect of κ OR agonists remains unknown.
Taken together, the available experimental data suggest that the heart is richly populated with ORs located on the sarcolemma of cardiomyocytes, cell membrane of ICA, and the coronary endothelial cells. In addition they are located on the sympathetic and parasympathetic nerve terminals in the heart, in the adrenal medula, and in the brain regions responsible for the regulation of the heart. Thus, it should come as no surprise that some of these can exert a cardioprotective effect against I/R injury.
3. ANTI‐INFARCT EFFECT OF PRETREATMENT WITH OPIOID RECEPTOR AGONISTS
A. δ1 Opioid Agonists
Rats given 0.3 mg/kg of morphine intravenously prior to coronary artery occlusion/reperfusion experienced a decrease in the IS/AAR by 4.5‐fold.10 A year later, the same group of researchers found that the infarct‐sparing effect of morphine depended upon δ OR activation.80 In 1998, Miki et al. reported that morphine reduced infarct size in rabbits.81 Morphine was tested at doses of 0.3, 0.8, and 3 mg/kg but only the highest dose protected suggesting a species difference between ORs in rats and rabbits. Morphine also increases the tolerance of isolated rat cardiomyocytes to a 90‐min hypoxia.82 Wu et al.83 confirmed the cardioprotective properties of morphine. They administered 8 mg/kg intraperitoneally to rats. It was not mentioned why they selected such a high dosage but they found that morphine's protection could be prevented by blocking μ, δ, or κ ORs suggesting that all three OR subtypes seem to be involved in the cardioprotection of morphine. Lu et al. corroborated the infarct‐sparing effect of morphine in rats at 0.3 mg/kg intravenously.84
Bilir et al. showed that tramadol, an agonist and antagonist of ORs, increases the isolated rat heart's tolerance to I/R.85 In a clinical trial tramadol was given prior to coronary artery bypass surgery.86 Surprisingly, tramadol caused an increase in a marker of cardiomyocyte necrosis, cardiac troponin I (cTnI) in the blood of patients suggesting that this opioid actually exacerbates injury of the heart during coronary artery bypass surgery. This demonstrates why results of any animal study must be tested in clinical trials.
Irwine's group was the first to demonstrate remifentanil‐induced cardioprotection in both in vivo and isolated heart models.87, 88 The infarct‐reducing effect of remifentanil was abolished by pretreatment with the selective κ OR antagonist nor‐binaltorphimine and the selective δ OR antagonist naltrindole. Later, the cardioprotective effect of remifentanil was confirmed in an isolated perfused rat heart.89 In 2010, a clinical trial of remifentanil was carried out.90 Forty patients with on‐pump coronary artery bypass surgery were included in this trial. All patients were anesthetized with propofol and pretreated with fentanyl. Some of the patients (n = 20) received remifentanil (1 μg/kg intravenously and then infusion with rate of 0.5 μg/kg during 30 min) prior to surgery. Cardioprotection was determined 24 h postoperatively by assessing biochemical markers of myocardial necrosis: creatine kinase MB (CK‐MB) and cTnI. CK‐MB and cTnI levels were significantly lower in patients that received remifentanil.90 Thus, unlike tramadol, remifentanil appears to be cardioprotective not only in animals but also in patients with I/R injury of heart.
Pretreatment with the selective δ1 OR agonist TAN‐67 (10 mg/kg intravenously) decreased the IS/AAR in rats and the selective δ1 OR antagonist BNTX abolished the effect.91 This experiment indicated that the δ1 OR was protective and a year later, using isolated perfused hearts, it was shown that the δ OR‐selective agonist DADLE could also protect.92 More recent studies indicate that 10 mg/kg DADLE prior to coronary artery occlusion decreases IS/AAR and the highly selective δ OR antagonist naltrindole abolished this effect.93 The cardioprotective effect of DADLE was confirmed in later investigations.94, 95 In vivo, this peptide exhibited an infarct‐reducing effect in rats at a dose of 1 mg/kg.94 It was also found that the μ OR‐selective agonist methadone (0.3 mg/kg) shows an infarct‐reducing effect, which is actually mediated via δ OR activation.96
Takasaki et al. found that cardiomyocytes tolerance to hypoxia/reoxygenation is increased after addition the μ and δ OR agonist met‐enkephalin to the incubation buffer.97 Later, this team of investigators using naltrindole showed that the cytoprotective effect of met‐enkephalin is mediated via δ OR occupancy.98 Infusion of met‐enkephalin to rabbits starting 24 hr before coronary artery occlusion with an osmotic minipump promoted a decrease in the IS/AAR by 60%.99 However, a 24‐hr infusion of met‐enkephalin in mice failed to reduce the infarct size.100 This indicates again that there are species differences in the response to some opioids. This is most likely due to small but important differences in the genetic codes for these receptors among the species. In in vivo experiments with pigs, researchers could not demonstrate an infarct‐reducing effect of DADLE at a dose of 1 mg/kg intravenously.101
The ability of the δ1 OR agonist TAN‐67 to mimic the cardioprotective effect of preconditioning in rat heart was confirmed in the later studies both in vivo102 and in vitro.103, 104, 105 We established that perfusion of the isolated rat heart with the δ1 OR‐selective agonist DPDPE (154 nM) decreases reperfusion‐induced creatine kinase release.106 Pretreatment with the δ OR‐selective antagonist naltrindole (1 nM) completely abolished DPDPE's cardioprotective effect. In 2001, McPherson and Yao107 showed that the δ‐selective agonist BW373U86 (10 pM) increases tolerance of isolated cardiomyocytes to hypoxia/reoxygenation. The cardioprotective property of TAN‐67 and BW373U86 were confirmed in vivo at coronary artery occlusion and reperfusion.108, 109 In addition, it was established that the infarct‐sparing effect of BW373U86 (1 mg/kg) is a consequence of δ1 OR activation.109
In pigs, an infarct‐reducing effect of DPDPE was found at a dose of 1 mg/kg intravenously101 but this dose was not protective in rats.110, 111 Again, a species difference was present. DPDPE at the final concentration of 0.1 mg/L (154 nM) did protect the isolated perfused rat heart112, 113 and DPDPE's protection in rat heart can be blocked by naltrindole.104, 106, 114 The infarct‐sparing effect of DPDPE was confirmed in experiments in the isolated rat heart by Huang et al.52 In 2006, Watson et al. reported that the δ1 and δ2 OR agonist ARD‐353 (0.3 mg/kg) decreased the IS/AAR in rats.115 This effect disappeared after δ1 OR inhibition with BNTX. In addition, Watson et al. obtained data that ARD‐353 does not penetrate through the BBB.115 These authors concluded that the cardioprotective effect of ARD‐353 is a consequence of peripheral δ1 OR activation. In 2006, Gross et al.116 reported that the δ OR agonist fentanyl isothiocynate caused infarct reduction at a dose of 10 μg/kg intravenously. Hence, there is a reason to believe that the δ1 OR agonists are excellent candidates for cardioprotective drug development.
B. δ2 Opioid Agonists
In 2002, in experiments with pigs, we demonstrated the infarct‐reducing effect of the putative δ2 OR agonist deltorphin D at a dose of 1 mg/kg.101 In 2009, in experiments with rats, we confirmed these data.117 The selective δ2 OR agonist deltorphin II at a dose of 0.12 mg/kg can decrease the IS/AAR. The infarct‐sparing effect of deltorphin II was maintained in the presence of the δ1 OR antagonist BNTX but disappeared after δ2 OR block with naltriben.117 The cardioprotective effect of deltorphin II was abolished after blocking peripheral OR with naloxone methiodide (5 mg/kg). Hence, the δ2 OR also seemed to be protective.
C. κ Opioid Agonists
The κ1 OR‐selective agonist U50,488 increases the tolerance of isolated cardiomyocytes to sodium cyanide toxicity in the incubation buffer.118 They showed that U50,488 (10 μM) decreases the IS/AAR in the isolated perfused rat heart. We confirmed that pretreatment with U50,488 protects the isolated perfused rat heart against global I/R.113, 119, 120 Addition of κ OR‐selective agonist dynorphin to incubation buffer increases tolerance of rabbit cardiomyocytes to 3 hr of hypoxia and the κ OR‐selective antagonist GNTI abolished dynorphin's protection.98 In 2004, it was shown that pretreatment with the κ OR agonists U50,488; ICI 204,448; and BRL 52537 exhibit infarct‐reducing effect in vivo.121 The κ OR‐selective antagonist nor‐binaltorphimine abolished the infarct‐sparing effect of U50,488 and ICI 204,448 but did not affect the cardioprotective effect of BRL 52537.121 Since ICI 204,448 does not penetrate through the BBB,122 it is likely that the peripheral κ OR activation promotes the protection. The antinecrotic effect of U50,488 is seen in the isolated heart. Recently, we have found that the quaternary ammonium salt of U50,488 (Q‐U50,488), which is not able to pass the BBB, elicits a protective effect against cardiac I/R injury.123 The infarct‐sparing effect of Q‐U50,488 was abolished by nor‐binaltorphimine implicating a peripheral κ OR. It can safely be assumed that the κ OR regulating cardiac tolerance to I/R is located in the heart.
D. μ Opioid Agonists
Using isolated perfused rat heart we found that the μ OR‐selective agonist DAMGO reduces infarction after global I/R.124 The protection from DAMGO was confirmed in our later investigations.125, 126 In addition, we established that the μ OR‐selective agonist DALDA also can prevent cardiac cell death during global I/R. However, intravenous DAMGO (0.1 mg/kg) or DALDA (0.1 mg/kg) 15 min prior to heart isolation actually increased the injury from I/R ex vivo.125, 126, 127 Gross's group found that DAMGO (0.1 mg/kg intravenously) had no effect on the IS/AAR in rats after I/R in vivo.128 In our in vivo investigation in rats with coronary artery occlusion (45 min) and reperfusion (2 hr), we studied the μ OR‐selective agonist dermorphin H (0.12 mg/kg) and DAMGO (0.08 or 0.8 mg/kg).110, 111 Neither of these peptides had any effect on the IS/AAR. It remains unclear why DAMGO is so protective ex vivo but not in vivo.
Gross et al. found that the μ and δ OR‐selective agonist Eribis peptide 94 starting 10 min after coronary artery occlusion (30 min) and continuing during reperfusion decreases the IS/AAR in open‐chest rats. This protection persisted after inhibition of δ OR with naltrindole, δ1 OR with BNTX, and κ OR with nor‐binaltorphimine. However, it was abolished with the selective μ OR antagonist CTOP.129 This was the first convincing evidence that μ OR activation protects the heart from I/R.
E. ORL1 Opioid Agonists
Recently, we evaluated a fourth OR subtype that usually denoted as ORL1 receptor (opioid‐like receptor 1) in an in vivo rat model using the endogenous ORL1‐selective agonist nociceptin. Neither 0.4 or 2.2 mg/kg had any effect on the IS/AAR.110, 111 In our opinion, it is too early to draw a final conclusion that ORL1 receptors do not affect the heart's tolerance to I/R because we have not yet studied it in the isolated heart. The μ OR agonists are cardioprotective ex vivo but not in vivo and it is possible that nociceptin may act in a similar fashion.
4. TOXICITY OF HIGH‐DOSE OPIOIDS AND THE CARDIOPROTECTIVE EFFECTS OF OR ANTAGONISTS
The aforementioned studies demonstrate that OR activation can increase the heart's tolerance to I/R but there are some studies demonstrating that OR stimulation can also exacerbate I/R injury. Intravenous administration of morphine at a dose of 2.1 mg/kg can induce ST segment depression in patients with ischemic heart disease.130 The authors interpreted this effect as a manifestation of myocardial ischemia. In another study, it was shown that morphine at a dose of 1 mg/kg increases ST segment elevation in cats with coronary artery ligation, which they regarded as worsening of the heart's ischemia.131 In 1982, the same group of authors obtained data that morphine can increase the infarct size in rats.132 Morphine was administered at a dose of 3 mg/kg intravenously for 10 min prior to a 48‐h coronary artery occlusion without reperfusion. Permanent occlusion without reperfusion in rodent hearts is now considered as an invalid methodology for evaluating cardioprotection since cardiac muscle cannot survive in the complete absence of blood flow. In addition, these data contradict the results of the Chinese investigators, which showed that morphine at a dose of 8 mg/kg evokes a decrease in the IS/AAR.83 We tested 0.3, 0.8, and 3.0 mg/kg morphine pretreatment in open‐chest rabbits and found no effect of the two lower doses but greatly reduced infarct size with the high dose.81
In 1985, it was reported that 1.1 or 3.6 mM naloxone in the perfusion solution protects the isolated heart.133 Naloxone's IC50 toward μ and δ OR is 8.2 nmol.134 Similarly, the K i of naloxone toward μ OR is reported to be 3.4 nmol but that toward δ OR is 50 nmol.135 We would suggest that the cardioprotective effect of their very high‐dose naloxone is probably a nonspecific membrane stabilizing effect of the drug, rather than a consequence of the blockade of ORs. It should also be noted that in most of our experiments we have not observed infarct‐reducing effect of naloxone, naltrexone, and most of other OR antagonists.81, 110 Similarly, many other investigators failed to observe a cardioprotective effect of the OR antagonists in situ or in vitro. An exception is the work of Chen et al. They performed 45‐min global ischemia and 60‐min reperfusion of an isolated rat heart in which OR antagonists were added to perfusion buffer for the first 10 min of reperfusion. Necrosis was evaluated by IS/AAR and by monitoring CK‐MB levels in coronary effluent. Naloxone (10 nM), naltrindole (5 nM), or nor‐binaltorphimine (5 nM) decreased the IS/AAR and CK‐MB release.136 Hence, these OR antagonists mimic IPost phenomenon. The concentrations of antagonists indicators used approach their published K i and IC50.134, 135, 137 Therefore, we cannot easily dismiss the cardioprotective effect of OR antagonists as a nonspecific effect. We found that intravenous administration of the μ OR antagonist CTAP (1 mg/kg) to rats prior to coronary artery occlusion (20 min) and reperfusion (3 hr) promotes a decrease in the IS/AAR.138 However, protection may have been mediated via the somatostatin receptor for which this peptide exhibits moderate affinity.139 Somatostatin is known to limit the IS/AAR in in vivo studies.140
Our investigations do indicate the existence of an OR pool, or non‐ORs, whose activation with opioids negatively affects cardiac tolerance to I/R.104, 112 In isolated rat heart studies, we observed that the cardioprotective effect of the δ1 agonist DPDPE disappears if the concentration of peptide in the perfusion buffer is increased to 740 nM.104, 112 In isolated murine heart, 10 μM of morphine was not protective141 while 0.3 μM did protect isolated rabbit heart.81 It seems highly likely that concentration of 10 μM was so high that morphine began binding to a pool of receptors that negatively affected the heart's tolerance to I/R. Gross's group was unable to protect hearts with 1 μM BW373U86 in isolated murine hearts141 while BW373U86 did protect isolated chick cardiomyocytes but only at a concentration of 10 pM.107 Mixing species always complicates interpretation but an obvious explanation is that overdosing can lead to negative off target effects. We recommend that ex vivo and in vitro experiments should test agonists at a concentration tenfold higher than the K i or EC50.81 They should also be aware that the binding affinities of these drugs can vary widely among species.
Aitchison et al. reported that DADLE at 10 nM exhibits infarct‐sparing that was diminished at 1 μM. Inhibition of κ OR with nor‐binaltorphimine restored the full cardioprotective effect of high concentration DADLE.142 The authors concluded that the diminished effect of DADLE at high concentration is due to activation of κ OR. These data closely resemble our results with DPDPE.104, 112 In addition, Aitchison et al. established that the nonselective κ OR agonist bremazocine (30 nM) ex vivo increased infarct size.142 This negative effect of bremazocine disappeared after inhibition of κ OR. In this regard it should be noted that U50,488 is the selective κ1 OR agonist but bremazocine is an agonist of κ2 OR.143 It seems reasonable to assume that the activation of κ2 OR exacerbates injury from I/R.
In 2005, Meine et al. published the results of a prospective, nonrandomized study, which included patients with acute coronary syndrome (ACS) with non‐ST‐segment elevation (NSTE; n = 57,039).144 The authors evaluated the outcome of patients treated with morphine and those who were not. It was found that treatment with morphine was associated with increased risk of in‐hospital mortality. The authors raised concerns about the safety of using morphine in patients with ACS NSTE but pointed out that that could only be answered with a randomized trial.144 These data were in accord with a few other studies.130, 145, 146 In particular, Conahan et al. demonstrated that morphine (2 mg/kg) caused severe hypertension and an increase in systemic vascular resistance in patients undergoing heart valve replacement.146 Later Lappas et al. reported that the addition of 5% NO to morphine (2 mg/kg intravenously) decreased BP, cardiac index, stroke index and increased pulmonary capillary wedge pressure.146 Intravenous administration of morphine at a dose of 2.1 mg/kg can induce ST segment depression in patients with ischemic heart disease.130 However, we would like to draw readers’ attention to the fact that an extremely large dose of morphine (2 mg/kg) was used in these three studies.130, 145, 146 Indeed, in current cardiological guidelines, the recommended dose of morphine is 4–8 mg (0.05–0.1 mg/kg).147 Experimental studies suggest that morphine has the infarct‐limiting effect at a dose of 0.3 mg/kg.10 It comes as no surprise that morphine can provide an adverse effect on the cardiovascular system at a dose many times exceeding the therapeutic dose. In cardiological practice, morphine and fentanyl are used not only for pain relief in patients with AMI but also to ease anxiety, reduce preload, due to venodilation,147, 148, 149 and afterload, due to reducing systemic vascular resistance.148, 150 It has been established that morphine and fentanyl can decrease myocardial oxygen consumption and reduce lactate production by the left ventricle in human.151 Both of those effects may increase cardiac resistance to ischemia and improve the outcome in AMI. Morphine is also used for the prevention of pulmonary edema, and cardiogenic shock.152 Therefore, morphine and other opioids are prescribed in the most serious cases characterized by a higher mortality than in patients with mild AMI, like those included in the study of Meine et al.144 An overdose of opioids may cause a depression of respiration, hypotension, and vomiting.152
In summary, pretreatment with agonists of μ, δ1, δ2, and κ1 OR exhibit cardioprotective properties both in vivo and in vitro. These pharmacological agents mimic the preconditioning phenomenon. The role of the ORL1‐receptors in this regard remains open, however. A number of reports points to the existence of important species differences in the reaction of infarcted myocardium to opioids. Some receptors, such as κ2 ORs, may actually exacerbate the ischemic and reperfusion heart injury. But the agonists of μ, δ1, δ2, and κ1 ORs can be considered as the most promising group of agents able to induce cardioprotection. This opinion can be supported by numerous studies.80, 87, 93, 96, 98, 106, 109, 110, 115, 116, 121, 128, 129
5. ANTIAPOPTOTIC EFFECT OF THE OPIOID RECEPTOR AGONISTS
It is well known that reperfusion induces enhancement of production of reactive oxygen species (ROS)153 and Ca2+ overload in cardiomyocytes.154 Calcium ions and ROS evoke opening of MPT (mitochondria permeability transition pore), which is a protein supramolecular complex built into the inner mitochondrial membrane.155, 156 Opening of this pore collapses the potential across the inner mitochondrial membrane, which prevents ATP generation by the mitochondria. MPT also releases cytochrome c and AIF (apoptosis inducing factor) in the intermembrane space into the cytosol.155, 156 Cytochrome c together with APAF‐1 (apoptosis protease activating factor), procaspase‐9, and ATP form a supramolecular complex named apoptosome.156 The apoptosome catalyzes proteolysis of procaspase‐9 to become active caspase‐9, which in turn catalyzes the cleavage of other proteins ultimately leading to apoptosis, a process in which the cell is killed and digested from within over several days. Protein AIF activates translocation of endonuclease G from cytosol into nucleus where the latter catalyzes DNA fragmentation that is a characteristic of apoptotic cells.156 These events are developed mainly during the first minutes of reperfusion. Therefore, the opening of MPT is a major cause of death of cardiomyocytes after the restoration of coronary blood flow.155 Necrosis quickly ensues if too many mitochondria within the cell are lost to MPT and the cell becomes tetrazolium negative (popular marker for infarct size studies) minutes after reperfusion due to membrane failure. If only a small fraction of the mitochondria is involved, however, the cell may survive the initial I/R only to succumb to apoptosis a day or two later. Apoptotic cells are tetrazolium positive in the first hours of reperfusion. The evidence is strong that IP protects by inhibiting MPT formation at reperfusion.157 Generally, apoptosis and necrosis act in parallel and markers of apoptosis can be used to assess injury from I/R.
The first work indicating that opioids inhibit apoptosis of cardiomyocytes was published in 2001. Isolated chicken embryo cardiomyocytes were subjected to 12 h of hypoxia and 12 h of reoxygenation. Apoptosis was evaluated by the number of TUNEL‐positive cells (terminal deoxyribonucleotide transferase‐mediated dUTP nick end labeling). Fifty‐four percent of the cells were TUNEL‐positive. However, if BW373U86 (20 pM) was added to medium only 39% of the cells became apoptotic. The selective inhibition of δ1 OR with BNTX abolished the cytoprotective effect of BW373U86.158 Okubo et al. found that opioids can exert an anti‐apoptotic effect in vivo. Morphine at 0.3 mg/kg prior to coronary artery occlusion reduced the number of TUNEL‐positive cells in the heart from 12.4% in control to only 5.2%. The δ OR antagonist naltrindole (10 mg/kg intravenously) abolished the effect of morphine.159 These findings led the investigators to conclude that the antiapoptotic effect of morphine was dependent upon δ OR activation. The antiapoptotic effect of morphine on isolated cardiomyocytes was confirmed in later experiments.160 In isolated perfused rat hearts exposed to 30‐min global ischemia and 60‐min reperfusion 16% of the cells were TUNEL‐positive but 3 μM morphine in the perfusion solution decreased this index to 5%.161 In 2012, Kim et al. using isolated cardiomyocytes found that addition of remifentanil to the cell incubation medium prior to hypoxia/reoxygenation increased cell survival, decreased the concentration of Ca2+ in the cytoplasm, decreased activity of caspase‐3, increased anti‐apoptotic protein Bcl‐2 (B‐cell lymphoma protein‐2) over that in untreated cells.162 In 2009, it was noted that the selective κ1 OR agonist U50,488 causes an antiapoptotic effect.163 This study was performed in rats with coronary artery occlusion (45 min) and reperfusion (3 h). The κ1 OR agonist U50,488 was administered intravenously prior to ischemia. The number of TUNEL‐positive cells in the area of I/R was 21.3% but in animals receiving U50,488 this number dropped to12%. The selective κ OR antagonist nor‐binaltorphimine eliminated this effect indicating that the antiapoptotic effect of U50,488 was mediated via κ1 OR activation. We recently confirmed their hypothesis using Q‐U50,488, which does not crosses the BBB.123
The aforementioned studies suggest that δ and κ1 OR activation reduces the appearance of apoptosis of cardiomyocytes following reperfusion. It has not been determined whether agonists of μ OR and ORL1 receptors can prevent apoptosis of cardiomyocytes.
6. OPIOIDS CAN MIMIC DELAYED ISCHEMIC PRECONDITIONING
Fryer et al. found that 24 h after injection of TAN‐67, there was a return of protection against I/R. Combining TAN‐67 with the δ1 OR antagonist BNTX abolished this delayed protection. The authors concluded that the delayed protective effect of TAN‐67 is dependent upon δ1 OR activation.164 The delayed protective effect of TAN‐67 was confirmed in later works.165, 166 In 2004, it was found that the nonpeptide δ OR‐selective agonist SNC‐121 caused a delayed window of protection in rats and surprisingly its protection was retained after inhibition of ORs with naloxone.167 The authors concluded that the cardioprotective effect of SNC‐121 was not dependent on OR and illustrates the importance of testing with antagonists. Shinmura and colleagues found that the selective δ OR agonist BW‐373U86 can mimic delayed preconditioning.168 Other investigators found that the nonpeptide δ1 and δ2 OR agonist ARD‐353 (0.3 mg/kg) evoked delayed conditioning.115 Morphine (3 mg/kg) also triggered delayed conditioning169 as did morphine at a dose of 0.3 mg/kg.170 OR antagonists were not used in these two studies. Hence, the responsible for delayed protective effect OR was not identified.
A 30‐min incubation of isolated cardiomyocytes with U50,488 for 20 hr prior to hypoxia/reoxygenation increases cell tolerance to hypoxia/reoxygenation.171 This effect of U50,488 did not occur after κ OR inhibition with nor‐binaltorphimine. The delayed preconditioning phenomenon of U50,488 was confirmed in later works by the same authors.172, 173 Intravenous administration of remifentanil, a nonselective OR agonist, can induce a delayed cardioprotective effect174 and this was confirmed by other investigators.175 All three OR antagonists (CTOP, nor‐binaltorphimine, naltrindole) abolished infarct‐sparing effect of remifentanil.174 Participation of ORs in the delayed cardioprotective effect of remifentanil has been confirmed by Sun et al.175
7. INVOLVEMENT OF ENDOGENOUS OPIOIDS IN THE INFARCT‐REDUCING EFFECT OF REMOTE ISCHEMIC PRECONDITIONING
In 2001, Dickson et al. attempted to clarify the nature of the humoral factor(s) mediating the infarct‐reducing effect of remote ischemic preconditioning (RIPC).176 Preconditioning of isolated perfused rabbit hearts was reproduced by three 5‐min episodes of ischemia interspersed with 10 min of reperfusion. Coronary effluent was collected, purified, and concentrated using Sep‐Pak C‐18 columns. They demonstrated that concentrated coronary effluent introduced to other isolated rabbit hearts can protect these hearts against ischemia (40 min) and reperfusion (120 min). This protective effect was eliminated by pretreatment with naloxone.176 In the next study, isolated jejunal segments were subjected to 1 hr of simulated ischemia followed by 30 min of reoxygenation.177 Pretreatment with coronary effluent concentrate also improved contraction of the jejunal segments during reperfusion. Naloxone abolished the inotropic effect of the coronary effluent. Authors believe that coronary effluent contains opioids, which mediate a protective effect of RIPC.177 The authors hypothesized that the endogenous mediator of the cardioprotective action of RIPC is endogenous opioid peptide Met5‐enkephalin‐Arg6‐Phe7.178 Patel et al. hypothesized that mesenteric preconditioning evokes release of endogenous opioids that protect the heart against I/R.179 Rats were subjected to coronary artery occlusion (30 min) followed by reperfusion (2 hr). Experimental groups underwent occlusion of the mesenteric artery (15 min) followed by reperfusion (10 min). Pretreatment with naloxone abolished the protective effects of RIPC.179 These data indicate that mesenteric preconditioning evokes release of endogenous opioid peptides that protect the myocardium against I/R. Weinbrenner et al. assumed that infarct‐sparing effect mediated by infrarenal occlusion of the aorta (IOA) may be transmitted by endogenous opioids.180 They established that IAO protected against I/R and this was abolished by pretreatment with the selective δ1 OR antagonist BNTX (7‐benzylidenenaltrexone).180 These results indicate that the protection by RIPC is transmitted by δ1 OR occupancy. Another group induced RIPC in rats by three cycles of femoral artery occlusion (5 min) followed by reperfusion (5 min).181 They demonstrated that RIPC evokes increase in plasma dynorphin (a nonselective κ OR agonist), but not met‐enkephalin (a μ OR and δ OR agonist) level. Pretreatment with the selective κ OR antagonist nor‐binaltorphimine eliminated the infarct‐sparing effect of RIPC. The selective δ OR antagonist naltrindole had no effect on the remote preconditioning.181 Hence, endogenous κ OR agonists, apparently dynorphin, mediate the cardioprotective effect of RIPC. Later, Rehmi et al. reported the participation of endogenous opioids in RIPC.182 Rentoukas et al. showed that morphine in combination with RIPC reduced infarct size in patients with primary percutaneous coronary intervention while RIPC alone did not.183
Thus, today, there is no doubt that endogenous opioid peptides participate in the mechanism of the cardioprotective effect of RIPC. However, it remains unclear what kinds of ORs are involved in the RIPC phenomenon. The aforementioned Met5‐enkephalin‐Arg6‐Phe7 and dynorphin are unlikely mediators of RIPC since they are not resistant to enzymatic hydrolysis.184, 185
8. OPIOIDS MIMIC POSTCONDITIONING PHENOMENON
The aforementioned studies demonstrate the ability of opioids to protect when applied as a pretreatment. A major indication for cardioprotection is ACS where the patient presents with ischemia already in progress. That makes pretreatment impossible so a postconditioning drug intervention is needed. Because much of the cell death in the heart is from MPT that form at reperfusion, it is theoretically possible to protect against infarction right up to the time of reperfusion. IPost has been shown to limit infarct size.7 Is it possible that OR agonists at reperfusion might also protect?
There are a few publications indicating that the OR agonist can protect when administered at the end of the ischemic period. In one such study, rats were exposed to 1‐h coronary artery occlusion and 2‐h reperfusion. When 0.3 mg/kg morphine was administered intravenously 10 min prior to reperfusion it evoked a decrease in the IS/AAR from 45 to 30%.186 ARD‐353 administered after 30 min of ischemia at a dose of 0.3 mg/kg immediately before removing the ligature decrease the IS/AAR from 55 to 35%.115 Since ARD‐353 does not penetrate the BBB, these authors concluded that its infarct‐reducing effect is mediated via peripheral OR.115 Tsutsumi et al. studied mice with 30‐min coronary artery occlusion and 2‐h reperfusion. The δ OR agonist SNC‐121 (10 mg/kg) was administered intravenously 3 min before reperfusion. The control IS/AAR was 44% but only 24% in SNC‐12‐treated mice.187 This study did not evaluate the role of OR antagonists.
The OR agonists mimic IPost not only in vivo but also ex vivo. In one study, the isolated perfused rat heart was exposed to 45‐min global ischemia and 60‐min reperfusion.136 Morphine was added to the perfusion buffer at 0.3, 3, and 30 μM for the first 10 min of reperfusion. Necrosis was assessed by tetrazolium staining and by CK‐MB in coronary effluent. Morphine decreased the IS/AAR at 0.3 μM and more so at 30 μM. Pretreatment with naloxone or nor‐binaltorphimine attenuated the protection.136 Unfortunately, the OR antagonists (naloxone, naltrindole, and nor‐binaltorphimine) exerted a small but significant cardioprotective effect by themselves, which complicates the interpretation.
In 2008, Jang et al. studied isolated rat heart with 30‐min coronary artery branch occlusion and 2‐hr reperfusion. Either morphine (1 μM) or the δ OR agonist BW373U86 (1 μM) were added to the perfusion solution starting 5 min prior to reperfusion of the occluded coronary branch. The total duration of perfusion with agonists was 15 min. Both agonists decreased the IS/AAR by threefold.188 Pretreatment with naltrindole (100 μM) abolished the infarct‐sparing effect of both agonists. Unfortunately, the authors used naltrindole in a concentration sufficient to inhibit all OR subtypes.135 Using isolated perfused rat heart, Mourouzis et al. reported that 10 μM morphine can mimic IPost.189 The ability of morphine at 1 μM to postcondition was also reported elsewhere.190, 191 In vivo I/R experiments in rats showed that intravenous administration of U50,488 (0.1 mg/kg) 5 min prior to reperfusion promotes a decrease in the IS/AAR but U50,488 10 sec prior to reperfusion had no effect on the IS/AAR.192 They also studied U50,488 (100 nM) in an isolated murine heart. The κ OR agonist was added to Krebs‐Henseleit buffer at the beginning of reperfusion and it decreased the IS/AAR.192 They did not test OR antagonists. However, this does not invalidate their conclusion that U50,488 protected via κ1 OR because K i of U50,488 for κ1 OR is 7.4 nmol but the K i of U50,488 for μ OR is 256 nmol.134
Methadone administered to an in situ rat experiencing 30‐min ischemia at a dose of 0.3 mg/kg for 5 min prior to reperfusion reduced infarct size. But if the injection was performed 10 sec after removal of the ligature, no changes in the IS/AAR could be detected.96 If the duration of ischemia of the heart was 45 min, the injection of methadone 5 min before reperfusion also had no effect on the IS/AAR. The authors concluded that this opioid mimics IPost if it is administered 5 min before reperfusion.96 Remifentanil was infused intravenously for 5 min starting 5 min before reperfusion in rats with 30‐min coronary artery occlusion and 2‐hr reperfusion. The IS/AAR was reduced by a dose of 10 μg/kg. Blocking δ or κ OR but not the μ OR by the agonist CTOP eliminated the protection.193 The ability of remifentanil to simulate IPost phenomenon was confirmed in another study performed in the isolated perfused rat heart.194
In 2011, it was reported that 1 μg/kg of a tetrapeptide referred to by the authors as Eribis peptide 94 (EP94) decreased the IS/AAR in rats at reperfusion.195 These authors did not confirm a role of ORs in the infarct‐reducing effect of EP94 but the authors did note that EP94 is a μ and δ OR agonist. Such a high potency of EP94 is surprising. However, in a later study by the same authors, it was reported that EP94 had an infarct‐sparing effect at a dose of 25 μg/kg but had no effect on the infarct size at the dose of 1 μg/kg.196 A 2012 study indicated that sufentanil simulates the IPost phenomenon at a dose of 1 μg/kg.197 It is known that sufentanil is also a selective agonist of μ OR.198 A further increase in the dose of this opioid did not lead to an enhancement of the infarct‐reducing effect.197 These data were confirmed in a later paper by the same group.199 Unfortunately, these researchers did not test OR antagonists, therefore, it remains unclear whether the cardioprotective effect of sufentanil is depended upon μ OR activation. Most recently, in the experiments on isolated perfused rat heart, the nonselective OR agonist remifentanil at reperfusion was protective.200 The infarct‐reducing effect of this opioid was eliminated by naloxone but the investigators did not use any of the selective OR antagonists.
Thus, the aforementioned studies provide ample evidence that activation of δ and κ1 OR can postcondition the heart. It remains unclear whether agonists of μ OR and ORL1 are also protective at the time of reperfusion.
9. LOCALIZATION OF OPIOID RECEPTORS THAT PROTECT THE HEART FROM I/R
Studies on the isolated heart seem to indicate that the infarct‐limiting effect of opioids is associated with the occupancy of the cardiac ORs. However, one should pay attention to two facts: (i) most studies have used OR ligands that penetrate the BBB, and (ii) in some studies the OR agonists were used at very large doses.81, 91, 187 For example, TAN‐67 was used at a dose of 10 mg/kg.91 But according to Knapp et al. the K i of TAN‐67 for δ OR is 0.65 nmol.201 For comparison, the K i of morphine against δ OR is 49 nmol.135 One can assume that in order to limit the size of myocardial infarction, a larger dose of morphine would be required. However, it has been demonstrated that morphine is protective at a dose of only 0.3 mg/kg in rats.9 An interesting possible explanation of this paradox could be that TAN‐67 activates a central δ OR that remotely increases cardiac tolerance to I/R via neural pathways and the low penetration of the BBB for TAN‐67 requires a higher dose.
There is a direct evidence of participation of central ORs in cardioprotection. A rat study with 30‐min coronary artery occlusion and 90 min reperfusion showed that intrathecal administration of morphine (0.3 μg/kg) for 20 min prior to ischemia promotes a decrease in the IS/AAR.202 In a similar study in 2009, intrathecal pretreatment with morphine again protected rat hearts in a dose‐dependent manner.203 Intrathecal administration of CTOP, naltrindole, or nor‐binaltorphimine abolished the infarct‐sparing effect and the authors concluded that all three (μ, δ, and κ) ORs are involved in the cardioprotective effect of morphine.203 The infarct‐reducing effect of morphine during intrathecal administration was confirmed in 2010.204 A cardioprotective effect of morphine was seen when it was infused for the 5 min prior to reperfusion and could be blocked by inhibition of μ, δ, or κ OR.205
In a study in rats with coronary artery occlusion/reperfusion, morphine was administered intravenously at a dose of 0.3 mg/kg.84 Naloxone methiodide, which does not cross the BBB, was administered prior to morphine injection intravenously or intrathecally at a dose of 20 mg/kg or 20 μg/kg.84, 206 Regardless of the route of administration, naloxone methiodide abolished the infarct‐sparing effect of morphine. The authors concluded that morphine protected through both central and peripheral ORs.
In 2012, it was shown that intrathecal administration of morphine to rats decreased the IS/AAR by twofold and the autonomic ganglion blocker hexamethonium completely abolished the protection.207 They concluded that the cardioprotective effect of morphine was mediated via central OR stimulation and signaling through the autonomic nervous system. In 2014, it was found that intrathecal administration of μ OR agonist fentanyl evokes a decrease in the IS/AAR.208 These data also indicate that the infarct‐reducing effect of opioids following intravenous administration may not only be a consequence of activation of peripheral but also of central ORs. The infarct‐limiting effect of opioid peptide EP94 occurred after blockade of peripheral OR with naloxone methiodide but disappeared after blocking peripheral and central ORs with naloxone.129 These authors concluded that the infarct‐reducing effect of EP94 is mediated via central OR activation. This result was surprising because opioid peptides usually penetrate the BBB poorly. For example, the opioid peptide dalargin exerts central effect only at a dose of 500 μg/kg.209 But Gross et al. used EP94 at a dose of 1 μg/kg.129
Thus, central OR stimulation clearly can increase cardiac tolerance to I/R. On the other hand, there is ample data with isolated hearts that cardiac OR can also protect the heart. It remains unclear, therefore, to what extent the infarct‐limiting effect of opioids during intravenous administration is mediated via central OR activation.
10. EFFECT OF OPIOIDS ON RECOVERY OF CARDIAC CONTRACTILITY DURING REPERFUSION
The above studies primarily concentrated on myocardial necrosis as the endpoint. Cardiac injury also manifests itself as a reduction in postreperfusion cardiac contractility. That reduction can be from loss of muscle to necrosis or it can be due to stunning, which is a transient loss of contractility following I/R. Preservation of mechanical function after I/R is paramount in the setting of cardiac surgery. Therefore, some studies used cardiac contractility as their endpoint rather than infarction.
A. μ OR
We found that intravenous administration of the μ OR‐selective agonists DALDA (0.1 mg/kg) or DAMGO (1 mg/kg) for 15 min prior to heart isolation promotes better recovery of ventricular developed pressure (LVDP) after I/R in the isolated rat heart. The μ OR‐selective antagonist CTAP (0.1 mg/kg) completely abolished DAMGO's protective effect. In contrast, perfusion of the isolated rat heart with DAMGO (0.1 mg/L or 195 nM) for 10 min prior to ischemia did not improve recovery of function.127 Only activation of the μ OR in vivo preserves postischemic contractility ex vivo. It is known that K i of DAMGO for μ OR is 1.23110 or 27 nmol.210 Therefore, we cannot explain an absence of inotropic effect of DAMGO ex vivo by a too low concentration of peptide. The protective effect of the μ OR agonist must be dependent upon μ OR activation somewhere outside the heart.47, 64, 211
B. κ OR versus δ OR
In a study on isolated rat heart, it was seen that perfusion with 200 μM DADLE prior to hypothermic cardiac arrest decreases the postischemic rise in end diastolic pressure (EDP) but not the decline in left ventricular developed pressure (LVDP).92 In 1999, Benedict et al. subjected the isolated rabbit heart to cardioplegic arrest (2 h of 34°C ischemia) followed by reperfusion. Perfusion of the isolated heart with morphine prior to ischemia promotes an increase in contractility during reperfusion.212, 213 The selective μ OR agonist fentanyl198 did not have a similar effect. Consequently, it may be concluded that positive inotropic effect of morphine was depended upon δ or κ OR activation. The British physiologists Kato and Foex subjected the isolated perfused rat heart to 30‐min global ischemia and 60‐min reperfusion. The heart was perfused with the μ OR agonist fentanyl (740 nM). Fentanyl increased LVDP, the rate of contraction, and the rate of relaxation of heart during reperfusion and pretreatment with naloxone abolished fentanyl's protective effect. These authors concluded that the inotropic effect of fentanyl was dependent upon δ OR activation.214 Kato and Foex gave fentanyl at a concentration sufficient to occupancy of μ, δ, and κ OR.214 Therefore, in our opinion, the presented data do not allow one to make a conclusion that the protective effect of fentanyl is mediated via δ OR stimulation. To further complicate the issue, the same authors published a paper in 2000, which reported that there was no improvement of contractility in the reperfusion period after 740 nM fentanyl.215 It is unclear, which study is correct.
Exposing the isolated rat heart to the selective κ1 OR agonist U50,488 (1 μM) for 2 min starting 10 min prior to global I/R promoted an increase in LVDP in the reperfusion period.216 The κ1 OR agonist had no effect on the EDP when given only during reperfusion. The inotropic effect of U50,488 was blocked by pretreatment with the selective κ OR antagonist nor‐binaltorphimine (1 μM during 4 min). These authors concluded that the protective effect of U50,488 was dependent upon κ OR activation. Their work could be criticized because they did not use U50,488 and nor‐binaltorphimine at receptor‐selective doses. The K i of U50,488 for κ1 OR is 0.89 nmol217 and the K i of nor‐binaltorphimine for κ OR is 0.18 nmol.218 Nor‐binaltorphimine at the final concentration of 100 nmol will also inhibit δ OR.219 In 2001, Genade et al. found that perfusion of the isolated heart with 10 nmol DADLE prior to ischemia improved mechanical function after reperfusion.220 Since DADLE at 10 nmol should only interact with δ OR,221 it may be assumed that the inotropic effect of DADLE was dependent upon δ OR activation.
We perfused isolated rabbit heart with 2 mM DADLE for 15 min before cardioplegic arrest and a 2‐h global ischemia followed by reperfusion.222 DADLE promoted an increase in LVDP over those hearts that were subjected to only cardioplegia.222 DADLE at the concentration of 2 mM activates all ORs.221, 223 Therefore, it is not clear what OR subtype was involved in the protective effect of DADLE. We continued this study with swine hearts. After pretreatment with DADLE (1 mg/kg intravenously), morphine (1 mg/kg intravenously), or saline, hearts were excised and kept for 75 min at 4⁰C, then reperfused them in a four‐chamber isolated working heart apparatus.224 We found that pretreatment with either DADLE or morphine promoted an increase in cardiac output during reperfusion. Since neither DADLE nor morphine are the δ OR‐selective agonists, it remains unclear what OR subtype was involved. We suspect that the protective effect of DADLE was dependent upon μ OR stimulation as noted in another of our studies.127 Similar data were obtained by Shinmura et al. They injected the δ OR‐selective agonist BW‐373U86 (1 mg/kg) into rats subcutaneously either for 1‐ or 24‐hr before the heart isolation. Isolated perfused rat hearts were subjected to 20 min of global ischemia followed by 20 min of reperfusion. Pretreatment with BW‐373U86 improved LVDP during reperfusion.225 Such evidence indicated that BW‐373U86 mimics both preconditioning and delayed preconditioning. It was not determined as to what OR subtype(s) were involved.
In 2002, Wu et al. subjected isolated perfused rat hearts to 30‐min global ischemia and 2‐h reperfusion. Hearts were perfused with 100 nM [Dmt1]DALDA or 1 μM morphine for 30 min and then subjected to 30‐min global ischemia. Reperfusion was performed using the same solutions. Both opioids increased contractile force during reperfusion over that seen with buffer only. The protection was present even when hearts were only perfused with [Dmt1]DALDA during reperfusion, whereas reperfusion with morphine only during reperfusion had no effect on the contractility.226 It is known that the peptide [Dmt1]DALDA is an agonist of μ and κ OR.227 Therefore, it remains open as to what OR was involved.
Peart and Gross presented evidence that either δ or κ OR stimulation improves cardiac function during reperfusion.141 Isolated murine heart was subjected to 20 min global ischemia followed by 45 min reperfusion. The OR agonists were infused for 10 min prior to ischemia, and then throughout reperfusion. Infusion of 10 μM morphine induced an improvement in postischemic recovery. Infusion with the selective δ OR agonist BW373U86 (1 μM) also improved recovery of LVDP. Pretreatment with the selective δ1 OR antagonist BNTX (1 μM) completely abolished this effect of BW373U86. Infusion of the selective κ1 OR agonist U50,488 (1 μM) produced a marked improvement in contractile recovery.141 This effect was blocked by the selective κ OR antagonist nor‐binaltorphimine (1 μM).
Gross's group showed that pretreatment with the selective δ OR agonist DPDPE (1 μM) also improves mechanical recovery of murine hearts following ischemia.228 In another study, cardioplegic arrest during global ischemia (2 hr at 34°C) was induced and followed by reperfusion. Hearts that were pretreated with either the preferential δ OR agonist DADLE or the κ OR agonist U50,488 demonstrated significantly improved functional recovery versus controls. The selective μ OR agonist fentanyl had no effect on recovery.229 Selective antagonists were not tested. An improvement of contractility during reperfusion after U50,488 was confirmed in isolated rat hearts.230 This effect was abolished after pretreatment with nor‐binaltorphimine indicating the protective effect of U50,488 is mediated via κ OR occupancy. Perfusion of the isolated rat heart with a solution containing the nonselective κ OR agonist pentazocine before or after 15‐min global ischemia improved cardiac contractility during reperfusion.231 These authors did not test with the OR antagonists. It was also shown that preliminary perfusion of the isolated heart with 1 μM morphine for 15 min before global ischemia promoted an increase in LVDP in the reperfusion period.161 These authors also did not test any OR antagonist. It should be noted that some investigators did not find a positive effect of morphine or U50,488 on cardiac contractility although they did decrease infarct size.194, 232
11. WORSENING OF POSTISCHEMIC MECHANICAL RECOVERY BY OPIOID LIGANDS
In the above studies, we presented data that the OR agonist can prevent an appearance of reperfusion contractile dysfunction. However, there are reports that some opioids can also exacerbate contractile dysfunction. The κ OR agonist bremazocine exacerbates reperfusion contractile dysfunction of the isolated heart.142 It is known that bremazocine is a potent κ2 OR agonist.143 Therefore, the above‐presented data on the effects of the κ1 OR agonist U50,488 and bremazocine do not contradict each other.
We observed that intravenous administration of the δ1 OR‐selective agonist DPDPE (0.1 or 0.5 mg/kg) 15 min prior to the heart isolation exacerbates reperfusion contractile dysfunction.112 If we added 0.1 or 0.5 mg/L DPDPE (154 or 771 nM) 15 min before global ischemia (45 min) and reperfusion (30 min), we also observed exacerbation of contractile dysfunction. DPDPE peptide can interact with only δ1 OR at the final concentration of 154 nM.198 Pretreatment with the selective δ OR antagonist naltrindole (1 nM) completely abolished the negative inotropic effect of DPDPE (154 nM).106 We later found that the selective δ1 OR agonist TAN‐67 (178 nM) also exacerbates dysfunction during reperfusion.103 Pretreatment with the selective δ OR antagonist naltrindole (1 nM) abolished this effect of TAN‐67. Our above result is drastically different from the data of Gross's group141, 228 where they generally found protection from δ1 OR agonists. It worth mentioning that their schedule of drug administration was quite different from that used in the above studies, however.
In most of the above studies, the ischemic period was long enough to cause some necrosis of the heart. In those studies, the postischemic recovery is influenced by a combination of stunning and infarction; so it is not clear which was contributing to an enhanced postischemic improvement in mechanical function. This is important in that the mechanisms of the two forms of injury differ drastically. In 2006, Grosse Hartlage et al. employed a pure stunning model where a coronary branch of a chronically instrumented dog is given a 10‐min coronary occlusion, which is too short to cause any infarction but does depress postischemic function. Function completely recovers spontaneously in a day proving that the segment was only stunned. They gave the selective κ OR receptor antagonist nor‐binaltorphimine (2.5 mg/kg intravenously). Pretreatment with the κ OR blocker prevented the decrease in ventricular wall function after ischemia. They found evidence that the endogenous opioid dynorphin was elevated in the plasma after the ischemic insult and concluded that this opioid was exacerbating the dysfunction in the untreated dogs.233 These data contradict the abovementioned data on positive inotropic effect of the κ OR agonist U50,488 during reperfusion141, 230 but those studies used isolated hearts with long ischemic periods where the agonist was confined to the pretreatment period. We found that perfusion of the isolated rat heart with solution containing U50,488 (0.1 μM) starting 10 min before global ischemia (45 min) decreases creatine kinase release during reperfusion but depresses the recovery of contractile dysfunction.120 If we used U50,488 at the final concentration of 1 μM, the cardioprotective effect disappeared but the negative inotropic effect was enhanced.
Thus, results of studies of the inotropic effects of opioids on cardiac stunning are very contradictory. Some studies indicated that pretreatment with opioids improves cardiac contractility in reperfusion period.92, 127, 161, 213, 214, 216, 220, 222, 225, 226, 230, 231 Other studies showed that pretreatment with opioids exacerbate contractile dysfunction.103, 106, 112, 120, 142 Other investigators could not find any alteration of postischemic recovery after pretreatment with the OR agonists.127, 194, 214, 232 Much of this confusion no doubt arises from heterogeneity in the models (isolated vs. in situ), the schedule of drug administration (pretreatment vs. post treatment vs. continuous treatment), and the type of injury (infarction vs. stunning). Therefore, the resolution of possible inotropic effects of opioids during myocardial reperfusion remains to be determined.
12. ANTIARRHYTHMIC EFFECT OF THE OPIOID RECEPTOR LIGANDS
The most frequent causes of death from myocardial infarction are cardiogenic shock (52%), arrhythmias (25%), thromboembolism of the pulmonary artery (10%), and rupture of the left ventricle (5%).234 These findings indicate that an antiarrhythmic drug could dramatically reduce mortality in this population. Opioids are potential candidates for developing such drugs.
The first report that an OR agonist has an antiarrhythmic effect was with meptazinol during coronary artery occlusion in rats in 1983.235 In 1989, it was shown that the selective μ OR agonist fentanyl (60 μg/kg intravenously) increased the ventricular fibrillation threshold (VFT) in dogs with coronary artery occlusion.236 The μ and κ OR agonist buprenorphine had the same effect.236 The antifibrillatory activity of the μ OR agonists fentanyl, sufentanil, and carfentanil in dogs with coronary artery occlusion was demonstrated by Hess et al. in 1989.237 Clinical observations established that fentanyl (60 μg/kg intravenously) could prevent the appearance of intraoperative ventricular fibrillation during cardiosurgery intervention in neonates.238 These studies indicate that opiates can increase cardiac tolerance to the arrhythmogenic effect of I/R.
Unfortunately, these historical studies were performed before highly selective OR antagonists were widely available; so none of these publications contained this approach aimed to confirm a receptor‐mediated effect and identify which subtype was responsible. Morphine is a μ OR‐selective agonist as is fentanyl.198 However, it was later shown that the cardioprotective effects of morphine10 and fentanyl214 are actually dependent upon δ OR stimulation. Furthermore, both narcotic analgesics easily penetrate through the BBB. Therefore, it was unclear whether their antiarrhythmic effect was dependent upon the central or peripheral OR occupancy.
A. μ OR agonists
In order to find out whether the peripheral ORs are involved in the arrhythmogenesis, we used D‐Ala2,Leu5,Arg6‐enkephalin (dalargin). This compound can penetrate the BBB at a dose of 0.5 mg/kg and higher.209 We found that intravenous administration of dalargin (0.1 mg/kg) decreases the incidence of ventricular fibrillation during coronary artery occlusion in rats.239 Other investigators confirmed our data in experiments on cats.240 According to the data of Grekova et al. dalargin (0.1 mg/kg) exhibits an antiarrhythmic effect when administered intravenously to dogs 5 min prior to coronary artery occlusion.241 Dalargin can prevent both ischemic and reperfusion arrhythmias. However, this opioid peptide was ineffective if it was administered after coronary artery ligation.241 Dalargin not only prevented the appearance of arrhythmias during ischemia, it also evoked an increase in the VFT in rats with postinfarction fibrosis.242 We therefore reasoned that the antiarrhythmic effect of dalargin is mediated via peripheral OR activation. However, it is still unknown what OR subtypes are involved in antiarrhythmic effect of dalargin because this peptide is a μ and δ OR agonist.243, 244
We found that the injection of the selective μ OR agonist DALDA prior to a 10‐min coronary artery occlusion and reperfusion in rats does not affect the incidence of ventricular arrhythmias.138 Nor could we find an antiarrhythmic effect of the selective μ OR agonist DMGO (150 or 1500 nmol/kg) or the μ OR agonist dermorphin H (150 nmol/kg) during coronary artery occlusion.245 In experiments with postinfarction cardiac fibrosis we obtained data that the nonselective μ OR agonists morphine and dalargin and the μ OR agonist DALDA increase the threshold for fibrillation.242, 246, 247 The antifibrillatory effect of DALDA (0.1 mg/kg) was not present after inhibition of the peripheral ORs with naloxone methiodide.247 We theorize that the antifibrillatory effect of DALDA is depended upon peripheral OR occupancy. Blockade with the μ OR‐selective antagonist CTAP (0.5 mg/kg) also abolished the antifibrillatory effect of DALDA.247 Therefore, it can be argued that DALDA induces an increase in cardiac electrical stability via the peripheral μ OR activation. Thus, peripheral μ OR activation increases cardiac electrical stability in animals with postinfarction cardiac fibrosis but, based on our data, μ OR agonists do not suppress acute I/R arrhythmias.
B. δ OR and Arrhythmias from Acute I/R
In more recent experiments with the selective δ1 OR agonists TAN‐67 (0.08 mg/kg), DPDPE (0.1, 0.2 and 0.5 mg/kg), and the selective peptide agonist DSLET (0.11 mg/kg), we found that these ligands after intravenous administration had no effect on the incidence of ventricular arrhythmias during a 10‐min coronary artery occlusion and reperfusion in rats.138 However, we found that selective δ2 OR activation with intravenous administration of deltorphin II (0.12 mg/kg or 150 nmol/kg) did reduce arrhythmias from I/R.138 We suspect that the strong antiarrhythmic effect of deltorphin II and the absence thereof, in DSLET is a consequence of different affinities of these ligands to δ OR. Deltorphin II exceeds DSLET in two‐fold in its affinity for δ OR.248 Therefore, it is not surprising that when using both peptides in equimolar doses only deltorphin II was antiarrhythmic. Recently, we have shown that the selective δ2 OR agonist deltorphin II (150 nmol/kg), the putative δ2‐selective agonist deltorphin Dvar (150 nmol/kg) and deltorphin E (150 nmol/kg) had antiarrhythmic properties during coronary artery occlusion/reperfusion.245 We performed further studies on Deltorphin II, which exerted the most pronounced antiarrhythmic effect. Pretreatment with the nonselective OR antagonist naltrexone (5 mg/kg), the nonselective peripheral OR antagonist naloxone methiodide (5 mg/kg), or the selective δ2 OR antagonist naltriben (0.3 mg/kg) completely abolished deltorphin II's antiarrhythmic effect. But pretreatment with the δ1‐selective antagonist BNTX (0.7 mg/kg) did not abrogate deltorphin II's antiarrhythmic effect. Therefore, we concluded that peripheral δ2 OR stimulation enhances cardiac tolerance to arrhythmogenic impact of ischemia and reperfusion.245
We found that intravenous administration of the δ1 OR‐selective peptide DPDPE at a dose of 150 and 1500 nmol/kg had no effect on the incidence of ischemic and reperfusion ventricular arrhythmias in rats.245 Most of the opioid peptides poorly penetrate the BBB.23, 24, 209 We also tested the δ1 OR‐selective agonist TAN‐67 and found that this opioid also was not antiarrhythmogenic.249 Therefore, we concluded that peripheral δ1 OR agonists are not antiarrhythmogenic. However, Fryer et al. demonstrated an antiarrhythmic effect of TAN‐67 in open‐chest rats.102 Unlike our study, they gave TAN‐67 at a dose of 10 mg/kg (125‐fold higher the 0.08 mg/kg dose used by us). The higher dose would have penetrated the BBB. The antiarrhythmic effect of TAN‐67 was no longer seen after selective blocking of δ OR.102 We suggest that the antiarrhythmic effect of TAN‐67 is mediated via central OR stimulation. We conclude that peripheral δ2 OR activation increases cardiac tolerance to the arrhythmogenic effect of I/R while occupancy of the central δ1 OR seems to have the same effect.
C. δ OR and Arrhythmias from Postinfarction Cardiac Fibrosis
The picture is different with arrhythmias generated by postinfarction cardiac fibrosis. Intravenous administration of the selective δ 1 OR agonist DPDPE (0.1 mg/kg) increased the fibrillation threshold by 36% in rats with postinfarction cardiac fibrosis.250 The antifibrillatory effect of DPDPE was lost after blockade of peripheral OR with naloxone methiodide or after selective inhibition of δ OR with ICI 174,864. The selective δ 2 OR agonist DSLET (0.5 mg/kg) did not exert any antifibrillatory effect. In this study, we did not test selective δ 1 and δ 2 OR antagonists. Nevertheless, we suspect that the antiarrhythmic effect depended upon peripheral δ 1 OR stimulation.
13. CONTROVERSY OVER κ OR AND ARRHYTHMIAS
In 1992, it was found that the selective κ OR agonist U‐50,488 (7.5 mg/kg, intravenously) enhanced cardiac tolerance to the arrhythmias from a 30‐min coronary artery occlusion. Because pretreatment with naloxone (2.5 mg/kg) did not eliminate U‐50,488's effect, these authors concluded that the protection was not dependent upon activation of κ OR.251 They proposed that the antiarrhythmic effect was caused by an off‐target blockade of fast Na+ channels because after injection of U‐50,488 they observed bradycardia and prolongation of the QRS duration (effects that are typical for I class antiarrhythmic drugs).251 However, our results did not concur with these data. They gave U‐50,488 at a dose of 7.5 mg/kg and naloxone was administered at a dose of only 2.5 mg/kg, a dose that is not high enough for inhibition of κ OR.198 We found that a lower dose of U‐50,488 (1 mg/kg intravenously) prevents ventricular arrhythmias during coronary artery occlusion and reperfusion.252 We believe that the dose of U‐50488 (7.5 mg/kg) used by Pugsley et al. was too high, causing it to exhibit nonreceptor effects.
The above researchers tried to compare antiarrhythmic properties of the κ OR agonist PD 129290 and its R,R (+)‐enantiomer that has a low affinity to κ OR.253 Both enantiomers at a dose of 3 mg/kg decreased arrhythmias from a 30‐min coronary artery occlusion. Since, naloxone at a dose of 2.5 mg/kg did not abolish this effect, these authors concluded again that the antiarrhythmic effect of both enantiomers was independent of κ OR. Furthermore, both enantiomers increased the QRS duration and in isolated cardiomyocytes, both compounds did inhibit Na+ current.253 They again concluded that the antiarrhythmic effect of these opioids is due to blocking Na+ channels. In a study with isolated cardiomyocytes, the same research group found that U‐50488 and PD 129290 also inhibit Na+ current. The κ OR antagonist itself MR2266 did not produce any change in the Na+ or K+ currents, nor did it alter the channel blocking properties of U‐50,488.254 The electrophysiological effects of U‐50,488 were compared with those of the class Ib antiarrhythmic agent lidocaine in rat heart and the sodium currents expressed in Xenopus laevis oocytes by using two‐electrode voltage clamp.255 Both U‐50,488H and lidocaine produced a concentration‐dependent tonic block of Na+ current but U‐50,488H was approximately fourfold more potent than lidocaine. These authors maintain that the antiarrhythmic properties of the κ OR agonists do not depend on OR activation and is an outcome of nonspecific Na+ channel blocking.253, 254, 255
We continued the study using U‐50488 enantiomers, which differ in affinity to κ1 OR.217 It appeared that (–)‐trans‐(1S,2S)‐U‐50,488 (1 mg/kg intravenously) with high affinity to κ1 OR can reduce arrhythmias from 10 min of coronary artery occlusion and reperfusion in rats.252, 256, 257 An enantiomer (+)‐trans‐(1R,2R)‐U‐50,488 with low affinity to κ1 OR did not exert similar effect.252, 256, 257 Others have shown that the preferential κ1 OR agonist dynorphin A1‐13 at a dose of 40 μg/kg intravenously also exerts antiarrhythmic effect in cats with coronary artery occlusion.258 Furthermore, we established that pretreatment with the selective κ OR antagonist nor‐binaltorphimine (9 mg/kg, intravenously) completely abolished antiarrhythmic effect of (–)‐U‐50,488.256 Pretreatment with the κ2 OR antagonist quadazocine (3 mg/kg, intravenously) did not alter antiarrhythmic effect of (–)‐U‐50,488.256 The selective κ2 OR agonist GR‐89696 (25 μg/kg, intravenously) also had no effect on the reperfusion arrhythmias in rats.259 However, inhibition of peripheral ORs with naloxone methiodide (5 mg/kg, intravenously) completely abolished antiarrhythmic effect of (–)‐U‐50,488.259
Comparison of the above observations convinced us that peripheral κ1 OR activation can enhance cardiac tolerance to I/R‐generated arrhythmias. The basis for this assertion are our data that the selective κ2 OR agonist GR‐89696 does not exhibit antiarrhythmic properties and that the κ2 OR antagonist quadazocine did not abolish the antiarrhythmic effect of (–)‐U‐50,488.259 But pretreatment with naloxone methiodide or nor‐binaltorphimine does abolish the antiarrhythmic effect of (–)‐U‐50,488.259 At high concentration, (–)‐U‐50,488 clearly does block sodium channels. But at a low dose, the antiarrhythmic effect of U‐50,488 is mediated via κ1 OR occupancy alone.
We determined that intravenous administration of the selective ORL1 agonist nociceptin at a dose of 220 or 1500 nmol/kg had no effect on the incidence of ischemic and reperfusion‐induced ventricular arrhythmias in vivo.245 However, nociceptin is not resistant to enzymatic hydrolysis. Therefore, we cannot completely exclude the possibility that a selective ORL1 agonist will exhibit antiarrhythmic properties.
14. ARRHYTHMOGENIC AND PROARRHYTHMIC EFFECTS OF OPIOIDS
It has been shown that injection of the nonselective OR agonist β‐endorphin into perfusion solution caused only atrial fibrillation and atrioventricular block in isolated rat heart.260, 261 It should be noted that in vivo β‐endorphin exhibits antiarrhythmic properties in anesthetized cats with coronary artery occlusion.262 In 1987, Lee and Wong demonstrated that injection of the preferential κ OR agonist dynorphin1‐13 (20 μg/heart) caused both atrial and ventricular arrhythmias in isolated rat heart.263 This effect was antagonized by naloxone. In 1990, Wong et al. published results of their experiments on isolated Langendorff‐perfused rat heart.264 The OR agonists and antagonists were injected directly into the aorta through cannulas. The OR antagonists were administered 1 min before the administration of OR agonist or 20‐min global ischemia and 60‐min reperfusion. The selective μ OR agonist DAMGO evoked atrial arrhythmias and at a higher dose caused frequent premature ventricular contractions (PVC).264 The selective κ OR agonist U50488 caused both atrial and ventricular arrhythmias. At a high dose (132 nmol/heart), this opioid induced frequent PVC and ventricular tachycardia. The δ OR agonists DPDPE and DADLE evoked only atrial arrhythmias. The arrhythmogenic effects of U50488 were attenuated by pretreatment with the κ OR antagonist MR 2266 in a dose‐related manner whilst the proarrhythmic effect of DAMGO was abolished by the preferential μ OR antagonist naloxone. Naloxone itself exhibited a weak antiarrhythmic effect manifested in prevention of only ventricular tachycardia during reperfusion. Authors concluded that the cardiac κ OR are the most likely receptors involved in arrhythmogenesis during ischemia and reperfusion.264 The limitation of this study was the fact that the bolus administration of opioids did not allow them to compare the concentrations of the opioids with their respective Kds and Kis. Consequently, this work does not allow evaluating the role of OR subtypes in the arrhythmogenic effects of opioids.
MR 2266 also exhibits an antiarrhythmic effect. Therefore, the combined use of MR 2266 and U50488 could eliminate the arrhythmogenic effect of U50488 regardless of OR blockade. Later it was shown that intravenous administration of dynorphin (300 nmol/kg) diminished the arrhythmogenic effect of coronary artery occlusion in rats.265 This effect was abolished by pretreatment with naloxone (1 mg/kg). However, since naloxone itself exhibits an antiarrhythmic effect, it remains unclear whether the inhibition of the proarrhythmic effect of dynorphin by naloxone occurred due to the blockade of ORs or the antiarrhythmic effect of naloxone overshadowed the proarrhythmic effect of dynorphin. Interestingly, according to others, dynorphin A1‐13 (25 nmol/kg) prevents the occurrence of ventricular fibrillation in anesthetized cats subjected to occlusion of the left coronary artery.266 In 2003, Coles et al. published a comparative study of the cardiovascular effects of opioids in pigs with coronary artery occlusion (45 min) and reperfusion (3 hr).267 They found that the preferential δ OR agonist DADLE (1 mg/kg) and the preferential κ OR agonist pentazocine (5 mg/kg) aggravated the arrhythmogenic effect of coronary artery occlusion. The selective κ OR antagonist nor‐binaltorphimine (1.5 mg/kg) exhibited the same effect. However, pretreatment with nor‐binaltorphimine completely abolished the proarrhythmic effect of both DADLE and pentazocine. The authors concluded that κ OR activation during ischemia exhibits proarrhythmic effect in pigs.267 Indeed, it is known that DADLE can activate κ ORs in isolated hearts and we cannot exclude the possibility that its proarrhythmic effect is mediated by activation of these receptors.143 However, we would like to draw the readers’ attention to the following paradox: the κ OR antagonist nor‐binaltorphimine also had a proarrhythmic effect that disappeared when treatment with the κ OR antagonist was combined with the κ OR agonist during ischemia. It is worth mentioning that the authors used high doses of the OR agonists. Meanwhile it is well known that opioid peptides at high concentration may interact with non‐ORs.268, 269, 270 Therefore, there is a possibility that the toxic effects of DALDA and pentazocine are unrelated to the ORs but mediated via stimulation of other receptors.
It is worth mentioning that we have never observed an arrhythmogenic or proarrhythmic effect of opioids administered intravenously to rats or in the experiments on isolated perfused rat heart. We noticed a proarrhythmic effect of DADLE only in pigs during coronary artery occlusion at a high dose of 1 mg/kg.101 However, we do not exclude the possibility that opioids may have arrhythmogenic and proarrhythmic effects associated with the activation of central ORs when used at a high dose. We have established that the κ1 OR agonist U‐50488, the κ OR agonist [D‐Ala2]‐Dynorphin A(1‐13) and the preferential κ2 OR agonist (–)‐bremazocine administered intracerebroventricularly potentiate the arrhythmogenic effect of intravenous epinephrine.271, 272 Pretreatment with N‐cholinergic receptor antagonist hexamethonium prevented proarrhythmic effects of the intracerebroventricular administration of U50488 and dynorphin.271 In contrast, intravenous administration of the preferential κ2 OR agonist (–)‐bremazocine and intraperitoneal injection of the selective κ OR agonist spiradoline blunted the arrhythmogenic impact of epinephrine.272 This effect was abolished by pretreatment with nor‐binaltorphimine but not hexamethonium or atropine. These data indicate that stimulation of the central κ OR may promote a proarrhythmic effect mediated by the autonomic nervous system. Stimulation of peripheral κ OR may have an antiarrhythmic effect that is independent of the autonomic regulation of heart rhythm.
Thus, we do not exclude the possibility that high doses of opioids may have proarrhythmic and arrhythmogenic effects in humans and animals associated with the activation of non‐ORs or activation of central κ ORs. There is also a possibility that activation of a cardiac κ OR subtype can also contribute to the appearance of ventricular arrhythmias.
15. ANTIARRHYTHMIC ACTIONS OF OPIOID ANTAGONISTS
There are reports that the OR antagonists can also exhibit antiarrhythmic properties during I/R of heart.273, 274, 275, 276, 277 These investigations showed that intravenous administration of naloxone (1 mg/kg) before coronary artery occlusion in anaesthetized dogs reduced the incidence and severity of cardiac arrhythmias during coronary artery occlusion and reperfusion.273 Studies also indicate that pretreatment with the κ2 OR antagonist quadazocine (3 mg/kg) or the OR antagonist (–)‐Mr 1452 (4 mg/kg) prevents the appearance of arrhythmias induced by coronary artery occlusion in rats.274 These investigators established that naloxone (0.5 mg/kg), the preferential κ OR antagonist Mr 2266 (4 mg/kg), and the nonselective OR antagonist MrZ 2593 (a quarternary complex of naloxone which does not readily cross the BBB at 1 mg/kg) all prevent the appearance of arrhythmias evoked with regional cardiac ischemia in rats.275 It has been reported that intravenous administration of the nonselective OR antagonist nalmephene (1 mg/kg) prevents reperfusion‐induced arrhythmias in dogs.276 There was also a report that pretreatment with naltrexone (2 mg/kg) or methylnaltrexone (2 mg/kg), a quaternary derivative of naltrexone that does not cross the BBB, prevents ventricular fibrillation induced with coronary artery occlusion in rabbits.277
Our studies found that the OR antagonists (naltrexone, naloxone methiodide, naltriben, BNTX, quadazocine, nor‐binaltorphimine, CTAP, β‐funaltrexamine, ICI‐174,864) had no effect on the incidence of ventricular arrhythmias during coronary artery occlusion/reperfusion in rats.256, 257, 259 It is unclear why our data contradict the data of the other researchers. It should be noted that many of the aforementioned studies used antagonists (Mr 1452, Mr 2266, nalmephene, methylnaltrexone) that are no longer used in OR studies because they exhibit OR independent effects. All our studies were performed with rats and perhaps there were also species differences in the response to the OR antagonist. The authors who found an antiarrhythmic effect of naloxone and naltrexone were performed in investigations with dogs and rabbits.273, 277 The nonselective OR agonist pentazocine reportedly prevented the appearance of ventricular reperfusion arrhythmias in isolated rat hearts.231
16. THE EFFECT OF COMORBIDITIES ON THE CARDIOVASCULAR EFFECTS OF OPIOIDS
Pathological processes may significantly change the cardiovascular system response to exogenous opioids. For example Bolte et al.278 found augmented negative inotropic and lusitropic response to administration of the selective δ OR and κ OR agonists in the failing hamster heart. However, Kasper et al. could not find any difference in the negative inotropic effect of κ OR agonist U‐50,488H on control and cardiomyopathic hamster cardiomyocytes.279 The inhibitory action of the selective κ OR agonist U50 488H on β‐adrenoceptor augmentation of voltage‐dependent [Ca2+]i transients in the isolated cardiomyocytes appeared to be significantly reduced in spontaneously hypertensive rats.280 In 2001, Pei et al. demonstrated that the effect of U50,488H on the [Ca2+]i transient in the isolated cardiomyocytes was significantly attenuated due to right ventricular hypertrophy induced by chronic hypoxia.281 The authors established that κ OR signaling was impaired in the hypertrophied cardiomyocytes due to a defect in the coupling between κ OR and PKC. It was also found that high fat‐induced obesity alters cardiovascular response to the administration of opioids in conscious rats.282
The pathological process itself may change the state of the endogenous opioid system. It has been shown that cardiomyopathy evoked an increase of the preproenkephalin A mRNA level in ventricles of hamsters.283 In spontaneously hypertensive rats, the heart content of dynorphin A was increased by 6.5‐fold compared to Wistar rats.284 Plasma β‐endorphin levels are also elevated in dogs with pacing‐induced congestive heart failure. Naloxone injection increased HR, mean aortic pressure, first derivative of left ventricular pressure and cardiac output in these dogs while in the intact animals, naloxone did not affect the hemodynamics.285 These data suggest the involvement of endogenous opioids in the pathogenesis of pacing‐induced congestive heart failure. This hypothesis can be supported by the data of Imai et al.286 They found that not only naloxone but also the selective δ OR antagonist ICI‐154,129 increased mean aortic pressure, cardiac output and positive first derivative of left ventricular pressure in dogs with artificially induced right heart failure. Constriction of the aorta induced an elevation of β‐endorphin level in blood plasma of rats.287 In 1995, Oldroyd et al. found that plasma β‐endorphin was increase by 29% in patients with acute heart failure and by 71% in patients with cardiogenic shock.288 However, the level of this opioid in plasma was decreased in spontaneously hypertensive hamsters.289 It was demonstrated that the decrease in left ventricular systolic pressure after administration of the κ OR agonist U50488H was attenuated in these hamsters.
These results show that pathological process can exert a significant effect on the endogenous opioid system. Can the accompanying pathological process change the cardioprotective effect of opioids? It has been demonstrated that aging does not alter a cardioprotective effect of BW373U86, a selective δ OR agonist.168 However, in 2007, Peart et al. reported that the selective δ OR agonist DPDPE only improves contractile recovery after reperfusion of isolated mouse hearts from young animals.228 They found that aging‐related loss of δ‐opioid‐mediated cardioprotection involves failure to activate p38 MAPK (mitogen‐activated protein kinase) and HSP27 (heat shock proteins). Gross's group demonstrated that morphine did not limit the infarct size in rats with streptozotocin‐induced diabetes. This lack of protective effect was associated with the loss of coupling between ORs and glycogen synthase kinase 3β (GSK‐3β).290 It was also shown that remifentanil reduced myocardial infarct size and prevented apoptosis of cardiomyocytes evoked by I/R in nondiabetic rats but not in rats with streptozotocin‐induced diabetes.89 Gross's group's data were confirmed by Chen et al.199 They demonstrated that sufentanil reduced myocardial infarct size in the nondiabetic rats, but not those with streptozotocin‐induced diabetes. The GSK‐3β inhibitor SB216763 reduced infarct size in both nondiabetic and diabetic rats. The authors concluded that absence of opioid‐induced tolerance of rat heart to reperfusion injury in diabetic animals is the result of impaired interaction of ORs and GSK‐3β.199
The results of these studies cannot be mechanically applied to the humans because the most common form of diabetes in humans is a type 2 diabetes mellitus characterized by insulin resistance. Streptozotocin causes damage to the β cells of Langerhans islets and leads to decreased insulin secretion. Therefore, streptozotocin‐induced diabetes is most similar to insulin‐dependent type 1 diabetes mellitus in humans. However, Tsang et al. showed in experiments using a rat model of type 2 diabetes mellitus (Goto‐Kakizaki rats) that diabetes depresses the PI3K/Akt pathway during IP causing loss of protection. But the elevated threshold for Akt phosphorylation and protection can be reached by simply increasing the number of IP cycles.291 Since this same signaling pathway is involved in the protective mechanism of opioids, as we discuss in the Section 18, it seems likely that an increasing the dose of an OR agonist may still be able to protect the diabetic heart against I/R injury. Taken together, the presented data suggest that diabetes and age may undermine the efficacy of opioid‐induced cardioprotective effects. It remains unknown whether atherosclerosis, arterial and pulmonary hypertension, myocardial hypertrophy, or heart failure might also alter their protection. The phenomenon of heart resistance to cardioprotective stimuli due to comorbid diseases is potentially a serious problem in the translation of cardioprotective interventions to clinical practice.292 However, this problem may be solved by either increasing the stimulus or by direct activation of the downstream components of the signaling pathways such as p38 MAPK/HSP27 or inhibition of GSK‐3β, which remain relatively intact in these conditions.
17. PROSPECTS FOR THE USE OF OPIOID RECEPTOR AGONISTS IN CARDIOLOGICAL PRACTICE
It has been found that the chronic administration of morphine increases cardiac tolerance to ischemia and reperfusion.293, 294, 295, 296, 297 The signaling mechanism of the cardioprotective effect of chronic morphine administration differs from that of acute administration of morphine.296 Chronic κ‐OR stimulation prevents isoprenaline‐induced cardiac hypertrophy and fibrosis.298 Unfortunately, morphine and heroin in chronic administration cause rapid formation of drug dependence.299 The formation of opioid dependence is associated with activation of central μ OR.300 The ability to form a dependence is much less pronounced in the δ OR or κ OR agonists.300 Therefore, δ OR and κ OR agonists are not on the DEA‐controlled substances list. However, since the κ OR agonists cause dysphoria, their indication for chronic use is also unlikely.301 In our opinion, the most promising agents for chronic use are peptide OR agonists, that poorly penetrate the blood–brain barrier.22, 23, 24 These include the selective μ OR agonist DALDA (NH2‐Tyr‐D‐Arg‐Phe‐Lys‐NH2)227 and nonselective μ OR and δ OR agonist dalargin (H‐Tyr‐D‐Ala‐Gly‐Phe‐Leu‐Arg‐OH).302 Our experiments have shown that dalargin exhibits antifibrillatory properties.239 In Russia, this drug is used to treat stomach ulcers.303 We have shown that a quaternary analogue of the κ opioid agonist U‐50488 is another opioid that does not penetrate the BBB and is capable of increasing cardiac tolerance to I/R.123 Therefore, we suggest that those opioids, which are not able to activate the central ORs after systemic administration, might be most amenable to chronic use as cardioprotectants in high‐risk cases.
Based on the data accumulated by our and Gross's group,129 the most promising compounds for development of drugs, which acutely increase the heart's tolerance to the detrimental effects of ischemia and reperfusion are deltorphin II, ICI 199,441 and Eribis peptide 94. Following clinical trials, these compounds or their analogs may find use in the treatment of acute myocardial infarction or in surgergical patients receiving coronary bypass.
18. SIGNALING MECHANISM OF THE CARDIOPROTECTIVE EFFECT OF OPIOIDS
The data described below are summarized in the signaling scheme for the cardioprotective effect of opioids presented in Figure 1. The phenomenon of IP was found to be triggered by activation of Gi/o‐coupled adenosine receptors that activate PKC and ultimately protects by inhibiting MPT early in reperfusion.304 Soon it was discovered that bradykinin and OR act in parallel with the adenosine receptors as all three are populated during a preconditioning ischemia. Occupation of any one receptor type can put the heart into a conditioned state. It is hypothesized that ORs couple with Gi/o proteins, which then inhibit adenylyl cyclase and activate phospholipase C, which, in turn, synthesizes diacylglycerols stimulating protein kinase C.248 Therefore, investigators have attempted to determine if the cardioprotective effect of opioids is associated with Gi/o protein activation. In a study with rats, it was shown that the Gi/o protein inhibitor pertussis toxin eliminated the infarct‐reducing effect of the δ1 OR agonist TAN‐67.91 Later experiments with isolated perfused murine heart demonstrated that pertussis toxin, which rybosylates the αi subunit of the Gi/o protein, eliminated the cardioprotective effect of morphine.296 Currently it is hypothesized that Gi/o proteins serve as an intermediary link between OR and the protein kinases that perform the protective signaling.
Figure 1.
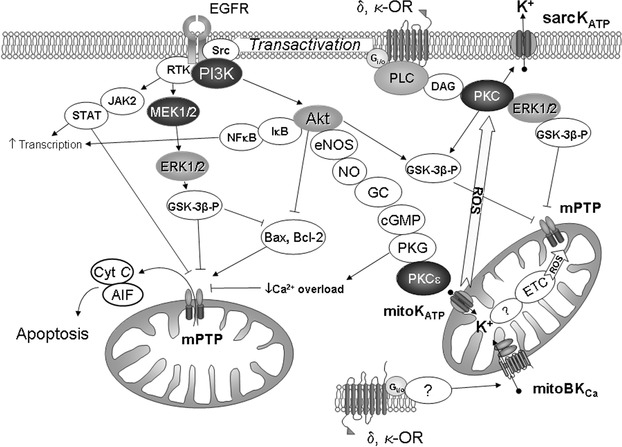
Proposed signaling scheme for the cardioprotective effect of opioids.OR, opioid receptor; PLC, phospholipase C; DAG, diacylglycerol; PKC, protein kinase C; Src, sarcoma tyrosine kinase; PI3K, phosphatidylinositol 3‐kinase; JAK2, Janus kinase 2; STAT, signal transducer and activator of transcription; Akt, kinase isolated from AKR thymoma cell line; MEK1/2, mitogen‐activated protein kinase kinase; ERK1/2, extracellular‐signal‐regulated kinase; NO, nitric oxide; NOS, NO‐synthase; GC, guanylyl cyclase; cGMP, cyclic guanosine monophosphate; PKG, protein kinase G; mitoKATP, mitochondrial ATP‐sensitive K+ channel; sarcKATP, sarcolemmal ATP‐sensitive K+ channel; GSK‐3β, glycogen synthase kinase‐3β; mPTP, mitochondrial permeability transition pore; EGFR, epidermal grows factor receptor; RTK, receptor tyrosine kinase; Cyt C, cytochrome C; AIF, apoptosis inducing factor; Bcl‐2, B‐cell lymphoma protein‐2; Bax, Bcl‐2‐associated X‐protein; ROS, reactive oxygen species; NFκB, nuclear factor κB; IκB, IκB kinase; mitoBKCa, mitochondrial big conductance Ca2+‐dependent K+ channel; ETC, electron transport chain.
A. OR Protect through PKC
In 1998, a study with isolated perfused rabbit heart it was shown that the protein kinase C (PKC) inhibitor chelerythrine abolishes the infarct‐sparing effect of morphine.81 In the following investigation performed with the isolated perfused rat heart it was found that pretreatment with chelerythrine eliminates the cardioprotective effects evoked by δ and κ1 OR in vitro stimulation.95, 105, 173, 215
In 2001, Gross's group sought to identify which PKC isoforms are involved in the infarct‐limiting effect of opioids.305 They established that the cardioprotective effect of TAN‐67 and DADLE did not occur after inhibition of all PKCs with chelerythrine and after pretreatment with the selective PKC‐δ inhibitor rottlerin. They concluded that the cardioprotective effect of δ OR agonists is mediated via PKC‐δ activation. This result surprised us because PKC δ activation and translocation to mitochondria also promotes cardiomyocyte apoptosis306 and inhibition of PKC δ enhances cardiac tolerance to reperfusion injury.307 Also preconditioning's protection of adult rabbit cardiomyocytes could be blocked by a PKC ε‐specific antagonist but not one for PKC δ.308 However, more recent studies revealed that rottlerin is not a specific PKC‐δ inhibitor.309 The key role of PKC in signaling mechanism of antinecrotic effect of opioids was confirmed in later works.110, 296, 310
In a 2001 study, isolated cardiomyocytes were assessed for apoptosis after exposing the cells to hypoxia (12 h) and reoxygenation (12 h). This study indicated that the selective δ agonist BW373U86 prevented apoptosis of cardiomyocytes. Pretreatment with Go‐6976, an inhibitor PKC‐α and PKC‐β, abolished this protective effect of BW373U86.158 This study showed the antiapoptotic effect of the δ OR agonist can be attributed to PKC‐α and PKC‐β activation.
A number of studies have demonstrated that PKC is involved in the antinecrotic and antiapoptotic effects of opioids. Debate still revolves around the question of which PKC isoforms are involved in the opioid‐induced enhancement of cardiac tolerance to I/R.
B. PI3 kinase, Akt, and MAPK—the RISK Pathway
Another set of kinases shown to be in the conditioning pathway are PI3 kinase, Akt, and p42/p44 MAPK. These kinases are collectively called the Reperfusion Injury Survival Kinase (RISK) pathway and are downstream of PKC and are involved in mediating the protection early in reperfusion.311 Isolated rabbit cardiomyocytes were subjected to hypoxia and reoxygenation and the endogenous μ and δ OR agonist met‐enkephalin reduced cardiomyocytes death. Pretreatment with the PI3 kinase inhibitor LY‐294002 abolished the cytoprotective effect.312 We have found that intravenous U‐50,488 decreases the IS/AAR in rats with coronary artery occlusion and the PI3 kinase inhibitor wortmannin abolished the infarct‐sparing effect.313 PI3 kinase phosphorylates membrane phosphoninositide at the 3 position and this product activates phosphoinositide‐dependent kinases (PDKs), which activates Akt by phosporylating it. Recently, it has been shown that the infarct‐limiting effect of sufentanil in vivo also was abolished by pretreatment with wortmannin.314
Gross's group found that intravenous administration of morphine (0.3 mg/kg) in rats induces an increase in the phosphorylation of cardiac Akt early in reperfusion.108, 116 The same group utilizing an isolated murine heart found that DPDPE evoked phosphorylation of Akt.228 These data were confirmed by Huang et al.52 It was also shown that the phosphorylation of Akt was involved in the cardioprotective effect of remifentanil162 and morphine.315 Based on the aforementioned studies it appears that PI3 kinase and Akt are involved in cardioprotective effect of opioids.
MAPK comprise a family of kinases involved in growth and cytoprotection. They include p38 MAPK, JNK, p42 MAPK, and p44 MAPK. The last two isoforms are also known as Extracellular Receptor Kinase (ERK). Like all MAPKs, ERK1/2 are activated by two upstream kinase kinases, MEK1/2 which can be inhibited by PD 098059. PD 098059 completely abolishes the anti‐infarct effect of TAN‐67. Furthermore, TAN‐67 increased the phosphorylation of ERKs during reperfusion and this was prevented by PD 098059.316 Ikeda and colleagues found that intravenous administration of the δ OR agonist DADLE (1 mg/kg) increased the phosphorylation of both isoforms of ERK in the myocardium.94 PD 098059 abolished both ERK's phosphorylation and the protection. van Winkle's group published a study where isolated rabbit cardiomyocytes subjected to hypoxia and reoxygenation were protected with met‐enkephaline and experienced increased phosphorylation of ERK1/2. Pretreatment with PD 098059 or the selective MEK1/2 inhibitor U‐0126 abolished both effects.312 The anti‐infarct effect of U50488H could be blocked by an ERK inhibitor but not by one for the PI3K‐Akt pathway.317 Ha and colleagues also found that the nonselective OR agonist remifentanil before reperfusion promotes an increase in the phosphorylation of ERK1/2, which could be reversed by naloxone or by a nonselective adenosine receptor inhibitor.200 The A2b adenosine receptor has been proposed to be upstream of ERK in the preconditioning pathway.304
C. Other Protective Kinases
In 2011, a study by Li et al. indicated that morphine can ameliorate myocardial contractile dysfunction and limit infarct size following ischemia and reperfusion by a mechanism involving activation of AMPK (AMP‐activated protein kinase).161 JNK and P38 MAPK are also members of the MAPK family that have been implicated in cardioprotection. Despite an increase in phosphorylation of both p38 MAPK and JNK by TAN 67, its cardioprotective effect could not be blocked by the p38 MAPK inhibitor SB‐203580.203.228, 305 Other groups also find an increased phosphorylation of p38 MAPK after morphine use.189, 318
Morphine induces phosphorylation of GSK‐3β and JAK2.116, 290 Phosphorylation of GSK‐3β has been proposed to directly inhibit the opening of MPT and is currently regarded as a critical component in the conditioning pathway.304 Phosphorylation of GSK‐3β inhibits its kinase activity and the GSK‐3β inhibitor SB‐216763 mimicks morphine's protection. Morphine's protection is lost after inhibition of JAK2. JAK2 is a member of the SAFE pathway and has been implicated in the mechanism of the heart's conditioning phenomenon.304 It is unclear whether JAK directly phosphorylates GSK‐3β kinase or acts indirectly through other kinases. DPDPE contributes to phosphorylation p70S6 kinase and GRK2 in isolated rat heart228 however, it is not known whether these effects have any relevance to its protective effect.
The endothelial nitric oxide synthase (eNOS) has been implicated in the preconditioning's trigger pathway between Gi/o coupled receptors and PKC.304 We have found that the infarct‐sparing and antiarrhythmic effects of deltorphin II disappears after pretreatment with the eNOS inhibitor L‐NAME.117 We have shown that pretreatment with L‐NAME abolished the infarct‐reducing effect of U‐50,488.313 In a study of the cardioprotective effect of Eribis peptide 94 similar data were noted by Gross et al.129
D. Tyrosine Kinases
The kinases discussed above phosphorylate serines or threonines in their target proteins. Another family of kinases only phosphorylate tyrosine residues. Tyrosine kinases are proposed to be involved in the transactivation of the epidermal growth factor (EGF) receptor, a step in the OR's trigger pathway for preconditioning.304 A 2001 study indicated that the nonselective tyrosine kinase genistein completely abolished the infarct‐sparing effect of TAN‐67 and DADLE but lavendustin, an inhibitor of Src kinase and the EGF receptor, did not affect the cardioprotective effect of TAN‐67 or DADLE.305 These investigators concluded that neither Src kinase nor the EGF receptor are involved in the cardioprotective effect of δ agonists but that some tyrosine kinase is involved. Quite opposite findings were obtained by Cao and colleagues.312 They subjected isolated cardiomyocytes to hypoxia and reoxygenation and the Src kinase inhibitor herbimycin A completely abolished the cytoprotective effect of met‐enkephalin. Finally, we have also shown that the infarct‐limiting effect of deltorphin II is maintained after pretreatment with genistein.117
Studies have shown that the infarct‐sparing effect of morphine and δ agonist FIT disappeared after pretreatment with AG‐490, an inhibitor of the tyrosine kinase JAK2, but not with the JAK3 inhibitor ZM‐449829.116 Morphine induced phosphorylation of JAK2 in the area at risk.116, 290 It is possible that JAK2 may be the tyrosine kinase that is involved in opioid induced enhancement of cardiac tolerance to I/R.
E. Interactions between Adenosine Receptors and ORs
Isolated perfused rat hearts were subjected to I/R and fentanyl improved the post ischemic recovery. Naloxone abolished the protection from fentanyl as did the selective adenosine A1 receptor antagonist DPCPX.214 These authors concluded that the protective effect of fentanyl involved both ORs and the A1 receptor. Similar data were obtained by Peart and Gross, they administered either morphine or the selective adenosine A1 receptor agonist CCPA to rats subjected to coronary artery occlusion and either compound evoked a decrease in the IS/AAR. The infarct‐limiting effect of morphine was eliminated by DPCPX and after selective blocking of δ1 OR with BNTX. Furthermore, the cardioprotective effect of CCPA was blocked after injection of either DPCPX or BNTX.320 Coadministration of morphine and CCPA did not offer any additive protection. These authors concluded that there is an interaction between δ1 OR and A1 receptor at the intracellular signaling level (cross‐talk).
F. Transactivation of the EGF Receptor
In 2005, Gross's group published data on the cardioprotective effect of endogenous adenosine.321 The level of adenosine was elevated with intravenous administration the adenosine kinase inhibitor 5‐iodotubercidin (1 mg/kg). This compound decreased the IS/AAR twofold. Pretreatment with the adenosine A1 receptor‐selective antagonist DPCPX, the adenosine A3 receptor‐selective antagonist MRS‐1523, or the δ1 OR‐selective antagonist BNTX abolished the infarct‐reducing effect of 5‐iodotubercidin.321 These data led these researchers to conclude that the cardioprotective effect of endogenous adenosine is mediated via simultaneous A1 receptor, A3 receptor and δ1 OR activation. Recently, Ha et al. found that the nonselective OR agonist remifentanil started 5 min before reperfusion in the isolated rat heart decreases the IS/AAR and evokes the phosphorylation of ERK1/2. These effects were blocked by pretreatment with naloxone or the nonselective adenosine receptor antagonist 8‐(p‐sulfophenyl) theophylline.200 It is our opinion that the aforementioned studies provide evidence of interaction of adenosine and OR at some level. It has been proposed that transactivation may be involved as both receptors converge through transactivation of the EGF receptor.322, 323 However, it is not clear how inhibition of one could turn off signaling of the other through this transactivation.
EGF receptor activation protects against I/R.312, 324, 325 Met‐enkephalin reduces cell death in isolated cardiomyocytes subjected to hypoxia and reoxygenation and phosphorylates the EGF receptor. The EGF tyrosine kinase inhibitor AG‐1478, the Src kinase inhibitor herbimycin A or naloxone eliminated phosphorylation of EGF receptor and the cytoprotective effect.312
The aforementioned data indicate that there is an important role of transactivation of opioid and adenosine receptors in cardiac tolerance to the impact of ischemia and reperfusion. Opioid transactivation of EGF receptor tyrosine kinase is a binder link between ORs and ERK1/2 and the downstream signaling pathways including ERK and PI3 kinase. Thus, EGF receptor transactivation is an important mechanism in implementing the protective effect of opioids is now shared by other physiologists.322, 323
G. KATP and BK Channels
Pretreatment with the KATP channel blocker glibenclamide abolished anti‐infarct effect of morphine.10 Two years later, the same group demonstrated that glibenclamide abolished the infarct‐reducing effect of TAN‐67.91 It was also shown that the cardioprotective effect of DADLE in vitro disappeared after KATP channel blockade with this inhibitor.92 Further studies were designed to determine what types of KATP channels are involved in the cardioprotective effect of opioids. It was observed that incubation of cardiomyocytes with morphine prevents cell death and the selective inhibitor of the mitochondrial KATP channel (mitoKATP) 5‐hydroxydecanoate (5‐HD) completely abolished the cytoprotective effect of morphine.82 Pretreatment with fentanyl improved post‐ischemic recovery of function and that was also dependent on the opening of mitoKATP.214 In 2000, it was found that the antiarrhythmic and infarct‐sparing effect of TAN‐67 in vivo was abolished by blocking mitoKATP with 5‐HD but not after blocking sarcolemmal KATP channels (sarcKATP) with HMR 1098.102 The cytoprotective effect of TAN‐67 in isolated cardiomyocytes disappeared after blocking mitoKATP with 5‐HD.105 5‐HD also blocked the cardioprotective effect of U‐50,488 in isolated perfused rat heart,326 abolished the cytoprotective effect of morphine and δ agonist BW373U86 in isolated chicken cardiomyocytes.107 In an in vivo study we found that the infarct‐reducing effect of deltorphin II did not occur after blockade of mitoKATP with 5‐HD.117 A large number of similar publications now confirm the key role of mitoKATP in the cardioprotective effect of opioids.
A few studies also show a role of sarcKATP in opioid‐induced cardioprotection. The cytoprotective effect of met‐enkephalin disappeared after selective blockade of mitoKATP with 5‐HD or after selective blockade of sarcKATP with HMR 1098.98, 99 The anti‐infarct effect of morphine and BW373U86 in vivo was no longer observed after either mitoKATP or sarcKATP blockade.108 In another study it was demonstrated that the cardioprotective effect of Eribis peptide 94 was dependent on both mitoKATP and sarcKATP opening.129
In 2005, it was proposed that the mitochondrial Ca2+‐dependent big conductance K+ channel (mitoBKCa channel) was involved in the cardioprotective mechanism of opioids. The κ1 OR agonist U‐50,488 provided a decrease in the IS/AAR in isolated rat heart and prevented cell death of isolated myocytes subjected to simulated I/R. The mitoBKCa channel inhibitor paxilline abolished both effects of U‐50488.327
H. Redox Signaling
The above studies indicate that mitoKATP and perhaps sarcKATP are involved in the cardioprotective effect of opioids. It was originally thought that KATP must be an end effectors of the protection, however, evidence now indicates that their role is primarily one of signal transduction. Studies with preconditioning provided strong evidence that opening of mito KATP activate PKC through redox signaling with free radicals304 and that seems to include signaling from the ORs. The anti‐infarct effect of morphine in rabbit heart could be blocked with the free radical scavenger N‐2‐mercaptopropionyl glycine (MPG).328 10 min preconditioning with morphine or BW373U86 increased cell survival of isolated chicken cardiomyocytes subjected to hypoxia and reoxygenation. Morphine‐induced protection and free radical production was abolished by MPG, naloxone, BNTX, a selective δ1 OR antagonist; or 5‐HD. The superoxide dismutase inhibitor diethyldithiocarbamic acid exhibited the same effect. Finally, the increase in oxygen radicals was abolished by the mitochondrial electron transport inhibitor myxothiazol.329 It was also shown later that the infarct‐sparing effect of morphine does not occur after blocking ROS production with MPG.320 In a study with isolated cardiomyocytes subjected to hypoxia/reoxygenation, it was shown that the cytoprotective effect of morphine disappeared after pretreatment with MPG.315 Thus, the above data indicate that opioid induced opening of mitoKATP leads to increased production of ROS, which through redox signaling enhances cardiac tolerance to I/R.
I. MPT
Elevated cytosolic ROS and Ca2+ in the first minutes of reperfusion are thought to open MPT (mitochondrial permeability transition pores).156 MPT can destroy the mitochondria and either kill the cardiomyocyte outright (necrosis) or release proapoptotic substances depending on how many mitochondria are lost. A 2005 study indicated that MPT are involved in the cardioprotective effect of opioids.327 The regional ischemia and reperfusion was carried out in the isolated perfused rat heart. U‐50488 decreased the IS/AAR and lactate dehydrogenase activity in the coronary effluent of isolated hearts. The MPT opener atractyloside abolished the cardioprotective effect of U‐50488.327 It is assumed that OR stimulation inhibited MPT opening at reperfusion and opening MPT directly with atractyloside overrode any inhibition from protective signaling. Mitochondria isolated from the ischemic zone of rat hearts receiving morphine have an elevated threshold for opening MPT with Ca2+ and this resistance to MPT opening was lost if PI3 kinase was inhibited with wortmannin.330 In our in vivo study we demonstrated that U‐50,488 induces an infarct‐reducing effect as well as an antiapoptotic effect. It also decreases the activity of caspases, which become activated after MPT opening.163 The infarct‐sparing and antiapoptotic effects disappeared after blockade of mitoKATP with 5‐HD. U‐50,488 evoked an enhancement of expression of antiapoptotic protein Bcl‐2 and decreased the expression of proapoptotic protein Bax.163 Presumably, the κ1 OR agonist prevented MPT opening with signaling through mitoKATP as discussed above. Kim et al. also found that remifentanil not only limits infarction but also increases Bcl‐2 and decreased Bax.89 We conclude that the current evidence indicates that the end effector of the cardioprotective effect of opioids is inhibition of the MPT early in reperfusion.
19. WILL AN OR AGONIST PROVIDE PROTECTION TO TODAY'S PATIENT POPULATION?
The ORs appear to protect through a signaling pathway similar if not identical to that of IP and IPost. Evidence suggests that the loading doses of P2Y12 receptor inhibitors that are now routinely given to all patients undergoing reperfusion therapy to prevent platelet aggregation in their stents may also trigger this same protection, which could make the OR agonist redundant. The P2Y12 blocker cangrelor limited infarct size when present at reperfusion in open‐chest rabbits by an amount similar to that with IPost. Protection from cangrelor could be blocked by 5‐HD, wortmannin, adenosine receptor inhibitors, an ERK inhibitor, or the antioxidant N‐2‐mercaptopropionyl glycine. These same signaling inhibitors will block protection from both IP and IPost. None of those agents restored the ability of platelets to aggregate indicating that protective signaling rather than prevention of thrombi was responsible for the protection. When cangrelor and IPost were combined in rabbits, IPost caused no additional protection, probably because both protect by the same mechanism.331
Recent attempts to translate IPost to clinical practice in the setting of acute myocardial infarction have been disappointing. The first IPost trial was performed just before P2Y12 blockers came into widespread use and was very positive but all of those performed thereafter had P2Y12 blockers present in all patients and showed minimal or no protection from adding IPost.332 These data indicate that all of today's patients receiving primary angioplasty to reperfuse their coronary arteries are already in a postconditioned state from their P2Y12 blocker loading dose. These drugs were quickly added to the guidelines because they greatly improved clinical outcomes. Their benefit was assumed to result from preventing intracoronary thrombi and the possibility of a direct anti‐infarct effect was not considered. Of course, today it would be impossible to do a clinical trial to measure their ability to reduce infarct size in man because it would be unethical to deny platelet inhibitors to a control group. Any clinically effective agent today must, therefore, be able to provide additional protection when combined with a P2Y12 inhibitor. To date, no OR agonists have been tested to see if any of them can provide additional protection in an animal model treated with a P2Y12 inhibitor. That screening should be done before considering a large scale clinical trial of any cardioprotectant.
20. CONCLUDING REMARKS
Stimulation of central μ OR promotes reduction of infarct size during coronary artery occlusion and reperfusion. Occupancy of peripheral μ OR by opioids promotes better recovery of cardiac contractile function after ischemia. Activation of peripheral and possibly central δ1 ORs prevents cardiomyocyte necrosis, apoptosis and arrhythmias caused by I/R of the heart. Stimulation of peripheral OR δ2 reduces infarct size and inhibits arrhythmogenesis during coronary artery occlusion and reperfusion. Activation of peripheral κ1 ORs also prevents cardiomyocyte necrosis, apoptosis and arrhythmias induced by I/R. Stimulation of peripheral δ and κ ORs contributes to increase in cardiac contractility during reperfusion. No data are available on the involvement of ORL1 receptor in the increased cardiac tolerance to I/R by opioids. It should be noted, however, that at high dose opioids may contribute to cardiac arrhythmias and this effect is probably mediated by non‐OR activation.
The data show that the μ, δ1, δ2, and κ1 OR agonists all are promising candidates for a drug, which would enhance the cardiac tolerance to I/R. The OR agonists exert infarct‐reducing effects both with prophylactic administration and with acute treatment just prior to reperfusion. Furthermore, opioids are also effective in preventing ischemia‐induced arrhythmias.
ACKNOWLEDGMENTS
The authors thank Professor Saadeh Suleiman and Professor Raimondo Ascione from the University of Bristol (UK) for useful advice. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: L.N.M. and N.N. are currently supported by a grant from the Russian National Foundation (grant 14‐15‐00008); I.K. is currently supported by a grant from the British Heart Foundation (grant no. PG/15/66/31710). The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
Biographies
Leonid N. Maslov was born in Batkat (Tomsk Region, Russia) in 1957 and studied at the Medical Faculty of Tomsk State Medical Institute in Tomsk (Russia). He was graduated as a Medical Doctor. In 1980 he became a Ph.D. degree in Pathological Physiology in 1991. In 1997, he obtained his degree of a Doctor of Science in Pathological Physiology and in 2003 became a Professor in Pathological Physiology in 2003. He is the head of the Laboratory of Experimental Cardiology, Federal State Budgetary Scientific Institution “Research Institute for Cardiology,” Tomsk, Russia. He is currently an editorial board member of Sib. Med. J. (Tomsk).
Igor Khaliulin was born in 1961 in Kemerovo (Russia). He graduated as a Biologist from Kemerovo State University in 1983. In 1992, he became a Ph.D. in Normal and Pathological Physiology. Dr. Khaliulin is a Research Fellow at the University of Bristol (Bristol, UK). He is a member of the Editorial Board of American Association for Science and Technology and a member of the Working Group on Cellular Biology of the Heart of the European Society of Cardiology. The main research interest of Dr. Khaliulin is cardioprotection and cell signaling.
Peter R. Oeltgen was born in Ottendorf (Germany) in 1942. He obtained his Ph.D. in Biochemistry from Loyola University, Stritch School of Medicine, Chicago (USA), in 1972. In 1994, he was promoted to Professor of Pathology and Toxicology at the University Of Kentucky College of Medicine, Lexington, Kentucky (USA). In 2008, he was awarded an honorary doctorate degree from the Agricultural University of Crackow (Poland). Since 2013, he is now Professor Emeritus of Pathology and Toxicology at the University of Kentucky College of Medicine.
Natalia V. Naryzhnaia was born in Taiga (Kemerovo Region, Russia) in 1970 and studied at the Medico‐Biological Faculty of Tomsk State Medical Institute in Tomsk (Russia). In 1993 she graduated as a Medical Doctor. In 1998, she became a Ph.D. degree in Pathological Physiology. She is the Senior Researcher of the Laboratory of Experimental Cardiology, Federal State Budgetary Scientific Institution “Research Institute for Cardiology,” Tomsk, Russia.
Jian‐Ming Pei was born in 1967 in Ningxia province (China). He studied at the Fourth Military Medical University from 1984 to 1990 in China. He obtained his Ph.D. degree in Physiology in 1997. He obtained title of Professor in Physiology in 2003. Since 2003, he is a head of Department of Physiology at the Fourth Military Medical University in China.
Stephen A. Brown was born in St. Louis, Missouri, in 1948 and studied at Southern Illinois University and the Department of Biochemistry and Biophysics at Iowa State University and obtained his PhD in Immunology in 1996 at the University of Kentucky. He retired as a Principal Investigator and an Assistant Research Professor at the University of Kentucky Department of Internal Medicine and a Principal Investigator at the Department of Veterans Affairs Medical Center, Lexington, Kentucky.
Yury B. Lishmanov was born in Sol‐Iletsk City (Orenburg Region, Russia) in 1951 and studied at the Medical Faculty of Mordovia State University (Russia). He graduated as a Medical Doctor in 1974, and became a Ph.D. degree in Pathological Physiology in 1977. He obtained his degree of Doctor of Science in Pathological Physiology in 1988. In 1996, he was awarded title of Professor in Pathological Physiology, after he became Corresponding Member of Russian Academy of Medical Science in 1996. In 2015, he is a Corresponding Member of Russian Academy of Science. Now he is a former Vice‐Director of Federal State Budgetary Scientific Institution “Research Institute for Cardiology,” Tomsk, Russia. He is currently an editorial board member of Sib. Med. J. (Tomsk).
James M. Downey was born in Wabash, Indiana, in 1944. He obtained his PhD from the University of Illinois in 1970 in mammalian physiology, and did a postdoctoral fellowship in cardiac pathophysiology at Peter Bent Brigham Hospital (Harvard) under the direction of Edmund Sonnenblick. He joined the faculty at University of South Alabama College of Medicine in 1975 and remains there today as professor emeritus. He is a past president of the International Society for Heart Research (2001–2004), was an NIH study section regular member and is currently an editorial board member of Basic Res. Cardiol., J. Cell. Mol. Cardiol., J. Cardiovsc. Pharm. Ther. He has published extensively on protecting the ischemic myocardium.
REFERENCES
- 1. Girn HR, Ahilathirunayagam S, Mavor AI, Homer‐Vanniasinkam S. Reperfusion syndrome: Cellular mechanisms of microvascular dysfunction and potential therapeutic strategies. Vasc Endovascular Surg 2007;41:277–293. [DOI] [PubMed] [Google Scholar]
- 2. Monassier JP. Reperfusion injury in acute myocardial infarction. From bench to cath lab. Part I: Basic considerations. Arch Cardiovasc Dis 2008;101:491–500. [DOI] [PubMed] [Google Scholar]
- 3. Ostadal B, Kolar F. Cardiac Ischemia: From Injury to Protection. Boston, Dordrecht, London: Kluwer Academic Publishers; 1999. [Google Scholar]
- 4. Sharma V, Bell RM, Yellon DM. Targeting reperfusion injury in acute myocardial infarction: A review of reperfusion injury pharmacotherapy. Expert Opin Pharmacother 2012;13:1153–1175. [DOI] [PubMed] [Google Scholar]
- 5. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986;74:1124–1136. [DOI] [PubMed] [Google Scholar]
- 6. Yellon DM, Baxter GF. A “second window of protection” or delayed preconditioning phenomenon: Future horizons for myocardial protection? J Mol Cell Cardiol 1995;27:1023–1034. [DOI] [PubMed] [Google Scholar]
- 7. Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten‐Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 2003;285:H579–H588. [DOI] [PubMed] [Google Scholar]
- 8. Liu GS, Thornton J, van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 1991;84:350–356. [DOI] [PubMed] [Google Scholar]
- 9. Schultz JE, Rose E, Yao Z, Gross GJ. Evidence for involvement of opioid receptors in ischemic preconditioning in rat hearts. Am J Physiol 1995;268:H2157–H2161. [DOI] [PubMed] [Google Scholar]
- 10. Schultz JE, Hsu AK, Gross GJ. Morphine mimics the cardioprotective effect of ischemic preconditioning via a glibenclamide‐sensitive mechanism in the rat heart. Circ Res 1996;78:1100–1104. [DOI] [PubMed] [Google Scholar]
- 11. Kin H, Zatta AJ, Jiang R, Reeves JG, Mykytenko J, Sorescu G, Zhao ZQ, Wang NP, Guyton RA, Vinten‐Johansen J. Activation of opioid receptors mediates the infarct size reduction by postconditioning. J Mol Cell Cardiol 2005;38:827. [Google Scholar]
- 12. Marson L, Kiritsy‐Roy JA, van Loon GR. μ‐Opioid peptide modulation of cardiovascular and sympathoadrenal responses to stress. Am J Physiol 1989;257:R901–R908. [DOI] [PubMed] [Google Scholar]
- 13. Panksepp J, Bishop P. An autoradiographic map of (3H)diprenorphine binding in rat brain: Effects of social interaction. Brain Res Bull 1981;7:405–410. [DOI] [PubMed] [Google Scholar]
- 14. Bunzow JR, Saez C, Mortrud M, Bouvier C, William JT, Low M, Grandy DK. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta, or kappa opioid receptor type. FEBS Lett 1994;347:284–288. [DOI] [PubMed] [Google Scholar]
- 15. Borghi V, Przewlocka B, Labuz D, Maj M, Ilona O, Pavone F. Formalin‐induced pain and μ‐opioid receptor density in brain and spinal cord are modulated by A1 and A2a adenosine agonists in mice. Brain Res 2002;956:339–348. [DOI] [PubMed] [Google Scholar]
- 16. Bailey A, Ledent C, Kelly M, Hourani SM, Kitchen I. Changes in spinal δ and κ opioid systems in mice deficient in the A2A receptor gene. J. Neurosci. 2002;22:9210–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bridge KE, Wainwright A, Reilly K, Oliver KR. Autoradiographic localization of 125I [Tyr14] nociceptin/orphanin FQ binding sites in macaque primate CNS. Neuroscience 2003;118:513–523. [DOI] [PubMed] [Google Scholar]
- 18. Pradhan AA, Clarke PB. Comparison between δ‐opioid receptor functional response and autoradiographic labeling in rat brain and spinal cord. J Comp Neurol 2005;481:416–426. [DOI] [PubMed] [Google Scholar]
- 19. Diaz SL, Barros VG, Antonelli MC, Rubio MC, Balerio GN. Morphine withdrawal syndrome and its prevention with baclofen: Autoradiographic study of mu‐opioid receptors in prepubertal male and female mice. Synapse 2006;60:132–140. [DOI] [PubMed] [Google Scholar]
- 20. Homberg JR, Mul JD, de Wit E, Cuppen E. Complete knockout of the nociceptin/orphanin FQ receptor in the rat does not induce compensatory changes in μ, δ and κ opioid receptors. Neuroscience 2009;163:308–315. [DOI] [PubMed] [Google Scholar]
- 21. Lalanne L, Ayranci G, Kieffer BL, Lutz PE. The kappa opioid receptor: From addiction to depression, and back. Front Psychiatry 2014;5:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cornford EM, Braun LD, Crane PD, Oldendorf WH. Blood‐brain barrier restriction of peptides and the low uptake of enkephalins. Endocrinology 1978;103:1297–1303. [DOI] [PubMed] [Google Scholar]
- 23. Samii A, Bickel U, Stroth U, Pardridge WM. Blood‐brain barrier transport of neuropeptides: Analysis with a metabolically stable dermorphin analogue. Am J Physiol 1994;267:E124–E131. [DOI] [PubMed] [Google Scholar]
- 24. Lishmanov IuB, Maslov LN, Rice K. Blood‐brain barrier permeability for the ligands of opioid receptors. Eksp Klin Farmakol 2002;65:71–77. (in Russian) [PubMed] [Google Scholar]
- 25. Freye E, Hartung E, Schenk GK. Perfusion of the fourth cerebral ventricle with the synthetic opioid peptide, FK 33‐824, induces dose‐related bradycardia and naloxone‐reversible respiratory depression in the awake dog. Pharmacology 1982;25(1):6–11. [DOI] [PubMed] [Google Scholar]
- 26. Inoue K, Nashan B, Arndt JO. [D‐Met2,Pro5]enkephalinamide activates cardioinhibitory efferents in anaesthetized dogs. Eur J Pharmacol 1985;110:233–239 [DOI] [PubMed] [Google Scholar]
- 27. Haddad GG, Jeng HJ, Lai TL. Effect of endorphins on heart rate and blood pressure in adult dogs. Am J Physiol 1986;250:H796–H805. [DOI] [PubMed] [Google Scholar]
- 28. Kiritsy‐Roy JA, Marson L, van Loon GR. Sympathoadrenal, cardiovascular and blood gas responses to highly selective mu and delta opioid peptides. J Pharmacol Exp Ther 1989;251:1096–1103. [PubMed] [Google Scholar]
- 29. Wittert G, Hope P, Pyle D. Tissue distribution of opioid receptor gene expression in the rat. Biochem Biophys Res Commun 1996;218:877–881. [DOI] [PubMed] [Google Scholar]
- 30. Krazinski BE, Koziorowski M, Brzuzan P, Okrasa S. The expression of genes encoding opioid precursors and the influence of opioid receptor agonists on steroidogenesis in porcine adrenocortical cells in vitro. J Physiol Pharmacol 2011;62:461–468. [PubMed] [Google Scholar]
- 31. Wang HB, Guan JS, Bao L, Zhang X. Distinct subcellular distribution of δ‐opioid receptor fused with various tags in PC12 cells. Neurochem Res 2008;33:2028–2034. [DOI] [PubMed] [Google Scholar]
- 32. Burnie J. Naloxone in shock. Lancet 1981;1:942. [DOI] [PubMed] [Google Scholar]
- 33. Dumont M, Lemaire S. Increased content of immunoreactive Leu‐enkephalin and alteration of δ opioid receptor in hearts of spontaneously hypertensive rats. Neurosci Lett 1988;94:114–118. [DOI] [PubMed] [Google Scholar]
- 34. Ventura C, Bastagli L, Bernardi P, Caldarera CM, Guarnieri C. Opioid receptors in rat cardiac sarcolemma: Effect of phenylephrine and isoproterenol. Biochim Biophys Acta 1989;987:69–74. [DOI] [PubMed] [Google Scholar]
- 35. Tai KK, Jin WQ, Chan TK, Wong TM. Characterization of [3H]U69593 binding sites in the rat heart by receptor binding assays. J Mol Cell Cardiol 1991;23:1297–1302. [DOI] [PubMed] [Google Scholar]
- 36. Jin WQ, Tai KK, Chan TK, Wong TM. Further characterization of [3H]U69593 binding sites in the rat heart. J Mol Cell Cardiol 1995;27:1507–1511. [DOI] [PubMed] [Google Scholar]
- 37. Zimlichman R, Gefel D, Eliahou H, Matas Z, Rosen B, Gass S, Ela C, Eilam Y, Vogel Z, Barg J. Expression of opioid receptors during heart ontogeny in normotensive and hypertensive rats. Circulation 1996;93:1020–1025. [DOI] [PubMed] [Google Scholar]
- 38. Ela C, Barg J, Vogel Z, Hasin Y, Eilam Y. Distinct components of morphine effects on cardiac myocytes are mediated by the κ and δ opioid receptors. J Mol Cell Cardiol 1997;29:711–720. [DOI] [PubMed] [Google Scholar]
- 39. Weil J, Zolk O, Griepentrog J, Wenzel U, Zimmermann WH, Eschenhagen T. Alterations of the preproenkephalin system in cardiac hypertrophy and its role in atrioventricular conduction. Cardiovasc Res 2006;69:412–422. [DOI] [PubMed] [Google Scholar]
- 40. Stefano GB, Hartman A, Bilfinger TV, Magazine HI, Liu Y, Casares F, Goligorsky MS. Presence of the mu3 opiate receptor in endothelial cells. Coupling to nitric oxide production and vasodilation. J Biol Chem 1995;270:30290–30293. [DOI] [PubMed] [Google Scholar]
- 41. Stefano GB, Salzet M, Hughes TK, Bilfinger TV. δ2 opioid receptor subtype on human vascular endothelium uncouples morphine stimulated nitric oxide release. Int J Cardiol 1998;64:S43–S51. [DOI] [PubMed] [Google Scholar]
- 42. Saeed RW, Stefano GB, Murga JD, Short TW, Qi F, Bilfinger TV, Magazine HI. Expression of functional delta opioid receptors in vascular smooth muscle. Int J Mol Med 2000;6:673–677. [DOI] [PubMed] [Google Scholar]
- 43. Dumont M, Lemaire S. Characterization of the high affinity [3H]nociceptin binding site in membrane preparations of rat heart: correlations with the non‐opioid dynorphin binding site. J Mol Cell Cardiol 1998;30:2751–2760. [DOI] [PubMed] [Google Scholar]
- 44. Kim KW, Chung YJ, Han JH, Woo RS, Park EY, Seul KH, Kim SZ, Cho KW, Kim SH. Nociceptin/orphanin FQ increases ANP secretion in neonatal cardiac myocytes. Life Sci 2002;70:1065–1074. [DOI] [PubMed] [Google Scholar]
- 45. Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG, Insel PA. G‐protein‐coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin‐3‐associated microdomains in adult cardiac myocytes. J Biol Chem 2005;280:31036–31044. [DOI] [PubMed] [Google Scholar]
- 46. McDonald J, Leonard AD, Serrano‐Gomez A, Young SP, Swanevelder J, Thompson JP, Lambert DG. Assessment of nociceptin/orphanin FQ and μ‐opioid receptor mRNA in the human right atrium. Br J Anaesth 2010;104:698–704. [DOI] [PubMed] [Google Scholar]
- 47. Sobanski P, Krajnik M, Shaqura M, Bloch‐Boguslawska E, Schafer M, Mousa SA. The presence of mu‐, delta‐, and kappa‐opioid receptors in human heart tissue. Heart Vessels 2014;29(6):855–863. [DOI] [PubMed] [Google Scholar]
- 48. Mousa SA, Shaqura M, Schдper J, Huang W, Treskatsch S, Habazettl H, Abdul‐Khaliq H, Schдfer M. Identification of μ‐ and κ‐opioid receptors as potential targets to regulate parasympathetic, sympathetic, and sensory neurons within rat intracardiac ganglia. J Comp Neurol 2010;518:3836–3847. [DOI] [PubMed] [Google Scholar]
- 49. Mousa SA, Shaqura M, Schaper J, Treskatsch S, Habazettl H, Schafer M, Abdul‐Khaliq H. Developmental expression of δ‐opioid receptors during maturation of the parasympathetic, sympathetic, and sensory innervations of the neonatal heart: Early targets for opioid regulation of autonomic control. J Comp Neurol 2011;519:957–971. [DOI] [PubMed] [Google Scholar]
- 50. Huang MH, Friend DS, Sunday ME, Singh K, Haley K, Austen KF, Kelly RA, Smith TW. An intrinsic adrenergic system in mammalian heart. J Clin Invest 1996;98:1298–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang MH, Wang HQ, Roeske WR, Birnbaum Y, Wu Y, Yang NP, Lin Y, Ye Y, McAdoo DJ, Hughes MG, Lick SD, Boor PJ, Lui CY, Uretsky BF. Mediating δ‐opioid‐initiated heart protection via the β2‐adrenergic receptor: role of the intrinsic cardiac adrenergic cell. Am J Physiol Heart Circ Physiol 2007;293:H376–H384. [DOI] [PubMed] [Google Scholar]
- 52. Huang MH, Nguyen V, Wu Y, Rastogi S, Lui CY, Birnbaum Y, Wang HQ, Ware DL, Chauhan M, Garg N, Poh KK, Ye L, Omar AR, Tan HC, Uretsky BF, Fujise K. Reducing ischaemia/reperfusion injury through δ‐opioid‐regulated intrinsic cardiac adrenergic cells: adrenopeptidergic co‐signalling. Cardiovasc Res 2009;84:452–460. [DOI] [PubMed] [Google Scholar]
- 53. Kett A, Omoniyi AT, Kim H, Olariu N, Wu D, Szeto HH, Clapp JF. Baroreflex‐mediated bradycardia but not tachycardia is blunted peripherally by intravenous mu‐opioid agonists. Am J Obstet Gynecol 1998;178:950–955. [DOI] [PubMed] [Google Scholar]
- 54. Omoniyi AT, Wu D, Soong Y, Szeto HH. Baroreflex‐mediated bradycardia is blunted by intravenous μ‐ but not κ‐opioid agonists. J Cardiovasc Pharmacol 1998;31:954–959. [DOI] [PubMed] [Google Scholar]
- 55. Urthaler F, Isobe JH, Gilmour KE, James TN. Morphine and autonomic control of the sinus node. Chest 1973;64:203–212. [DOI] [PubMed] [Google Scholar]
- 56. Weihe E, McKnight AT, Corbett AD, Hartschuh W, Reinecke M, Kosterlitz HW. Characterizations of opioid peptides in guinea‐pig heart and skin. Life Sci 1983;33:711–714. [DOI] [PubMed] [Google Scholar]
- 57. Gautret B, Schmitt H. Cardiac slowing induced by peripheral κ‐opiate receptor stimulation in rats. Eur J Pharmacol 1984;102:159–163. [DOI] [PubMed] [Google Scholar]
- 58. Weitzell R, Illes P, Starke K. Inhibition via opioid μ‐ and δ‐receptors of vagal transmission in rabbit isolated heart. Naunyn Schmiedebergs Arch Pharmacol 1984;328:186–190. [DOI] [PubMed] [Google Scholar]
- 59. Musha T, Satoh E, Koyanagawa H, Kimura T, Satoh S. Effects of opioid agonists on sympathetic and parasympathetic transmission to the dog heart. J Pharmacol Exp Ther 1989;250:1087–1091. [PubMed] [Google Scholar]
- 60. Malinowska B, Piszcz J, Koneczny B, Hryniewcz A, Schlicker E. Modulation of the cardiac autonomic transmission of the pithed rats by presynaptic opioid OP4 and cannabinoid CB1 receptors. Naunyn Schmiedebergs Arch Pharmacol 2001;364:233–241. [DOI] [PubMed] [Google Scholar]
- 61. Jackson KE, Farias M, Stanfill AS, Caffrey JL. Transient arterial occlusion raises enkephalin in the canine sinoatrial node and improves vagal bradycardia. Auton Neurosci 2001;94:84–92. [DOI] [PubMed] [Google Scholar]
- 62. Farias M, Jackson KE, Yoshishige D, Caffrey JL. Cardiac enkephalins interrupt vagal bradycardia via δ2‐opioid receptors in sinoatrial node. Am J Physiol Heart Circ Physiol 2003;284:H1693–H1701. [DOI] [PubMed] [Google Scholar]
- 63. Deo SH, Johnson‐Davis S, Barlow MA, Yoshishige D, Caffrey JL. Repeated δ1‐opioid receptor stimulation reduces δ2‐opioid receptor responses in the SA node. Am J Physiol Heart Circ Physiol. 2006;291:H2246–H2254. [DOI] [PubMed] [Google Scholar]
- 64. Deo SH, Barlow MA, Gonzalez L, Yoshishige D, Caffrey JL. Cholinergic location of δ‐opioid receptors in canine atria and SA node. Am J Physiol Heart Circ Physiol 2008;294:H829–H838. [DOI] [PubMed] [Google Scholar]
- 65. Ledda F, Mantelli L. Possible presynaptic inhibitory effect of etorphine on synaptic nerve terminals of guinea‐pig heart. Eur J Pharmacol 1982;85:247–250. [DOI] [PubMed] [Google Scholar]
- 66. Ledda F, Mantelli L, Corti V, Fantozzi R. Inhibition of cardiac response to sympathetic nerve stimulation by opioid peptides and its potentiation by morphine and methadone. Eur J Pharmacol 1984;102:443–450. [DOI] [PubMed] [Google Scholar]
- 67. Ledda F, Mantelli L, Corti V. Sensitivity to dynorphin‐(1‐13) of the presynaptic inhibitory opiate receptors of the guinea‐pig heart. Eur J Pharmacol 1985;117:377–380. [DOI] [PubMed] [Google Scholar]
- 68. Starke K, Schoffel E, Illes P. The sympathetic axons innervating the sinus node of the rabbit possess presynaptic opioid κ‐ but not μ‐ or δ‐receptors. Naunyn Schmiedebergs Arch Pharmacol 1985;329:206–209. [DOI] [PubMed] [Google Scholar]
- 69. Fuder H, Buder M, Riers HD, Rothacher G. On the opioid receptor subtype inhibiting the evoked release of 3H‐noradrenaline from guinea‐pig atria in vitro. Naunyn Schmiedebergs Arch Pharmacol 1986;332:148–155. [DOI] [PubMed] [Google Scholar]
- 70. Hung CF, Chang WL, Liang HC, Su MJ. Identification of opioid receptors in the sympathetic and parasympathetic nerves of guinea‐pig atria. Fundam Clin Pharmacol 2000;14:387–394. [DOI] [PubMed] [Google Scholar]
- 71. Ensinger H, Hedler L, Schurr C, Starke K. Ethylketocyclazocine decreases noradrenaline release and blood pressure in the rabbit at a peripheral opioid receptor. Naunyn Schmiedebergs Arch Pharmacol 1984;328:20–23. [DOI] [PubMed] [Google Scholar]
- 72. Ensinger H, Hedler L, Szabo B, Starke K. Bremazocine causes sympatho‐inhibition and hypotension in rabbits by activating peripheral kappa‐receptors. J Cardiovasc Pharmacol 1986;8:470–475. [DOI] [PubMed] [Google Scholar]
- 73. Gu H, Barron BA, Gaugl JF, Caffrey JL. Dynorphin, naloxone, and overflow of norepinephrine during cardiac nerve stimulation in dogs. Am J Physiol 1992;263:H153–H161. [DOI] [PubMed] [Google Scholar]
- 74. Feuerstein G, Zukowska‐Grojec Z. Effect of dermorphin and morphine on the sympathetic and cardiovascular system of the pithed rat. Neuropeptides 1987;9:139–150. [DOI] [PubMed] [Google Scholar]
- 75. Arndt ML, Wu D, Soong Y, Szeto HH. Nociceptin/orphanin FQ increases blood pressure and heart rate via sympathetic activation in sheep. Peptides 1999;20:465–470. [DOI] [PubMed] [Google Scholar]
- 76. Sander GE, Given MB, Lowe RF, Wolf RH, Brizzee KR, Giles TD. The effect of area postrema lesions on hemodynamic responses to systemic methionine‐enkephalin in conscious dogs. Am J Hypertens 1988;1:1S–3S. [DOI] [PubMed] [Google Scholar]
- 77. Sander GE, Lowe RF, Given MB, Giles TD. Interactions between circulating peptides and the central nervous system in hemodynamic regulation. Am J Cardiol 1989;64:44C–50C. [DOI] [PubMed] [Google Scholar]
- 78. Giuliani S, Maggi CA, Meli A. Opioid receptors and prejunctional modulation of capsaicin‐sensitive sensory nerves in guinea‐pig left atrium. Gen Pharmacol 1990;21:417–421. [DOI] [PubMed] [Google Scholar]
- 79. Giuliani S, Maggi CA. Prejunctional modulation by nociceptin of nerve‐mediated inotropic responses in guinea‐pig left atrium. Eur J Pharmacol 1997;332:231–236. [DOI] [PubMed] [Google Scholar]
- 80. Gulati A, Bhargava HN. Cardiovascular responses to κ opioid agonists in intact and adrenal demedullated rats. Eur J Pharmacol 1988;156:247–257. [DOI] [PubMed] [Google Scholar]
- 81. Schultz JE, Hsu AK, Nagase H, Gross GJ. Ischemic preconditioning and morphine‐induced cardioprotection involve the delta (δ)‐opioid receptor in the intact rat heart. J Mol Cell Cardiol 1997;29:2187–2195. [DOI] [PubMed] [Google Scholar]
- 82. Miki T, Cohen MV, Downey JM. Opioid receptors contributes to ischemic preconditioning through protein kinase C activation in rabbits. Mol Cell Biochem 1998;186:3–12. [PubMed] [Google Scholar]
- 83. Liang BT, Gross GJ. Direct preconditioning of cardiac myocytes via opioid receptors and KATP channels. Circ Res 1999;84:1396–1400. [DOI] [PubMed] [Google Scholar]
- 84. Wu S, Wong MC, Chen M, Cho CH, Wong TM. Role of opioid receptors in cardioprotection of cold‐restraint stress and morphine. J Biomed Sci 2004;11:726–731. [DOI] [PubMed] [Google Scholar]
- 85. Lu Y, Dong C, Yu J, Li L. Role of central and peripheral opioid receptors in the cardioprotection of intravenous morphine preconditioning. Ir J Med Sci 2011;180:881–885. [DOI] [PubMed] [Google Scholar]
- 86. Bilir A, Erkasap N, Koken T, Gulec S, Kaygisiz Z, Tanriverdi B, Kurt I. Effects of tramadol on myocardial ischemia‐reperfusion injury. Scand Cardiovasc J 2007;41:242–247. [DOI] [PubMed] [Google Scholar]
- 87. Wagner R, Piler P, Bedanova H, Adamek P, Grodecka L, Freiberger T. Myocardial injury is decreased by late remote ischaemic preconditioning and aggravated by tramadol in patients undergoing cardiac surgery: A randomised controlled trial. Interact Cardiovasc Thorac Surg 2010;11:758–762. [DOI] [PubMed] [Google Scholar]
- 88. Zhang Y, Irwin MG, Wong TM. Remifentanil preconditioning protects against ischemic injury in the intact rat heart. Anesthesiology 2004;101:918–923. [DOI] [PubMed] [Google Scholar]
- 89. Zhang Y, Irwin MG, Wong TM, Chen M, Cao CM. Remifentanil preconditioning confers cardioprotection via cardiac kappa‐ and delta‐opioid receptors. Anesthesiology 2005;102:371–388. [DOI] [PubMed] [Google Scholar]
- 90. Kim HS, Cho JE, Hwang KC, Shim YH, Lee JH, Kwak YL. Diabetes mellitus mitigates cardioprotective effects of remifentanil preconditioning in ischemia‐reperfused rat heart in association with anti‐apoptotic pathways of survival. Eur J Pharmacol 2010;628:132–139. [DOI] [PubMed] [Google Scholar]
- 91. Wong GT, Huang Z, Ji S, Irwin MG. Remifentanil reduces the release of biochemical markers of myocardial damage after coronary artery bypass surgery: A randomized trial. J Cardiothorac Vasc Anesth 2010;24:790–796. [DOI] [PubMed] [Google Scholar]
- 92. Schultz JE, Hsu AK, Nagase H, Gross GJ. TAN‐67, a δ1‐opioid receptor agonist, reduces infarct size via activation of Gi/o proteins and KATP channels. Am J Physiol 1998;274:H909–H914. [DOI] [PubMed] [Google Scholar]
- 93. Kevelaitis E, Peynet J, Mouas C, Launay JM, Menasche P. Opening of potassium channels: The common cardioprotective link between preconditioning and natural hibernation? Circulation 1999;99:3079–3085. [DOI] [PubMed] [Google Scholar]
- 94. Valtchanova‐Matchouganska A, Ojewole JA. Involvement of opioid delta (δ)‐ and kappa (κ)‐receptors in ischemic preconditioning in a rat model of myocardial infarction. Methods Find Exp Clin Pharmacol 2002;24:139–144. [DOI] [PubMed] [Google Scholar]
- 95. Ikeda Y, Miura T, Sakamoto J, Miki T, Tanno M, Kobayashi H, Ohori K, Takahashi A, Shimamoto K. Activation of ERK and suppression of calcineurin are interacting mechanisms of cardioprotection afforded by δ‐opioid receptor activation. Basic Res Cardiol 2006;101:418–426. [DOI] [PubMed] [Google Scholar]
- 96. Seymour EM, Wu SY, Kovach MA, Romano MA, Traynor JR, Claycomb WC, Bolling SF. HL‐1 myocytes exhibit PKC and KATP channel‐dependent delta opioid preconditioning. J Surg Res 2003;114:187–194. [DOI] [PubMed] [Google Scholar]
- 97. Gross ER, Hsu AK, Gross GJ. Acute methadone treatment reduces myocardial infarct size via the δ‐opioid receptor in rats during reperfusion. Anesth Analg 2009;109:1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Takasaki Y, Wolff RA, Chien GL, van Winkle DM. Met5‐enkephalin protects isolated adult rabbit cardiomyocytes via δ‐opioid receptors. Am J Physiol 1999;277:H2442–H2450. [DOI] [PubMed] [Google Scholar]
- 99. Cao Z, Liu L, van Winkle DM. Activation of δ‐ and κ‐opioid receptors by opioid peptides protects cardiomyocytes via KATP channels. Am J Physiol Heart Circ Physiol 2003;285:H1032–H1039. [DOI] [PubMed] [Google Scholar]
- 100. Kuzume K, Wolff RA, Amakawa K, Kuzume K, van Winkle DM. Sustained exogenous administration of Met5‐enkephalin protects against infarction in vivo. Am J Physiol Heart Circ Physiol 2003;285:H2463–H2470. [DOI] [PubMed] [Google Scholar]
- 101. Kuzume K, Kuzume K, Cao Z, Liu L, van Winkle DM. Long‐term infusion of Met5‐enkephalin fails to protect murine hearts against ischemia‐reperfusion injury. Am J Physiol Heart Circ Physiol 2005;288:H1717–H1723. [DOI] [PubMed] [Google Scholar]
- 102. Sigg DC, Coles JA, Oeltgen PR, Iaizzo PA. Role of δ‐opioid receptor agonists on infarct size reduction in swine. Am J Physiol Heart Circ Physiol 2002;282:H1953–H1960. [DOI] [PubMed] [Google Scholar]
- 103. Fryer RM, Hsu AK, Nagase H, Gross GJ. Opioid‐induced cardioprotection against myocardial infarction and arrhythmias: mitochondrial versus sarcolemmal ATP‐sensitive potassium channels. J Pharmacol Exp Ther 2000;294:451–457. [PubMed] [Google Scholar]
- 104. Maslov LN, Platonov AA, Lasukova TV, IuB Lishmanov, Oeltgen P, Nagase H, Podoksenov IuK, Podoksenov AIu. Delta‐opioid receptor activation prevents appearance of irreversible damages of cardiomyocytes and exacerbates myocardial contractility dysfunction during ischemia and reperfusion. Patol Fiziol Eksp Ter 2006;4:13–17. [PubMed] [Google Scholar]
- 105. Platonov AA, Lasukova TV, Maslov LN, Downey JM, Nagaze H, Ovchinnikov MV. Delta‐opioid receptor agonists prevent the irreversible damage of cardiomyocytes in ischemised‐reperfused isolated rat heart. Eksp Klin Farmakol 2004;67:26–29. [PubMed] [Google Scholar]
- 106. Huh J, Gross GJ, Nagase H, Liang BT. Protection of cardiac myocytes via δ1‐opioid receptors, protein kinase C, and mitochondrial KATP channels. Am J Physiol Heart Circ Physiol 2001;280:H377–H383. [DOI] [PubMed] [Google Scholar]
- 107. Lasukova TV, Maslov LN, Platonov AA, Lishmanov IuB, Oeltgen P. The effect of stimulation of cardiac δ1‐opioid receptors on the resistance of isolated heart to the action of ischemia and reperfusion. Eksp Klin Farmakol 2005;68:9–24. [PubMed] [Google Scholar]
- 108. McPherson BC, Yao Z. Morphine mimics preconditioning via free radical signals and mitochondrial KATP channels in myocytes. Circulation 2001;103:290–295. [DOI] [PubMed] [Google Scholar]
- 109. Gross ER, Hsu AK, Gross GJ. GSK3β inhibition and KATP channel opening mediate acute opioid‐induced cardioprotection at reperfusion. Basic Res Cardiol 2007;102:341–349. [DOI] [PubMed] [Google Scholar]
- 110. Peart JN, Patel HH, Gross GJ. Delta‐opioid receptor activation mimics ischemic preconditioning in the canine heart. J Cardiovasc Pharmacol 2003;42:78–81. [DOI] [PubMed] [Google Scholar]
- 111. Maslov LN, Barzakh EI, Krylatov AV, Brown SA, Oeltgen PR, Govindaswami M, Chernysheva GA, Solenkova NV, Lishmanov AIu, Tsibul'nikov SIu, Krieg T, Zhang E. Significance of opioid receptors in regulation of cardiac tolerance in pathogenic impact of long‐term ischemia‐reperfusion in vivo. Ross Fiziol Zh Im I M Sechenova 2009;95:563–572. [PubMed] [Google Scholar]
- 112. Maslov LN, Lishmanov YB, Oeltgen PR, Barzakh EI, Krylatov AV, Naryzhnaya NV, Pei JM, Brown SA. Comparative analysis of the cardioprotective properties of opioid receptor agonists in a rat model of myocardial infarction. Acad Emerg Med 2010;17:1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lasukova TV, Maslov LN, Lishmanov YuB, Gross GJ. Role of δ1‐opiate receptors in regulation of contractility in isolated rat heart during normal oxygenation and ischemia/reperfusion. Bull Exp Biol Med 2004;31:80–86. [PubMed] [Google Scholar]
- 114. Lasukova TV, Maslov LN, Gorbunov AC, Tsibul'nuikov SIu. Intracellular mechanisms of opioidergic regulation of the myocardial function during normoxia and postischemic reperfusion. Ross Fiziol Zh Im I M Sechenova 2009;95:376–386. [PubMed] [Google Scholar]
- 115. Lishmanov YuB, Maslov LN, Lasukova TV, Platonov AA, Oeltgen P. Activation of δ‐opioid receptors increases resistance of isolated heart to ischemia/reperfusion: The role of cAMP and intracellular calcium. Bull Exp Biol Med 2005;32:45–51. [Google Scholar]
- 116. Watson MJ, Holt JD, O'Neill SJ, Wei K, Pendergast W, Gross GJ, Gengo PJ, Chang KJ. ARD‐353 [4‐((2R,5S)‐4‐(R)‐(4‐diethylcarbamoylphenyl)(3‐hydroxyphenyl)methyl)‐2,5‐dimethylpiperazin‐1‐ylmethyl)benzoic acid], a novel nonpeptide delta receptor agonist, reduces myocardial infarct size without central effects. J Pharmacol Exp Ther 2006;316:423–430. [DOI] [PubMed] [Google Scholar]
- 117. Gross ER, Hsu AK, Gross GJ. The JAK/STAT pathway is essential for opioid‐induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK‐3β;. Am J Physiol Heart Circ Physiol 2006;291:H827–H834. [DOI] [PubMed] [Google Scholar]
- 118. Maslov LN, Lishmanov YB, Oeltgen PR, Barzakh EI, Krylatov AV, Govindaswami M, Brown SA. Activation of peripheral δ2 opioid receptors increases cardiac tolerance to ischemia/reperfusion injury. Involvement of protein kinase C, NO‐synthase, KATP channels and the autonomic nervous system. Life Sci 2009;84:657–663. [DOI] [PubMed] [Google Scholar]
- 119. Wu S, Li H.Y, Wong TM. Cardioprotection of preconditioning by metabolic inhibition in the rat ventricular myocyte. Involvement of κ‐opioid receptor. Circ Res 1999;84:1388–1395. [DOI] [PubMed] [Google Scholar]
- 120. Lasukova TV, Maslov LN, Platonov AA, Guzarova NV, Lishmanov YuB. Cardioprotective effect of κ1‐opioid receptor activation and role of cAMP in its realization. Bull Exp Biol Med 2007;143:22–25. [DOI] [PubMed] [Google Scholar]
- 121. Lasukova TV, Maslov LN, Platonov AA, Guzarova NV, Lishmanov IuB. Role of κ1 opioid receptors and cAMP in regulation of cardiac tolerance to ischemia and reperfusion. Bull Exp Biol Med 2008;35:499–506. [Google Scholar]
- 122. Peart JN, Gross ER, Gross GJ. Effect of exogenous kappa‐opioid receptor activation in rat model of myocardial infarction. J Cardiovasc Pharmacol 2004;43:410–415. [DOI] [PubMed] [Google Scholar]
- 123. Schindler CW, Graczyk Z, Gilman JP, Negus SS, Bergman J, Mello NK, Goldberg SR. Effects of kappa opioid agonists alone and in combination with cocaine on heart rate and blood pressure in conscious squirrel monkeys. Eur J Pharmacol 2007;576:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wang Q, Sun Y, Li J, Xing W, Zhang S, Gu X, Feng N, Zhao L, Fan R, Wang Y, Yin W, Pei J. Quaternary ammonium salt of U50488H, a new κ‐opioid receptor agonist, protects rat heart against ischemia/reperfusion injury. Eur J Pharmacol 2014;737:177–184. [DOI] [PubMed] [Google Scholar]
- 125. Lasukova TV, Rebrova TY, Tam SV. Activation of mu‐opiate receptors as a factor of regulation of heart resistance to ischemia‐reperfusion and oxidative stress. Bull Exp Biol Med 2000;130:752–755. [DOI] [PubMed] [Google Scholar]
- 126. Lasukova TV, Maslov LN, Lishmanov IuB, Tam SV, Gross GJ. Effect of stimulation of μ‐opiate receptors on contractility of the isolated heart in normal oxygenation and during ischemia‐reperfusion. Ross Fiziol Zh Im I M Sechenova 2001;87:649–658. [PubMed] [Google Scholar]
- 127. Maslov LN, Lasukova TV, Solenkova NV, Lishmanov AIu, Bogomaz SA, Tam SV, Gross GJ. Participation of KATP‐channels in cardioprotective effect of mu‐opioid receptor agonists in acute ischemia and reperfusion of the isolated heart. Eksp Klin Farmakol 2001;64:23–27. [PubMed] [Google Scholar]
- 128. Lishmanov IuB, Maslov LN, Naumova AV, Bogomaz SA. μ‐Opiate receptor activation and increase in heart resistance to ischemic and reperfusion injury. Ross Fiziol Zh Im I M Sechenova 1998;84:1223–1230. [PubMed] [Google Scholar]
- 129. Schultz JE, Hsu AK, Gross GJ. Ischemic preconditioning in the intact rat heart is mediated by δ1‐ but not μ‐ or κ‐opioid receptors. Circulation 1998;97:1282–1289. [DOI] [PubMed] [Google Scholar]
- 130. Gross GJ, Hsu A, Nithipatikom K, Bobrova I, Bissessar E. Eribis peptide 94 reduces infarct size in rat hearts via activation of centrally located μ opioid receptors. J Cardiovasc Pharmacol 2012;59:194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kistner JR, Miller ED, Lake CL, Ross WT. Indices of myocardial oxygenation during coronary‐artery revascularization in man with morphine versus halothane anesthesia. Anesthesiology 1979;50:324–330. [DOI] [PubMed] [Google Scholar]
- 132. Kisin I, Markiewicz W, Birkhahn J. Effect of large doses of morphine on experimental myocardial ischemia in cats. Isr J Med Sci 1979;15:588–591. [PubMed] [Google Scholar]
- 133. Markiewicz W, Finberg JPM, Lichtig C. Morphine increases myocardial infarct size in rats. Anesth Analg 1982;61:843–846. [PubMed] [Google Scholar]
- 134. Lee AY, Wong TM. Naloxone reduces release of creatine kinase in the isolated ischemic rat heart. Proc Soc Exp Biol Med 1985;179:219–221. [DOI] [PubMed] [Google Scholar]
- 135. Lahti RA, Mickelson MM, McCall JM, von Voigtlander PF [3H]U‐69593 a highly selective ligand for the opioid κ receptor. Eur J Pharmacol 1985;109:281–284. [DOI] [PubMed] [Google Scholar]
- 136. Roques BP, Gacel G, Dauge V, Baamonde A, Calenco G, Turcaud S, Coric P, Fournie‐Zaluski MC. Novel approaches in the development of new analgesics. Neurophysiol Clin 1990;20:369–387. [DOI] [PubMed] [Google Scholar]
- 137. Chen Z, Li T, Zhang B. Morphine postconditioning protects against reperfusion injury in the isolated rat hearts. J Surg Res 2008;145: 287–294. [DOI] [PubMed] [Google Scholar]
- 138. Heijna MH, Padt M, Hogenboom F, Portoghese PS, Mulder AH, Schoffelmeer AN. Opioid receptor‐mediated inhibition of dopamine and acetylcholine release from slices of rat nucleus accumbens, olfactory tubercle and frontal cortex. Eur J Pharmacol 1990;181:267–278. [DOI] [PubMed] [Google Scholar]
- 139. Maslov LN, Lishmanov IuB, Headrick JP, Pei J‐M, Hanus L, Krylatov AV, Naryzhnaia NV. Perspective of creation of drugs on basis of opioids increasing cardiac tolerance to pathogenic impact of ischemia reperfusion. Eksp Klin Farmakol 2012;75:22–28. [PubMed] [Google Scholar]
- 140. Kazmierski W, Wire WS, Lui GK, Knapp RJ, Shook JE, Burks TF, Yamamura HI, Hruby VJ. Design and synthesis of somatostin analogues with topographical properties that lead to highly potent and specific μ opioid receptor antagonists with greatly reduced binding at somatostatin receptors. J Med Chem 1988;31:2170–2177. [DOI] [PubMed] [Google Scholar]
- 141. Wang TL, Huang YH, Chang H. Somatostatin analogue mimics acute ischemic preconditioning in a rat model of myocardial infarction. J Cardiovasc Pharmacol 2005;5:327–332. [DOI] [PubMed] [Google Scholar]
- 142. Peart JN, Gross GJ. Exogenous activation of δ‐ and κ‐opioid receptors affords cardioprotection in isolated murine heart. Basic Res Cardiol 2004;99:29–37. [DOI] [PubMed] [Google Scholar]
- 143. Aitchison KA, Baxter GF, Awan MM, Smith RM, Yellon DM, Opie LH. Opposing effects on infarction of delta and kappa opioid receptor activation in the isolated rat heart: Implications for ischemic preconditioning. Basic Res Cardiol 2000;95:1–10. [DOI] [PubMed] [Google Scholar]
- 144. Rothman RB, Bykov V, Xue BG, Xu H, De Costa BR, Jacobson AE, Rice KC, Kleiman JE, Brady LS. Interaction of opioid peptides and other drugs with multiple kappa receptors in rat and human brain. Evidence for species differences. Peptides 1992;13:977–987. [DOI] [PubMed] [Google Scholar]
- 145. Meine TJ, Roe MT, Chen AY, Patel MR, Washam JB, Ohman EM, Peacock WF, Pollack CV, Gibler WB, Peterson ED; CRUSADE Investigators . Association of intravenous morphine use and outcomes in acute coronary syndromes: Results from the CRUSADE Quality Improvement Initiative. Am Heart J 2005;149:1043–1049. [DOI] [PubMed] [Google Scholar]
- 146. Conahan TJ, Ominsky AJ, Wollman H, Stroth RA. A prospective random comparison of halothane and morphine for open‐heart anesthesia: One year's experience. Anesthesiology 1973;38:528–535. [DOI] [PubMed] [Google Scholar]
- 147. Lappas DG, Buckley MJ, Laver MB, Daggett WM, Lowenstein E. Left ventricular performance and pulmonary circulation following addition of nitrous oxide to morphine during coronary‐artery surgery. Anesthesiology 1975;43:61–69. [DOI] [PubMed] [Google Scholar]
- 148. v an de Werf F, Ardissino D, Betriu A, Cokkinos DV, Falk E, Fox KA, Julian D, Lengyel M, Neumann FJ, Ruzyllo W, Thygesen C, Underwood SR, Vahanian A, Verheugt FW, Wijns W. Task Force on the Management of Acute Myocardial Infarction of the European Society of Cardiology. Management of acute myocardial infarction in patients presenting with ST‐segment elevation. The Task Force on the Management of Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J 2003;24:28–66. [DOI] [PubMed] [Google Scholar]
- 149. Metelitsa VI. Handbook on Clinical Pharmacology of Cardiovascular Drugs. Moscow: Medpraktika; 1996. [Google Scholar]
- 150. Stanley TH, Gray NH, Stanford W, Armstrong R. The effects of high‐dose morphine on fluid and blood requirements in open‐heart operations. Anesthesiology 1973;38:536–541. [DOI] [PubMed] [Google Scholar]
- 151. Kulbertus H, Pierard L. Myocardial infarction. In: Jeffers JD, Fulvio B, Eds. Cardiology: McGraw‐Hill International; 1999. p 386. [Google Scholar]
- 152. Kettler D, Hilfiker O, Sonntag H. Narcotics and the coronary circulation In: Estafanous FG, Ed. Opioids in Anesthesia. Boston, London: Butterworth Publishers; 1991. p 194–201. [Google Scholar]
- 153. Khan MG. Cardiac Drug Therapy, 7th ed Totowa, NJ: Humana Press; 2007. p 186, 187, 236. [Google Scholar]
- 154. Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res 2006;70:181–190. [DOI] [PubMed] [Google Scholar]
- 155. Webster KA. Programmed death as a therapeutic target to reduce myocardial infarction. Trends Pharmacol Sci 2007;28:492–499. [DOI] [PubMed] [Google Scholar]
- 156. Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta 2007;1767:1007–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007;87:99–163. [DOI] [PubMed] [Google Scholar]
- 158. Hausenloy DJ, Ong SB, Yellon DM. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol 2009;104:189–202. [DOI] [PubMed] [Google Scholar]
- 159. Liu H, Zhang HY, McPherson BC, Baman T, Roth S, Shao Z, Zhu X, Yao Z. Role of opioid δ1 receptors, mitochondrial KATP channels, and protein kinase C during cardiocyte apoptosis. J Mol Cell Cardiol 2001;33:2007–2014. [DOI] [PubMed] [Google Scholar]
- 160. Okubo S, Tanabe Y, Takeda K, Kitayama M, Kanemitsu S, Kukreja RC, Takekoshi N. Ischemic preconditioning and morphine attenuate myocardial apoptosis and infarction after ischemia‐reperfusion in rabbits: Role of δ‐opioid receptor. Am J Physiol Heart Circ Physiol 2004;287:H1786–H1791. [DOI] [PubMed] [Google Scholar]
- 161. Barrere‐Lemaire S, Combes N, Sportouch‐Dukhan C, Richard S, Nargeot J, Piot C. Morphine mimics the antiapoptotic effect of preconditioning via an Ins(1,4,5)P3 signaling pathway in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 2005;288:H83–H88. [DOI] [PubMed] [Google Scholar]
- 162. Li L, Zhang H, Li T, Zhang B. Involvement of adenosine monophosphate‐activated protein kinase in morphine‐induced cardioprotection. J Surg Res 2011;169:179–187. [DOI] [PubMed] [Google Scholar]
- 163. Kim HS, Kim SY, Kwak YL, Hwang KC, Shim YH. Hyperglycemia attenuates myocardial preconditioning of remifentanil. J Surg Res 2012;174:231–237. [DOI] [PubMed] [Google Scholar]
- 164. Rong F, Peng Z, Ye MX, Zhang QY, Zhao Y, Zhang SM, Guo HT, Hui B, Wang YM, Liang C, Gu CH, Tao C, Cui Q, Yu SQ, Yi DH, Pei JM. Myocardial apoptosis and infarction after ischemia/reperfusion are attenuated by κ‐opioid receptor agonist. Arch Med Res 2009;40:227–234. [DOI] [PubMed] [Google Scholar]
- 165. Fryer RM, Hsu AK, Eells JT, Nagase H, Gross GJ. Opioid‐induced second window of cardioprotection: Potential role of mitochondrial KATP channels. Circ Res 1999;84:846–851. [DOI] [PubMed] [Google Scholar]
- 166. Fryer RM, Hsu AK, Gross GJ. ERK and p38 MAP kinase activation are components of opioid‐induced delayed cardioprotection. Basic Res Cardiol 2001;96:136–142. [DOI] [PubMed] [Google Scholar]
- 167. Guo Y, Stein AB, Wu WJ, Zhu X, Tan W, Li Q, Bolli R. Late preconditioning induced by NO donors, adenosine A1 receptor agonists, and δ1‐opioid receptor agonists is mediated by iNOS. Am J Physiol Heart Circ Physiol 2005;289:H2251–H2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Patel HH, Hsu A, Gross GJ. Delayed cardioprotection is mediated via a non‐peptide δ opioid agonist, SNC‐121, independent of opioid receptor stimulation. Basic Res Cardiol 2004;99:38–45. [DOI] [PubMed] [Google Scholar]
- 169. Shinmura K, Nagai M, Tamaki K, Bolli R. Gender and aging do not impair opioid‐induced late preconditioning in rats. Basic Res Cardiol 2004;99:46–55. [DOI] [PubMed] [Google Scholar]
- 170. Frassdorf J, Weber NC, Obal D, Toma O, Mullenheim J, Kojda G, Preckel B, Schlack W. Morphine induces late cardioprotection in rat hearts in vivo: the involvement of opioid receptors and nuclear transcription factor κB. Anesth Analg 2005;101:934–941. [DOI] [PubMed] [Google Scholar]
- 171. Jiang X, Shi E, Nakajima Y, Sato S. COX‐2 mediates morphine‐induced delayed cardioprotection via an iNOS‐dependent mechanism. Life Sci 2006;78:2543–2549. [DOI] [PubMed] [Google Scholar]
- 172. Zhou JJ, Pei JM, Wang GY, Wu S, Wang WP, Cho CH, Wong TM. Inducible HSP70 mediates delayed cardioprotection via U‐50488H pretreatment in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 2001;281:H40–H47. [DOI] [PubMed] [Google Scholar]
- 173. Chen M, Zhou JJ, Kam KW, Qi JS, Yan WY, Wu S, Wong TM. Roles of KATP channels in delayed cardioprotection and intracellular Ca2+ in the rat heart as revealed by κ‐opioid receptor stimulation with U50488H. Br J Pharmacol 2003;140:750–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Wang GY, Zhou JJ, Shan J, Wong TM. Protein kinase C‐epsilon is a trigger of delayed cardioprotection against myocardial ischemia of kappa‐opioid receptor stimulation in rat ventricular myocytes. J Pharmacol Exp Ther 2001;299(2):603–610. [PubMed] [Google Scholar]
- 175. Yu CK, Li YH, Wong GT, Wong TM, Irwin MG. Remifentanil preconditioning confers delayed cardioprotection in the rat. Br J Anaesth 2007;99:632–638. [DOI] [PubMed] [Google Scholar]
- 176. Sun HT, Xue FS, Liu KP, Sun L, Xu YC, Liao X, Yang QY, Zhang YM. Effect of remifentanil preconditioning on myocardial ischemia‐reperfusion injury. Acta Acad Med Sci 2009;31:612–615. [PubMed] [Google Scholar]
- 177. Dickson EW, Blehar DJ, Carraway RE, Heard SO, Steinberg G, Przyklenk K. Naloxone blocks transferred preconditioning in isolated rabbit hearts. J Mol Cell Cardiol 2001;33:1751–1756. [DOI] [PubMed] [Google Scholar]
- 178. Dickson EW, Tubbs RJ, Porcaro WA, Lee WJ, Blehar DJ, Carraway RE, Darling CE, Przyklenk K. Myocardial preconditioning factors evoke mesenteric ischemic tolerance via opioid receptors and KATP channels. Am J Physiol Heart Circ Physiol 2002;283(1):H22–H28. [DOI] [PubMed] [Google Scholar]
- 179. Dickson EW, Ludwig PS, Ackermann LW, Buresh CT, Denning GM. Met5‐enkephalin‐Arg6‐Phe7 (MEAP): A cardioprotective hormonal opioid. Acad Emerg Med 2006;13:813–819. [DOI] [PubMed] [Google Scholar]
- 180. Patel HH, Moore J, Hsu AK, Gross GJ. Cardioprotection at a distance: Mesenteric artery occlusion protects the myocardium via an opioid sensitive mechanism. J Mol Cell Cardiol 2002;34:1317–1323. [DOI] [PubMed] [Google Scholar]
- 181. Weinbrenner C, Schulze F, Sarvary L, Strasser RH. Remote preconditioning by infrarenal aortic occlusion is operative via δ1‐opioid receptors and free radicals in vivo in the rat heart. Cardiovasc Res 2004;61:591–599. [DOI] [PubMed] [Google Scholar]
- 182. Zhang SZ, Wang NF, Xu J, Gao Q, Lin GH, Bruce IC, Xia Q. Kappa‐opioid receptors mediate cardioprotection by remote preconditioning. Anesthesiology 2006;105:550–556. [DOI] [PubMed] [Google Scholar]
- 183. Rehni AK, Singh N, Jaggi AS. Possible involvement of insulin, endogenous opioids and calcitonin gene‐related peptide in remote ischaemic preconditioning of the brain. Yakugaku Zasshi 2007;127:1013–1020. [DOI] [PubMed] [Google Scholar]
- 184. Rentoukas I, Giannopoulos G, Kaoukis A, Kossyvakis C, Raisakis K, Driva M, Panagopoulou V, Tsarouchas K, Vavetsi S, Pyrgakis V, Deftereos S. Cardioprotective role of remote ischemic periconditioning in primary percutaneous coronary intervention: Enhancement by opioid action. JACC Cardiovasc Interv 2010;3:49–55. [DOI] [PubMed] [Google Scholar]
- 185. Hiranuma T, Kitamura K, Taniguchi T, Kanai M, Arai Y, Iwao K, Oka T. Protection against dynorphin‐(1‐8) hydrolysis in membrane preparations by the combination of amastatin, captopril and phosphoramidon. J Pharmacol Exp Ther 1998;286:863–869. [PubMed] [Google Scholar]
- 186. Hiranuma T, Kitamura K, Taniguchi T, Kobayashi T, Tamaki R, Kanai M, Akahori K, Iwao K, Oka T. Effects of three peptidase inhibitors, amastatin, captopril and phosphoramidon, on the hydrolysis of [Met5]‐enkephalin‐Arg6‐Phe7 and other opioid peptides. Naunyn Schmiedebergs Arch Pharmacol 1998;357:276–282. [DOI] [PubMed] [Google Scholar]
- 187. Chang WL, Lee SS, Su MJ. Attenuation of post‐ischemia reperfusion injury by thaliporphine and morphine in rat hearts. J Biomed Sci 2005;12:611–619. [DOI] [PubMed] [Google Scholar]
- 188. Tsutsumi YM, Yokoyama T, Horikawa Y, Roth DM, Patel HH. Reactive oxygen species trigger ischemic and pharmacological postconditioning: in vivo and in vitro characterization. Life Sci 2007;81:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Jang Y, Xi J, Wang H, Mueller RA, Norfleet EA, Xu Z. Postconditioning prevents reperfusion injury by activating δ‐opioid receptors. Anesthesiology 2008;108:243–250. [DOI] [PubMed] [Google Scholar]
- 190. Mourouzis I, Saranteas T, Perimenis P, Tesseromatis C, Kostopanagiotou G, Pantos C, Cokkinos DV. Morphine administration at reperfusion fails to improve postischaemic cardiac function but limits myocardial injury probably via heat‐shock protein 27 phosphorylation. Eur J Anaesthesiol 2009;26:572–581. [DOI] [PubMed] [Google Scholar]
- 191. Huhn R, Heinen A, Weber NC, Schlack W, Preckel B, Hollmann MW. Ischaemic and morphine‐induced post‐conditioning: Impact of mKCa channels. Br J Anaesth 2010;105:589–595. [DOI] [PubMed] [Google Scholar]
- 192. Kim JH, Chun KJ, Park YH, Kim J, Kim JS, Jang YH, Lee MY, Park JH. Morphine‐induced postconditioning modulates mitochondrial permeability transition pore opening via delta‐1 opioid receptors activation in isolated rat hearts. Korean J Anesthesiol 2011;61:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Peart JN, Gross ER, Reichelt ME, Hsu A, Headrick JP, Gross GJ. Activation of kappa‐opioid receptors at reperfusion affords cardioprotection in both rat and mouse hearts. Basic Res Cardiol 2008;103:454–463. [DOI] [PubMed] [Google Scholar]
- 194. Wong GT, Li R, Jiang LL, Irwin MG. Remifentanil post‐conditioning attenuates cardiac ischemia‐reperfusion injury via κ or δ opioid receptor activation. Acta Anaesthesiol Scand 2010;54(4):510–518. [DOI] [PubMed] [Google Scholar]
- 195. Chun KJ, Park YH, Kim JS, Jang Y, Kim JH, Kim J, Lee MY. Comparison of 5 different remifentanil strategies against myocardial ischemia‐reperfusion injury. J Cardiothorac Vasc Anesth 2011;25:926–930. [DOI] [PubMed] [Google Scholar]
- 196. Karlsson LO, Grip L, Bissessar E, Bobrova I, Gustafsson T, Kavianipour M, Odenstedt J, Wikstrom G, Gonon AT. Opioid receptor agonist Eribis peptide 94 reduces infarct size in different porcine models for myocardial ischaemia and reperfusion. Eur J Pharmacol 2011;651:146–151. [DOI] [PubMed] [Google Scholar]
- 197. Karlsson LO, Bergh N, Li L, Bissessar E, Bobrova I, Gross GJ, Akyurek LM, Grip L. Dose‐dependent cardioprotection of enkephalin analogue Eribis peptide 94 and cardiac expression of opioid receptors in a porcine model of ischaemia and reperfusion. Eur J Pharmacol 2012;674:378–383. [DOI] [PubMed] [Google Scholar]
- 198. Wu Y, Gu EW, Zhu Y, Zhang L, Liu XQ, Fang WP. Sufentanil limits the myocardial infarct size by preservation of the phosphorylated connexin 43. Int Immunopharmacol 2012;13:341–346. [DOI] [PubMed] [Google Scholar]
- 199. Emmerson PJ, Liu M‐R, Woods JH, Medzihradsky F. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther 1994;271:1630–1637. [PubMed] [Google Scholar]
- 200. Chen QL, Gu EW, Zhang L, Cao YY, Zhu Y, Fang WP. Diabetes mellitus abrogates the cardioprotection of sufentanil against ischaemia/reperfusion injury by altering glycogen synthase kinase‐3β. Acta Anaesthesiol Scand 2013;57:236–242. [DOI] [PubMed] [Google Scholar]
- 201. Ha JY, Lee YC, Park SJ, Jang YH, Kim JH. Remifentanil postconditioning has cross talk with adenosine receptors in the ischemic‐reperfused rat heart. J Surg Res 2015;195:37–43. [DOI] [PubMed] [Google Scholar]
- 202. Knapp RJ, Landsman R, Waite S, Malatynska E, Varga E, Haq W, Hruby V, Roeske WR, Nagase H, Yamamura HJ. Properties of TAN‐67, a nonpeptidic δ‐opioid receptor agonist, at cloned human δ‐ and μ‐opioid receptors. Eur J Pharmacol 1995;291:129–134. [DOI] [PubMed] [Google Scholar]
- 203. Groban L, Vernon JC, Butterworth J. Intrathecal morphine reduces infarct size in a rat model of ischemia‐reperfusion injury. Anesth Analg 2004;98:903–909. [DOI] [PubMed] [Google Scholar]
- 204. Li R, Wong GT, Wong TM, Zhang Y, Xia Z, Irwin MG. Intrathecal morphine preconditioning induces cardioprotection via activation of delta, kappa, and mu opioid receptors in rats. Anesth Analg 2009;108:23–29. [DOI] [PubMed] [Google Scholar]
- 205. Wong GT, Ling Ling J, Irwin MG. Activation of central opioid receptors induces cardioprotection against ischemia‐reperfusion injury. Anesth Analg 2010;111:24–28. [DOI] [PubMed] [Google Scholar]
- 206. Ling Ling J, Wong GT, Yao L, Xia Z, Irwin MG. Remote pharmacological post‐conditioning by intrathecal morphine: cardiac protection from spinal opioid receptor activation. Acta Anaesthesiol Scand 2010;54:1097–1104. [DOI] [PubMed] [Google Scholar]
- 207. Lu Y, Dong C, Yu J, Li L. Erratum to: Role of central and peripheral opioid receptors in the cardioprotection of intravenous morphine preconditioning. Ir J Med Sci 2016;185(2):545. [DOI] [PubMed] [Google Scholar]
- 208. Wong GT, Yao L, Xia Z, Irwin MG. Intrathecal morphine remotely preconditions the heart via a neural pathway. J Cardiovasc Pharmacol 2012;60:172–178. [DOI] [PubMed] [Google Scholar]
- 209. Lu Y, Hu J, Zhang Y, Dong C. Spinal neuronal NOS activation mediates intrathecal fentanyl preconditioning induced remote cardioprotection in rats. Int Immunopharmacol 2014;19:127–131. [DOI] [PubMed] [Google Scholar]
- 210. Polonskii VM, Yarygin KN, Krovosheev OG, Moskovkin GN, Vinogradov VA. Effect of the antiulcerative action (central or peripheral) of the synthetic enkaphalin analog dalargin in experimental cysteamine‐induced duodenal ulcer in rats. Bull Exp Biol Med 1987;103(4):488–490. [PubMed] [Google Scholar]
- 211. Varga EV, Li X, Stropova D, Zalewska T, Landsman RS, Knapp RJ, Malatynska E, Kawai K, Mizusura A, Nagase H, Calderon SN, Rice K, Hruby VJ, Roeske WR, Yamamura HI. The third extracellular loop of the human δ‐opioid receptor determines the selectivity of δ‐opioid agonists. Mol Pharmacol 1996;50:1619–1624. [PubMed] [Google Scholar]
- 212. Szabo B, Hedler L, Ensinger H, Starke K. Opioid peptides decrease noradrenaline release and blood pressure in the rabbit at peripheral receptors. Naunyn Schmiedebergs Arch Pharmacol 1986;332:50–56. [DOI] [PubMed] [Google Scholar]
- 213. Benedict PE, Benedict MB, Su TP, Bolling SF. Opiate drugs and δ‐receptor‐mediated myocardial protection. Circulation 1999;100:II357–II360. [DOI] [PubMed] [Google Scholar]
- 214. Shi E, Jiang X, Bai H, Gu T, Chang Y, Wang J. Cardioprotective effects of morphine on rat heart suffering from ischemia and reperfusion. Chin Med J (Engl) 2003;116:1059–1062. [PubMed] [Google Scholar]
- 215. Kato R, Foex P. Fentanyl protects the heart against ischaemic injury via opioid receptors, adenosine A1 receptors and KATP channel linked mechanisms in rats. Br J Anaesth 2000;84:204–214. [DOI] [PubMed] [Google Scholar]
- 216. Kato R, Foex P. Fentanyl reduces infarction but not stunning via δ‐opioid receptors and protein kinase C in rats. Br J Anaesth 2000;84:608–614. [DOI] [PubMed] [Google Scholar]
- 217. Pyle WG, Smith TD, Hofmann PA. Cardioprotection with κ‐opioid receptor stimulation is associated with a slowing of cross‐bridge cycling. Am J Physiol Heart Circ Physiol 2000;297:H1941–H1948. [DOI] [PubMed] [Google Scholar]
- 218. De Costa BR, Bowen WD, Hellewell SB, George C, Rothman RB, Reid AA, Walker JM, Jacobson AE, Rice KC. Alterations in the stereochemistry of the κ‐selective opioid agonist U50,488 results in high‐affinity σ ligands. J Med Chem 1989;32:1996–2002. [DOI] [PubMed] [Google Scholar]
- 219. Townsend D, Brown DR. Characterization of specific δ‐opioid binding sites in the distal small intestine of swine. Eur J Pharmacol 2003;482:111–116. [DOI] [PubMed] [Google Scholar]
- 220. Poonyachoti S, Portoghese PS, Brown DR. Characterization of opioid receptors modulating neurogenic contractions of circular muscle from porcine ileum and evidence that delta‐ and kappa‐opioid receptors are coexpressed in myenteric neurons. J Pharmacol Exp Ther 2001;297:69–77. [PubMed] [Google Scholar]
- 221. Genade S, Moolman JA, Lochner A. Opioid receptor stimulation acts as mediator of protection in ischaemic preconditioning. Cardiovasc J S Afr 2001;12:8–16. [PubMed] [Google Scholar]
- 222. Gacel G, Fournie‐Zaluski M‐C, Fellion E, Roques BP. Evidence of the preferential involvement of μ receptors in analgesia using enkephalins highly selective for peripheral μ or δ receptors. J Med Chem 1981;24:1119–1124. [DOI] [PubMed] [Google Scholar]
- 223. Bolling SF, Badhwar V, Schwartz CF, Oeltgen PR, Kilgore K, Su TP. Opioids confer myocardial tolerance to ischemia: Interaction of delta opioid agonists and antagonists. J Thorac Cardiovasc Surg 2001;122:476–481. [DOI] [PubMed] [Google Scholar]
- 224. Rothman RB, Xu H, Char GU, Kim A, De Costa BR, Rice KC, Zimmerman DM. Phenylpiperidine opioid antagonists that promote weight loss in rats have high affinity for the κ2B (enkephalin‐sensitive) binding site. Peptides 1993;14:17–20. [DOI] [PubMed] [Google Scholar]
- 225. Sigg DC, Coles JA, Gallagher WJ, Oeltgen PR, Iaizzo PA. Opioid preconditioning: Myocardial function and energy metabolism. Ann Thorac Surg 2001;72:1576–1582. [DOI] [PubMed] [Google Scholar]
- 226. Shinmura K, Nagai M, Tamaki K, Tani M, Bolli R. COX‐2‐derived prostacyclin mediates opioid‐induced late phase of preconditioning in isolated rat hearts. Am J Physiol Heart Circ Physiol 2002;283:H2534–H2543. [DOI] [PubMed] [Google Scholar]
- 227. Wu D, Soong Y, Zhao GM, Szeto HH. A highly potent peptide analgesic that protects against ischemia‐reperfusion‐induced myocardial stunning. Am J Physiol Heart Circ Physiol 2002;283:H783–H791. [DOI] [PubMed] [Google Scholar]
- 228. Schiller PW, Nguyen TM, Berezowska I, Dupuis S, Weltrowska G, Chung NN, Lemieux C. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur J Med Chem 2000;35:895–901. [DOI] [PubMed] [Google Scholar]
- 229. Peart JN, Gross ER, Headrick JP, Gross GJ. Impaired p38 MAPK/HSP27 signaling underlies aging‐related failure in opioid‐mediated cardioprotection. J Mol Cell Cardiol 2007;42:972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Romano MA, McNish R, Seymour EM, Traynor JR, Bolling SF. Differential effects of opioid peptides on myocardial ischemic tolerance. J Surg Res 2004;119:46–50. [DOI] [PubMed] [Google Scholar]
- 231. Chen BP, Mao HJ, Fan FY, Bruce IC, Xia Q. Delayed uncoupling is related to cardioprotection induced by κ‐agonist U‐50,488H in rat heart. Scand Cardiovasc J 2005;39:375–382. [DOI] [PubMed] [Google Scholar]
- 232. Shan Y, Sun S, Yang X, Weil MH, Tang W. Opioid receptor agonist reduces myocardial ischemic injury when administered during early phase of myocardial ischemia. Resuscitation 2010;81:761–765. [DOI] [PubMed] [Google Scholar]
- 233. Cheng L, Ma S, Wei LX, Guo HT, Huang LY, Bi H, Fan R, Li J, Liu YL, Wang YM, Sun X, Zhang QY, Yu SQ, Yi DH, Ma XL, Pei JM. Cardioprotective and antiarrhythmic effect of U50,488H in ischemia/reperfusion rat heart. Heart Vessels 2007;22:335–344. [DOI] [PubMed] [Google Scholar]
- 234. Grosse Hartlage MA, Theisen MM, Monteiro de Oliveira NP, van Aken H, Fobker M, Weber TP. κ‐Opioid receptor antagonism improves recovery from myocardial stunning in chronically instrumented dogs. Anesth Analg 2006;103:822–832. [DOI] [PubMed] [Google Scholar]
- 235. Panova EI. Short‐term and long‐term prognosis in patients with myocardial infarction. Klin Med 2008;86:19–23. [PubMed] [Google Scholar]
- 236. Fagbemi O, Kane KA, Lepran J, Parratt JR, Szekeres L. Anti‐arrhythmic actions of meptazinol, a partial agonist at opiate receptors, in acute myocardial ischemia. Br J Pharmacol 1983;78:455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Saini V, Carr DB, Verrier RL. Comparative effects of the opioids fentanyl and buprenorphine on ventricular vulnerability during acute coronary artery occlusion. Cardiovasc Res 1989;23:1001–1006. [DOI] [PubMed] [Google Scholar]
- 238. Hess L, Vrana M, Vranova Z, Fejfar Z. The antifibrillatory effect of fentanyl, sufentanil and carfentanil in acute phase of local myocardial ischemia in the dog. Acta Cardiol 1989;44:303–311. [PubMed] [Google Scholar]
- 239. Hansen DD, Hickey PR. Anesthesia for hypoplastic left heart syndrome: Use of high dose fentanyl in 30 naonatans. Anesth Analg 1986;65:127–132. [PubMed] [Google Scholar]
- 240. Maslov LN, Lishmanov YuB. The anti‐arrhythmic effect of D‐Ala2,Leu5,Arg6‐enkephalin and its possible mechanism. Int J Cardiol 1993;40:89–94. [DOI] [PubMed] [Google Scholar]
- 241. Mikhailova SD, Semushkina TM, Bebiakova NA. Effect of dalargin on the course of myocardial ischemia. Kardiologiia 1991;31:13–15. [PubMed] [Google Scholar]
- 242. Grekova TI, Reznikov KM, Vinokurova OV, Kireeva AA, Taratinova TI, Nikolaevskii VA, Shchetinkina NA. The effect of dalargin on the course of experimental cardiac arrhythmias. Eksp Klin Farmakol 1994;57:24–26. [PubMed] [Google Scholar]
- 243. Lishmanov YuB, Maslov LN, Naryzhnaya NV, Tam SW. Ligands for opioid and σ‐receptors improve cardiac electrical stability in rat models of post‐infarction cardiosclerosis and stress. Life Sci 1999;65:PL13–PL17. [DOI] [PubMed] [Google Scholar]
- 244. Korobov NV. Dalargin is an opioid‐like peptide with peripheral action. Farmakol Toksikol 1988;51:35–38. [PubMed] [Google Scholar]
- 245. Pencheva N, Pospisek J, Hauzerova L, Barth T, Milanov P. Activity profiles of dalargin and its analogues in mu‐, delta‐ and kappa‐opioid receptor selective bioassays. Br J Pharmacol 1999;128:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Maslov LN, Oeltgen PR, Lishmanov YuB, Brown SA, Barzakh EI, Krylatov AV, Pei J‐M. Activation of peripheral opioid receptors increases cardiac tolerance to arrhythmogenic effect of ischemia/reperfusion. Acad Emer Med 2014;21:31–39. [DOI] [PubMed] [Google Scholar]
- 247. Maslov LN, Lishmanov IuB, Naryzhnaia NV, Krylatov AV, Tam SV. Ligands of opioid and sigma receptors and correction of cardiac electrical instability in post‐infarction cardiosclerosis. Eksp Klin Farmakol 2001;64:38–41. [PubMed] [Google Scholar]
- 248. Maslov LN, Krylatov AV, Naryzhnaia NV, Solenkova NV, AIu Lishmanov, Bogomaz SA, Gross GJ, Stefano JB, Loktiushina BA. Interactions of peripheral μ‐opioid receptors and KATP‐channels in regulation of cardiac electrical stability in ischemia, reperfusion, and postinfarction cardiosclerosis. Ross Fiziol Zh Im I M Sechenova 2002;88:842–850. [PubMed] [Google Scholar]
- 249. Dhawan BN, Cesselin F, Raghubir R, Reisine T, Bradley PB, Portoghese PS, Hamon M. International union of pharmacology. XII. Classification of opioid receptors. Pharmacol Rev 1996;48:567–592. [PubMed] [Google Scholar]
- 250. Solenkova NV, Maslov LN, Budankova EV, Lishmanov AIu, Oeltgen P, Govindaswami M, Bespalova ZhD, Ovchinnikov MV, Nagaze H. Comparative study of the antiarrhythmic activity of mu‐ and delta‐opioid receptor agonists during acute cardiac ischemia and reperfusion models in rats. Eksp Klin Farmakol 2005;68:25–29. [PubMed] [Google Scholar]
- 251. Maslov LN, Lishmanov YB, Solenkova NV, Gross GJ, Stefano GB, Tam SW. Activation of peripheral delta opioid receptors eliminates cardiac electrical instability in a rat model of post‐infarction cardiosclerosis via mitochondrial ATP‐dependent K+ channels. Life Sci 2003;73:947–952. [DOI] [PubMed] [Google Scholar]
- 252. Pugsley MK, Penz WP, Walker MJ, Wong TM. Antiarrhythmic effects of U‐50,488H in rats subject to coronary artery occlusion. Eur J Pharmacol 1992;212:15–19. [DOI] [PubMed] [Google Scholar]
- 253. Ugdyzhekova DS, Maslov LN, Krylatov AV, Lishmanov IuB, Tam SV. Specificity of the anti‐arrhythmic effect of κ1‐opioid receptor agonists. Eksp Klin Farmakol 2001;64:17–20. (in Russian) [PubMed] [Google Scholar]
- 254. Pugsley MK, Saint DA, Penz MP, Walker MJ. Electrophysiological and antiarrhythmic actions of the kappa agonist PD 129290, and its R,R(+)‐enantiomer, PD 129289. Br J Pharmacol 1993;110:1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Pugsley MK, Saint DA, Walker MJA. An electrophysiological basis for the antiarrhythmic actions of the κ‐opioid receptor agonist U‐50,488H. Eur J Pharmacol 1994;261:303–309. [DOI] [PubMed] [Google Scholar]
- 256. Pugsley MK, Yu EJ, Goldin AL. Potent and use‐dependent block of cardiac sodium channels by U‐50,488H, a benzeneacetamide kappa opioid receptor agonist. Exp Clin Cardiol 2001;6:61–71. [PMC free article] [PubMed] [Google Scholar]
- 257. Maslov LN, AYu Lishmanov, Solenkova NV, Budankova EV, Crawford D, Wong TM, Chang WC, Bray LX. The antiarrhythmic effect of (‐)‐U‐50,488 in rats with acute ischemia and reperfusion of heart is mediated by κ1‐opioid receptor activation. Eksp Klin Farmakol 2005;68:25–29. [PubMed] [Google Scholar]
- 258. Maslov LN, Lishmanov AYu, Budankova EV, Stakheev DL, Solenkova NV, Barzakh EI, Oeltgen PR, Gross GJ, Chang WC. Contribution of the endogenous opioid system to regulation of heart resistance to the arrhythmogenic effect of short‐term ischemia and reperfusion. Bull Exp Biol Med 2005;32:375–380. [DOI] [PubMed] [Google Scholar]
- 259. Mikhailova SD, Vasil'eva TV, Semushkina TM, Storozhakov GI. Role of sympathetic nervous system in protective effects of selective κ‐opiate receptor agonist dynorphin A1–13 on the incidence of cardiac arrhythmia during myocardial ischemia. Bull Exp Biol Med 2000;129:27–29. [Google Scholar]
- 260. Lishmanov AYu, Lasukova TV, Maslov LN, Platonov AA. The role of kappa‐opioid receptors in regulation of cardiac resistance against arrhythmogenic action of ischemia and reperfusion. Ross Fiziol Zh Im I M Sechenova 2006;92:1419–1428. [PubMed] [Google Scholar]
- 261. Lee AYS, Zhan CY, Wong TM. Effects of β‐endorphin on the contraction and electrical activity of the isolated perfused rat heart. Int J Peptide Protein Res 1984;24:525–528. [DOI] [PubMed] [Google Scholar]
- 262. Lee AYS. Endogenous opioid peptides and cardiac arrhythmias. Int J Cardiol 1990;27(2):145–151. [DOI] [PubMed] [Google Scholar]
- 263. Mikhailova SD, Storozhakov GI, Kudinova AV, Semushkina TM. Different antiarrhythmic effects of dalargin and β‐endorphin in severe myocardial ischemia during stimulation of the sensorimotor cortex. Bull Exp Biol Med 1997;124:645–647. [PubMed] [Google Scholar]
- 264. Lee AYS, Wong TM. Effects of dynorphin1‐13 on cardiac rhythm and cyclic adenosine monophosphate (cAMP) levels in the isolated perfused rat heart. Neurosci Lett 1987;80:289–292. [DOI] [PubMed] [Google Scholar]
- 265. Wong TM, Lee AYS, Tai KK. Effect of drugs interacting with opioid receptors during normal perfusion or ischemia and reperfusion in the isolated rat heart – An attempt to identify cardiac opioid receptors subtype(s) involved in arrhythmogenesis. J Mol Cell Cardiol 1990;22:1167–1175. [DOI] [PubMed] [Google Scholar]
- 266. Wu J‐P, Chen Y‐T, Lee AY‐S opioids in myocardial ischemia: Potentiating effects of dynorphin on ischaemic arrhythmia, bradycardia and cardiogenic shock following coronary artery occlusion in the rat. Eur Heart J 1993;14(9):1273–1277. [DOI] [PubMed] [Google Scholar]
- 267. Mikhailova SD, Glushchenko NV, Semushkina TM, Storozhakov GI. Peculiarities of ischemic cardiac arrhythmias in cats against the background of stimulation of sensorimotor cortex and administration of selective opiate receptor agonists. Bull Exp Biol Med 2000;129:423–424. [DOI] [PubMed] [Google Scholar]
- 268. Coles JA, Sigg DC, Iaizzo PA. The role of κ‐opioid receptor activation in pharmacological preconditioning of swine. Am J Physiol Heart Circ Physiol 2003;284:H2091–H2099. [DOI] [PubMed] [Google Scholar]
- 269. Dumont M, Lemaire S. Characterization of non‐opioid [3H]Dynorphin A‐(1‐13) binding sites in the rat heart. J Mol Cell Cardiol 1993;25:983–991. [DOI] [PubMed] [Google Scholar]
- 270. Dumont M, Lemaire S. Interactions of dynorphin A‐(1‐13) and nociceptin with cardiac D2 binding sites: Inhibition of ischemia‐evoked release of noradrenaline from synaptosomal‐mitochondrial fractions. J Mol Cell Cardiol 2000;32:1567–1574. [DOI] [PubMed] [Google Scholar]
- 271. Nekrasova YN, Zolotarev YA, Navolotskaya EV. Detection of nonopioid β‐endorphin receptor in the rat myocardium. J Pept Sci 2012;18:83–87. [DOI] [PubMed] [Google Scholar]
- 272. Lishmanov YB, Maslov LN, Ugdyzhekova DS, Smagin GN. Participation of central kappa‐opioid receptor in arrhythmogenesis. Life Sci 1997;61:PL33–PL38. [DOI] [PubMed] [Google Scholar]
- 273. Lishmanov YB, Maslov LN, Ugdyzhekova DS. Participation of central and peripheral κ1 and κ2 opioid receptors in arrhythmogenesis. Clin Exp Pharmacol Physiol 1999;26:716–723. [DOI] [PubMed] [Google Scholar]
- 274. Huang XD, Lee AYS, Wong TM. Naloxone inhibits arrhythmias induced by coronary artery occlusion and reperfusion in anaesthetized dogs. Br J Pharmacol 1986;87:475–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Parratt JR, Sitsapesan R stereospecific antiarrhythmic effect of opioid receptor antagonist in myocardial ischemia. Br J Pharmacol 1986;87:621–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Sitsapesan R, Parratt JR. The effects of drugs interacting with opioid receptors on the early ventricular arrhythmias arising from myocardial ischaemia. Br J Pharmacol 1989;97:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Caldwell RW, Nagarajan R, Chryssanthis A, Tuttle RR. Actions of the opioid antagonist, nalmefene, and congers on reperfusion cardiac arrhythmias and regional left coronary blood flow. Pharmacology 1990;41:161–166. [DOI] [PubMed] [Google Scholar]
- 278. Murphy DB, Murphy MB. Opioid antagonist modulation of ischemia‐induced ventricular arrhythmias: A peripheral mechanism. J Cardiovasc Pharmacol 1999;33:122–125. [DOI] [PubMed] [Google Scholar]
- 279. Bolte C, Newman G, Schultz JEJ. Kappa and delta opioid receptor signaling is augmented in the failing heart. J Mol Cell Cardiol 2009;47(4):493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Kasper E, Ventura C, Ziman BD, Lakatta EG, Weisman H, Capogrossi MC. Effect of U‐50,488H on the contractile response of cardiomyopathic hamster ventricular myocytes. Life Sci 1992;50:2029–2035. [DOI] [PubMed] [Google Scholar]
- 281. Yu XC, Wang HX, Wong TM. Reduced inhibitory actions of adenosine A1 and κ1‐opioid receptor agonists on β‐adrenoceptors in spontaneously hypertensive rat heart. Clin Exp Pharmacol Physiol 1997;24:976–977. [DOI] [PubMed] [Google Scholar]
- 282. Pei JM, Wang YM, Zhu YL, Chen M, Wong TM. Signaling pathway mediated by kappa‐opioid receptor is impaired in cardiac hypertrophy. Acta Pharmacol Sin 2001;22:887–895. [PubMed] [Google Scholar]
- 283. Hill‐Pryor C, Dunbar JC. The effect of high fat‐induced obesity on cardiovascular and physical activity and opioid responsiveness in conscious rats. Clin Exp Hypertens 2006;28:133–145. [DOI] [PubMed] [Google Scholar]
- 284. Ouellette M, Brakier‐Gingras L. Increase in the relative abundance of proenkephalin: A messenger RNA in the ventricles of cardiomiopathic hamsters. Biochem Biophys Res Commun 1988;155:449–454. [DOI] [PubMed] [Google Scholar]
- 285. Dumont M, Lemaire S. Alterations of heart dynorphyn‐A in the development of sponatneously hypertensive rats. Neuropeptides 1990;15:43–48. [DOI] [PubMed] [Google Scholar]
- 286. Himura Y, Liang CS, Imai N, Delehanty JM, Woolf PD, Hood WB. Short‐term effects of naloxone on hemodynamics and baroreflex function in conscious dogs with pacing‐induced congestive heart failure. J Am Coll Cardiol 1994;23:194–200. [DOI] [PubMed] [Google Scholar]
- 287. Imai N, Kashiki M, Woolf PD, Liang CS. Comparison of cardiovascular effects of μ‐ and δ‐opioid receptor antagonists in dogs with congestive heart failure. Am J Physiol 1994;267:H912–H917. [DOI] [PubMed] [Google Scholar]
- 288. Forman LJ, Hock CE, Harwell M, Estilow‐Isabell S. Comparison of the effects of immobilization and pressure overload induced cardiac hypertrophy on immunoreactive beta‐endorphin. Life Sci 1995;57:2041–2047. [DOI] [PubMed] [Google Scholar]
- 289. Oldroyd KG, Gray CE, Carter R, Harvey K, Borland W, Beastall G, Cobbe SM. Activation and inhibition of the endogenous opioid system in human heart failure. Br Heart J 1995;73:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Bolte C, Newman G, Schultz JEJ. Hypertensive state, independent of hypertrophy, exhibits an attenuated decrease in systolic function on cardiac κ‐opioid receptor stimulation. Am J Physiol Heart Circ Physiol 2009;296:H967–H975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Gross ER, Hsu AK, Gross GJ. Diabetes abolishes morphine‐induced cardioprotection via multiple pathways upstream of glycogen synthase kinase‐3β. Diabetes 2007;56:127–136. [DOI] [PubMed] [Google Scholar]
- 292. Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM. Preconditioning the diabetic heart. The importance of Akt phosphorylation. Diabetes 2005;54:2360–2364. [DOI] [PubMed] [Google Scholar]
- 293. Ludman AJ, Yellon DM, Hausenloy DJ. Cardiac preconditioning for ischaemia: Lost in translation. Dis Models Mech 2010;3:35–38. [DOI] [PubMed] [Google Scholar]
- 294. Wong TM, Lee AY. Chronic morphine treatment reduces the incidence of ventricular arrhythmias in the isolated rat heart induced by dynorphin1‐13 or myocardial ischemia and reperfusion. Neurosci Lett 1987;77:61–65. [DOI] [PubMed] [Google Scholar]
- 295. Peart JN, Gross GJ. Chronic exposure to morphine produces a marked cardioprotective phenotype in aged mouse hearts. Exp Gerontol 2004;39:1021–1026. [DOI] [PubMed] [Google Scholar]
- 296. Peart JN, Gross GJ. Morphine‐tolerant mice exhibit a profound and persistent cardioprotective phenotype. Circulation 2004;109:1219–1122. [DOI] [PubMed] [Google Scholar]
- 297. Peart JN, Gross GJ. Cardioprotective effects of acute and chronic opioid treatment are mediated via different signaling pathways. Am J Physiol Heart Circ Physiol 2006;291:H1746–H1753. [DOI] [PubMed] [Google Scholar]
- 298. Skrabalova J, Neckar J, Hejnova L, Bartonova I, Kolar F, Novotny J. Antiarrhythmic effect of prolonged morphine exposure is accompanied by altered myocardial adenylyl cyclase signaling in rats. Pharmacol Rep 2012;64:351–359. [DOI] [PubMed] [Google Scholar]
- 299. Yin W, Zhang P, Huang JH, Zhang QY, Fan R, Li J, Zhou JJ, Hu YZ, Guo HT, Zhang SM, Wang YM, Kaye AD, Gu CH, Liu JC, Cheng L, Cui Q, Yi DH, Pei JM. Stimulation of κ‐opioid receptor reduces isoprenaline‐induced cardiac hypertrophy and fibrosis. Eur J Pharmacol 2009;607:135–142. [DOI] [PubMed] [Google Scholar]
- 300. Robson P. Human studies of cannabinoids and medicinal cannabisHandb Exp Pharmacol. 2005;168:719–756. [DOI] [PubMed] [Google Scholar]
- 301. Cowan A, Zhu XZ, Mosberg HI, Omnaas JR, Porreca F. Direct dependence studies in rats with agents selective for different types of opioid receptor. J Pharmacol Exp Ther 1988;246:950–955. [PubMed] [Google Scholar]
- 302. Hilal‐Dandan R, Brunton L. Section II: Neuropharmacology; opioids, analgesia, and pain management. In Goodman and Gilman Manual of Pharmacology and Therapeutics. 2nd ed. New York, Chicago, San Francisco: McGraw‐Hill Education; 2013.
- 303. Polonskii VM, Iarygin KN, Krivosheev OG, Moskovkin GN, Vinogradov VA. The site (central or peripheral) of the anti‐ulcer action of dalargin, a synthetic analog of endogenous opioids in an experimental model of cysteamine‐induced duodenal ulcer in rats. Bull Exp Biol Med 1987;103:433–434. [PubMed] [Google Scholar]
- 304. Mashkovsky MD. Drugs. Manual. Moscow: Novaya Volna; 2002.
- 305. Cohen MV, Downey JM. Signalling pathways and mechanisms of protection in pre‐ and postconditioning: Historical perspective and lessons for the future. Br J Pharmacol 2015;172(8):1913–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Fryer RM, Patel HH, Hsu AK, Gross GJ. Stress‐activated protein kinase phosphorylation during cardioprotection in the ischemic myocardium. Am J Physiol Heart Circ Physiol 2001;281:H1184–H1192. [DOI] [PubMed] [Google Scholar]
- 307. Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly‐Rosen D. Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem 2004;279:47985–47991. [DOI] [PubMed] [Google Scholar]
- 308. Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly‐Rosen D. Inhibition of δ‐protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation 2003;108:2304–2307. [DOI] [PubMed] [Google Scholar]
- 309. Liu GS, Cohen MV, Mochly‐Rosen D, Downey JM. Protein kinase C‐ε is responsible for the protection of preconditioning in rabbit cardiomyocytes. J Mol Cell Cardiol 1999;31:1937–1948. [DOI] [PubMed] [Google Scholar]
- 310. Soltoff SP. Rottlerin: An inappropriate and ineffective inhibitor of PKCδ. Trends Pharmacol Sci 2007;28:453–458. [DOI] [PubMed] [Google Scholar]
- 311. Zhang Y, Chen ZW, Girwin M, Wong TM. Remifentanil mimics cardioprotective effect of ischemic preconditioning via protein kinase C activation in open chest of rats. Acta Pharmacol Sin 2005;26:546–550. [DOI] [PubMed] [Google Scholar]
- 312. Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: Taking a RISK for cardioprotection. Heart Fail Rev 2007;12:217–234. [DOI] [PubMed] [Google Scholar]
- 313. Cao Z, Liu L, van Winkle DM. Met5‐enkephalin‐induced cardioprotection occurs via transactivation of EGFR and activation of PI3K. Am J Physiol Heart Circ Physiol 2005;288:H1955–H1964. [DOI] [PubMed] [Google Scholar]
- 314. Wu X, Zhang B, Fan R, Zhao L, Wang Y, Zhang S, Kaye AD, Huang L, Pei J. U50,488H inhibits neutrophil accumulation and TNF‐α induction induced by ischemia‐reperfusion in rat heart. Cytokine 2011;56:503–507. [DOI] [PubMed] [Google Scholar]
- 315. Liu X, Jing G, Bai J, Yuan H. Effect of sufentanil preconditioning on myocardial P‐Akt expression in rats during myocardial ischemia‐reperfusion. Nan Fang Yi Ke Da Xue Xue Bao 2014;34:335–340. [PubMed] [Google Scholar]
- 316. Xu J, Tian W, Ma X, Guo J, Shi Q, Jin Y, Xi J, Xu Z. The molecular mechanism underlying morphine‐induced Akt activation: Roles of protein phosphatases and reactive oxygen species. Cell Biochem Biophys 2011;61:303–311. [DOI] [PubMed] [Google Scholar]
- 317. Fryer RM, Pratt PF, Hsu AK, Gross GJ. Differential activation of extracellular signal regulated kinase isoforms in preconditioning and opioid‐induced cardioprotection. J Pharmacol Exp Ther 2001;296:642–649. [PubMed] [Google Scholar]
- 318. Kim JH, Jang YH, Chun KJ, Kim J, Park YH, Kim JS, Kim JM, Lee MY. Kappa‐opioid receptor activation during reperfusion limits myocardial infarction via ERK1/2 activation in isolated rat hearts. Korean J Anesthesiol 2011;60:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Zhang Y, Gu EW, Zhang J, Chen ZW. Role of p38 mitogen‐activated protein kinases in cardioprotection of morphine preconditioning. Chin Med J (Engl) 2007;120:777–781. [PubMed] [Google Scholar]
- 320. Fryer RM, Wang Y, Hsu AK, Nagase H, Gross GJ. Dependence of δ1‐opioid receptor‐induced cardioprotection on a tyrosine kinase‐dependent but not a Src‐dependent pathway. J Pharmacol Exp Ther 2001;299:477–482. [PubMed] [Google Scholar]
- 321. Peart JN, Gross GJ. Adenosine and opioid receptor‐mediated cardioprotection in the rat: Evidence for cross‐talk between receptors. Am J Physiol Heart Circ Physiol 2003;285:H81–H89. [DOI] [PubMed] [Google Scholar]
- 322. Peart JN, Gross GJ. Cardioprotection following adenosine kinase inhibition in rat hearts. Basic Res Cardiol 2005;100:328–336. [DOI] [PubMed] [Google Scholar]
- 323. Headrick JP, See Hoe LE, Du Toit EF, Peart JN. Opioid receptors and cardioprotection – ‘Opioidergic conditioning’ of the heart. Br J Pharmacol 2015;172:2026–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Maslov LN, Headrick JP, Mechoulam R, Krylatov AV, Lishmanov AYu, Barzakh EI, Naryzhnaya NV, Zhang Y. The role of receptor transactivation in the cardioprotective effects of preconditioning and postconditioning. Neurosci Behav Physiol 2013;43:1015–1022. [PubMed] [Google Scholar]
- 325. Forster K, Kuno A, Solenkova N, Felix SB, Krieg T. The δ‐opioid receptor agonist DADLE at reperfusion protects the heart through activation of pro‐survival kinases via EGF receptor transactivation. Am J Physiol Heart Circ Physiol 2007;293:H1604–H1608. [DOI] [PubMed] [Google Scholar]
- 326. Krieg T, Cui L, Qin Q, Cohen MV, Downey JM. Mitochondrial ROS generation following acetylcholine‐induced EGF receptor transactivation requires metalloproteinase cleavage of proHB‐EGF. J Mol Cell Cardiol 2004;36:435–443. [DOI] [PubMed] [Google Scholar]
- 327. Wang GY, Wu S, Pei JM, Yu XC, Wong TM. K‐ but not δ‐opioid receptors mediate effects of ischemic preconditioning on both infarct and arrhythmia in rats. Am J Physiol Heart Circ Physiol 2001;280:H384–H391. [DOI] [PubMed] [Google Scholar]
- 328. Cao CM, Chen M, Wong TM. The KCa channel as a trigger for the cardioprotection induced by kappa‐opioid receptor stimulation – Its relationship with protein kinase C. Br J Pharmacol 2005;145:984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM. Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial KATP channels. Circ Res 2001;89:273–278. [DOI] [PubMed] [Google Scholar]
- 330. McPherson BC, Yao Z. Signal transduction of opioid‐induced cardioprotection in ischemia‐reperfusion. Anesthesiology 2001;94:1082–1088. [DOI] [PubMed] [Google Scholar]
- 331. Obame FN, Plin‐Mercier C, Assaly R, Zini R, Dubois‐Rande JL, Berdeaux A, Morin D. Cardioprotective effect of morphine and a blocker of glycogen synthase kinase 3β, SB216763 [3‐(2,4‐dichlorophenyl)‐4(1‐methyl‐1H‐indol‐3‐yl)‐1H‐pyrrole‐2,5‐dione], via inhibition of the mitochondrial permeability transition pore. J Pharmacol Exp Ther 2008;326:252–258. [DOI] [PubMed] [Google Scholar]
- 332. Yang XM, Liu Y, Cui L, Yang X, Liu Y, Tandon N, Kambayashi J, Downey JM, Cohen MV Platelet P2Y12 blockers confer direct postconditioning‐like protection in reperfused rabbit hearts. J Cardiovasc Pharmacol Ther 2013;18:251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Iliodromitis EK, Cohen MV, Dagres N, Andreadou I, Kremastinos DT, Downey JM. What is wrong with cardiac conditioning? We may be shooting at moving targets. J Cardiovasc Pharmacol Ther 2015;20:357–369. [DOI] [PubMed] [Google Scholar]